BMS-986020, a Specific LPA1 Antagonist, Provides Neuroprotection against Ischemic Stroke in Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Induction of tMCAO Challenge in Mice
2.3. BMS Administration
2.4. Assessment of Neurological Deficits and Survival Rate
2.5. Assessment of Brain Infarction
2.6. Histological Analysis
2.6.1. Tissue Preparation
2.6.2. TUNEL Assay
2.6.3. Immunohistochemistry Against Iba1 or 4-HNE
2.6.4. Double Immunofluorescence Followed by 5-Bromo-2′-Deoxyuridine (BrdU) Incorporation
2.6.5. Image Preparation and Quantification
2.7. Statistical Analysis
3. Results
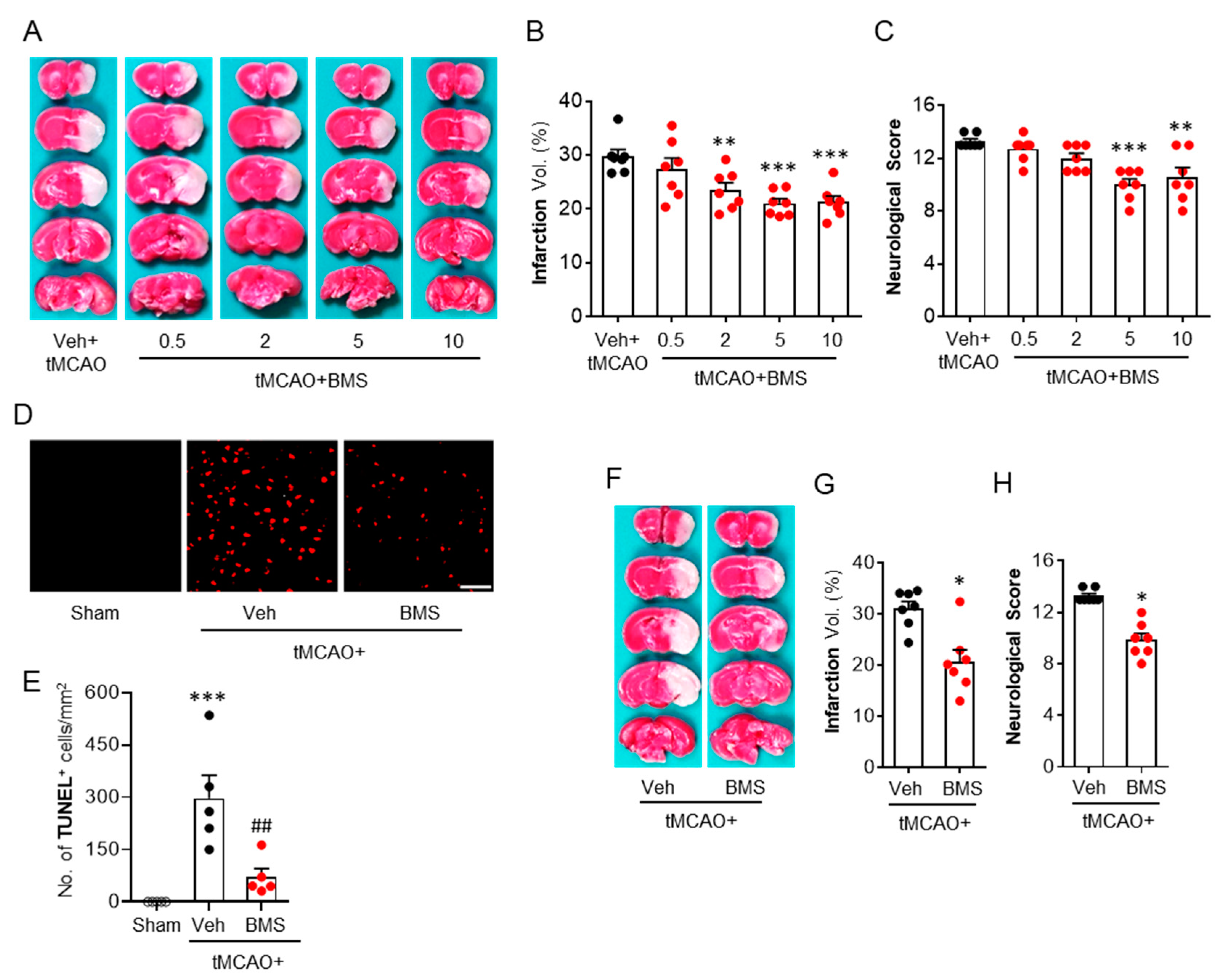
3.1. BMS Administration Attenuates tMCAO-Induced Brain Infarction and Neurological Deficits during the Acute Phase
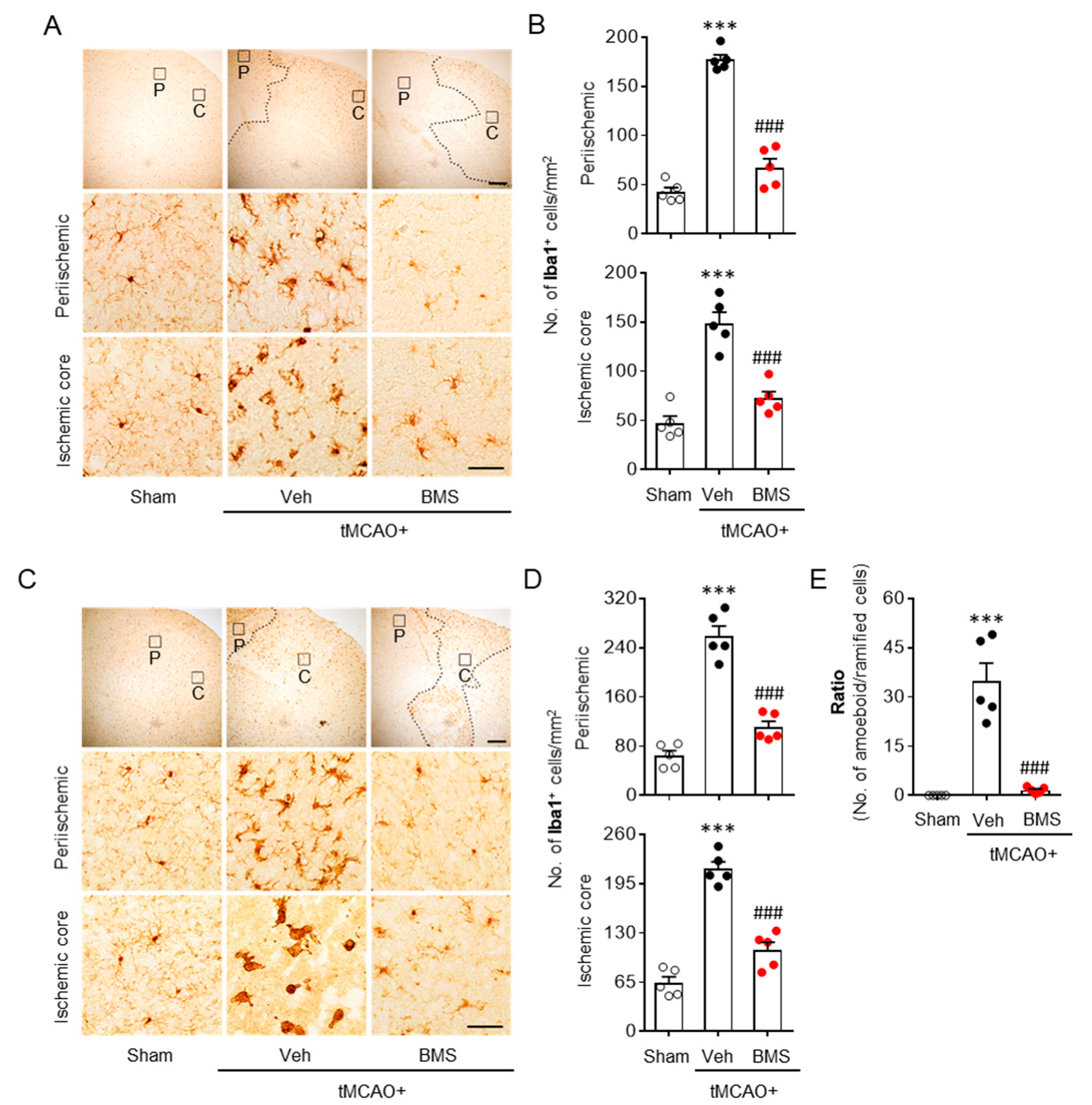
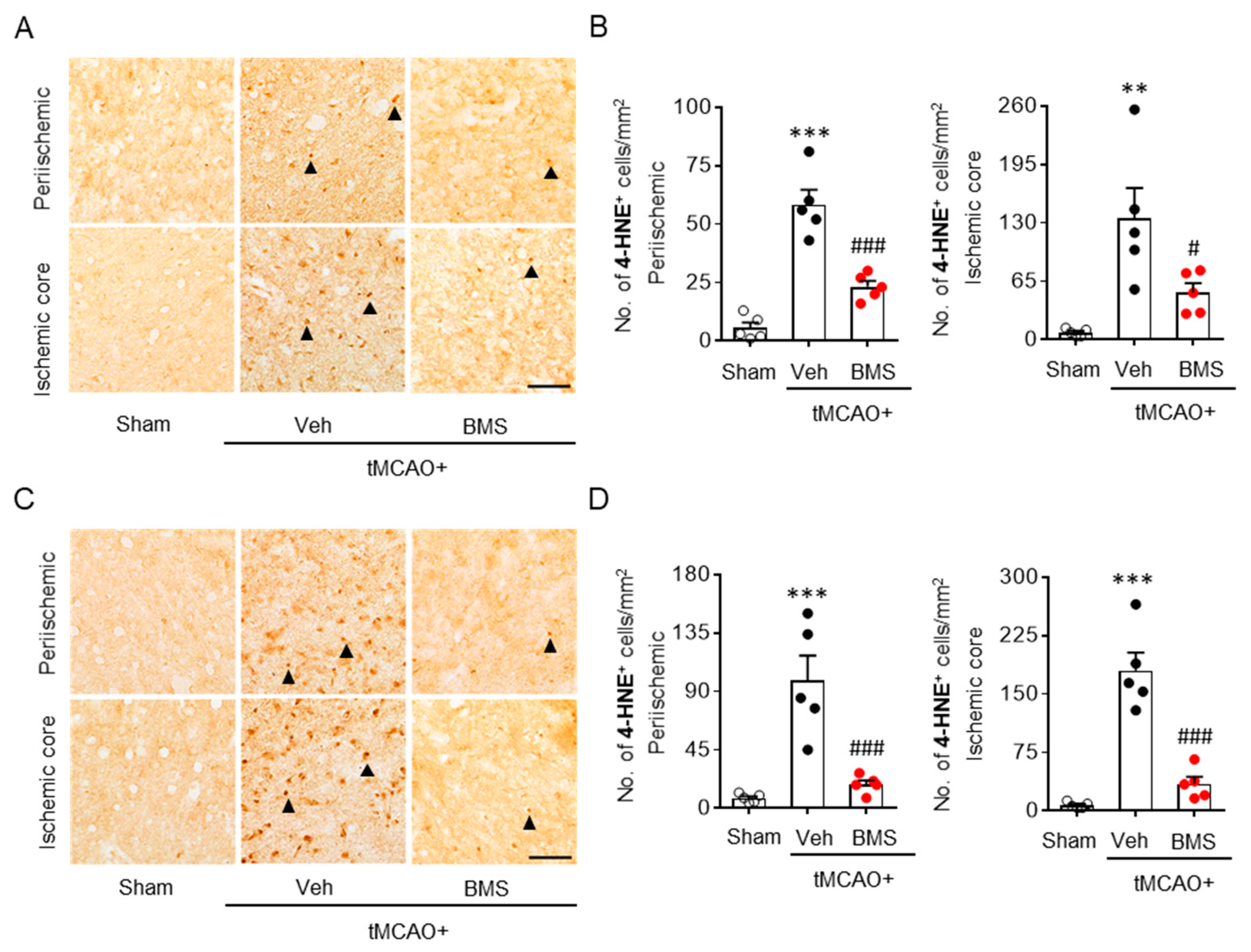
3.2. BMS Administration Attenuates tMCAO-Induced Microglial Activation and Lipid Peroxidation during the Acute Phase
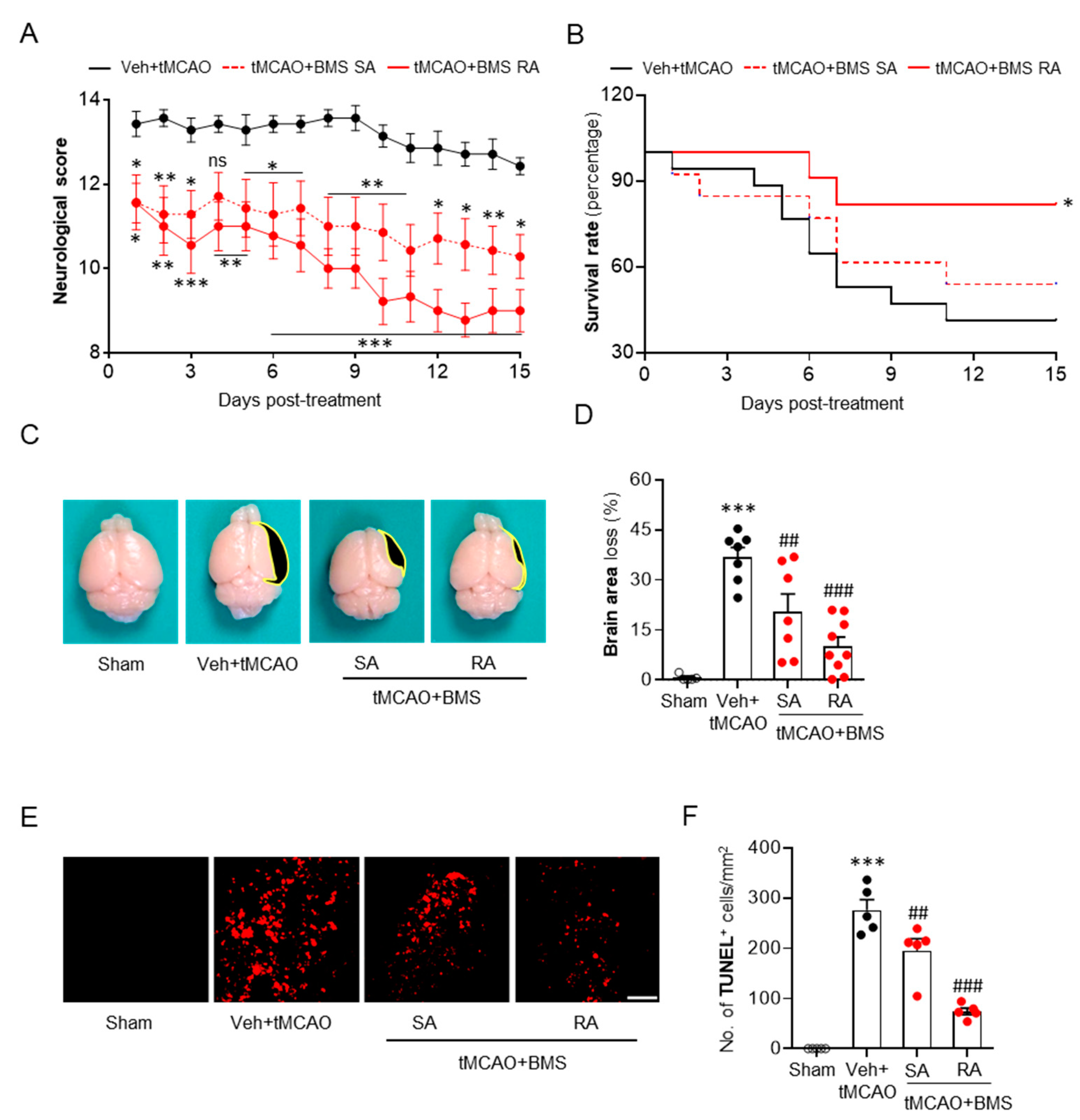
3.3. BMS Administration Attenuates tMCAO-Induced Neurological Deficits and Improves Survival Rate during the Sub-Acute Phase Along with Attenuation of Brain Tissue Loss and Cell Apoptosis
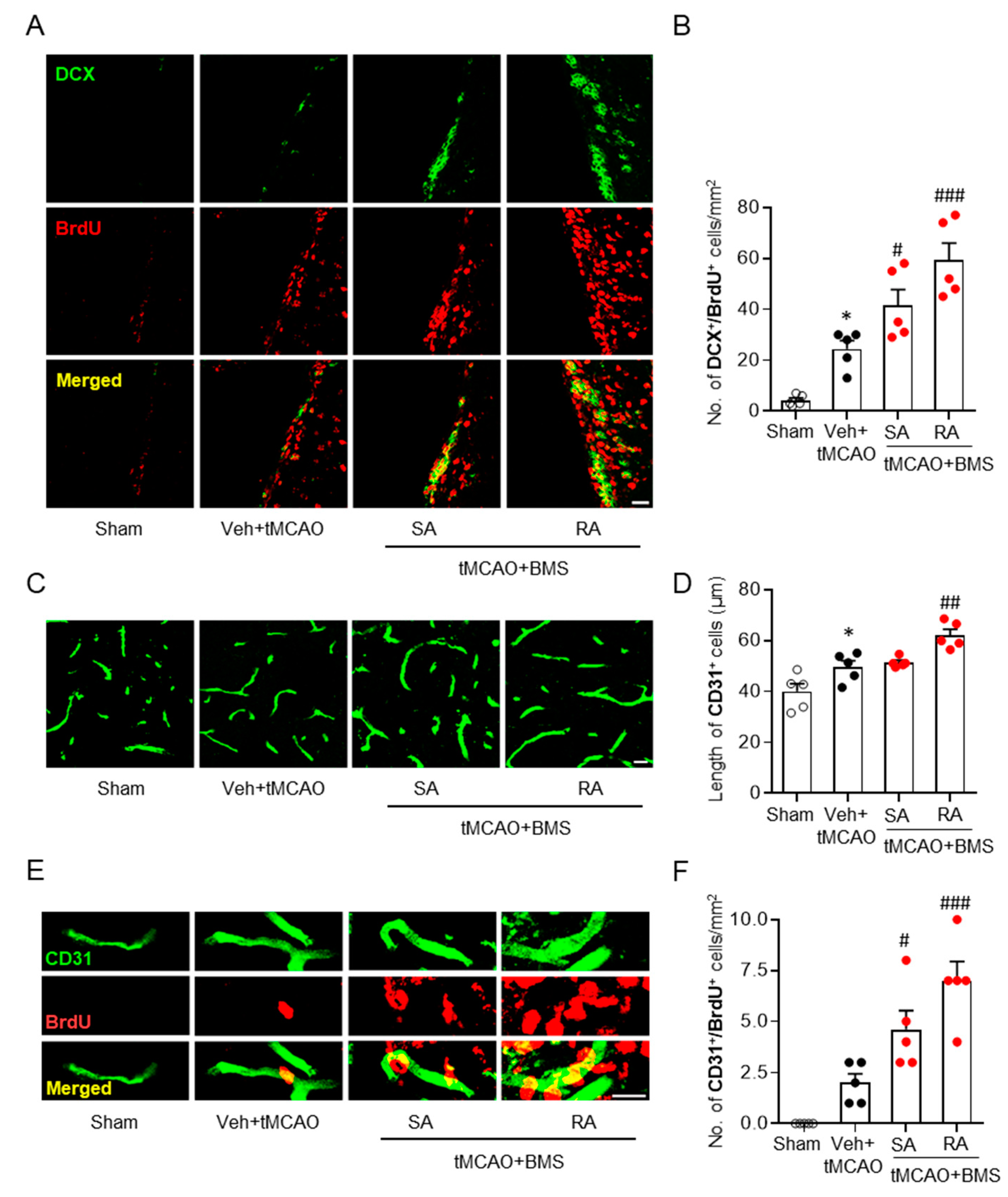
3.4. BMS Administration Enhances Neurogenesis and Angiogenesis in Post-Ischemic Brains
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.W.; Mutoh, T.; Lin, M.E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA receptors: Subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.E.; Herr, D.R.; Chun, J. Lysophosphatidic acid (LPA) receptors: Signaling properties and disease relevance. Prostaglandins Other Lipid Mediat. 2010, 91, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Kihara, Y.; Mizuno, H.; Chun, J. Lysophospholipid receptors in drug discovery. Exp. Cell Res. 2015, 333, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.L.; Lawler, M.J.; Kopp, N.; Mcleod, D.D.; Davulcu, A.H.; Lin, D.; Katipally, K.; Sfoggatakis, C. Development of a Concise Multikilogram Synthesis of LPA-1 Antagonist BMS-986020 via a Tandem Borylation–Suzuki Procedure. Org. Process. Res. Dev. 2017, 21, 1859–1863. [Google Scholar] [CrossRef]
- Hutchinson, J.H.; Seiders, T.J.; Zhao, L.; Wang, B.; Arruda, J.M.; Roppe, J.R.; Parr, T.; Stock, N.S. Polycyclic Antagonists of Lysophosphatidic Acid Receptors. U.S. Patent WO2010141768, 9 December 2010. [Google Scholar]
- Pena, A.; Kim, J.; Donnelly, D.; Murphy, B.; Shuster, D.; Watson, L.; Bonacrosi, S.; Hayes, W.; Chow, P.; Du, S. Autoradiographic evaluation of [18F]BMT-083133, a lysophosphatidic acid receptor 1 (LPA1) radioligand. J. Nucl. Med. 2014, 55, 1207. [Google Scholar]
- Bollong, M.J.; Yang, B.; Vergani, N.; Beyer, B.A.; Chin, E.N.; Zambaldo, C.; Wang, D.; Chatterjee, A.K.; Lairson, L.L.; Schultz, P.G. Small molecule-mediated inhibition of myofibroblast transdifferentiation for the treatment of fibrosis. Proc. Natl. Acad. Sci. USA 2017, 114, 4679–4684. [Google Scholar] [CrossRef]
- Sakai, N.; Bain, G.; Furuichi, K.; Iwata, Y.; Nakamura, M.; Hara, A.; Kitajima, S.; Sagara, A.; Miyake, T.; Toyama, T.; et al. The involvement of autotaxin in renal interstitial fibrosis through regulation of fibroblast functions and induction of vascular leakage. Sci. Rep. 2019, 9, 7414. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.M.; Snyder, L.; Todd, J.L.; Soule, B.; Christian, R.; Anstrom, K.; Luo, Y.; Gagnon, R.; Rosen, G. Randomized, Double-Blind, Placebo-Controlled, Phase 2 Trial of BMS-986020, a Lysophosphatidic Acid Receptor Antagonist for the Treatment of Idiopathic Pulmonary Fibrosis. Chest 2018, 154, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Rosen, G.S.L.; Cheng, P.; Murphy, B.; Chadwick, K.; Lehman-McKeeman, L.; Christian, R.; Gill, M. LPA1 antagonists BMS-986020 and BMS-986234 for idiopathic pulmonary fibrosis: Preclinical evaluation of hepatobiliary homeostasis. Eur. Respir. J. 2017, 50, PA1038. [Google Scholar] [CrossRef]
- Gaire, B.P.; Sapkota, A.; Song, M.R.; Choi, J.W. Lysophosphatidic acid receptor 1 (LPA1) plays critical roles in microglial activation and brain damage after transient focal cerebral ischemia. J. Neuroinflam. 2019, 16, 170. [Google Scholar] [CrossRef]
- Inoue, M.; Rashid, M.H.; Fujita, R.; Contos, J.J.; Chun, J.; Ueda, H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat. Med. 2004, 10, 712–718. [Google Scholar] [CrossRef]
- Kwon, J.H.; Gaire, B.P.; Park, S.J.; Shin, D.Y.; Choi, J.W. Identifying lysophosphatidic acid receptor subtype 1 (LPA1) as a novel factor to modulate microglial activation and their TNF-alpha production by activating ERK1/2. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Santos-Nogueira, E.; Lopez-Serrano, C.; Hernandez, J.; Lago, N.; Astudillo, A.M.; Balsinde, J.; Estivill-Torrus, G.; de Fonseca, F.R.; Chun, J.; Lopez-Vales, R. Activation of Lysophosphatidic Acid Receptor Type 1 Contributes to Pathophysiology of Spinal Cord Injury. J. Neurosci. 2015, 35, 10224–10235. [Google Scholar] [CrossRef]
- Stoddard, N.C.; Chun, J. Promising pharmacological directions in the world of lysophosphatidic Acid signaling. Biomol. Ther. 2015, 23, 1–11. [Google Scholar] [CrossRef]
- Uchida, H.; Nagai, J.; Ueda, H. Lysophosphatidic acid and its receptors LPA1 and LPA3 mediate paclitaxel-induced neuropathic pain in mice. Mol. Pain 2014, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Neyama, H.; Sasaki, K.; Miyama, C.; Iwamoto, R. Lysophosphatidic acid LPA1 and LPA3 receptors play roles in the maintenance of late tissue plasminogen activator-induced central poststroke pain in mice. Neurobiol. Pain 2019, 5, 100020. [Google Scholar] [CrossRef]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Varona, J.F.; Bermejo, F.; Guerra, J.M.; Molina, J.A. Long-term prognosis of ischemic stroke in young adults. Study of 272 cases. J. Neurol. 2004, 251, 1507–1514. [Google Scholar] [CrossRef]
- Katz, N.; Hartman-Maeir, A.; Ring, H.; Soroker, N. Functional disability and rehabilitation outcome in right hemisphere damaged patients with and without unilateral spatial neglect. Arch. Phys. Med. Rehabil. 1999, 80, 379–384. [Google Scholar] [CrossRef]
- Levine, D.A.; Davydow, D.S.; Hough, C.L.; Langa, K.M.; Rogers, M.A.; Iwashyna, T.J. Functional disability and cognitive impairment after hospitalization for myocardial infarction and stroke. Circ. Cardiovasc. Qual. Outcomes 2014, 7, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Yano, R.; Chun, J.; Ueda, H. Involvement of LPA1 receptor signaling in cerebral ischemia-induced neuropathic pain. Neuroscience 2013, 235, 10–15. [Google Scholar] [CrossRef]
- Gaire, B.P.; Lee, C.H.; Sapkota, A.; Lee, S.Y.; Chun, J.; Cho, H.J.; Nam, T.G.; Choi, J.W. Identification of Sphingosine 1-Phosphate Receptor Subtype 1 (S1P1) as a Pathogenic Factor in Transient Focal Cerebral Ischemia. Mol. Neurobiol. 2018, 55, 2320–2332. [Google Scholar] [CrossRef]
- Gaire, B.P.; Song, M.R.; Choi, J.W. Sphingosine 1-phosphate receptor subtype 3 (S1P3) contributes to brain injury after transient focal cerebral ischemia via modulating microglial activation and their M1 polarization. J. Neuroinflamm. 2018, 15, 284. [Google Scholar] [CrossRef] [PubMed]
- Gaire, B.P.; Kwon, O.W.; Park, S.H.; Chun, K.H.; Kim, S.Y.; Shin, D.Y.; Choi, J.W. Neuroprotective effect of 6-paradol in focal cerebral ischemia involves the attenuation of neuroinflammatory responses in activated microglia. PLoS ONE 2015, 10, e0120203. [Google Scholar] [CrossRef]
- Sapkota, A.; Gaire, B.P.; Kang, M.G.; Choi, J.W. S1P2 contributes to microglial activation and M1 polarization following cerebral ischemia through ERK1/2 and JNK. Sci. Rep. 2019, 9, 12106. [Google Scholar] [CrossRef]
- Sapkota, A.; Gaire, B.P.; Cho, K.S.; Jeon, S.J.; Kwon, O.W.; Jang, D.S.; Kim, S.Y.; Ryu, J.H.; Choi, J.W. Eupatilin exerts neuroprotective effects in mice with transient focal cerebral ischemia by reducing microglial activation. PLoS ONE 2017, 12, e0171479. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, A.; Lee, C.H.; Park, S.J.; Choi, J.W. Lysophosphatidic Acid Receptor 5 Plays a Pathogenic Role in Brain Damage after Focal Cerebral Ischemia by Modulating Neuroinflammatory Responses. Cells 2020, 9, 1446. [Google Scholar] [CrossRef]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef]
- Yang, C.; Lafleur, J.; Mwaikambo, B.R.; Zhu, T.; Gagnon, C.; Chemtob, S.; Di Polo, A.; Hardy, P. The role of lysophosphatidic acid receptor (LPA1) in the oxygen-induced retinal ganglion cell degeneration. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1290–1298. [Google Scholar] [CrossRef]
- Arvidsson, A.; Collin, T.; Kirik, D.; Kokaia, Z.; Lindvall, O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 2002, 8, 963–970. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, C.; Ghibaudi, M.; Bonfanti, L. Newly Generated and Non-Newly Generated “Immature” Neurons in the Mammalian Brain: A Possible Reservoir of Young Cells to Prevent Brain Aging and Disease? J. Clin. Med. 2019, 8, 685. [Google Scholar] [CrossRef]
- Dillen, Y.; Kemps, H.; Gervois, P.; Wolfs, E.; Bronckaers, A. Adult Neurogenesis in the Subventricular Zone and Its Regulation After Ischemic Stroke: Implications for Therapeutic Approaches. Transl. Stroke Res. 2020, 11, 60–79. [Google Scholar] [CrossRef]
- Rahman, A.A.; Amruta, N.; Pinteaux, E.; Bix, G.J. Neurogenesis After Stroke: A Therapeutic Perspective. Transl. Stroke Res. 2020. [Google Scholar] [CrossRef]
- Kanazawa, M.; Takahashi, T.; Ishikawa, M.; Onodera, O.; Shimohata, T.; Del Zoppo, G.J. Angiogenesis in the ischemic core: A potential treatment target? J. Cereb. Blood Flow Metab. 2019, 39, 753–769. [Google Scholar] [CrossRef]
- Slevin, M.; Kumar, P.; Gaffney, J.; Kumar, S.; Krupinski, J. Can angiogenesis be exploited to improve stroke outcome? Mechanisms and therapeutic potential. Clin. Sci. 2006, 111, 171–183. [Google Scholar] [CrossRef]
- Choi, J.W.; Chun, J. Lysophospholipids and their receptors in the central nervous system. Biochim. Biophys. Acta 2013, 1831, 20–32. [Google Scholar] [CrossRef]
- Ueda, H. LPA receptor signaling as a therapeutic target for radical treatment of neuropathic pain and fibromyalgia. Pain Manag. 2020, 10, 43–53. [Google Scholar] [CrossRef]
- Xue, J.; Gan, L.; Li, X.; Li, J.; Qi, G.; Wu, Y.; Fu, X.; Mao, Q.; Ao, R.; Lang, J.; et al. Effects of lysophosphatidic acid and its receptors LPA__AMB__frac13; on radiation pneumonitis. Oncol. Rep. 2010, 24, 1515–1520. [Google Scholar]
- Adams, H.P., Jr.; Nudo, R.J. Management of patients with stroke: Is it time to expand treatment options? Ann. Neurol. 2013, 74, 4–10. [Google Scholar] [CrossRef]
- Felling, R.J.; Levison, S.W. Enhanced neurogenesis following stroke. J. Neurosci. Res. 2003, 73, 277–283. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; He, X.; Tang, G.; Tang, Y.H.; Liu, Y.; Lin, X.; Lu, Y.; Yang, G.Y.; Wang, Y. Postacute stromal cell-derived factor-1alpha expression promotes neurovascular recovery in ischemic mice. Stroke 2014, 45, 1822–1829. [Google Scholar] [CrossRef]
- Li, J.; Tang, Y.; Wang, Y.; Tang, R.; Jiang, W.; Yang, G.Y.; Gao, W.Q. Neurovascular recovery via co-transplanted neural and vascular progenitors leads to improved functional restoration after ischemic stroke in rats. Stem Cell Rep. 2014, 3, 101–114. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaire, B.P.; Sapkota, A.; Choi, J.W. BMS-986020, a Specific LPA1 Antagonist, Provides Neuroprotection against Ischemic Stroke in Mice. Antioxidants 2020, 9, 1097. https://doi.org/10.3390/antiox9111097
Gaire BP, Sapkota A, Choi JW. BMS-986020, a Specific LPA1 Antagonist, Provides Neuroprotection against Ischemic Stroke in Mice. Antioxidants. 2020; 9(11):1097. https://doi.org/10.3390/antiox9111097
Chicago/Turabian StyleGaire, Bhakta Prasad, Arjun Sapkota, and Ji Woong Choi. 2020. "BMS-986020, a Specific LPA1 Antagonist, Provides Neuroprotection against Ischemic Stroke in Mice" Antioxidants 9, no. 11: 1097. https://doi.org/10.3390/antiox9111097
APA StyleGaire, B. P., Sapkota, A., & Choi, J. W. (2020). BMS-986020, a Specific LPA1 Antagonist, Provides Neuroprotection against Ischemic Stroke in Mice. Antioxidants, 9(11), 1097. https://doi.org/10.3390/antiox9111097