Susceptibility of Protein Methionine Oxidation in Response to Hydrogen Peroxide Treatment–Ex Vivo Versus In Vitro: A Computational Insight


Abstract

1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Intrinsically Disordered Regions and Protein Abundances
2.3. Empirical Null Distributions
2.4. Analysis of Subcellular Distribution of MetO-Containing Proteins
2.5. Gene Ontology Protein Network Analyses
2.6. Assortativity
2.7. Co-Regulated Protein Pairs Analysis
2.8. MetO Sites Sequence Environments
3. Results and Discussion
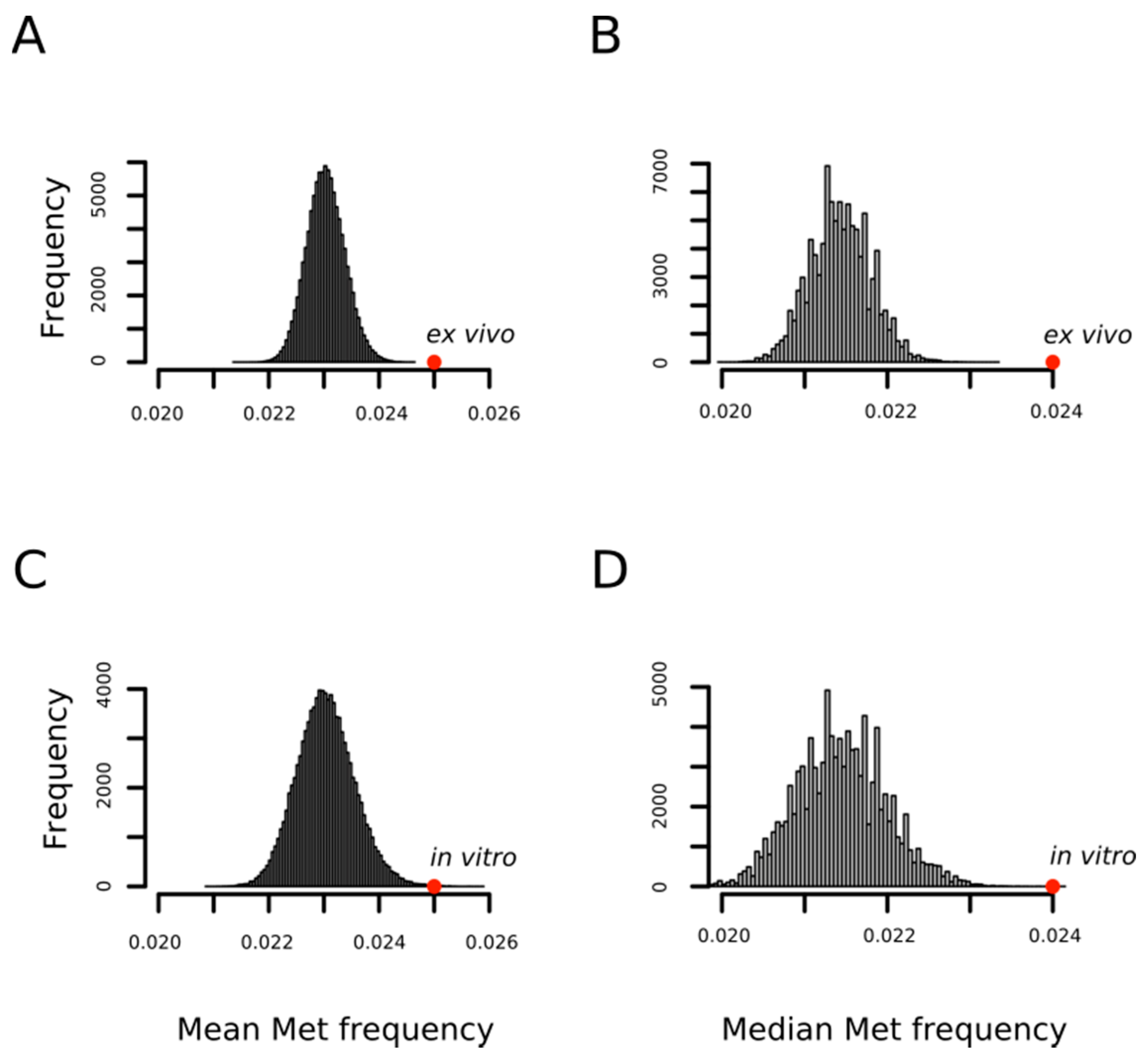
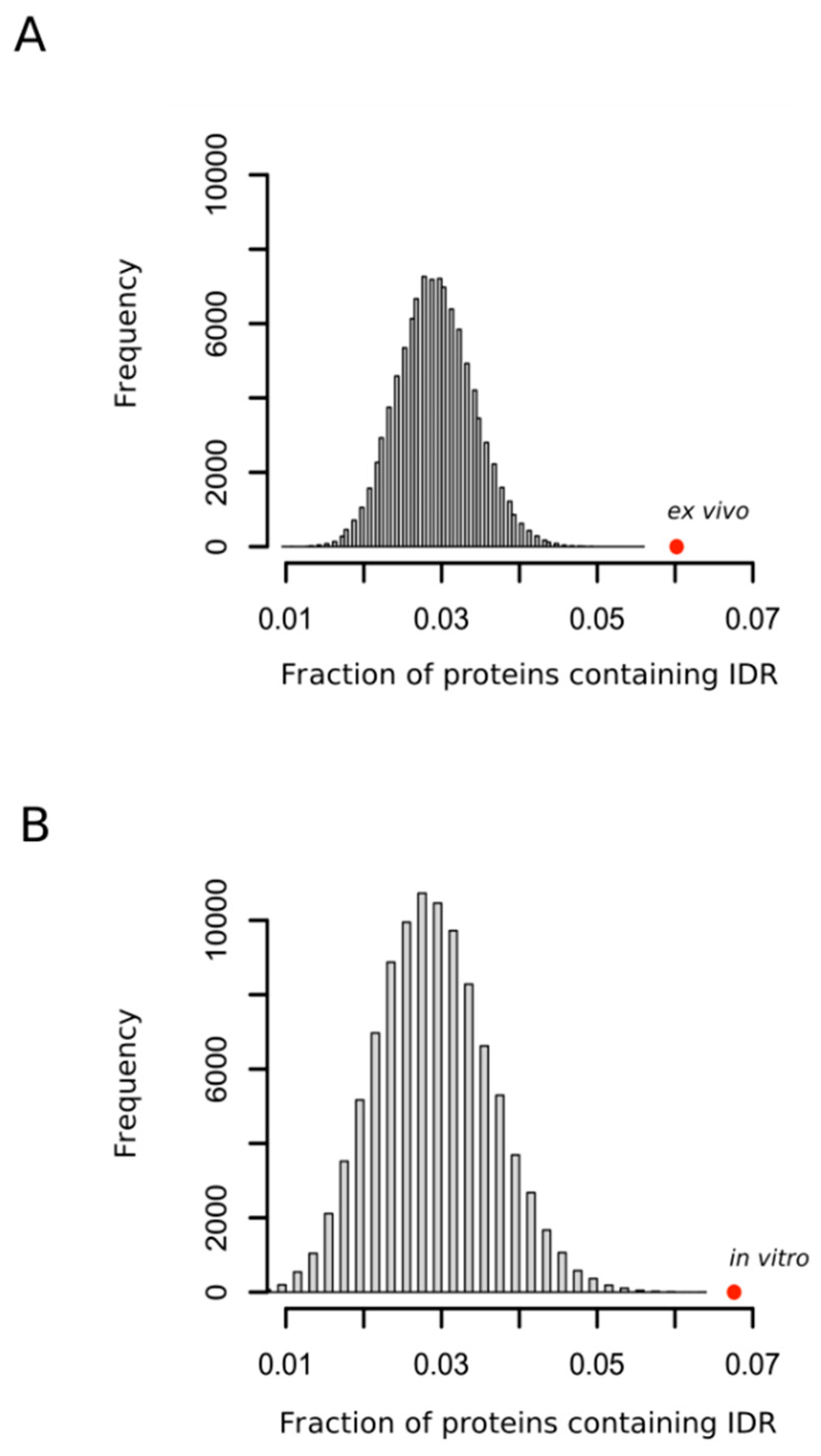
3.1. MetO-Containing Proteins Are Enriched in Methionine Residues and Intrinsically Disordered Regions
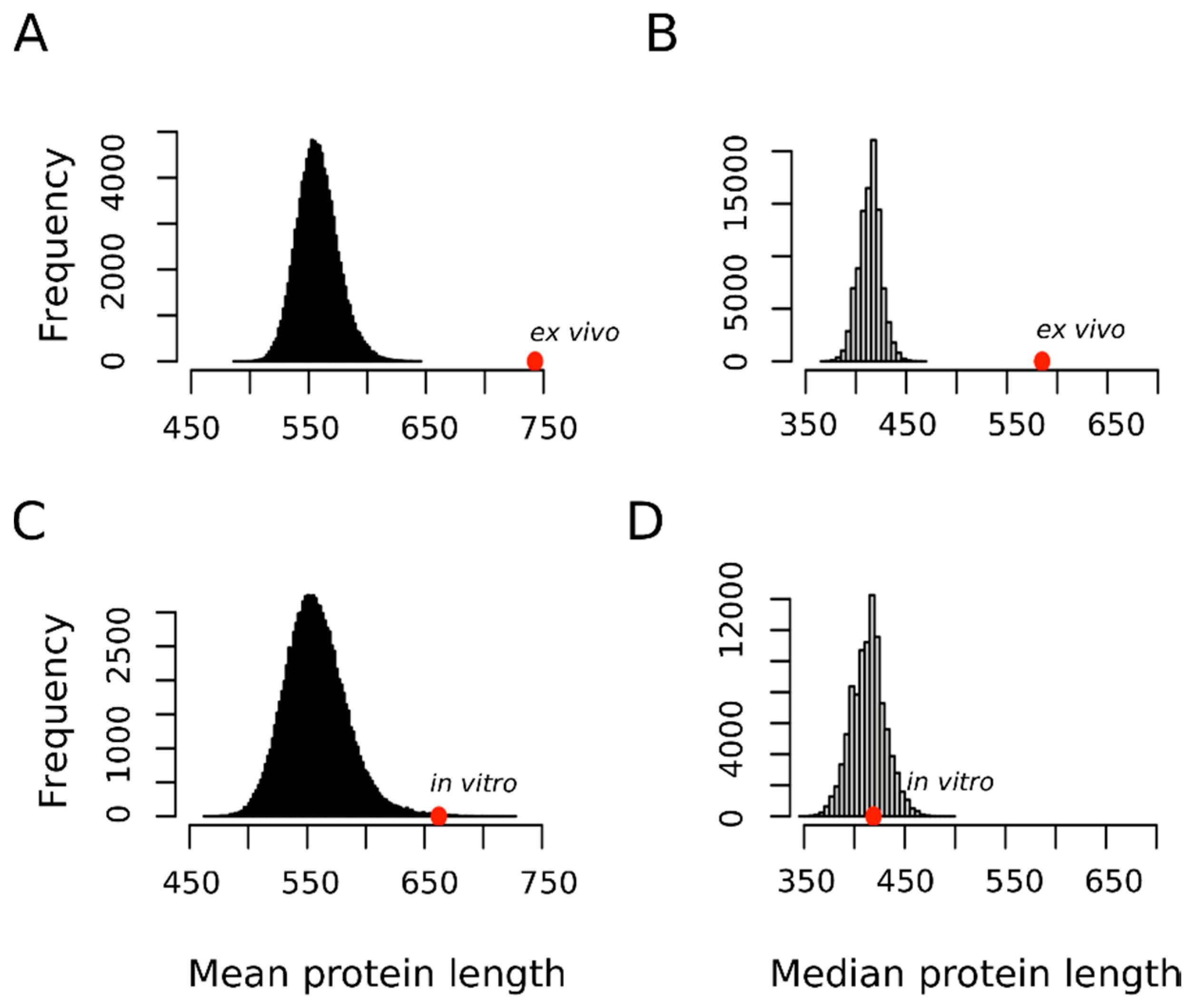
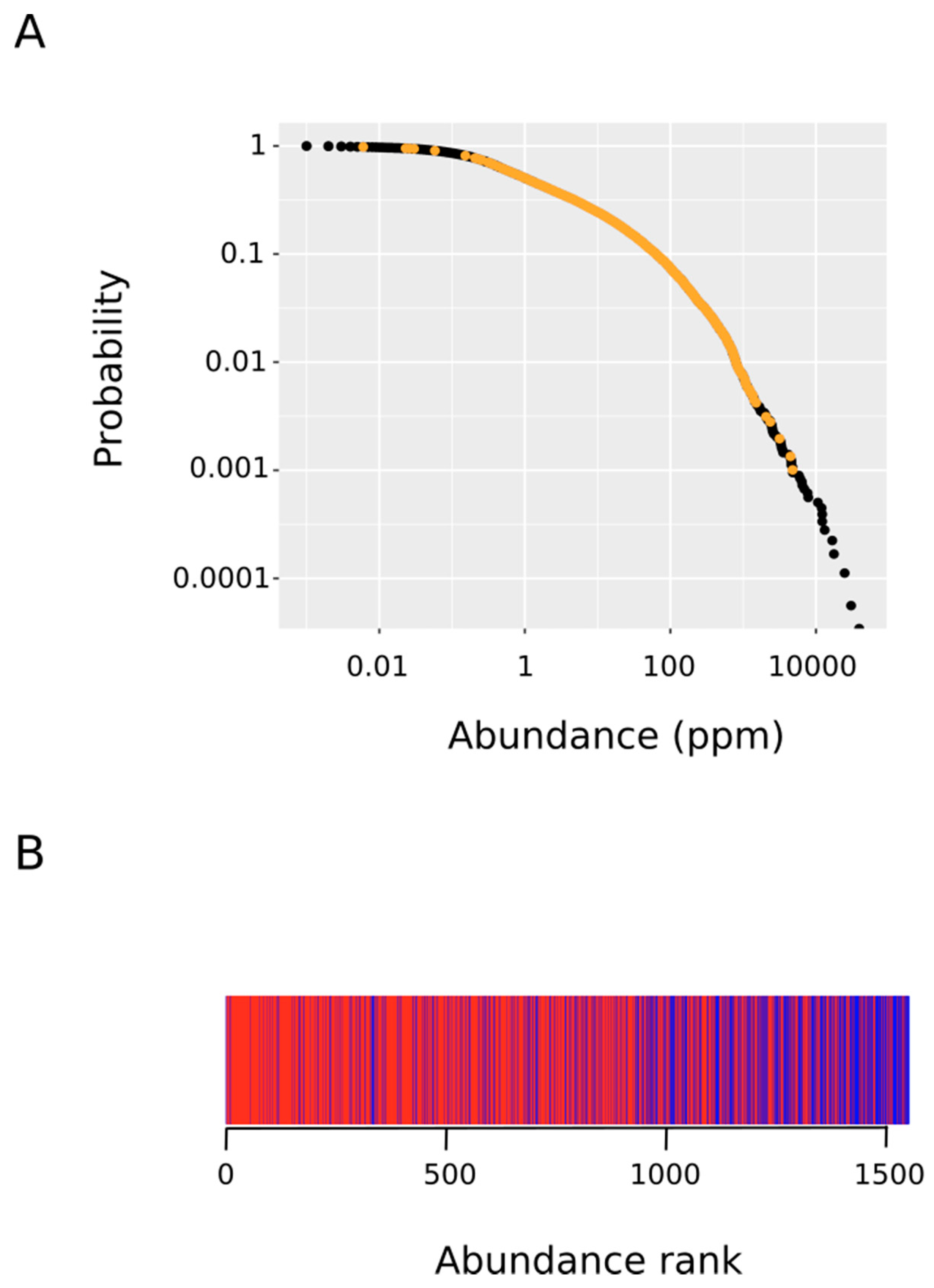
3.2. Proteins Oxidized Ex Vivo Are Bigger and Less Abundant Than Proteins Oxidized In Vitro
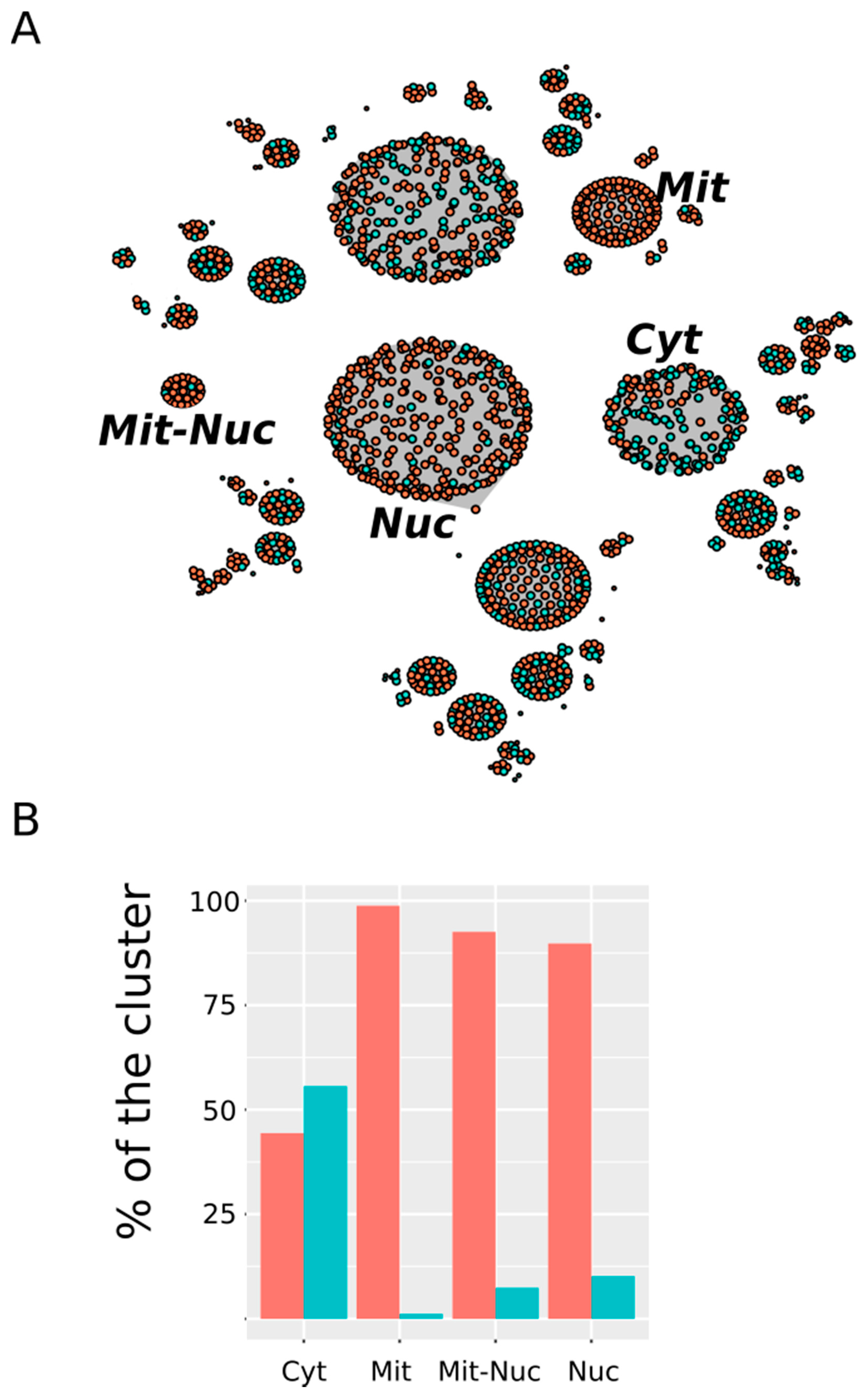
3.3. Nuclear and Mitochondrial Proteins Are Preferentially Oxidized Ex Vivo
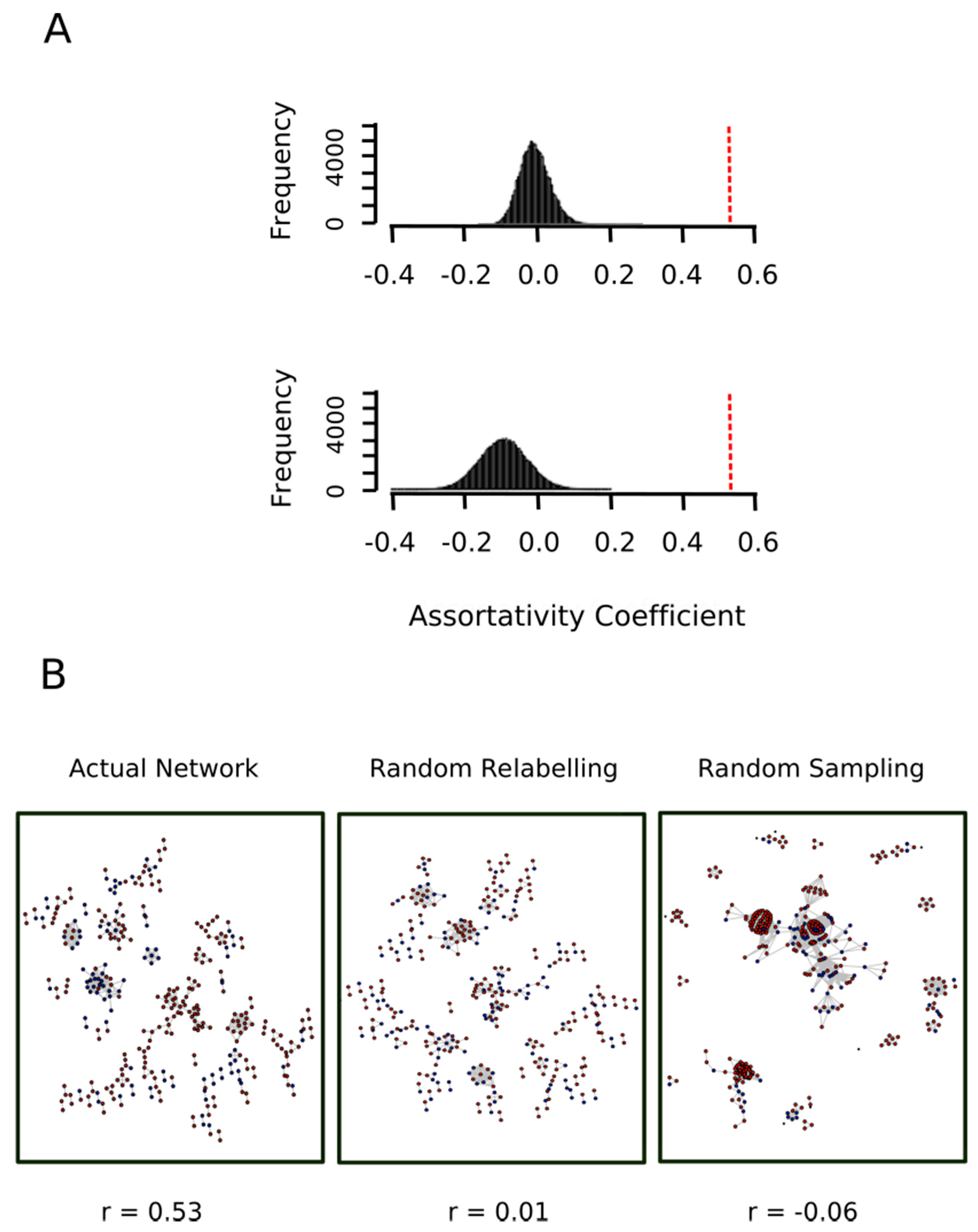
3.4. The Ex Vivo and In Vitro Sets Exhibit Assortative Mixing in GO Protein Networks
3.5. Proteins Oxidized Ex Vivo Are More Often Co-Regulated Than Expected by Chance
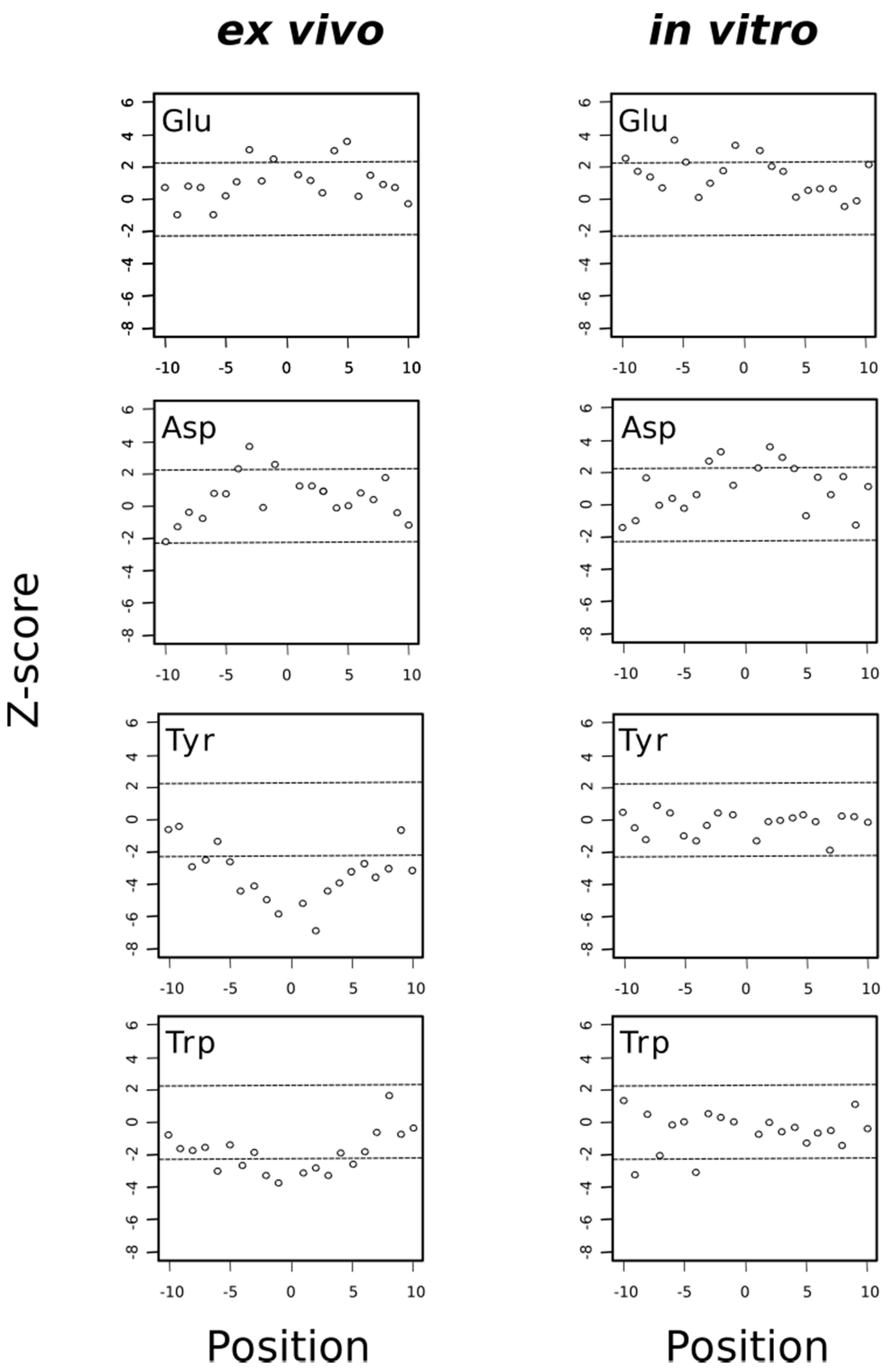
3.6. The Oxidation and Reduction of Methionine Residues Are Sequence Dependent
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aledo, J.C. Life-history constraints on the mechanisms that control the rate of ROS production. Curr. Genom. 2014, 15, 217–230. [Google Scholar] [CrossRef]
- Raymond, J.; Segrè, D. The effect of oxygen on biochemical networks and the evolution of complex life. Science 2006, 311, 1764–1767. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Aledo, J.C. Methionine in proteins: The Cinderella of the proteinogenic amino acids. Protein Sci. 2019, 28, 1785–1796. [Google Scholar] [CrossRef]
- Schöneich, C. Methionine oxidation by reactive oxygen species: Reaction mechanisms and relevance to Alzheimer’s disease. Biochim. Biophys. Acta Proteins Proteom. 2005, 1703, 111–119. [Google Scholar] [CrossRef]
- Vogt, W. Oxidation of methionyl residues in proteins: Tools, targets, and reversal. Free Radic. Biol. Med. 1995, 18, 93–105. [Google Scholar] [CrossRef]
- Zhang, X.H.; Weissbach, H. Origin and evolution of the protein-repairing enzymes methionine sulphoxide reductases. Biol. Rev. 2008, 83, 249–257. [Google Scholar] [CrossRef]
- Achilli, C.; Ciana, A.; Minetti, G. The discovery of methionine sulfoxide reductase enzymes: An historical account and future perspectives. BioFactors 2015, 41, 135–152. [Google Scholar] [CrossRef]
- Tarrago, L.; Grosse, S.; Siponen, M.I.; Lemaire, D.; Alonso, B.; Miotello, G.; Armengaud, J.; Arnoux, P.; Pignol, D.; Sabaty, M. Rhodobacter sphaeroides methionine sulfoxide reductase P reduces R- and S-diastereomers of methionine sulfoxide from a broad-spectrum of protein substrates. Biochem. J. 2018, 475, 3779–3795. [Google Scholar] [CrossRef]
- Drazic, A.; Miura, H.; Peschek, J.; Le, Y.; Bach, N.C.; Kriehuber, T.; Winter, J. Methionine oxidation activates a transcription factor in response to oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 9493–9498. [Google Scholar] [CrossRef]
- Tang, X.D.; Daggett, H.; Hanner, M.; Garcia, M.L.; McManus, O.B.; Brot, N.; Weissbach, H.; Heinemann, S.H.; Hoshi, T. Oxidative regulation of large conductance calcium-activated potassium channels. J. Gen. Physiol. 2001, 117, 253–273. [Google Scholar] [CrossRef]
- Erickson, J.R.; Joiner, M.A.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; Donnell, S.E.O.; Aykin-burns, N.; et al. A dynamic pathway for calcium-Independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of either methionine 351 or methionine 358 in α1-antitrypsin causes loss of anti-neutrophil elastase activity. J. Biol. Chem. 2000, 275, 27258–27265. [Google Scholar]
- Härndahl, U.; Kokke, B.P.A.; Gustavsson, N.; Linse, S.; Berggren, K.; Tjerneld, F.; Boelens, W.C.; Sundby, C. The chaperone-like activity of a small heat shock protein is lost after sulfoxidation of conserved methionines in a surface-exposed amphipathic α-helix. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2001, 1545, 227–237. [Google Scholar] [CrossRef]
- Hardin, S.C.; Larue, C.T.; Oh, M.; Jain, V.; Huber, S.C. Coupling oxidative signals to protein phosphorylation via methionine oxidation in Arabidopsis. Biochem. J. 2009, 422, 305–312. [Google Scholar] [CrossRef]
- Veredas, F.J.; Cantón, F.R.; Aledo, J.C. Methionine residues around phosphorylation sites are preferentially oxidized in vivo under stress conditions. Sci. Rep. 2017, 7, 40403. [Google Scholar] [CrossRef]
- Jiang, B.; Moskovitz, J. The functions of the mammalian methionine sulfoxide reductase system and related diseases. Antioxidants 2018, 7, 122. [Google Scholar] [CrossRef]
- Ghesquière, B.; Jonckheere, V.; Colaert, N.; Van Durme, J.; Timmerman, E.; Goethals, M.; Schymkowitz, J.; Rousseau, F.; Vandekerckhove, J.; Gevaert, K. Redox proteomics of protein-bound methionine oxidation. Mol. Cell. Proteom. 2011, 10, 6866. [Google Scholar] [CrossRef]
- Marondedze, C.; Turek, I.; Parrott, B.; Thomas, L.; Jankovic, B.; Lilley, K.S.; Gehring, C. Structural and functional characteristics of cGMP-dependent methionine oxidation in Arabidopsis thaliana proteins. Cell Commun. Signal. 2013, 11, 1. [Google Scholar] [CrossRef]
- Jacques, S.; Ghesquière, B.; De Bock, P.J.; Demol, H.; Wahni, K.; Willems, P.; Messens, J.; Van Breusegem, F.; Gevaert, K. Protein methionine sulfoxide dynamics in Arabidopsis thaliana under oxidative stress. Mol. Cell. Proteom. 2015, 14, 1217–1229. [Google Scholar] [CrossRef]
- Hsieh, Y.J.; Chien, K.Y.; Yang, I.F.; Lee, I.N.; Wu, C.C.; Huang, T.Y.; Yu, J.S. Oxidation of protein-bound methionine in Photofrin-photodynamic therapy-treated human tumor cells explored by methionine-containing peptide enrichment and quantitative proteomics approach. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Madeira, J.P.; Alpha-Bazin, B.M.; Armengaud, J.; Duport, C. Methionine residues in exoproteins and their recycling by methionine sulfoxide reductase AB serve as an antioxidant strategy in Bacillus cereus. Front. Microbiol. 2017, 8, 1342. [Google Scholar] [CrossRef]
- Madeira, J.P.; Alpha-Bazin, B.; Armengaud, J.; Duport, C. Time-course proteomics dataset to monitor protein-bound methionine oxidation in Bacillus cereus ATCC 14579. Data Br. 2018, 18, 394–398. [Google Scholar] [CrossRef]
- Bettinger, J.Q.; Welle, K.A.; Hryhorenko, J.R.; Ghaemmaghami, S. Quantitative analysis of in vivo methionine oxidation of the human proteome. J. Proteome Res. 2020, 19, 624–633. [Google Scholar] [CrossRef]
- Valverde, H.; Cantón, F.R.; Aledo, J.C. MetOSite: An integrated resource for the study of methionine residues sulfoxidation. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Hatos, A.; Hajdu-Soltész, B.; Monzon, A.M.; Palopoli, N.; Álvarez, L.; Aykac-Fas, B.; Bassot, C.; Benítez, G.I.; Bevilacqua, M.; Chasapi, A.; et al. DisProt: Intrinsic protein disorder annotation in 2020. Nucleic Acids Res. 2020, 48, D269–D276. [Google Scholar] [CrossRef]
- Wang, M.; Herrmann, C.J.; Simonovic, M.; Szklarczyk, D.; von Mering, C. Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 2015, 15, 3163–3168. [Google Scholar] [CrossRef]
- Newman, M.E.J. Mixing patterns in networks. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Top. 2003, 67, 13. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Kustatscher, G.; Grabowski, P.; Schrader, T.A.; Passmore, J.B.; Schrader, M.; Rappsilber, J. Co-regulation map of the human proteome enables identification of protein functions. Nat. Biotechnol. 2019, 37, 1361–1371. [Google Scholar] [CrossRef]
- Aledo, J.C.; Cantón, F.R.; Veredas, F.J. Sulphur atoms from methionines interacting with aromatic residues are less prone to oxidation. Sci. Rep. 2015, 5, 16955. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Uversky, N.V.; Xue, B. Local flexibility facilitates oxidization of buried methionine residues. Protein Pept. Lett. 2012, 19, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Milzani, A.; Rossi, R.; Simplicio, P.D.; Giustarini, D.; Colombo, R.; Dalledonne, I. The oxidation produced by hydrogen peroxide on Ca-ATP-G-actin. Protein Sci. 2000, 9, 1774–1782. [Google Scholar] [CrossRef] [PubMed]
- Aledo, J.C.; Cantón, F.R.; Veredas, F.J. A machine learning approach for predicting methionine oxidation sites. BMC Bioinform. 2017, 18. [Google Scholar] [CrossRef]
- Walker, E.J.; Bettinger, J.Q.; Welle, K.A.; Hryhorenko, J.R.; Ghaemmaghami, S. Global analysis of methionine oxidation provides a census of folding stabilities for the human proteome. Proc. Natl. Acad. Sci. USA 2019, 116, 6081–6090. [Google Scholar] [CrossRef]
- Brocchieri, L.; Karlin, S. Protein length in eukaryotic and prokaryotic proteomes. Nucleic Acids Res. 2005, 33, 3390–3400. [Google Scholar] [CrossRef]
- Bashton, M.; Chothia, C. The generation of new protein functions by the combination of domains. Structure 2007, 15, 85–99. [Google Scholar] [CrossRef]
- Milo, R.; Phillips, R. Cell Biology by the Numbers; Garland Science, Taylor & Francis Group, LLC: New York, NY, USA, 2016; ISBN 9786468600. [Google Scholar]
- Weiss, M.; Schrimpf, S.; Hengartner, M.O.; Lercher, M.J.; Von Mering, C. Shotgun proteomics data from multiple organisms reveals remarkable quantitative conservation of the eukaryotic core proteome. Proteomics 2010, 10, 1297–1306. [Google Scholar] [CrossRef]
- Laurent, J.M.; Vogel, C.; Kwon, T.; Craig, S.A.; Boutz, D.R.; Huse, H.K.; Nozue, K.; Walia, H.; Whiteley, M.; Ronald, P.C.; et al. Protein abundances are more conserved than mRNA abundances across diverse taxa. Proteomics 2010, 10, 4209–4212. [Google Scholar] [CrossRef]
- Vogel, C.; Marcotte, E.M. Insights into regulation of pr1. Vogel, C. & Marcotte, E.M. Insights into regulation of protein abundance from proteomics and transcriptomis analyses. Nat. Rev. Genet. 2013, 13, 227–232. [Google Scholar]
- Souza, J.M.; Daikhin, E.; Yudkoff, M.; Raman, C.S.; Ischiropoulos, H. Factors determining the selectivity of protein tyrosine nitration. Arch. Biochem. Biophys. 1999, 371, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Bayden, A.S.; Yakovlev, V.A.; Graves, P.R.; Mikkelsen, R.B.; Kellogg, G.E. Factors influencing protein tyrosine nitration—Structure-based predictive models. Free Radic. Biol. Med. 2011, 50, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zielonka, J.; Sikora, A.; Joseph, J.; Xu, Y.; Kalyanaraman, B. The effect of neighbouring methionine residue on tyrosine nitration & oxidation in peptides with MPO, H2O2, & NO2- or peroxynitrite and bicarbonate: Role of intramolecular electron-transfer mechanism. Arch. Biochem. Biophys. 2009, 484, 134–145. [Google Scholar] [PubMed]
- Wang, M.; Gao, J.; Müller, P.; Giese, B. Electron transfer in peptides with cysteine and methionine as relay amino acids. Angew. Chem. Int. Ed. 2009, 48, 4232–4234. [Google Scholar] [CrossRef] [PubMed]
- Bobrowskit, K.; Wierzchowskit, K.L.; Holcmanj, J.; Ciurak, M. Intramolecular Electron Transfer in Peptides Containing Methionine, Tryptophan and Tyrosine: A Pulse Radiolysis Study. Institute of Biochemistry and Biophysics; Polish Academy of Sciences: Warsaw, Poland, 1990. [Google Scholar]
- Warren, J.J.; Winkler, J.R.; Gray, H.B. Redox properties of tyrosine and related molecules. FEBS Lett. 2012, 586, 596–602. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subcellular Compartment | GO Cellular Component Term | Abbreviation |
|---|---|---|
| Mitochondrion | GO:0005739 | Mit |
| Nucleus | GO:0005634 | Nuc |
| Lysosome | GO:0005764 | Lys |
| Endoplasmic reticulum | GO:0005783 | ER |
| Plasma membrane | GO:0005886 | Plm |
| Golgi apparatus | GO:0005794 | Gol |
| Endosome | GO:0005768 | End |
| Cytoskeleton | GO:0005856 | Ske |
| Peroxisome | GO:0005777 | Per |
| Cytosol | GO:0005829 | Cyt |
| Ex Vivo | In Vitro | ||
|---|---|---|---|
| Nucleus | 255 | 29 | 284 |
| Others | 858 | 474 | 1332 |
| 1113 | 503 | 1616 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aledo, J.C.; Aledo, P. Susceptibility of Protein Methionine Oxidation in Response to Hydrogen Peroxide Treatment–Ex Vivo Versus In Vitro: A Computational Insight. Antioxidants 2020, 9, 987. https://doi.org/10.3390/antiox9100987
Aledo JC, Aledo P. Susceptibility of Protein Methionine Oxidation in Response to Hydrogen Peroxide Treatment–Ex Vivo Versus In Vitro: A Computational Insight. Antioxidants. 2020; 9(10):987. https://doi.org/10.3390/antiox9100987
Chicago/Turabian StyleAledo, Juan C., and Pablo Aledo. 2020. "Susceptibility of Protein Methionine Oxidation in Response to Hydrogen Peroxide Treatment–Ex Vivo Versus In Vitro: A Computational Insight" Antioxidants 9, no. 10: 987. https://doi.org/10.3390/antiox9100987
APA StyleAledo, J. C., & Aledo, P. (2020). Susceptibility of Protein Methionine Oxidation in Response to Hydrogen Peroxide Treatment–Ex Vivo Versus In Vitro: A Computational Insight. Antioxidants, 9(10), 987. https://doi.org/10.3390/antiox9100987