Abstract
Being characterized by progressive and severe damage in neuronal cells, neurodegenerative diseases (NDDs) are the major cause of disability and morbidity in the elderly, imposing a significant economic and social burden. As major components of the central nervous system, lipids play important roles in neural health and pathology. Disturbed lipid metabolism, particularly lipid peroxidation (LPO), is associated with the development of many NDDs, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS), all of which show elevated levels of LPO products and LPO-modified proteins. Thus, the inhibition of neuronal oxidation might slow the progression and reduce the severity of NDD; natural antioxidants, such as polyphenols and antioxidant vitamins, seem to be the most promising agents. Here, we summarize current literature data that were derived from human studies on the effect of natural polyphenols and vitamins A, C, and E supplementation in patients with AD, PD, and ALS. Although these compounds may reduce the severity and slow the progression of NDD, research gaps remain in antioxidants supplementation in AD, PD, and ALS patients, which indicates that further human studies applying antioxidant supplementation in different forms of NDDs are urgently needed.
1. Introduction
Neurodegenerative diseases (NDDs) have become the major cause of disability and morbidity among older people worldwide due to the ageing society and the increased average life expectancy. Suffering from severe memory and behavioral impairment (dementia) and the loss of movement control (ataxia and paralysis), these patients need constant and long-term care, which is connected with huge economic and societal costs.
Neurodegenerative diseases is a collective term for the clinical conditions characterized by gradual and progressive severe damage to neuronal cells, particularly in the central nervous system (CNS), which results in the loss of functions that are associated with the affected brain region [1,2]. Different in etiology and clinical symptomatology, NDDs share some common features at the cellular and molecular levels, such as protein misfolding, aggregation, and deposition; mitochondrial disfunction; chronic inflammation; and, oxidative damage of biomolecules, including lipids, proteins, and DNA (Figure 1). The most common type of NDD is Alzheimer’s disease (AD), accounting for approximately two-thirds of all cases [3]. Other NDDs include Parkinson’s disease (PD), Huntington’s disease, multiple sclerosis, amyotrophic lateral sclerosis (ALS), and many other rare conditions, such as: prion diseases, motor neuron diseases, spinocerebellar ataxia, spinal muscular atrophy, Friedreich’s ataxia, and Lewy body disease. All of these diseases, whether genetic or acquired, lead to the progressive decline or even the complete loss of sensory, motor, and cognitive function. AD, PD, and ALS are typically found in the elderly and are primarily classified as proteinopathies, meaning that they are associated with the aggregation and deposition of misfolded proteins that trigger neurotoxicity through cellular stress pathways [4,5]. In ALS, both upper and lower motor neurons are affected [6]. The disruption of proteostasis (protein homeostasis) can occur at any step of protein synthesis, including during transcription, translation, and post-translational modification.
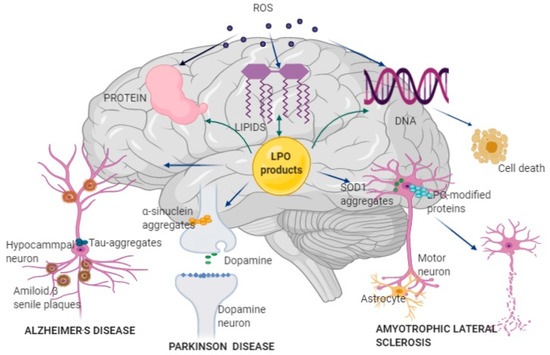
Figure 1.
Pathophysiological mechanisms of Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) development and progression. Reactive oxygen species (ROS) produced by mitochondrial Cyt p-450, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and lipoxygenase (LOX) attacks brain lipids, proteins, and DNA, further increasing oxidative stress. Oxidative damage of lipids results in the formation of lipid peroxidation (LPO) products, which further attack lipids, proteins, and DNA, impairing brain function. LPO can affect different types of neurons: in hippocampal neurons, LPO products bind to amyloid β peptide and form misfolded amyloid β peptide and amyloid β senile plaque, which disturbs nerve signaling and structure and induces AD; in dopaminergic neurons, LPO products induce generation and accumulation of misfolded α-synuclein, resulting in insufficient dopamine production and development of PD; in motor neurons, mutation of the superoxide dismutase 1 (SOD1) gene leads to the formation of misfolded SOD1 enzymes and abnormal production of ROS and LPO products, causing necrosis and death of the affected neurons in ALS; LPO-modified proteins are also associated with neural disruption in ALS. AD, Alzheimer’s disease; PD, Parkinson’s disease; ALS, amyotrophic lateral sclerosis; ROS, reactive oxygen species; NADPH, nicotinamide adenine dinucleotide phosphate; LOX, lipoxygenase; LPO, Lipid peroxidation; SOD1, superoxide dismutase 1.
Chaperons, which are proteins that facilitate the formation of correct and stable protein conformations, recognition and translocation of misfolded proteins into the cytosol, and cooperation with the ubiquitin/proteasome pathway (UPP) or the autophagy–lysosome pathway (ALP) to trigger degradation of misfolded proteins, regulate protein folding [7,8]. The mammalian targets of the rapamycin (mTOR) and sirtuin (SIRT) signaling pathways are key regulators of clearance mechanisms to prevent accumulation of misfolded proteins in neurons. mTOR regulates the ALP, which destroys transient proteins in the cytoplasm and core organelles, whereas sirtuin, specifically SIRT1, regulates the UPP [9].
The accumulation and aggregation of misfolded proteins in the brain and tissue, i.e., amyloid ß-peptide (Aß) in AD, α-synuclein in PD, ubiquitinated proteins in ALS, and their spread from cell to cell significantly contribute to the progression of NDDs [10]. The aggregation of these proteins in the endoplasmic reticulum (ER), a condition that is referred to as ER stress, activates a group of transcriptional signaling molecules, called the unfolded protein response, which aims to clear unfolded proteins, restore ER homeostasis, and ensure cell survival. In ER stress, reactive oxygen species (ROS) are generated, leading to chronic oxidative stress [11]. The accumulation of misfolded proteins in mitochondria also leads to their disfunction. Mitochondria play important roles in cell respiratory processes, metabolism, intracellular signaling, free radical production, apoptosis, and adenosine triphosphate (ATP) synthesis through oxidative phosphorylation. Mitochondrial disfunction leads to the increased production of ROS and oxidative phosphorylation defects and plays pivotal roles in ageing and the pathogenesis of NDDs, as neurons are especially vulnerable and susceptible to oxidative stress, because of their high energy requirements and high oxygen turnover [12].
Oxidative stress biomarkers have been developed due to the interaction of reactive oxygen and nitrogen species with major biomolecules, like carbohydrate, lipids, proteins, and nucleic acid. Besides the abovementioned cellular enzymes, endogenous ROS production is induced by the actions of lipoxygenase, myeloperoxidase, angiotensin II, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. Naturally, antioxidant enzymatic and non-enzymatic defense factors maintain a balance and alleviate oxidative stress. The main components of enzymatic antioxidant defense include superoxide dismutase, glutathione peroxidase and reductase, glutamyl transpeptidase, and catalase, whereas non-enzymatic endogenous antioxidants include glutathione (GSH), uric acid, ubiquinone, tocopherol, retinol, melatonin, and nuclear factor erythroid 2-related factor 2. In NDD, ROS production is increased and the defense system is weakened, which further aggravates the condition [13,14,15].
The onset of NDD is associated with a decline in autophagy activity, i.e., the incorporation of cargoes, such as proteins, organelles, and microbial invaders, into autophagosomes, as a result of genetic variation, ageing, or lifestyle [16]. Autophagosome formation and maturation depends on available lipids and lipid-binding proteins, thus indicating the significance of lipids in neural tissue health and pathology.
2. Lipids in the Central Nervous System (CNS)
Evidence is emerging that, besides disturbances in protein metabolism, the disturbances in lipid metabolism, particularly of phosphoinositols and sphingolipids, also play significant roles in neurodegeneration.
Lipids are key components of the structural and functional organization of the CNS, composing almost 60% of the dry mass of human brain. Lipid properties and the effects in the CNS are directly determined by the proportion of specific fatty acids in their molecular structure and, in particular, by the content of long-chain polyunsaturated fatty acids (PUFAs). PUFAs represent about 35% of total brain lipids and are mostly bonded in phospholipids; palmitic acid (16:0), stearic acid (18:0), and oleic acid (18:1n-9) together account for ~50% of the total, while all of the other fatty acids constitute less than 20% of the human brain [17,18]. The most abundant PUFAs in brain tissue are those that belong to the omega-3 (n-3) and omega-6 (n-6) series: arachidonic acid (20:4n-6; AA) and docosahexaenoic acid (22:6n-3; DHA) [19]. AA and DHA can both be produced from their precursors through the activity of the Δ5- and Δ6-desaturase and elongase in liver cell and brain cell endoplasmic reticulum and peroxisomes. Humans are relatively inefficient in performing this synthesis [20]. Thus, the majority of AA and DHA have to be consumed in the diet.
The three basic mechanisms of PUFA effects on the nervous system are: (1) modulation of the physical properties of the cell membrane, (2) secondary messenger activity, and (3) regulation of gene expression [21]. The presence of PUFAs in neural phospholipids favorably affects membrane permeability and fluidity, and it promotes endo- and exocytosis, ion channel activity, and activity of membrane-bound proteins including neurotransmitter receptors [19]. The effects of PUFAs on secondary messenger activity are related to the action of enzyme phospholipases. Phospholipases act directly on the membrane structure, liberate PUFAs from membrane phospholipids, and generate free fatty acids, which can serve as endogenous secondary messengers. In this way, AA that is released from the nerve cell membrane by the enzyme phospholipase A2 participates in the transmission of the signals responsible for the growth, activity, and maturation of neural branches into mature synaptic terminals [22]. PUFAs regulate gene expression by direct binding to the gene transcription factors or after being translated into biologically active compounds, such as eicosanoids and prostaglandins [23]. The products of the genes that are activated by AA and DHA participate in the interaction of nerve cells, enter the membrane ion channels, and contribute to neuro- and synaptogenesis [24,25]. DHA plays important roles in the regulation of the genes responsible for the glial response to CNS injury, as well as in the inhibition of proinflammatory and proapoptotic genes and the stimulation of antiapoptotic genes [26].
The amount of PUFAs, particularly n-3 PUFAs, in the brain decreases during ageing [27,28]. The n-3 PUFAs content of the aged brain largely depends on n-3 PUFAs intake during the life span. However, reduced activity of key enzymes involved in the biosynthesis of long-chain n-3 PUFAs from dietary precursors, Δ6- and Δ5-desaturases, is also found in the aged brain. Normal ageing is connected with decreased antioxidant capacity, an increased rate of lipid peroxidation (LPO), and the consequent decrease in n-3 PUFAs in brain tissue [29], which results in the altered chemical composition, structure, and function of the aged brain [19]. Johnson et al. reported a decrease in antioxidants and an increase in LPO in elderly people when compared with young adult controls [30]. Several reports confirmed the age-related weakening of the enzymatic antioxidant defense [31]. Moreover, lipid hydroperoxide and thiobarbituric acid-reactive substances have been identified as sensitive markers of normal ageing [32]. Therefore, LPO is associated with the development and progression of NDDs as well as with normal ageing.
3. Lipid Peroxidation
LPO is a complex non-enzymatic process that occurs in three distinct stages: initiation, propagation, and termination. The process is initiated when reactive oxygen metabolites cause hydrogen abstraction from the methylene group of the carbon–carbon double bond of PUFAs molecules, thus forming a fatty acid radical. These unstable compounds stabilize their molecular structure by forming conjugated dienes with the concomitant production of carbon-centered alkyl radicals [33]. The oxidation of the carbon-centered alkyl radical with para-magnetic molecular oxygen generates a lipid peroxyl radical that subsequently attacks another PUFAs. In the propagation stage, the process continues as an uncontrolled self-perpetuating chain reaction, which leads to the amplification of the initial oxidative event. Potentially, all PUFAs in the membrane might be oxidized [34]. Termination occurs when different types of radicals react mutually to form stable products, when radicals react with chain-breaking antioxidants (e.g., vitamin E) and produce non-radical products, or when the substrate is depleted [35]. In addition, the generation of LPO toxic products can be significantly decreased by the activity of antioxidant enzymes, such as catalase (CAT), superoxide dismutase 1 and 2 (SOD1 and SOD2, respectively), peroxiredoxin (Prx), glutathione peroxidase (GPx), glutathione reductase (GR), and heme oxygenase-1 (HO-1) [36,37].
4. Products of LPO
The primary products of LPO are unstable peroxides or hydroperoxides that can be further degraded to secondary products, including hydrocarbons, alcohols, ether, epoxides, and aldehydes. They react with proteins, lipids, nucleic acids, cofactors, and vitamins, thus influencing their structure and function [38,39]. Linoleic acid (LA), arachidonic acid (AA), and docosahexaenoic acid (DHA) are the fatty acids that are most commonly oxidized in the brain [40].
4.1. Oxidation Products from LA
Hydroperoxyoctadecadienoic acids (HPODE) are formed by radical-mediated and/or enzymatic oxidation via the lipoxygenase (LOX) of linoleic acid (LA). They exist as four isomers, 13-(E,E)-HPODE, 9-(E,E)-HPODE, 13-(Z,E)-HPODE, and 9-(Z,E)-HPODE, which can be further reduced to hydroxyoctadecadienoic acid (HODE) by glutathione peroxidase (Figure 2) [35]. 13-HODE may generate an anti-inflammatory 13-octadecadienoic acid (13-oxoODE) in a dehydrogenation process that is mediated by NADPH-dependent fatty acid dehydrogenases [41]. The biosyntheses of 13-HPODE and HODE are associated with the pathology of severe inflammatory based diseases [42,43]. 13-HODE incorporates into phospholipids and neutral lipids; as a major component of oxidized low-density lipoprotein (LDL) and atherosclerotic plaques, it plays a central role in the pathogenesis of atherosclerosis [44].
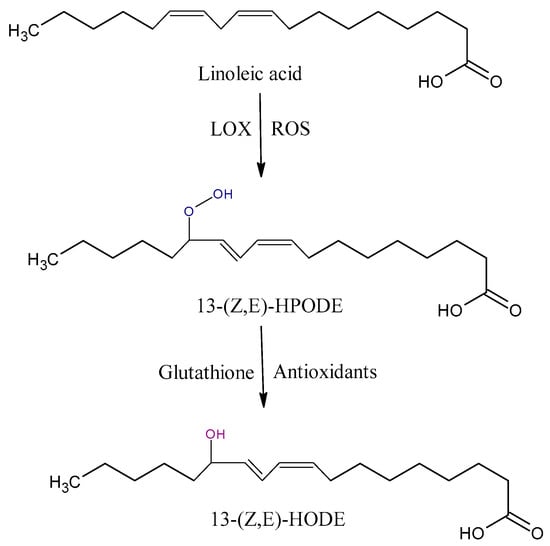
Figure 2.
Some of oxidation products from linoleic acid (LA). HPODE, hydroperoxyoctadecadienoic acid; HODE, hydroxyoctadecadienoic acid; LOX, lipoxygenase.
4.2. Oxidation Products from AA
Arachidonic acid is vulnerable to free-radical-mediated oxidation. As consequences of this process, six hydroperoxyeicosatetraenoic acid products (5-, 8-, 9-, 11-, 12-, and 15-hydroperoxyeicosatetraenoic acid (HPETE)) are formed (Figure 3). Some of them may be generated by LOX enzymes [45]. All the HPETE molecules may be further reduced, generating hydroxyeicosatetraenoic acid (HETE). Literature data indicate that 15-HETE incorporates into phospholipids, especially phosphatidylinositol, and thus changes its structure and function and indirectly influences signal transduction in the cells [46]. 20-HETE is another product of AA oxidation that is formed by mediation cytochrome P450 oxidoreductase.
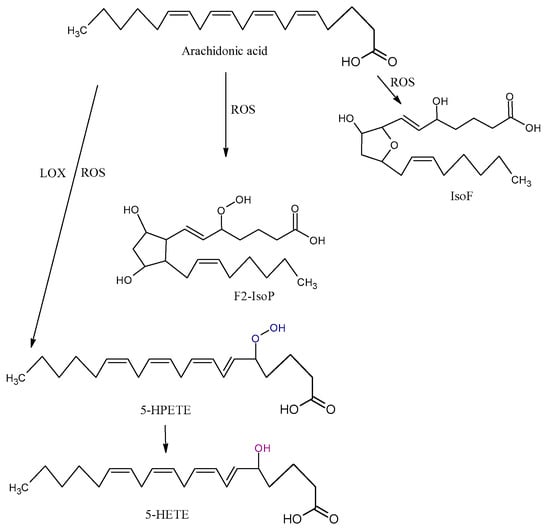
Figure 3.
Three types of oxidation products of arachidonic acid (AA): isofurans (IsoFs), isoprostanes (IsoP), hydroperoxyeicosatetraenoic acid (HPETE), hydroxyeicosatetraenoic acid (HETE), ROS, and LOX.
The isoprostanes (IsoPs) F, E, and D are a series of prostaglandin-like compounds formed via free-radical-initiated peroxidation of AA. As chemically very stable molecules, F2-IsoPs are considered to be the most reliable markers of oxidative damage in humans [47]. F2-IsoPs appear in four isomers, 5-, 12-, 8-, and 15-series, each of which comprises eight diastereomers, forming a total of 64 compounds [48]. The formation of IsoPs affects membrane fluidity and integrity. In addition, IsoPs may be released from cell membranes by phospholipases; they then circulate in plasma, where they further affect other molecules. Several studies demonstrated that the level of isoprostanes is higher in some NDDs. Although IsoPs are mainly generated from AA, F-ring IsoPs may be formed from the peroxidation of other PUFAs with three double bonds, such as alpha-linolenic acid, EPA, and DHA.
The oxidation of AA also leads to the formation of isofurans (IsoFs). IsoFs, as biomarkers of oxidative stress, are used in clinical settings when high concentrations of oxygen are used during treatments or procedures [40].
4.3. Oxidation Products from DHA
The peroxidation of DHA generates eight isomers with a total of 128 compounds named neuroprostanes (NPs) (Figure 4), which are abundantly concentrated in the neuronal membranes [49]. Although their biological roles are not entirely clear, some authors found that NPs have anti-inflammatory properties [50]. The neurofurans (NFs) are also oxidation products from DHA [51]. NPs and NFs are both sensitive and specific markers of neuronal oxidative damage, and their analysis could more accurately reflect the levels of lipid peroxidation in DHA-rich tissues, such as the brain.
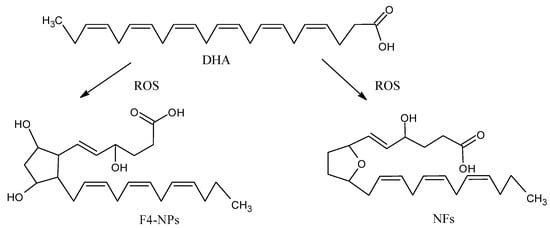
Figure 4.
Oxidation products from docosahecsaenoic acid (DHA): neurofurans (NFs) and neuroprostanes (NPs).
4.4. Short-Chain Aldehydes
The final products of lipid peroxidation of PUFAs are reactive short-chain aldehydes (Figure 5) [52]. These short-chain aldehydes are mainly classified into three families: 2-alkenals, 4-hydroxy-2-alkenals, and ketoaldehydes. Among them, acrolein (2-alkenal) is the most reactive, whereas 4-hydroxy-2-nonenal (HNE) and malondialdehyde (MDA) are the most abundant [53].
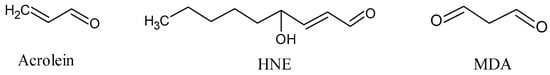
Figure 5.
Short-chain aldehydes: acrolein, 4-hydroxy-2-hexenal (HNE), and malondialdehyde (MDA).
Acrolein, which is an α, β-unsaturated aldehyde, is produced from the lipid peroxidation of PUFAs. As with other LPO peroxides, acrolein is capable of initiating a further process of lipid peroxidation: it attacks and deforms mitochondrial membranes, and then reacts with DNA and proteins [54]. Acrolein induces non-programmed necrosis and apoptosis [55], modification and aggregation of the protein, and inactivation of enzymes [56]. Generally, acrolein is capable of inducing cellular degeneration and death, and particularly the deterioration of hippocampal neurons [57]. In vitro studies have demonstrated the neurotoxic effects of acrolein on different types of cells line, thus confirming that acrolein plays a neurotoxic role in CNS neurodegeneration [58].
4-hydroxy-2-alkenal (HNE) is formed by peroxidation of n-6 PUFA, especially LA and AA, as a product of a non-enzymatic process in which an initial hydroperoxide undergoes fragmentation and form HNE [59]. n-3 PUFA, especially alpha-linoleic acid, may be attacked by free radicals, and its peroxidation generates 4-hydroxy-2-hexenal (HHE). Although HNE usually remains associated with the site where it is generated, HNE can diffuse to different cellular compartments and interact with many different substrates, including covalent binding to cysteine, histidine, and lysine residues [60]. Some plasma membrane ion and nutrient transporters, such as Na+/K+-ATPase, glucose, and glutamate transporters, several receptors for growth factors, neurotransmitters, mitochondrial, cytoskeletal, and proteasomal proteins, as well as proteins that repair oxidative damage may be targets for HNE [61]. As a signal molecule, HNE may suppress the activity of nuclear transcription factor κB [62] and activate the caspase pathways, leading to cell death in NDDs [63].
Malondialdehyde (MDA), a ketoaldehyde, which is an extremely reactive and toxic aldehyde, is generated by the decomposition of AA and larger PUFAs through enzymatic or non-enzymatic processes [64]. MDA can be generated during the enzymatic biosynthesis of thromboxane A2 [65]. It often has a relatively longer half-life and may, therefore, diffuse from the places of generation to other sites in vivo, further increasing oxidative and carbonyl stresses. MDA is capable of interacting or crosslinking on cellular and tissue proteins or DNA, resulting in the formation of adducts and biomolecular damage [66].
5. NDDs, LPO, and Antioxidants
Evidence shows that the high incidence of NDD may be attributed in part to the negative influence of daily risk factors, including stress, lack of physical exercise, and unhealthy nutrition. As such, as oxidative stress plays a crucial role in the process of neurodegeneration, numerous studies have documented the beneficial effects of exogenous nutritional antioxidants as neuroprotectors [67]. Nutritional antioxidants can modify oxidative stress on several levels: by decreasing the production of ROS and repairing the oxidized membranes, by neutralizing the free radicals, or through lipid metabolism in which cholesteryl esters and short-chain free fatty acids neutralize ROS [68]. Fruits, vegetables, beverages, green tea, coffee, spices, nuts, and cereal products are major sources of plant-derived antioxidants: polyphenols (phenolic acids, flavonoids, anthocyanins, lignans, and stilbenes), carotenoids (xanthophylls and carotenes), and vitamins (vitamins C and E) [69]. Table 1 summarizes the beneficial effects of the plant-derived antioxidants on NDD.

Table 1.
Dietary antioxidants intake and supplementation in prevention and adjuvant treatment in patients with neurodegenerative disease (NDD).
6. Alzheimer’s Disease
Alzheimer’s disease is a progressive brain disorder that is associated with neuronal degeneration and a loss of neurons in brain regions controlling memory, cognition, and emotional behaviors. AD patients experience rapid declines in the ability to learn, reason, maintain emotional stability, communicate, and perform common daily functions [101]. The onset of familial AD relates to genetic mutations in enzymes that are involved in amyloid precursor protein (APP) processing, whereas the etiology of sporadic AD is still unclear [102]. The complex and heterogeneous AD pathophysiology is dominated by two main hallmarks: overproduction and extracellular deposition of Aß and the formation of intracellular neurofibrillary tangles (NFTs), consisting of hyperphosphorylated tau protein [103]. The aberrant aggregations of Aβ and tau create an overall cytotoxic environment that results in the disturbance of neuronal cell shape and function, including the disturbance of ATP production, axonal transport, and synaptic signaling, together leading to severe cognitive and motor impairment characteristic for AD [104].
LPO is an important factor in the pathogenesis of AD (Figure 1) [105]. Initially, the oxidative stress environment that prevails in the AD brain induces LPO, which, in turn, further promotes disturbance of antioxidant capacity within the brain cells [106]. The elevated levels of LPO products and LPO-modified proteins, all of which are recognized as neurotoxic agents, have been found in AD-affected subjects. HNE occurs in several NDDs, such as AD, PD, ALS, Huntington disease, and Down syndrome [107,108,109,110]. Increased levels of HNE–protein adducts have been detected in the diseased brain regions and body fluids of AD patients. HNE commonly targets proteins involved in energy metabolism and the antioxidant response, leading to the disturbance of these important cell functions and, consequently, to neuronal dysfunction and death [111]. In addition, HNE–protein adducts add to the stimulation of the autoimmune response [112]. IsoPs and NPs are also increased in AD [113]. High levels of F2-IsoP were found in the hippocampus and the cerebrospinal fluid (CSF) of patients with AD [114]. Acrolein, which is predominantly localized within NFTs, directly attacks DNA, reacts with DNA base guanine, and forms acrolein–deoxyguanosine, which are excessively presented in the AD brain [101,113]. There are clinical studies reporting neurotoxicity due to increased acrolein levels in the brain and spinal cord of patients with AD and spinal cord injury [115,116]. An elevated level of circulating HODE, which is released from phospholipids by phospholipases, has been found in the plasma and erythrocytes of patients with AD [117]. However, MDA is the most abundant LPO product [118]. Significantly higher levels of MDA are found in AD patients when compared to healthy subjects [119]. MDA accumulated in the AD brain covalently binds to a variety of proteins and promotes the formation of aberrant protein adducts, which disturb nerve signaling and structure in AD-affected brain regions, such as the frontal, temporal, and occipital lobes and hippocampus [120].
AD and Antioxidants
Because oxidative stress and LPO lie at the basis of neuronal damage in AD, the potential benefits from dietary supplementation with antioxidants, such as polyphenols, and antioxidant vitamins tocopherol (vitamin E), ascorbic acid (vitamin C), and carotenoids (vitamin A), have become the subject of considerable scientific interest. The majority of studies were conducted in animal models and in vitro; reports on the effects of antioxidant supplementation in humans with AD are sparse (Table 1).
Polyphenols are phytochemicals that are widely present in plant drinks and foods [121]. Because of their small molecules and lipophilic nature, they can cross the blood-brain barrier and exert strong antioxidant and radical scavenging activity within the brain tissue [122,123]. Among others, the most investigated polyphenols are curcumin, epigallocatechin gallate (EGCG), and rosmarinic acid, which exert many beneficial effects on AD pathology, although the majority of the results were produced from studies that were conducted on animal models and in vitro [124,125]. In a mice model of familial AD, EGCG significantly decreased Aß production and induced marked increases in brain synapses; these effects were accompanied by improved spatial learning and memory [126,127]. A few human studies investigating supplementation with curcumin, a polyphenol from turmeric, have found improved cognitive status in AD patients [80,128]. The consumption of EGCG-rich green tea correlated with decreased risk of neurodegeneration and cognitive impairment [129,130], whereas resveratrol, a polyphenol from red wine, increased brain blood flow and oxygen uptake, and improved auditory, verbal, and learning memory in AD patients [79]. Similarly, pills containing polyphenols from blueberry and green tea increased cognitive processing in treated older adults when compared to placebo [131]. Mastroiacovo et al. reported improvement in cognitive function in the elderly due to regular cocoa flavanols consumption, which is in line with results of Nurk et al., who reported increases in the cognitive abilities in the elderly consuming a diet high in flavonoids-rich food, such as wine, chocolate, and tea in a dose-dependent manner [130,132]. The protective effects on cognition were also found in people that consumed a diet based on walnuts, which are rich in polyphenols [133].
In patients with MCI, supplementation with vitamin E slowed disease progression and reduced risk of dementia [90]. Parallel supplementation with vitamin C has been shown to improve the beneficial effect of vitamin E, leading to a decrease in AD incidence and prevalence [134,135]. In contrast, other groups reported no benefits of vitamin E in patients with MCI [87], or in the prevention of AD, even in combination with selenium [89]. Vitamin E overdose correlated with an increased risk of mortality [136]. Various in vivo and in vitro studies reported decreases in oxidative stress and Aß peptide oligomerization by vitamin C supplementation. Studies in humans also found reduced oxidative stress, systemic inflammation, and atherosclerosis in persons that were supplemented with vitamin C [137,138]. However, randomized clinical trials still failed to demonstrate any association between vitamin C and alleviation of AD pathology, which indicated that the prevention of deficiency seems to be more beneficial than vitamin C supplementation [139]. The in vitro results for vitamin A indicated reduction in Aβ plaques and a decrease in cognitive impairment due to vitamin A and β-carotene supplementation [140,141]. In AD patients, high levels of these vitamins are associated with better memory and learning performance.
7. Parkinson’s Disease
Parkinson’s disease is the second most common neurodegenerative disorder, with incidence being consistently higher in men than in women that increases over the age of 60 years [142]. The most common clinical manifestations in patients with PD are resting tremors, slowness of movement, rigidity, and postural instability, along with other symptoms, such as dementia, depression, insomnia, and anosmia. As a chronic and progressive neurodegenerative disorder, PD is associated with an increased turnover of dopamine and reduced levels of striatal dopamine and its metabolites in the brain (Figure 1). The persistent and diffuse degeneration of dopamine-producing neurons in the substantia nigra pars compacta (SNpc) is observed. The pathogenesis of PD is characterized by misfolding and aggregation of proteins, particularly small synaptic protein α-synuclein, which is the main component of Lewy bodies [143]. α-synuclein was found to aggregate and accumulate in the remaining neurons in the SNpc, locus coeruleus, cerebrospinal cord, enteric nervous system, and autonomic ganglia [144]. The role of α-synuclein is primarily connected with the effects on mitochondrial processes and the formation of synaptic vesicles [143]. It has been proven that reduced levels of glutathione, an antioxidant critical for protecting dopaminergic neurons in the SNpc from free radical damage, increase LPO, which is involved in the pathogenesis and progression of PD [145].
Clinical studies have reported significant acrolein levels in the brain. Acrolein promotes initiation of LPO and further elevation of oxidative stress, as indicated by acrolein-induced increases in HNE [146]. Additionally, acrolein acts on the modification of α-synuclein in dopaminergic neurons, leading to mitochondrial dysfunction [147]. This results in ROS-mediated apoptosis of the affected neurons [148]. There are three independent mutations in α-synuclein, including A53T, A30P, and E46K, which are involved in the development of familial PD. However, mutation and aggregation of α-synuclein can both cause Parkinsonism. The misfolded α-synuclein protein is soluble and it serves as a mediator of neurotoxicity in dopaminergic neurons. Clinically, the accumulation of acrolein-α-synuclein adducts was detected in the nigral dopaminergic neurons of PD patients [149], in parallel with results from in vivo studies that found that acrolein acts as a Parkinsonian neurotoxin in the nigrostriatal dopaminergic system of rat brain [58]. In addition, the role of DHA in α-synuclein oligomerization and aggregation has been suggested [113,150].
4-HNE and Nε-(carboxymethyl) lysine have been localized in Lewy bodies in post-mortem PD brain tissue [151]. HNE modification leads to conformational changes and the oligomerization of α-synuclein. The modified oligomers are toxic and may contribute to the deterioration of neurons [152]. It was found that HNE modifies the transport and possibly the loss of dopamine since its content increases proportionally to the severity stages of PD [153]. Elevated HNE, protein accumulation, and dopamine loss probably affect the physical capabilities and the process of learning in patients with PD [147]. Another actin-binding protein has been observed in cell lines and it acts to regulate the development of the actin microfilament [147]. Upon LPO, this protein decreased in the PD patients, which led to the reduced recovery of dynamic development of neurons [154]. Consequently, this results in the muscle damage widely observed in patients with PD. LPO grades are higher in PD patients; plasma levels of F2-IsoPs, HETEs, 7β-hydroxycholesterol (7β-OHCh), and 27-hydoroxycholesterol (27-OHCh) 7-ketocholesterol (7-KCh), and NPs are higher when compared to healthy subjects [155]. In particular, plasma F2-IsoPs and HETEs levels are elevated in the early stages of PD. Interestingly, IsoFs but not F2-IsoPs are increased in the SNpc of patients with PD [156]. Elevated levels of HNE, MDA, and acrolein have been reported in Lewy bodies in the brain stem and neocortical neurons, as well as in the CSF of living PD patients [147,148,154].
PD and Antioxidants
Evidence is increasing that oxidative stress plays a role in the pathogenesis of PD [157], and supplementation with the abovementioned antioxidants could produce beneficial effects in PD patients (Table 1).
Agents such as flavonoids that can target ROS and mitochondrial dysfunction are prime candidates for neuroprotection in PD [158,159]. The Mediterranean diet is a rich source of antioxidant bioflavonoids and polyphenols, which are associated with a decreased risk of PD [160,161]. Nevertheless, data on the polyphenol intake and the PD risk are contradictory. A large epidemiological study has shown that men with the highest quintile of flavonoid consumption (tea, berry fruits, apples, red wine, and orange or orange juice) had a 40% lower risk of developing PD when compared to those in the lowest quintile, but this was not observed in women [162]. However, 41-year follow-up Finnish data reported the association of berry consumption with increased risk of PD in men [163]. Other studies demonstrated that the consumption of green and black tea had beneficial effects in reducing the risk of PD [157,164]. In a rotenone model of PD, contrary to the previously reported neuroprotective effects that were observed in AD [165], pomegranate juice exacerbated oxidative stress and neurodegeneration [166]. Polyphenols, like curcumin, resveratrol, catechin, and oleuropein, inhibit the formation of Lewy bodies [167]. Overall, the preclinical and epidemiological data strongly support the further investigation of specific flavonoids for the treatment of PD [158].
The roles of vitamins in PD prevention and therapy are yet to be determined. So far, no link between vitamin A and PD has been established in few human studies [93,168,169,170], which was supported by a recent meta-analysis [170]. Only three case-control studies reported a significant association between lutein intake and PD risk, whereas two studies found a protective effect of dietary β-carotene intake [84,93] and risk of PD, one only in women [93]. The serum levels of some carotenoids—α-carotene, β-carotene, and lycopene—were lower in the PD patients, with evidence that carotenoids are inversely correlated with clinical variables that represent disease progression [84,171]. Decreased serum carotenoid levels are associated with poorer motor function [171].
As for vitamin A, despite numerous studies, no clear association between vitamin C and human PD [81,93,169,172] has been established. One study indicated that higher intake of fruits and vegetables containing vitamin C is associated with an increased risk of PD [173]. In contrast, in a case-controlled study, individuals consuming a diet rich in vitamin C showed a 40% reduction in PD risk [174]. It was suggested that supplementation with vitamin C may not affect disease development, because access to the brain is limited by high water solubility and the requirement for active transport at the choroid plexus to enter the brain [175]. The serum level of vitamin C in patients with PD remains controversial [176,177]. For instance, the vitamin C level in lymphocytes is significantly lower in patients with severe PD [178], suggesting that vitamin C supplementation may be beneficial for the treatment of PD. This was evident in a large cohort study, including patients with PD, which found that dietary vitamin C intake significantly reduced the risk of PD, but this effect was invalid for the four-year-lag analysis [85].
Based on literature data, among antioxidant vitamins, only vitamin E intake was found to be associated with a reduced risk of PD in three of the four studies [179]. A meta-analysis showed a protective effect against PD in humans with both moderate and high intake of vitamin E [169]. This effect is more pronounced in men than in women [94], and only higher vitamin E intake (>9.759 mg/day) is significantly associated with decreased risk of PD in women [93]. These protective effects may be achieved through preventing oxidative stress in cells and inhibiting apoptosis. Apart from PD risk, vitamin E has also been used in intervention studies, as PD patients were found to have lower serum levels of vitamin E than controls [180]. A high-dose supplementation (2000 IU/day) can significantly elevate the vitamin E level in CSF [92]. Vitamin E supplementation was tested as a therapeutic against PD in the Deprenyl and Tocopherol Antioxidative Therapy of Parkinsonism (DATATOP) study [181]. However, no beneficial effect of α-tocopherol was observed during follow-up evaluation of PD symptoms [181]. Recently published data indicate that vitamin E represents a potential therapeutic target for disease-modifying treatments in PD as a result of both the clinical retrospective analysis and electrophysiological experiments [88]. Additional trials are still needed in order to confirm the role of vitamin E in slowing the progressive deterioration of function in PD.
To summarize, a research gap exists in the effect of antioxidants supplementation on lipid peroxidation products in PD patients [182,183]. Data on supplementation in patients already diagnosed with PD has failed to show a disease-modifying effect.
8. Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis is an NDD that is characterized by progressive loss of motor neurons in the CNS, leading to muscular atrophy, paralysis, and death [184,185] ALS occurs in the sporadic form (sALS) in 90% of cases, and in the familial form due to inherited genetic mutations (fALS) with an estimated incidence of between one and two per 100,000 people [186]. Among all ALS patients, 50% die within 30 months of symptom onset, often from respiratory insufficiency, whereas about 10% of patients may survive for more than a decade [187,188]. Riluzole, which is the only Food and Drug Administration (FDA)-approved drug that is currently accessible for ALS, slows disease progression, and improves limb function and muscle strength, but, unfortunately, it increases life span by only 2–3 months [189]. The mean age at onset this disease is 40–60 years for fALS and 58–63 years for sALS, with a peak incidence at age 70–79 years [190]. Men have a higher risk of ALS than women, leading to a male-to-female ratio of 1.2–1.5 [191]. Genetic studies have shown that C9orf72, SOD1, TARDBP, and FUS are the most common mutated genes in ALS [192]. The first pathological mutation was identified in ALS patients in the SOD1 gene in 1993 [193]. To date, over 180 different mutations have been described in the SOD1 gene, which can be found in 10–20% of familial ALS cases and 1–5% of sporadic ALS [192,194]. The SOD1 gene encodes the Cu/Zn SOD1, which is one to three isoenzymes of SOD responsible for the conversion of the superoxide anion radical to molecular oxygen and hydrogen peroxide. Mutations in SOD1 lead to numerous alterations in the structure and function of motor neurons in ALS patients (Figure 1). Thus, the mutated enzymes result in misfolded protein chains, and they form small neurotoxic aggregates in the nuclei of glial cells (mostly astrocytes) of the spinal cord, which contributes to neuron degeneration [195]. The SOD enzyme may be post-translationally modified and hyper-oxidized in sALS patients; through this oxidation, altered SOD1 gains toxic properties [196]. In addition, the SOD mutant has reduced enzymatic activity, which results in an abnormal production of ROS, which causes an alteration in the cell function, apoptosis, and necrosis [197]. Previous pathological studies have reported evidence of increased LPO products in biological fluids of ALS patients compared with control samples. Several literature data demonstrate a significant increase in MDA leve-ls in the sera in ALS patients [198,199], thus strengthening the clinical evidence that prooxidative imbalances contribute to ALS pathophysiology [200]. In ALS, HNE is bound to three key proteins: dihydropyrimidinase-related protein 2 (DRP-2), heat shock protein 70, and α-enolase, which leads to their modification [201]. They are involved in axonal development, transmission, and modulation of extracellular signals; repair mechanism; and, maintaining of redox-homeostasis; thus, their modification leads to the loss of motor neuron function [53]. The elevated levels of HNE and HNE-modified proteins are observed in the spinal cord motor neurons of ALS patients [202]. In addition, when HNE attacks DNA, it may cause cellular damage and apoptosis [203]. Literature data demonstrate significantly elevated levels of HNE in the sera and spinal fluid of sALS patients when compared with control subjects, which were positively correlated with the extent of disease but not a rate of progression [204]. Moreover, these authors showed that HNE levels from sALS serum and CSF samples were significantly above those that were collected from fALS patients, suggesting that the familial and sporadic forms are qualitatively different concerning oxidative stress. In accordance, the same group of authors previously documented increased levels of HNE in the CSF and spinal cord motor neurons of ALS patients [109,202]. Several studies examined the level of IsoP in patients with ALS. Significantly higher levels of F2-IsoPs have been found in the urine of patients with sALS when compared with healthy subjects [205]. However, Montine et al. did not find differences in IsoP among ALS patients and healthy subjects [114].
ALS and Antioxidants
Evidence shows that neural oxidative damage contributes to neuronal oxidation, dysfunction, and degeneration in ALS, as discussed above. In line with this, inhibition or suppression of neuronal oxidation may slow or even stop disease progression. Among natural antioxidants, vitamins A, E, and C, as well as polyphenols-rich fruits, can be potential antioxidants whose effect could be investigated. However, the literature data about the effects of antioxidative therapy in ALS are scarce (Table 1) and they are mostly limited to in vitro studies and experimental model studies.
Despite a well-documented protective role against LPO [206], only a few studies examined the effect of vitamin E on ALS. One of them investigated the influence of vitamin E supplementation on survival and motor function in ALS. After three months of treatment with vitamin E (500 mg twice daily) and riluzole as standard drug therapy in ALS, a decrease in plasma MDA levels and an increase in plasma GSH levels were observed [96]. However, survival was not influenced by the treatment. Additionally, Ascherio et al. observed that regular use of vitamin E supplements for 10 years or more was associated with a lower risk of dying from ALS [97]. Similarly, Wang et al. found a positive trend for a decline in ALS progression in patients with long-term use of vitamin E supplements, but did not find an overall protective role for vitamin E [98]. In a randomized controlled trial (RCT) that was conducted in ex-Yugoslavia, a combination of methionine, vitamin E, and selenium led to a significant increase in the rate of survival and increase in activity of GPx in 28 patients with ALS after 12 months of supplementation [99]. However, the analysis of data from 10 different RCT studies with 1050 participants and shorter durations of the supplementation period revealed no significant beneficial effect of vitamin E on survival in ALS patients [207]. Studies on the impact of vitamin C on progression and duration of ALS are also rare. A recently-published original study, including 202 patients with ALS, showed lower serum level of vitamin C level in patients with ALS when compared to healthy controls [208]. Conversely, in a study with only 19 ALS patients, there was no significant difference in plasma and CSF vitamin C levels compared with controls [209]. The results from pooled analysis of five large prospective studies with 1093 cases of ALS indicated that high dietary intakes or supplemental use of vitamin C appear to not affect the risk of ALS [86]. One animal study indicated that a high dose of vitamin C administered before the onset of disease prolonged survival and motor function in fALS transgenic mice, whereas vitamin C that was administered after the onset disease did not have effect on survival [210].
A recently published study showed that the level of vitamin A is significantly higher in patients with ALS when compared to healthy controls [208]. Conversely, in a case-controlled study with 77 patients with ALS, β-carotene was found to decrease the risk of sporadic ALS [211]. Several studies have documented a beneficial association between ALS and the intake of carotenes [212,213], as well as the consumption of foods rich in carotenoids helping the prevention or even delay of the onset of ALS [86]. According to our knowledge, there are no data about the influence of vitamin A on the level of LPO and/or other parameters of oxidative stress in ALS patients. Additionally, long-term dietary supplementation with retinoic acid has been reported to shorten the lifespan in an ALS mouse model [214].
Flavonoids are among the natural substances that are present in fruits and vegetables and have a protective effect against ROS. However, few studies examined whether these molecules impact ALS. Among them, Korkmas et al. showed that chronic administration of 7,8-dihydroxyflavone significantly improved motor deficits and enhanced lower neuronal survival in the transgenic ALS mouse model [215], whereas Ip et al. found that quercetin and its derivative could be therapeutic inhibitors of the aggregation and misfolding of SOD1 that is noticed in ALS [216].
9. Future Perspectives
Searching the FDA registry, Cummings et al. found 121 pharmacologic agents currently being investigated in clinical trials for the treatment of AD. The majority of agents (17 out of 29) in 36 Phase III trials are disease modifiers that mainly target amyloid, inflammation/infection/immunity, and synaptic plasticity. Among them, gingko biloba extract, rich in flavonoids, is the only natural antioxidant [217]. Plant extracts may produce better antioxidant/disease-modifying activities than a single compound through the additive or synergistic effects of their different active ingredients and a variety of secondary metabolites [218]. Evidence is accumulating that combination of FDA-approved drugs with natural antioxidants, as well as combinations of appropriate antioxidants, may be more effective, considerably lower cost, and, therefore, more affordable and acceptable, especially for long-term prevention of NDDs [68,219].
Dietary habits and nutrition consumption affect cognitive functions. The peak of cognitive function is around 20 or 30 years of age, and then cognitive functions decline after 50 or 60 years of age. Evidence suggests that diet interventions show promise for dementia prevention [220]. Clinical trials for the prevention decline of cognitive function in adults with antioxidants are still in their infancy. Polyphenol berry fruit juice consumption was most beneficial for immediate verbal memory. Isoflavone-based interventions were associated with significant improvements for delayed spatial memory and executive function [221]. However, no clear evidence exists for an association between cognitive outcomes and polyphenol dose response, duration of intervention, or population studied. Therefore, further studies are needed in order to determine whether long-term antioxidants intake can reduce the risk of memory loss in adult population.
10. Conclusions
No cure exists for NDDs, particularly in advanced stages. The drugs that are approved by the FDA, such as acetylcholine esterase inhibitors (donepezil, rivastigmine) as well as levodopa for PD, which crosses the blood–brain barrier and restores dopamine levels in the substantia nigra, only ameliorate the symptoms and slow the progression of the diseases for several years [1]. Newer therapy approaches that are focused on neuroregeneration, i.e., structural and functional recovery of the damaged nervous system through immunomodulation, inhibition of formation of protein aggregates, disaggregation of misfolded proteins, and induction of autophagy, give hope that the degeneration process of afflicted neurons might be slowed and the recovery rates and longevity improved [222]. Because of the complex nature of NDDs, a multi-target drug approach is encouraged, as it may produce additional beneficial effects. When considering the role of lipid peroxidation in the development and progression of NDDs, further human studies applying antioxidant supplementation in different forms of NDD are urgently needed.
Author Contributions
S.P., A.A., D.R.-M., and Z.C., drafted the manuscript and approved the final manuscript; S.P., reviewed and edited drafts; V.V., conceptualized manuscript, edited draft, and approved the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
This work was supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia contract 451-03-68/2020-14/200015.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Malar, D.S.; Prasanth, M.I.; Brimson, J.M.; Sharika, R.; Sivamaruthi, B.S.; Chaiyasut, C.; Tencomnao, T. Neuroprotective Properties of Green Tea (Camellia sinensis) in Parkinson’s Disease: A Review. Molecules 2020, 25, 3926. [Google Scholar] [CrossRef] [PubMed]
- Prasansuklab, A.; Brimson, J.M.; Tencomnao, T. Potential Thai medicinal plants for neurodegenerative diseases: A review focusing on the anti-glutamate toxicity effect. J. Tradit. Complement. Med. 2020, 10, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Desikan, R.S.; Fan, C.C.; Wang, Y.; Schork, A.J.; Cabral, H.J.; Cupples, L.A.; Thompson, W.K.; Besser, L.; Kukull, W.A.; Holland, D.; et al. Genetic assessment of age-associated Alzheimer disease risk: Development and validation of a polygenic hazard score. PLoS Med. 2017, 14, e1002258. [Google Scholar] [CrossRef] [PubMed]
- Shafi, S.; Singh, A.; Gupta, P.; Chawla, P.A.; Fayaz, F.; Sharma, A.; Pottoo, F.H. Deciphering the role of aberrant protein post translational modification in the pathology of neurodegeneration. CNS Neurol. Disord. Drug Targets 2020, 19. [Google Scholar] [CrossRef]
- Gandhi, J.; Antonelli, A.C.; Afridi, A.; Vatsia, S.; Joshi, G.; Romanov, V.; Murray, I.V.J.; Khan, S.A. Protein misfolding and aggregation in neurodegenerative diseases: A review of pathogeneses, novel detection strategies, and potential therapeutics. Rev. Neurosci. 2019, 30, 339–358. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Abrahams, S.; Goldstein, L.H.; Woolley, S.; Mclaughlin, P.; Snowden, J.; Mioshi, E.; Roberts-South, A.; Benatar, M.; HortobáGyi, T.; et al. Amyotrophic lateral sclerosis—frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 153–174. [Google Scholar] [CrossRef] [PubMed]
- Tawo, R.; Pokrzywa, W.; Kevei, É.; Akyuz, M.E.; Balaji, V.; Adrian, S.; Höhfeld, J.; Hoppe, T. The Ubiquitin Ligase CHIP Integrates Proteostasis and Ageing by Regulation of Insulin Receptor Turnover. Cell 2017, 169, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, T.; Cohen, E. Organismal Protein Homeostasis Mechanisms. Genetics 2020, 215, 889–901. [Google Scholar] [CrossRef]
- Abdullah, A.; Mohd Murshid, N.; Makpol, S. Antioxidant Modulation of mTOR and Sirtuin Pathways in Age-Related Neurodegenerative Diseases. Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Tewari, D.; Sharma, G.; Kabir, M.T.; Barreto, G.E.; Bin-Jumah, M.N.; Perveen, A.; Abdel-Daim, M.M.; Ashraf, G.M. Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 2902–2919. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. 2017, 23, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Saleem, U.; Sabir, S.; Niazi, S.G.; Naeem, M.; Ahmad, B. Role of Oxidative Stress and Antioxidant Defense Biomarkers in Neurodegenerative Diseases. Crit. Rev. Eukaryot. Gene Expr. 2020, 30, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Warraich, U.-A.; Hussain, F.; Kayani, H.U.R. Ageing—Oxidative stress, antioxidants and computational modeling. Heliyon 2020, 6, e04107. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Hernandez-Diaz, S.; Soukup, S.-F. The role of lipids in autophagy and its implication in neurodegeneration. Cell Stress 2020, 4, 167–186. [Google Scholar] [CrossRef]
- Barón-Mendoza, I.; González-Arenas, A. Relationship between the effect of polyunsaturated fatty acids (PUFAs) on brain plasticity and the improvement on cognition and behavior in individuals with autism spectrum disorder. Nutr. Neurosci. 2020, 1–24. [Google Scholar] [CrossRef]
- Taha, A.Y. Linoleic acid–good or bad for the brain? NPJ Sci. Food 2020, 4, 1. [Google Scholar] [CrossRef]
- Bos, D.J.; van Montfort, S.J.T.; Oranje, B.; Durston, S.; Smeets, P.A.M. Effects of omega-3 polyunsaturated fatty acids on human brain morphology and function: What is the evidence? Eur. Neuropsychopharmacol. 2016, 26, 546–561. [Google Scholar] [CrossRef]
- Vucic, V. The role of dietary polyunsaturated fatty acids in inflammation. Serb. J. Exp. Clin. Res. 2013, 14, 93–99. [Google Scholar] [CrossRef]
- Ristic-Medic, D.; Vucic, V.; Takic, M.; Karadzic, I.; Glibetic, M. Polyunsaturated fatty acids in health and disease. J. Serb. Chem. Soc. 2013, 78, 1269–1289. [Google Scholar] [CrossRef]
- Cauli, B.; Hamel, E. Brain Perfusion and Astrocytes. Trends Neurosci. 2018, 41, 409–413. [Google Scholar] [CrossRef]
- Hopperton, K.E.; Trépanier, M.-O.; James, N.C.E.; Chouinard-Watkins, R.; Bazinet, R.P. Fish oil feeding attenuates neuroinflammatory gene expression without concomitant changes in brain eicosanoids and docosanoids in a mouse model of Alzheimer’s disease. Brain. Behav. Immun. 2018, 69, 74–90. [Google Scholar] [CrossRef] [PubMed]
- Echeverría, F.; Valenzuela, R.; Catalina Hernandez-Rodas, M.; Valenzuela, A. Docosahexaenoic acid (DHA), a fundamental fatty acid for the brain: New dietary sources. ProstaglandinsLeukot. Essent. Fat. Acids 2017, 124, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ugidos, I.F.; Santos-Galdiano, M.; Pérez-Rodríguez, D.; Anuncibay-Soto, B.; Font-Belmonte, E.; López, D.J.; Ibarguren, M.; Busquets, X.; Fernández-López, A. Neuroprotective effect of 2-hydroxy arachidonic acid in a rat model of transient middle cerebral artery occlusion. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1648–1656. [Google Scholar] [CrossRef]
- Hashimoto, M.; Hossain, S.; Al Mamun, A.; Matsuzaki, K.; Arai, H. Docosahexaenoic acid: One molecule diverse functions. Crit. Rev. Biotechnol. 2017, 37, 579–597. [Google Scholar] [CrossRef]
- Cutuli, D. Functional and Structural Benefits Induced by Omega-3 Polyunsaturated Fatty Acids During Ageing. Curr. Neuropharmacol. 2017, 15, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, L.; Brambilla, P.; Mazzocchi, A.; Harsløf, L.; Ciappolino, V.; Agostoni, C. DHA Effects in Brain Development and Function. Nutrients 2016, 8, 6. [Google Scholar] [CrossRef]
- Grimm, A.; Eckert, A. Brain ageing and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef]
- Johnson, A.A.; Stolzing, A. The role of lipid metabolism in, lifespan regulation, and age-related disease. Aging Cell 2019, 18. [Google Scholar] [CrossRef]
- Kozakiewicz, M.; Kornatowski, M.; Krzywińska, O.; Kędziora-Kornatowska, K. Changes in the blood antioxidant defense of advanced age people. Clin. Interv. Aging 2019, 14, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Yavuzer, H.; Yavuzer, S.; Cengiz, M.; Erman, H.; Doventas, A.; Balci, H.; Erdincler, D.S.; Uzun, H. Biomarkers of lipid peroxidation related to hypertension in aging. Hypertens. Res. 2016, 39, 342–348. [Google Scholar] [CrossRef]
- Tadokoro, K.; Ohta, Y.; Inufusa, H.; Loon, A.F.N.; Abe, K. Prevention of Cognitive Decline in Alzheimer’s Disease by Novel Antioxidative Supplements. Int. J. Mol. Sci. 2020, 21, 1974. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Mattson, M.P. Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer’s disease. Neurobiol. Dis. 2020, 138, 104795. [Google Scholar] [CrossRef]
- Shichiri, M. The role of lipid peroxidation in neurological disorders. J. Clin. Biochem. Nutr 2014, 54, 151–160. [Google Scholar] [CrossRef]
- Vasiljevic, D.; Veselinovic, M.; Jovanovic, M.; Jeremic, N.; Arsic, A.; Vucic, V.; Lucic-Tomic, A.; Zivanovic, S.; Djuric, D.; Jakovljevic, V. Evaluation of the effects of different supplementation on oxidative status in patients with rheumatoid arthritis. Clin. Rheumatol. 2016, 35, 1909–1915. [Google Scholar] [CrossRef]
- Levin, J.; Maaß, S.; Schuberth, M.; Giese, A.; Oertel, W.H.; Poewe, W.; Trenkwalder, C.; Wenning, G.K.; Mansmann, U.; Südmeyer, M.; et al. Safety and efficacy of epigallocatechin gallate in multiple system atrophy (PROMESA): A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2019, 18, 724–735. [Google Scholar] [CrossRef]
- Arsic, A.; Vucic, V.; Glibetic, M.; Popovic, T.; Debeljak-Martacic, J.; Cubrilo, D.; Ahmetovic, Z.; Peric, D.; Borozan, S.; Djuric, D.; et al. Redox balance in elite female athletes: Differences based on sport types. J. Sports Med. Phys. Fit. 2016, 56, 1–8. [Google Scholar]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef]
- Milne, G.L.; Yin, H.; Hardy, K.D.; Davies, S.S.; Roberts, L.J., 2nd. Isoprostane generation and function. Chem. Rev. 2011, 111, 5973–5996. [Google Scholar] [CrossRef]
- Vangaveti, V.; Baune, B.T.; Kennedy, R.L. Hydroxyoctadecadienoic acids: Novel regulators of macrophage differentiation and atherogenesis. Adv. Endocrinol. Metab. 2010, 1, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Narumiya, S. Prostaglandins and chronic inflammation. Trends Pharm. Sci. 2012, 33, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Tang, E.H.C.; Libby, P.; Vanhoutte, P.M.; Xu, A. Anti-inflammation therapy by activation of prostaglandin EP4 receptor in cardiovascular and other inflammatory diseases. J. Cardiovasc. Pharm. 2012, 59, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.E.; Ringel, A.; Feldstein, A.E.; Taha, A.Y.; MacIntosh, B.A.; Hibbeln, J.R.; Majchrzak-Hong, S.F.; Faurot, K.R.; Rapoport, S.I.; Cheon, Y.; et al. Lowering dietary linoleic acid reduces bioactive oxidized linoleic acid metabolites in humans. Prostaglandins. Leukot. Essent. Fat. Acids 2012, 87, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Hanna, V.S.; Hafez, E.A.A. Synopsis of arachidonic acid metabolism: A review. J. Adv. Res. 2018, 11, 23–32. [Google Scholar] [CrossRef]
- Hammond, V.J.; O’Donnell, V.B. Esterified eicosanoids: Generation, characterization and function. Biochim. Biophys. Acta 2012, 1818, 2403–2412. [Google Scholar] [CrossRef]
- Halliwell, B.; Lee, C.Y.J. Using isoprostanes as biomarkers of oxidative stress: Some rarely considered issues. Antioxid. Redox Signal. 2010, 13, 145–156. [Google Scholar] [CrossRef]
- Galano, J.-M.; Lee, Y.Y.; Oger, C.; Vigor, C.; Vercauteren, J.; Durand, T.; Giera, M.; Lee, J.C.-Y. Isoprostanes, neuroprostanes and phytoprostanes: An overview of 25years of research in chemistry and biology. Prog. Lipid Res. 2017, 68, 83–108. [Google Scholar] [CrossRef]
- Galano, J.-M.; Lee, J.C.-Y.; Gladine, C.; Comte, B.; Le Guennec, J.-Y.; Oger, C.; Durand, T. Non-enzymatic cyclic oxygenated metabolites of adrenic, docosahexaenoic, eicosapentaenoic and α-linolenic acids; bioactivities and potential use as biomarkers. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 446–455. [Google Scholar] [CrossRef]
- Musiek, E.; Brooks, J.; Joo, M.; Brunoldi, E.; Porta, A.; Zanoni, G.; Vidari, G.; Blackwell, T.; Montine, T.; Milne, G.; et al. Electrophilic Cyclopentenone Neuroprostanes Are Anti-inflammatory Mediators Formed from the Peroxidation of the -3 Polyunsaturated Fatty Acid Docosahexaenoic Acid. J. Biol. Chem. 2008, 283, 19927–19935. [Google Scholar] [CrossRef]
- Song, W.-L.; Lawson, J.A.; Reilly, D.; Rokach, J.; Chang, C.-T.; Giasson, B.; FitzGerald, G.A. Neurofurans, novel indices of oxidant stress derived from docosahexaenoic acid. J. Biol. Chem. 2008, 283, 6–16. [Google Scholar] [CrossRef]
- Uchida, K. 4-Hydroxy-2-nonenal: A product and mediator of oxidative stress. Prog. Lipid Res. 2003, 42, 318–343. [Google Scholar] [CrossRef]
- Perluigi, M.; Coccia, R.; Butterfield, D.A. 4-Hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: A toxic combination illuminated by redox proteomics studies. Antioxid. Redox Signal. 2012, 17, 1590–1609. [Google Scholar] [CrossRef] [PubMed]
- Moghe, A.; Ghare, S.; Lamoreau, B.; Mohammad, M.; Barve, S.; McClain, C.; Joshi-Barve, S. Molecular mechanisms of acrolein toxicity: Relevance to human disease. Toxicol. Sci. 2015, 143, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Rickett, T.; Sun, W. Acrolein-mediated injury in nervous system trauma and diseases. Mol. Nutr. Food Res. 2011, 55, 1320–1331. [Google Scholar] [CrossRef]
- Taso, O.V.; Philippou, A.; Moustogiannis, A.; Zevolis, E.; Koutsilieris, M. Lipid peroxidation products and their role in neurodegenerative diseases. Ann. Res. Hosp. 2019, 3, 2. [Google Scholar] [CrossRef]
- Erejuwa, O.O.; Sulaiman, S.A.; Ab Wahab, M.S. Evidence in support of potential applications of lipid peroxidation products in cancer treatment. Oxid. Med. Cell. Longev. 2013, 2013, 931251. [Google Scholar] [CrossRef]
- Wang, Y.-T.; Lin, H.-C.; Zhao, W.-Z.; Huang, H.-J.; Lo, Y.-L.; Wang, H.-T.; Lin, A.M.-Y. Acrolein acts as a neurotoxin in the nigrostriatal dopaminergic system of rat: Involvement of α-synuclein aggregation and programmed cell death. Sci. Rep. 2017, 7, 45741. [Google Scholar] [CrossRef] [PubMed]
- Pratt, D.A.; Tallman, K.A.; Porter, N.A. Free radical oxidation of polyunsaturated lipids: New mechanistic insights and the development of peroxyl radical clocks. Acc. Chem. Res. 2011, 44, 458–467. [Google Scholar] [CrossRef]
- Mattson, M.P. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp. Gerontol. 2009, 44, 625–633. [Google Scholar] [CrossRef]
- Poli, G.; Biasi, F.; Leonarduzzi, G. 4-Hydroxynonenal-protein adducts: A reliable biomarker of lipid oxidation in liver diseases. Mol. Asp. Med. 2008, 29, 67–71. [Google Scholar] [CrossRef]
- Lim, J.H.; Lee, J.-C.; Lee, Y.H.; Choi, I.Y.; Oh, Y.-K.; Kim, H.-S.; Park, J.-S.; Kim, W.-K. Simvastatin prevents oxygen and glucose deprivation/reoxygenation-induced death of cortical neurons by reducing the production and toxicity of 4-hydroxy-2E-nonenal. J. Neurochem. 2006, 97, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Liang, Q.; Johnson, M.S.; Redmann, M.; Fineberg, N.; Darley-Usmar, V.M.; Zhang, J. Inhibition of glycolysis attenuates 4-hydroxynonenal-dependent autophagy and exacerbates apoptosis in differentiated SH-SY5Y neuroblastoma cells. Autophagy 2013, 9, 1996–2008. [Google Scholar] [CrossRef]
- Reed, T.T. Lipid peroxidation and neurodegenerative disease. Free Radic. Biol. Med. 2011, 51, 1302–1319. [Google Scholar] [CrossRef]
- Tsikas, D.; Suchy, M.-T.; Niemann, J.; Tossios, P.; Schneider, Y.; Rothmann, S.; Gutzki, F.-M.; Frölich, J.C.; Stichtenoth, D.O. Glutathione promotes prostaglandin H synthase (cyclooxygenase)-dependent formation of malondialdehyde and 15(S)-8-iso-prostaglandin F2α. Febs Lett. 2012, 586, 3723–3730. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Dumitrascu, D.; Capitanescu, B.; Petcu, E.; Surugiu, R.; Fang, W.-H.; Dumbrava, D.-A. Dietary habits, lifestyle factors and neurodegenerative diseases. Neural Regen. Res. 2020, 15, 394. [Google Scholar] [CrossRef] [PubMed]
- Pohl, F.; Lin, P.K.T. The Potential Use of Plant Natural Products and Plant Extracts with Antioxidant Properties for the Prevention/Treatment of Neurodegenerative Diseases: In Vitro, In Vivo and Clinical Trials. Molecules 2018, 23, 3283. [Google Scholar] [CrossRef]
- Xu, D.-P.; Li, Y.; Meng, X.; Zhou, T.; Zhou, Y.; Zheng, J.; Zhang, J.-J.; Li, H.-B. Natural Antioxidants in Foods and Medicinal Plants: Extraction, Assessment and Resources. Int. J. Mol. Sci. 2017, 18, 96. [Google Scholar] [CrossRef]
- Calapai, G.; Bonina, F.; Bonina, A.; Rizza, L.; Mannucci, C.; Arcoraci, V.; Laganà, G.; Alibrandi, A.; Pollicino, C.; Inferrera, S.; et al. A Randomized, Double-Blinded, Clinical Trial on Effects of a Vitis vinifera Extract on Cognitive Function in Healthy Older Adults. Front. Pharm. 2017, 8. [Google Scholar] [CrossRef]
- Holland, T.M.; Agarwal, P.; Wang, Y.; Leurgans, S.E.; Bennett, D.A.; Booth, S.L.; Morris, M.C. Dietary flavonols and risk of Alzheimer dementia. Neurology 2020. [Google Scholar] [CrossRef]
- Assogna, M.; Casula, E.P.; Borghi, I.; Bonnì, S.; Samà, D.; Motta, C.; Di Lorenzo, F.; D’Acunto, A.; Porrazzini, F.; Minei, M.; et al. Effects of Palmitoylethanolamide Combined with Luteoline on Frontal Lobe Functions, High Frequency Oscillations, and GABAergic Transmission in Patients with Frontotemporal Dementia. J. Alzheimer’s Dis. 2020, 76, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Tomata, Y.; Sugiyama, K.; Sugawara, Y.; Tsuji, I. Citrus consumption and incident dementia in elderly Japanese: The Ohsaki Cohort 2006 Study. Br. J. Nutr. 2017, 117, 1174–1180. [Google Scholar] [CrossRef]
- Bowtell, J.L.; Aboo-Bakkar, Z.; Conway, M.E.; Adlam, A.-L.R.; Fulford, J. Enhanced task-related brain activation and resting perfusion in healthy older adults after chronic blueberry supplementation. Appl. Physiol. Nutr. Metab. 2017, 42, 773–779. [Google Scholar] [CrossRef]
- Gleason, C.E.; Fischer, B.L.; Dowling, N.M.; Setchell, K.D.R.; Atwood, C.S.; Carlsson, C.M.; Asthana, S. Cognitive Effects of Soy Isoflavones in Patients with Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 47, 1009–1019. [Google Scholar] [CrossRef]
- Ozawa, M.; Ninomiya, T.; Ohara, T.; Doi, Y.; Uchida, K.; Shirota, T.; Yonemoto, K.; Kitazono, T.; Kiyohara, Y. Dietary patterns and risk of dementia in an elderly Japanese population: The Hisayama Study. Am. J. Clin. Nutr. 2013, 97, 1076–1082. [Google Scholar] [CrossRef]
- Tomata, Y.; Sugiyama, K.; Kaiho, Y.; Honkura, K.; Watanabe, T.; Zhang, S.; Sugawara, Y.; Tsuji, I. Dietary Patterns and Incident Dementia in Elderly Japanese: The Ohsaki Cohort 2006 Study. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2016, 71, 1322–1328. [Google Scholar] [CrossRef]
- Yang, L.; Jin, X.; Yan, J.; Jin, Y.; Yu, W.; Wu, H.; Xu, S. Prevalence of dementia, cognitive status and associated risk factors among elderly of Zhejiang province, China in 2014. Age Ageing 2016, 45, 707–710. [Google Scholar] [CrossRef]
- Sawda, C.; Moussa, C.; Turner, R.S. Resveratrol for Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2017, 1403, 142–149. [Google Scholar] [CrossRef]
- Baum, L.; Lam, C.W.K.; Cheung, S.K.-K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; et al. Six-Month Randomized, Placebo-Controlled, Double-Blind, Pilot Clinical Trial of Curcumin in Patients With Alzheimer Disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef]
- Sanmukhani, J.; Satodia, V.; Trivedi, J.; Patel, T.; Tiwari, D.; Panchal, B.; Goel, A.; Tripathi, C.B. Efficacy and Safety of Curcumin in Major Depressive Disorder: A Randomized Controlled Trial. Phyther. Res. 2014, 28, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Small, G.W.; Siddarth, P.; Li, Z.; Miller, K.J.; Ercoli, L.; Emerson, N.D.; Martinez, J.; Wong, K.-P.; Liu, J.; Merrill, D.A.; et al. Memory and Brain Amyloid and Tau Effects of a Bioavailable Form of Curcumin in Non-Demented Adults: A Double-Blind, Placebo-Controlled 18-Month Trial. Am. J. Geriatr. Psychiatry 2018, 26, 266–277. [Google Scholar] [CrossRef]
- Nagayama, H.; Hamamoto, M.; Ueda, M.; Nito, C.; Yamaguchi, H.; Katayama, Y. The Effect of Ascorbic Acid on the Pharmacokinetics of Levodopa in Elderly Patients with Parkinson Disease. Clin. Neuropharmacol. 2004, 27, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wolk, A.; Håkansson, N.; Pedersen, N.L.; Wirdefeldt, K. Dietary antioxidants and risk of Parkinson’s disease in two population-based cohorts. Mov. Disord. 2017, 32, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.C.; Gao, X.; Kim, I.Y.; Rimm, E.B.; Wang, M.; Weisskopf, M.G.; Schwarzschild, M.A.; Ascherio, A. Intake of antioxidant vitamins and risk of Parkinson’s disease. Mov. Disord. 2016, 31, 1909–1914. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.C.; O’Reilly, É.J.; Fondell, E.; Falcone, G.J.; McCullough, M.L.; Park, Y.; Kolonel, L.N.; Ascherio, A. Intakes of vitamin C and carotenoids and risk of amyotrophic lateral sclerosis: Pooled results from 5 cohort studies. Ann. Neurol. 2013, 73, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C.; Thomas, R.G.; Grundman, M.; Bennett, D.; Doody, R.; Ferris, S.; Galasko, D.; Jin, S.; Kaye, J.; Levey, A.; et al. Vitamin E and Donepezil for the Treatment of Mild Cognitive Impairment. N. Engl. J. Med. 2005, 352, 2379–2388. [Google Scholar] [CrossRef]
- Schirinzi, T.; Martella, G.; Imbriani, P.; Di Lazzaro, G.; Franco, D.; Colona, V.L.; Alwardat, M.; Sinibaldi Salimei, P.; Mercuri, N.B.; Pierantozzi, M.; et al. Dietary Vitamin E as a Protective Factor for Parkinson’s Disease: Clinical and Experimental Evidence. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef]
- Kryscio, R.J.; Abner, E.L.; Caban-Holt, A.; Lovell, M.; Goodman, P.; Darke, A.K.; Yee, M.; Crowley, J.; Schmitt, F.A. Association of Antioxidant Supplement Use and Dementia in the Prevention of Alzheimer’s Disease by Vitamin E and Selenium Trial (PREADViSE). JAMA Neurol. 2017, 74, 567. [Google Scholar] [CrossRef]
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; Malphurs, J.; et al. Effect of Vitamin E and Memantine on Functional Decline in Alzheimer Disease. JAMA 2014, 311, 33. [Google Scholar] [CrossRef]
- The Parkinson study group. Effects of Tocopherol and Deprenyl on the Progression of Disability in Early Parkinson’s Disease. N. Engl. J. Med. 1993, 328, 176–183. [Google Scholar] [CrossRef]
- Vatassery, G.T.; Fahn, S.; Kuskowski, M.A. Alpha tocopherol in CSF of subjects taking high-dose vitamin E in the DATATOP study. Neurology 1998, 50, 1900–1902. [Google Scholar] [CrossRef]
- Miyake, Y.; Fukushima, W.; Tanaka, K.; Sasaki, S.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; Kawamura, N.; et al. Dietary intake of antioxidant vitamins and risk of Parkinson’s disease: A case-control study in Japan. Eur. J. Neurol. 2011, 18, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M.; Hernan, M.A.; Chen, H.; Spiegelman, D.; Willett, W.C.; Ascherio, A. Intakes of vitamins E and C, carotenoids, vitamin supplements, and PD risk. Neurology 2002, 59, 1161–1169. [Google Scholar] [CrossRef]
- De Rijk, M.C. Dietary Antioxidants and Parkinson Disease. Arch. Neurol. 1997, 54, 762. [Google Scholar] [CrossRef] [PubMed]
- Desnuelle, C.; Dib, M.; Garrel, C.; Favier, A. A double-blind, placebo-controlled randomized clinical trial of alpha-tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis. ALS riluzole-tocopherol Study Group. Amyotroph. Lateral Scler. 2001, 2, 9–18. [Google Scholar] [CrossRef]
- Ascherio, A.; Weisskopf, M.G.; O’Reilly, E.J.; Jacobs, E.J.; McCullough, M.L.; Calle, E.E.; Cudkowicz, M.; Thun, M.J. Vitamin E intake and risk of amyotrophic lateral sclerosis. Ann. Neurol. 2005, 57, 104–110. [Google Scholar] [CrossRef]
- Wang, H.; O’Reilly, É.J.; Weisskopf, M.G.; Logroscino, G.; McCullough, M.L.; Schatzkin, A.; Kolonel, L.N.; Ascherio, A. Vitamin E Intake and Risk of Amyotrophic Lateral Sclerosis: A Pooled Analysis of Data From 5 Prospective Cohort Studies. Am. J. Epidemiol. 2011, 173, 595–602. [Google Scholar] [CrossRef]
- Stevic, Z.; Nicolic, A.; Blagjevic, D.; Saicic, Z.S.; Kocev, N.; Apostolski, S.; Spaic, M. A controlled trial of combination of methionine and antioxidants in ALS patients. Yugosl. Med. Biochem. 2001, 20, 223–228. [Google Scholar]
- Nolan, J.M.; Mulcahy, R.; Power, R.; Moran, R.; Howard, A.N. Nutritional Intervention to Prevent Alzheimer’s Disease: Potential Benefits of Xanthophyll Carotenoids and Omega-3 Fatty Acids Combined. J. Alzheimer’s Dis. 2018, 64, 367–378. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.R.; Mill, J.; Lunnon, K. The Molecular Etiology of Alzheimer’s disease. Brain Pathol. 2020, 12879. [Google Scholar] [CrossRef] [PubMed]
- Ebanks, B.; Ingram, T.L.; Chakrabarti, L. ATP synthase and Alzheimer’s disease: Putting a spin on the mitochondrial hypothesis. Aging 2020, 12, 16647–16662. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Esteras, N.; Abramov, A.Y. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: Finding ways for prevention. Med. Res. Rev. 2020. [Google Scholar] [CrossRef]
- Yadav, R.S.; Tiwari, N.K. Lipid Integration in Neurodegeneration: An Overview of Alzheimer’s Disease. Mol. Neurobiol. 2014, 50, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Bader Lange, M.L.; Sultana, R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 924–929. [Google Scholar] [CrossRef]
- Ruipérez, V.; Darios, F.; Davletov, B. Alpha-synuclein, lipids and Parkinson’s disease. Prog. Lipid Res. 2010, 49, 420–428. [Google Scholar] [CrossRef]
- Smith, R.G.; Henry, Y.K.; Mattson, M.P.; Appel, S.H. Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann. Neurol. 1998, 44, 696–699. [Google Scholar] [CrossRef]
- Shichiri, M.; Yoshida, Y.; Ishida, N.; Hagihara, Y.; Iwahashi, H.; Tamai, H.; Niki, E. α-Tocopherol suppresses lipid peroxidation and behavioral and cognitive impairments in the Ts65Dn mouse model of Down syndrome. Free Radic. Biol. Med. 2011, 50, 1801–1811. [Google Scholar] [CrossRef]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Clemen, R.; Bekeschus, S. Oxidatively Modified Proteins: Cause and Control of Diseases. Appl. Sci. 2020, 10, 6419. [Google Scholar] [CrossRef]
- Peña-Bautista, C.; Vigor, C.; Galano, J.-M.; Oger, C.; Durand, T.; Ferrer, I.; Cuevas, A.; López-Cuevas, R.; Baquero, M.; López-Nogueroles, M.; et al. Plasma lipid peroxidation biomarkers for early and non-invasive Alzheimer Disease detection. Free Radic. Biol. Med. 2018, 124, 388–394. [Google Scholar] [CrossRef]
- Montine, T.J.; Montine, K.S.; McMahan, W.; Markesbery, W.R.; Quinn, J.F.; Morrow, J.D. F2-isoprostanes in Alzheimer and other neurodegenerative diseases. Antioxid. Redox Signal. 2005, 7, 269–275. [Google Scholar] [CrossRef]
- Bradley, M.A.; Markesbery, W.R.; Lovell, M.A. Increased levels of 4-hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free Radic. Biol. Med. 2010, 48, 1570–1576. [Google Scholar] [CrossRef]
- Park, J.; Muratori, B.; Shi, R. Acrolein as a novel therapeutic target for motor and sensory deficits in spinal cord injury. Neural Regen. Res. 2014, 9, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Yoshikawa, A.; Kinumi, T.; Ogawa, Y.; Saito, Y.; Ohara, K.; Yamamoto, H.; Imai, Y.; Niki, E. Hydroxyoctadecadienoic acid and oxidatively modified peroxiredoxins in the blood of Alzheimer’s disease patients and their potential as biomarkers. Neurobiol. Aging 2009, 30, 174–185. [Google Scholar] [CrossRef]
- Arslan, J.; Jamshed, H.; Qureshi, H. Early Detection and Prevention of Alzheimer’s Disease: Role of Oxidative Markers and Natural Antioxidants. Front. Aging Neurosci. 2020, 12. [Google Scholar] [CrossRef]
- Dare, L.R.; Garcia, A.; Soares, C.B.; Lopes, L.; Neves, B.-H.S.; Dias, D.V.; Mello-Carpes, P.B. The Reversal of Memory Deficits in an Alzheimer’s Disease Model Using Physical and Cognitive Exercise. Front. Behav. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- López-Riquelme, N.; Alom-Poveda, J.; Viciano-Morote, N.; Llinares-Ibor, I.; Tormo-Díaz, C. Apolipoprotein E ε4 allele and malondialdehyde level are independent risk factors for Alzheimer’s disease. Sage Open Med. 2016, 4, 205031211562673. [Google Scholar] [CrossRef]
- Petrovic, S.; Arsic, A.; Glibetic, M.; Cikiriz, N.; Jakovljevic, V.; Vucic, V. The effects of polyphenol-rich chokeberry juice on fatty acid profiles and lipid peroxidation of active handball players: Results from a randomized, double-blind, placebo-controlled study. Can. J. Physiol. Pharm. 2016, 94, 1058–1063. [Google Scholar] [CrossRef]
- Sarubbo, F.; Moranta, D.; Pani, G. Dietary polyphenols and neurogenesis: Molecular interactions and implication for brain ageing and cognition. Neurosci. Biobehav. Rev. 2018, 90, 456–470. [Google Scholar] [CrossRef]
- Vučić, V.; Grabež, M.; Trchounian, A.; Arsić, A. Composition and Potential Health Benefits of Pomegranate: A Review. Curr. Pharm. Des. 2019, 25, 1817–1827. [Google Scholar] [CrossRef]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Epigallocatechin-3-gallate and curcumin suppress amyloid beta-induced beta-site APP cleaving enzyme-1 upregulation. Neuroreport 2008, 19, 1329–1333. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, J.; Derreumaux, P.; Mu, Y. Molecular mechanism of the inhibition of EGCG on the Alzheimer Aβ(1-42) dimer. J. Phys. Chem. B 2013, 117, 3993–4002. [Google Scholar] [CrossRef]
- Ettcheto, M.; Cano, A.; Manzine, P.R.; Busquets, O.; Verdaguer, E.; Castro-Torres, R.D.; García, M.L.; Beas-Zarate, C.; Olloquequi, J.; Auladell, C.; et al. Epigallocatechin-3-Gallate (EGCG) Improves Cognitive Deficits Aggravated by an Obesogenic Diet Through Modulation of Unfolded Protein Response in APPswe/PS1dE9 Mice. Mol. Neurobiol. 2020, 57, 1814–1827. [Google Scholar] [CrossRef]
- Cano, A.; Ettcheto, M.; Chang, J.-H.; Barroso, E.; Espina, M.; Kühne, B.A.; Barenys, M.; Auladell, C.; Folch, J.; Souto, E.B.; et al. Dual-drug loaded nanoparticles of Epigallocatechin-3-gallate (EGCG)/Ascorbic acid enhance therapeutic efficacy of EGCG in a APPswe/PS1dE9 Alzheimer’s disease mice model. J. Control. Release 2019, 301, 62–75. [Google Scholar] [CrossRef]
- Belkacemi, A.; Doggui, S.; Dao, L.; Ramassamy, C. Challenges associated with curcumin therapy in Alzheimer disease. Expert Rev. Mol. Med. 2011, 13, e34. [Google Scholar] [CrossRef]
- Weinreb, O.; Mandel, S.; Amit, T.; Youdim, M.B.H. Neurological mechanisms of green tea polyphenols in Alzheimer’s and Parkinson’s diseases. J. Nutr. Biochem. 2004, 15, 506–516. [Google Scholar] [CrossRef]
- Nurk, E.; Refsum, H.; Drevon, C.A.; Tell, G.S.; Nygaard, H.A.; Engedal, K.; Smith, A.D. Intake of Flavonoid-Rich Wine, Tea, and Chocolate by Elderly Men and Women is Associated with Better Cognitive Test Performance. J. Nutr. 2009, 139, 120–127. [Google Scholar] [CrossRef]
- Small, B.J.; Rawson, K.S.; Martin, C.; Eisel, S.L.; Sanberg, C.D.; McEvoy, C.L.; Sanberg, P.R.; Shytle, R.D.; Tan, J.; Bickford, P.C. Nutraceutical Intervention Improves Older Adults’ Cognitive Functioning. Rejuvenation Res. 2014, 17, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Mastroiacovo, D.; Kwik-Uribe, C.; Grassi, D.; Necozione, S.; Raffaele, A.; Pistacchio, L.; Righetti, R.; Bocale, R.; Lechiara, M.C.; Marini, C.; et al. Cocoa flavanol consumption improves cognitive function, blood pressure control, and metabolic profile in elderly subjects: The Cocoa, Cognition, and Aging (CoCoA) Study—A randomized controlled trial. Am. J. Clin. Nutr. 2015, 101, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Rajaram, S.; Valls-Pedret, C.; Cofán, M.; Sabaté, J.; Serra-Mir, M.; Pérez-Heras, A.M.; Arechiga, A.; Casaroli-Marano, R.P.; Alforja, S.; Sala-Vila, A.; et al. The Walnuts and Healthy Aging Study (WAHA): Protocol for a Nutritional Intervention Trial with Walnuts on Brain Aging. Front. Aging Neurosci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. Why Vitamin E Therapy Fails for Treatment of Alzheimer’s Disease. J. Alzheimer’s Dis. 2010, 19, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Xu, W.; Kivipelto, M.; Costanzi, E.; Ercolani, S.; Pigliautile, M.; Cecchetti, R.; Baglioni, M.; Simmons, A.; Soininen, H.; et al. Tocopherols and tocotrienols plasma levels are associated with cognitive impairment. Neurobiol. Aging 2012, 33, 2282–2290. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-Analysis: High-Dosage Vitamin E Supplementation May Increase All-Cause Mortality. Ann. Intern. Med. 2005, 142, 37. [Google Scholar] [CrossRef]
- Harrison, F.E. A Critical Review of Vitamin C for the Prevention of Age-Related Cognitive Decline and Alzheimer’s Disease. J. Alzheimer’s Dis. 2012, 29, 711–726. [Google Scholar] [CrossRef]
- Kim, S.-M.; Lim, S.-M.; Yoo, J.-A.; Woo, M.-J.; Cho, K.-H. Consumption of high-dose vitamin C (1250 mg per day) enhances functional and structural properties of serum lipoprotein to improve anti-oxidant, anti-atherosclerotic, and anti-aging effects via regulation of anti-inflammatory microRNA. Food Funct. 2015, 6, 3604–3612. [Google Scholar] [CrossRef]
- Monacelli, F.; Acquarone, F.M.E.; Giannotti, C.; Borghi, R.; Nencioni, A. Vitamin C, Aging and Alzheimer’s Disease. Nutrients 2017, 9, 670. [Google Scholar] [CrossRef]
- Mehta, V.; Desai, N.; Perwez, A.; Nemade, D.; Dawoodi, S.; Zaman, S. Bin ACE Alzheimer’s: The Role of Vitamin A, C and E (ACE) in Oxidative Stress induced Alzheimer’s Disease. J. Med. Res. Innov. 2017, 2, e000086. [Google Scholar] [CrossRef]
- Ono, K.; Yamada, M. Vitamin A and Alzheimer’s disease. Geriatr. Gerontol. Int. 2012, 12, 180–188. [Google Scholar] [CrossRef]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Pu, J. Alpha-Synuclein in Parkinson’s Disease: From Pathogenetic Dysfunction to Potential Clinical Application. Parkinsons. Dis. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Goswami, P.; Joshi, N.; Singh, S. Neurodegenerative signaling factors and mechanisms in Parkinson’s pathology. Toxicol. Vitr. 2017, 43, 104–112. [Google Scholar] [CrossRef]
- Smeyne, M.; Smeyne, R.J. Glutathione metabolism and Parkinson’s disease. Free Radic. Biol. Med. 2013, 62, 13–25. [Google Scholar] [CrossRef]
- Schaur, R.; Siems, W.; Bresgen, N.; Eckl, P. 4-Hydroxy-nonenal—A Bioactive Lipid Peroxidation Product. Biomolecules 2015, 5, 2247–2337. [Google Scholar] [CrossRef]
- Pizzimenti, S.; Ciamporcero, E.; Daga, M.; Pettazzoni, P.; Arcaro, A.; Cetrangolo, G.; Minelli, R.; Dianzani, C.; Lepore, A.; Gentile, F.; et al. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front. Physiol. 2013, 4, 242. [Google Scholar] [CrossRef]
- Guéraud, F.; Atalay, M.; Bresgen, N.; Cipak, A.; Eckl, P.M.; Huc, L.; Jouanin, I.; Siems, W.; Uchida, K. Chemistry and biochemistry of lipid peroxidation products. Free Radic. Res. 2010, 44, 1098–1124. [Google Scholar] [CrossRef]
- Shamoto-Nagai, M.; Maruyama, W.; Hashizume, Y.; Yoshida, M.; Osawa, T.; Riederer, P.; Naoi, M. In parkinsonian substantia nigra, α-synuclein is modified by acrolein, a lipid-peroxidation product, and accumulates in the dopamine neurons with inhibition of proteasome activity. J. Neural Transm. 2007, 114, 1559–1567. [Google Scholar] [CrossRef]
- Csala, M.; Kardon, T.; Legeza, B.; Lizák, B.; Mandl, J.; Margittai, É.; Puskás, F.; Száraz, P.; Szelényi, P.; Bánhegyi, G. On the role of 4-hydroxynonenal in health and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 826–838. [Google Scholar] [CrossRef]
- Castellani, R.J.; Perry, G.; Siedlak, S.L.; Nunomura, A.; Shimohama, S.; Zhang, J.; Montine, T.; Sayre, L.M.; Smith, M.A. Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci. Lett. 2002, 319, 25–28. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.-S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, T.N.; Logvinenko, A.A.; Poleshchuk, V.V.; Muzychuk, O.A.; Shabalina, A.A.; Illarioshkin, S.N. Lipid Peroxidation Products in the Blood Plasma of Patients with Parkinson’s Disease as Possible Biomarkers of Different Stages of the Disease. Neurochem. J. 2019, 13, 391–395. [Google Scholar] [CrossRef]
- Visanji, N.P.; Brooks, P.L.; Hazrati, L.-N.; Lang, A.E. The prion hypothesis in Parkinson’s disease: Braak to the future. Acta Neuropathol. Commun. 2013, 1, 2. [Google Scholar] [CrossRef]
- Seet, R.C.S.; Lee, C.-Y.J.; Lim, E.C.H.; Tan, J.J.H.; Quek, A.M.L.; Chong, W.-L.; Looi, W.-F.; Huang, S.-H.; Wang, H.; Chan, Y.-H. Oxidative damage in Parkinson disease: Measurement using accurate biomarkers. Free Radic. Biol. Med. 2010, 48, 560–566. [Google Scholar] [CrossRef]
- Fessel, J.P.; Hulette, C.; Powell, S.; Roberts, L.J.; Zhang, J. Isofurans, but not F2-isoprostanes, are increased in the substantia nigra of patients with Parkinson’s disease and with dementia with Lewy body disease. J. Neurochem. 2003, 85, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Xie, S.P.; Saw, W.T.; Ho, P.G.H.; Wang, H.; Lei, Z.; Yi, Z.; Tan, E.K. The Therapeutic Implications of Tea Polyphenols Against Dopamine (DA) Neuron Degeneration in Parkinson’s Disease (PD). Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Kujawska, M.; Jodynis-Liebert, J. Polyphenols in Parkinson’s Disease: A Systematic Review of In Vivo Studies. Nutrients 2018, 10, 642. [Google Scholar] [CrossRef]
- Maher, P. The Potential of Flavonoids for the Treatment of Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3056. [Google Scholar] [CrossRef]
- Mischley, L.K.; Lau, R.C.; Bennett, R.D. Role of Diet and Nutritional Supplements in Parkinson’s Disease Progression. Oxid. Med. Cell. Longev. 2017, 2017, 1–9. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Gu, Y.; Mejia-Santana, H.; Cote, L.; Marder, K.S.; Scarmeas, N. The association between Mediterranean diet adherence and Parkinson’s disease. Mov. Disord. 2012, 27, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Chen, H.; Fung, T.T.; Logroscino, G.; Schwarzschild, M.A.; Hu, F.B.; Ascherio, A. Prospective study of dietary pattern and risk of Parkinson disease. Am. J. Clin. Nutr. 2007, 86, 1486–1494. [Google Scholar] [CrossRef]
- Sääksjärvi, K.; Knekt, P.; Lundqvist, A.; Männistö, S.; Heliövaara, M.; Rissanen, H.; Järvinen, R. A cohort study on diet and the risk of Parkinson’s disease: The role of food groups and diet quality. Br. J. Nutr. 2013, 109, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Checkoway, H.; Powers, K.; Smith-Weller, T.; Franklin, G.M.; Longstreth, W.T.; Swanson, P.D. Parkinson’s Disease Risks Associated with Cigarette Smoking, Alcohol Consumption, and Caffeine Intake. Am. J. Epidemiol. 2002, 155, 732–738. [Google Scholar] [CrossRef]
- Hartman, R.E.; Shah, A.; Fagan, A.M.; Schwetye, K.E.; Parsadanian, M.; Schulman, R.N.; Finn, M.B.; Holtzman, D.M. Pomegranate juice decreases amyloid load and improves behavior in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2006, 24, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Tapias, V.; Cannon, J.R.; Greenamyre, J.T. Pomegranate juice exacerbates oxidative stress and nigrostriatal degeneration in Parkinson’s disease. Neurobiol. Aging 2014, 35, 1162–1176. [Google Scholar] [CrossRef]
- Park, H.-A.; Ellis, A.C. Dietary Antioxidants and Parkinson’s Disease. Antioxidants 2020, 9, 570. [Google Scholar] [CrossRef]
- Logroscino, G.; Marder, K.; Cote, L.; Tang, M.-X.; Shea, S.; Mayeux, R. Dietary lipids and antioxidants in Parkinson’s disease: A population-based, case-control study. Ann. Neurol. 1996, 39, 89–94. [Google Scholar] [CrossRef]
- Etminan, M.; Gill, S.S.; Samii, A. Intake of vitamin E, vitamin C, and carotenoids and the risk of Parkinson’s disease: A meta-analysis. Lancet Neurol. 2005, 4, 362–365. [Google Scholar] [CrossRef]
- Takeda, A.; Nyssen, O.P.; Syed, A.; Jansen, E.; Bueno-de-Mesquita, B.; Gallo, V. Vitamin A and Carotenoids and the Risk of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Neuroepidemiology 2014, 42, 25–38. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Abbott, R.D.; Webster Ross, G.; White, L.R.; Sanderson, W.T.; Burchfiel, C.M.; Kashon, M.; Sharp, D.S.; Masaki, K.H.; Curb, J.D.; Petrovitch, H. Environmental, life-style, and physical precursors of clinical Parkinson?s disease: Recent findings from the Honolulu-Asia Aging Study. J. Neurol. 2003, 250, iii30–iii39. [Google Scholar] [CrossRef]
- Scheider, W.L.; Hershey, L.A.; Vena, J.E.; Holmlund, T.; Marshall, J.R.; Freudenheim, J.L. Dietary antioxidants and other dietary factors in the etiology of Parkinson’s disease. Mov. Disord. 1997, 12, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Hellenbrand, W.; Boeing, H.; Robra, B.-P.; Seidler, A.; Vieregge, P.; Nischan, P.; Joerg, J.; Oertel, W.H.; Schneider, E.; Ulm, G. Diet and Parkinson’s disease II: A possible role for the past intake of specific nutrients: Results from a self-administered food-frequency questionnaire in a case-control study. Neurology 1996, 47, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Férnandez-Calle, P.; Jiménez-Jiménez, F.J.; Molina, J.; Cabrera-Valdivia, F.; Vázquez, A.; Urra, D.G.; Bermejo, F.; Matallana, M.C.; Codoceo, R. Serum levels of ascorbic acid (vitamin C) in patients with Parkinson’s disease. J. Neurol. Sci. 1993, 118, 25–28. [Google Scholar] [CrossRef]
- Sudha, K.; Rao, A.V.; Rao, S.; Rao, A. Free radical toxicity and antioxidants in Parkinson’s disease. Neurol. India 2003, 51, 60–62. [Google Scholar]
- Ide, K.; Yamada, H.; Umegaki, K.; Mizuno, K.; Kawakami, N.; Hagiwara, Y.; Matsumoto, M.; Yoshida, H.; Kim, K.; Shiosaki, E.; et al. Lymphocyte vitamin C levels as potential biomarker for progression of Parkinson’s disease. Nutrition 2015, 31, 406–408. [Google Scholar] [CrossRef]
- Boulos, C.; Yaghi, N.; El Hayeck, R.; Heraoui, G.N.; Fakhoury-Sayegh, N. Nutritional Risk Factors, Microbiota and Parkinson’s Disease: What Is the Current Evidence? Nutrients 2019, 11, 1896. [Google Scholar] [CrossRef]
- Abbott, R.A.; Cox, M.; Markus, H.; Tomkins, A. Diet, body size and micronutrient status in Parkinson’s disease. Eur. J. Clin. Nutr. 1992, 46, 879–884. [Google Scholar]
- Shoulson, I.; Oakes, D.; Fahn, S.; Lang, A.; Langston, J.W.; LeWitt, P.; Olanow, C.W.; Penney, J.B.; Tanner, C.; Kieburtz, K.; et al. Impact of sustained deprenyl (selegiline) in levodopa-treated Parkinson’s disease: A randomized placebo-controlled extension of the deprenyl and tocopherol antioxidative therapy of parkinsonism trial. Ann. Neurol. 2002, 51, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Seidl, S.E.; Santiago, J.A.; Bilyk, H.; Potashkin, J.A. The emerging role of nutrition in Parkinson’s disease. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef]
- Agim, Z.S.; Cannon, J.R. Dietary Factors in the Etiology of Parkinson’s Disease. Biomed Res. Int. 2015, 2015, 672838. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.J.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Gros-Louis, F.; Gaspar, C.; Rouleau, G.A. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 956–972. [Google Scholar] [CrossRef]
- Talbot, K. Motor neuron disease: The bare essentials. Pr. Neurol. 2009, 9, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Del Aguila, M.A.; Longstreth, W.T.J.; McGuire, V.; Koepsell, T.D.; van Belle, G. Prognosis in amyotrophic lateral sclerosis: A population-based study. Neurology 2003, 60, 813–819. [Google Scholar] [CrossRef]
- Miller, R.G.; Mitchell, J.D.; Lyon, M.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2007. [Google Scholar] [CrossRef]
- Chiò, A.; Mora, G.; Calvo, A.; Mazzini, L.; Bottacchi, E.; Mutani, R. Epidemiology of ALS in Italy: A 10-year prospective population-based study. Neurology 2009, 72, 725–731. [Google Scholar] [CrossRef]
- Manjaly, Z.R.; Scott, K.M.; Abhinav, K.; Wijesekera, L.; Ganesalingam, J.; Goldstein, L.H.; Janssen, A.; Dougherty, A.; Willey, E.; Stanton, B.R.; et al. The sex ratio in amyotrophic lateral sclerosis: A population based study. Amyotroph. Lateral Scler. 2010, 11, 439–442. [Google Scholar] [CrossRef]
- Zou, Z.; Zhou, Z.; Che, C.; Liu, C.; He, R. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Saccon, R.A.; Bunton-Stasyshyn, R.K.A.; Fisher, E.M.C.; Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013, 136, 2342–2358. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.; Andersen, P.M.; Marklund, S.L.; Brännström, T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2011, 121, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Guareschi, S.; Cova, E.; Cereda, C.; Ceroni, M.; Donetti, E.; Bosco, D.A.; Trotti, D.; Pasinelli, P. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl. Acad. Sci. USA 2012, 109, 5074–5079. [Google Scholar] [CrossRef]
- Rakhit, R.; Crow, J.P.; Lepock, J.R.; Kondejewski, L.H.; Cashman, N.R.; Chakrabartty, A. Monomeric Cu,Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J. Biol. Chem. 2004, 279, 15499–15504. [Google Scholar] [CrossRef]
- Blasco, H.; Garcon, G.; Patin, F.; Veyrat-Durebex, C.; Boyer, J.; Devos, D.; Vourc’h, P.; Andres, C.R.; Corcia, P. Panel of Oxidative Stress and Inflammatory Biomarkers in ALS: A Pilot Study. Can. J. Neurol. Sci. 2017, 44, 90–95. [Google Scholar] [CrossRef]
- Fang, L.; Huber-Abel, F.; Teuchert, M.; Hendrich, C.; Dorst, J.; Schattauer, D.; Zettlmeissel, H.; Wlaschek, M.; Scharffetter-Kochanek, K.; Tumani, H.; et al. Linking neuron and skin: Matrix metalloproteinases in amyotrophic lateral sclerosis (ALS). J. Neurol. Sci. 2009, 285, 62–66. [Google Scholar] [CrossRef]
- Wang, Z.; Bai, Z.; Qin, X.; Cheng, Y. Aberrations in Oxidative Stress Markers in Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Oxid. Med. Cell. Longev. 2019, 2019, 1712323. [Google Scholar] [CrossRef]
- Perluigi, M.; Fai Poon, H.; Hensley, K.; Pierce, W.M.; Klein, J.B.; Calabrese, V.; De Marco, C.; Butterfield, D.A. Proteomic analysis of 4-hydroxy-2-nonenal-modified proteins in G93A-SOD1 transgenic mice—a model of familial amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2005, 38, 960–968. [Google Scholar] [CrossRef]
- Pedersen, W.A.; Fu, W.; Keller, J.N.; Markesbery, W.R.; Appel, S.; Smith, R.G.; Kasarskis, E.; Mattson, M.P. Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann. Neurol. 1998, 44, 819–824. [Google Scholar] [CrossRef]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.; Henry, Y.K.; Henkel, J.; Smith, R.; Appel, S. Increased lipid peroxidation in sera of ALS patients. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef] [PubMed]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.-C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative stress biomarkers in sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Castegna, A.; Drake, J.; Scapagnini, G.; Calabrese, V. Vitamin E and neurodegenerative disorders associated with oxidative stress. Nutr. Neurosci. 2002, 5, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Orrell, R.W.; Lane, R.J.M.; Ross, M. Antioxidant treatment for amyotrophic lateral sclerosis / motor neuron disease. Cochrane Database Syst. Rev. 2007. [Google Scholar] [CrossRef]
- Wang, M.; Liu, Z.; Sun, W.; Yuan, Y.; Jiao, B.; Zhang, X.; Shen, L.; Jiang, H.; Xia, K.; Tang, B.; et al. Association Between Vitamins and Amyotrophic Lateral Sclerosis: A Center-Based Survey in Mainland China. Front. Neurol. 2020, 11, 488. [Google Scholar] [CrossRef]
- Paraskevas, G.P.; Kapaki, E.; Libitaki, G.; Zournas, C.; Segditsa, I.; Papageorgiou, C. Ascorbate in healthy subjects, amyotrophic lateral sclerosis and Alzheimer’s disease. Acta Neurol. Scand. 1997, 96, 88–90. [Google Scholar] [CrossRef]
- Nagano, S.; Fujii, Y.; Yamamoto, T.; Taniyama, M.; Fukada, K.; Yanagihara, T.; Sakoda, S. The efficacy of trientine or ascorbate alone compared to that of the combined treatment with these two agents in familial amyotrophic lateral sclerosis model mice. Exp. Neurol. 2003, 179, 176–180. [Google Scholar] [CrossRef]
- Jin, Y.; Oh, K.; Oh, S.; Baek, H.; Kim, S.H.; Park, Y. Dietary intake of fruits and beta-carotene is negatively associated with amyotrophic lateral sclerosis risk in Koreans: A case-control study. Nutr. Neurosci. 2014, 17, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kihira, T.; Kobashi, G.; Washio, M.; Sasaki, S.; Yokoyama, T.; Miyake, Y.; Sakamoto, N.; Inaba, Y.; Nagai, M. Fruit and vegetable intake and risk of amyotrophic lateral sclerosis in Japan. Neuroepidemiology 2009, 32, 251–256. [Google Scholar] [CrossRef]
- Nieves, J.W.; Gennings, C.; Factor-Litvak, P.; Hupf, J.; Singleton, J.; Sharf, V.; Oskarsson, B.; Fernandes Filho, J.A.M.; Sorenson, E.J.; D’Amico, E.; et al. Association Between Dietary Intake and Function in Amyotrophic Lateral Sclerosis. JAMA Neurol. 2016, 73, 1425–1432. [Google Scholar] [CrossRef]
- Crochemore, C.; Virgili, M.; Bonamassa, B.; Canistro, D.; Pena-Altamira, E.; Paolini, M.; Contestabile, A. Long-term dietary administration of valproic acid does not affect, while retinoic acid decreases, the lifespan of G93A mice, a model for amyotrophic lateral sclerosis. Muscle Nerve 2009, 39, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, O.T.; Aytan, N.; Carreras, I.; Choi, J.-K.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. 7,8-Dihydroxyflavone improves motor performance and enhances lower motor neuronal survival in a mouse model of amyotrophic lateral sclerosis. Neurosci. Lett. 2014, 566, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Ip, P.; Sharda, P.R.; Cunningham, A.; Chakrabartty, S.; Pande, V.; Chakrabartty, A. Quercitrin and quercetin 3-β-d-glucoside as chemical chaperones for the A4V SOD1 ALS-causing mutant. Protein Eng. Des. Sel. 2017, 30, 431–440. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Lankatillake, C.; Dias, D.A.; Docea, A.O.; Mahomoodally, M.F.; Lobine, D.; Chazot, P.L.; Kurt, B.; Boyunegmez Tumer, T.; Catarina Moreira, A.; et al. Impact of Natural Compounds on Neurodegenerative Disorders: From Preclinical to Pharmacotherapeutics. J. Clin. Med. 2020, 9, 1061. [Google Scholar] [CrossRef]
- Veurink, G.; Perry, G.; Singh, S.K. Role of antioxidants and a nutrient rich diet in Alzheimer’s disease. Open Biol. 2020, 10, 200084. [Google Scholar] [CrossRef]
- Rakesh, G.; Szabo, S.T.; Alexopoulos, G.S.; Zannas, A.S. Strategies for dementia prevention: Latest evidence and implications. Adv. Chronic Dis. 2017, 8, 121–136. [Google Scholar] [CrossRef]
- Lamport, D.J.; Dye, L.; Wightman, J.D.; Lawton, C.L. The effects of flavonoid and other polyphenol consumption on cognitive performance: A systematic research review of human experimental and epidemiological studies. Nutr. Aging 2012, 1, 5–25. [Google Scholar] [CrossRef]
- Hussain, R.; Zubair, H.; Pursell, S.; Shahab, M. Neurodegenerative Diseases: Regenerative Mechanisms and Novel Therapeutic Approaches. Brain Sci. 2018, 8, 177. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).