Abstract
Neuromuscular diseases (NMDs) are a heterogeneous group of acquired or inherited rare disorders caused by injury or dysfunction of the anterior horn cells of the spinal cord (lower motor neurons), peripheral nerves, neuromuscular junctions, or skeletal muscles leading to muscle weakness and waste. Unfortunately, most of them entail serious or even fatal consequences. The prevalence rates among NMDs range between 1 and 10 per 100,000 population, but their rarity and diversity pose difficulties for healthcare and research. Some molecular hallmarks are being explored to elucidate the mechanisms triggering disease, to set the path for further advances. In fact, in the present review we outline the metabolic alterations of NMDs, mainly focusing on the role of mitochondria. The aim of the review is to discuss the mechanisms underlying energy production, oxidative stress generation, cell signaling, autophagy, and inflammation triggered or conditioned by the mitochondria. Briefly, increased levels of inflammation have been linked to reactive oxygen species (ROS) accumulation, which is key in mitochondrial genomic instability and mitochondrial respiratory chain (MRC) dysfunction. ROS burst, impaired autophagy, and increased inflammation are observed in many NMDs. Increasing knowledge of the etiology of NMDs will help to develop better diagnosis and treatments, eventually reducing the health and economic burden of NMDs for patients and healthcare systems.
1. Introduction
1.1. Neuromuscular Diseases (NMDs)
NMDs are a heterogeneous group of acquired or inherited rare disorders caused by injury or dysfunction of the anterior horn cells of the spinal cord (lower motor neurons), peripheral nerves, neuromuscular junctions (NMJ), or skeletal muscles, resulting in muscle weakness and waste, swallowing and breathing difficulties, and cardiac failure [1]. NMDs share some common features: weakness, twitching, torpidity, and cramps but signs and symptoms of various NMDs might be very different [2]. Unfortunately, most of them entail serious or even fatal consequences.
NMDs exhibit a diversity of symptoms due to large genetic and clinical heterogeneity, as more than 500 genes are implicated in their pathogenesis, setting difficulties for healthcare and research [2,3]. Most NMDs’ prevalence ranges between 1 and 10 per 100,000 population [4]. Given the rarity and diversity of NMDs, patients experience long delays in diagnosis (typically 7 years) and 30% are never achieved [1], probably because many of them lack non-invasive diagnostic and prognostic biomarkers. Likewise, models of disease or therapeutic options are infrequent, but growing interests and knowledge are focused in depicting the etiology of these disorders [5].
Currently, diagnosing NMD starts with clinical observation, which can identify atrophy or loss of muscle bulk or tone. Blood tests can determine abnormal levels of various common metabolites and antigens in certain NMD. In addition, electromyography (EMG) helps to assess the health of muscles and lower motor neuron nerve cells that control those muscles to diagnose a number of NMD [6]. Once muscle affection is assured, multitude of diagnostic algorithms are developed to establish the specific disorder. Usually, muscle biopsy takes part in the diagnostic clues. Depending on the clinical and pathological presentation, the study of mitochondrial DNA (mtDNA) genome and mitochondrial respiratory chain (MRC) function is also required [7]. This fact indicates the mitochondrial etiology of some NMDs and why mitochondrial deregulation may play a role in the rest.
Despite the challenges in NMD diagnosis, genetic diagnosis of these diseases has improved over the last 20 years [8]. Advances in sequencing and understanding the human genome has helped to detect monogenic NMD. Improved diagnosis and increased life expectancy may rise the prevalence of NMD worldwide while massive genome sequencing helps to depict complex disease inheritance and etiology. In addition, research in pathways affected in NMD is essential to improve the treatments and guide toward more personalized therapies in NMDs.
Classification of NMDs
The knowledge about new NMDs is continually increasing and changing. Currently, there are 7 main groups of NMDs, represented in Figure 1, which are [6,9,10]:
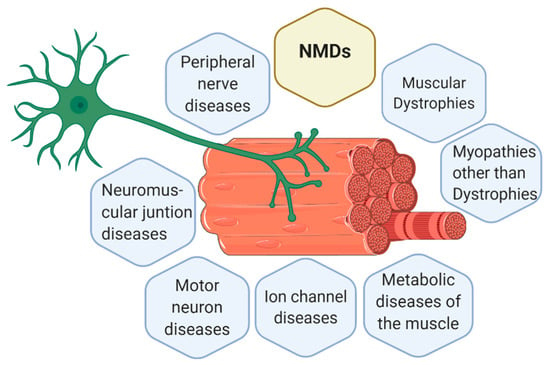
Figure 1.
Representation of a motor unit according to the classification of the main neuromuscular diseases (NMDs). Muscle cells and neurons interact in the neuromuscular junction (NMJ) where muscle cells receive the instructions from the lower motor neurons to perform the actions required. Thus, body movement is actioned by muscle fibers but controlled by the nervous system. Consequently, alterations in muscle fibers or nerves can both trigger a NMD. Figure created with BioRender.com.
- Muscular dystrophies (MD): affect the structure of the muscle cells, causing weakness and degeneration of the skeletal muscles. MD subtypes are described in Table 1.
 Table 1. Examples of studies reporting altered mitochondrial functions in muscular dystrophies.
Table 1. Examples of studies reporting altered mitochondrial functions in muscular dystrophies. - Myopathies other than dystrophies: affect tone and contraction of muscles controlling voluntary movements; may include inflammation of muscles or related tissues, resulting in muscular weakness. Numerous myopathies have been described and classified in different groups (Table 2).
Table 2. Examples of studies reporting altered mitochondrial functions in myopathies other than dystrophies.
- Neuromuscular junction (NMJ) diseases: result from the destruction, dysfunction, or absence of one or more key proteins involved in the transmission of signals between nerves and muscles (Table 3).
Table 3. Examples of studies reporting altered mitochondrial functions in Neuromuscular Junction Diseases (NMJ) and Motor Neuron Diseases.
- Motor neuron diseases: involve nerve cells in the spinal cord (lower motor neurons). Lower motor neurons progressively lose their function, causing the muscles they control to become weak and eventually non-functional (Table 3).
- Peripheral nerve diseases: involve motor and sensory nerves that connect the brain and spinal cord to the rest of the body causing impaired sensations, movement or other functions (Table 4).
Table 4. Examples of studies reporting altered mitochondrial functions in peripheral nerve diseases and mitochondrial diseases.
- Mitochondrial diseases: involve errors in metabolism that affect energy production in muscle cells (Table 4).
- Ion channel diseases: diseases associated with defects in proteins forming ion channels, leading to muscular weakness, absent muscle tone, or episodic muscle paralysis (Table 5).
Table 5. Examples of studies reporting altered mitochondrial functions in ion channel diseases.
1.2. Muscle and Nerve System
As outlined in the classification of NMDs, either muscle or nerve dysfunction can result in a NMD. Both tissues are formed by highly specialized cells; neurons in the peripheral nervous system and muscle cells (organized into sarcomeres) in skeletal muscles. Although nerve and muscle tissues contain a reservoir of stem cells for tissue remodeling and regeneration (called satellite cells in muscles), the rate of cell and tissue renewal is quite low, and neurons and sarcomeres, once formed, never proliferate. This is one of the features that causes nerve and muscle tissue disease in case of cell injury.
One of the reasons for cell injury is the strong dependence that nerve and muscle tissue have on mitochondrial function. Their high energetic needs are almost entirely sustained by oxidative metabolism (high-energy demands provided by mitochondrial oxidative respiration). Thus, any deficiency at mitochondrial level may endanger cell and tissue function, being the base of NMDs.
At structural level, the interaction of nervous and musculoskeletal system occurs in the NMJ, also called motor end plate [54]. Muscles are organized in bundles, called muscle fibers, made up of myocytes (muscle cells). Muscle fibers together with lower motor neurons form the motor unit. To reach motor units, nerve impulses travel from the central nervous system (CNS) to the spinal cord and to the whole body within peripheral nerves [86]. In Figure 1, a motor unit is represented together with the classification of the main NMDs.
Muscle fibers are innervated with motor neuron axon terminals. The communication of muscle cells with lower motor neurons is mainly based in changes in the membrane potential and ion channel opening and closing. Briefly, when lower motor neurons are stimulated, their membrane potential changes due to exchange of sodium (Na+) and potassium (K+) across the cell membrane, prompting the depolarization of the neuron. Inside the neuron, Na+ entrance through specific channels promotes the release of calcium (Ca2+) ions from the endoplasmic reticulum (ER) into the cytosol, triggering the further release of neurotransmitters out of nerve cells. These neurotransmitters are sensed by muscle cells, promoting the contraction of muscle fibers [87]. Muscle contraction is mainly controlled by Ca2+ fluxes.
Ca2+ increase in the cytosol of muscle cells allows actin and myosin (contractile proteins), to interact, causing muscle contraction. Right after, Ca2+ are pumped back into the ER causing muscle relaxation [87]. Overall, body movement is actioned by muscle fibers controlled by the nervous system.
Energetic supply is essential for ion exchange and NMJ functioning. Based on energetic criteria and mitochondrial metabolism, muscle fibers can be divided into two categories: type I (slow-twitch) fibers and type II (fast-twitch) fibers. Type I fibers contain more mitochondria and myoglobin than type II fibers. Mitochondria generate ATP by cellular respiration, for which oxygen is required and provided by myoglobin, which stores and carries oxygen in muscle cells. Type II fibers synthesize the majority of their energy through anaerobic respiration, without the need of oxygen, a faster process than mitochondrial respiration but that provides lower amount of ATP [88].
Overall, the CNS and muscle require high amount of energy, mostly produced in mitochondria, and that is why mitochondrial defects are causal or remarkable in the development of NMDs [14]. As mitochondria are essential in nerve and muscle tissue function, primary mitochondrial diseases, caused by mutations in a mitochondrial gene, generally manifest as encephalomyopathies [89], mainly affecting brain and muscle tissues. This is the case of mitochondrial encephalomyopathy with lactic acidosis syndrome (MELAS) or myoclonic epilepsy with ragged-red fibers (MERRF). NMD such as Charcot-Marie-Tooth neuropathy type 2A (CMT2A) [74] or amyotrophic lateral sclerosis (ALS) can also be caused by mitochondrial mutations (CMT2A can be caused by mutations in MFN2 and ALS by mutations in SOD1) [59]. Interestingly, mitochondria can also play a secondary role in the development of the rest of NMD when the mutation or deficiency is not directly related or located in the mitochondria, since affected cells will need additional ATP to support homeostatic mechanisms disbalanced (anti-stress or antioxidant responses), while minimizing the production of ROS. If mitochondria are unable to counterbalance cell dysfunction, a secondary mitochondrial disease will appear, like spinal muscular atrophy (SMA) (usually caused by a mutation in SMN1) [71] or Duchenne muscular dystrophy (DMD) due to mutation in DMD) [90]. Therefore, mitochondrial function is key in the onset or progression of most of NMDs, regardless of their genetic origin.
1.3. Mitochondrial Function
Despite their clinical and pathological diversity, some overlaps can arise among all NMDs. One of them are molecular events revealing metabolic alterations related to the mitochondria. Although originally considered the “powerhouse of the cell” [91], mitochondria are organelles with a central role in multiple cellular processes: ATP production, apoptosis, β-oxidation of fatty acids, iron-sulfur cluster synthesis and in a broader range of signaling pathways. Additionally, in case of troubleshooting, mitochondria are the main center of ROS production and cell death through the control of apoptosis [92]. Thus, mitochondrial health is essential for cell function and any misbalance in bioenergetics can endanger cell, tissue, and patient survival [93].
Mitochondria can multiply their number in case of energy need or survival of cells, through a process called mitochondrial biogenesis [94]. In addition, mitochondria are dynamic organelles that constantly fuse and divide to increase their energy supply (fusion) or target the mitochondria for degradation (fission). This process is called mitochondrial dynamics and triggers changes in mitochondrial number, morphology, and size [92].
Even if the number of mitochondria rises in a cell, impairment of MRC increases the production of ROS [95], which increases the oxidative stress levels in cells, activating mechanisms to counterbalance this excessive ROS. These repair mechanisms start with autophagy and mitophagy. Autophagy is the clearance of damaged organelles and protein aggregates by lysosomal degradation; while mitophagy is a specialized autophagy that degrades dysfunctional mitochondria to maintain mitochondrial integrity [96]. Both processes remove the excess of ROS produced in mitochondria. Excessive ROS can also affect the ER, as it is one of the main sensors of cellular stress. ER and mitochondria are connected through mitochondria-ER membrane contact sites, called mitochondria-associated membranes (MAMs). When ER senses ROS, ER redox status can change, and might induce ER stress [97]. To defend against ER stress and overcome potential protein misfolding, cells can activate an adaptive unfolded protein response (UPR), which increases the size and capacity of ER to reduce ER stress and increase the removal of misfolded proteins by autophagy [98].
In addition, excessive ROS can trigger an intracellular inflammatory response [99] which has an impact in mitochondrial genomic instability and respiratory chain dysfunction [100]. ROS burst, impaired autophagy, and increased inflammation are observed in many NMDs [101], whose prevalence increases with aging. However, these mechanisms have not been thoroughly revised in all groups of NMD and have not been compared before.
Overall, the aim of this review is to point out the role of excessive oxidative stress in mitochondrial function and its impact in autophagy and inflammation in NMDs (Table 1, Table 2, Table 3, Table 4 and Table 5). Increasing knowledge of the etiology of NMDs will help to develop better diagnosis and treatments to modify the natural history of these chronic disabling diseases, eventually reducing both health and economic burdens of NMDs for patients and healthcare systems [9].
2. Mitochondrial Pathways Altered in NMD
2.1. Mitochondrial Genome and mtDNA Mutations
Mitochondria contain their own genome, which encodes 13 proteins, 22 tRNAs, and 2 rRNAs. The remaining proteins involved in mitochondrial structure and function are encoded by the nuclear DNA, translated in the cytosol and translocated into mitochondria [92]. Although mtDNA only encodes ±1% of the mitochondrial proteins, these elements are critical components of the MRC, essential for mitochondrial function [94]. Nuclear and mitochondrial intergenomic communication is essential to build the MRC complexes that enables cell bioenergetics and promotes healthy organ function [102].
If mtDNA mutates, these mutations can be present in all mtDNA copies in the cell (homoplasmy) or only in a fraction (heteroplasmy). Almost all humans carry low levels of heteroplasmic mtDNA point mutations, which could be pathogenic [103]. Both the subcellular distribution of mutated mtDNA and the intrinsic pathogenicity of the mutation itself may also be important in determining whether a mtDNA defect is expressed phenotypically [7]. mtDNA mutations are also related to the aging process or age-associated diseases [94].
The distribution of a mutation in different tissues may change with time depending on the mitotic activity of the tissue [92]. In tissues with ongoing cell division, there is a chance that a defect in mtDNA may be selected in or out. This may explain the predilection for mitochondrial diseases to manifest clinically in postmitotic tissues such as the central nervous system and skeletal muscle. This mechanism may also explain why some mitochondrial phenotypes change with time [7,104].
Pathogenic mtDNA mutations must be present in 60–90% of mitochondria in a cell to trigger MRC dysfunction [105]. Computational models estimate that only mtDNA mutations generated in early life could expand to reach the threshold and cause MRC dysfunction in aging human adults [106]. One of the reasons is because mtDNA is organized into mitochondrial nucleoids [107] and coupled with mitochondrial transcription factor A (TFAM), to make mtDNA less accessible to mutations than naked DNA. In fact, lower amount of TFAM is found in hypothyroid myopathy (endocrine myopathy, Table 2), triggering reduced mtDNA copy number and mitochondrial dysfunction [38].
Other NMDs associated to mtDNA mutations are metabolic myopathies, ALS, CMT2, mitochondrial myopathies, and Friedreich’s ataxia (Table 2, Table 3 and Table 4).
Apart from mtDNA point mutations, single large deletion in mtDNA at high levels cause multisystem diseases in children and NMDs associated with chronic progressive external ophthalmoplegia in adults and proximal myopathy [76,79]. Other NMDs with mtDNA deletions are sporadic inclusion body myositis (sIBM) (reported in 67% of sIBM patients) [108,109] and giant axonal neuropathy (GAN). These deletions are generally spontaneously acquired [105], probably as accidents during mtDNA replication in the oocyte, in the early embryo or later on, along lifetime. Some multiple large mtDNA deletions are inherited, as they are caused by mutations in nuclear genes involved in mtDNA replication, nucleotide synthesis or transport [92]. Spontaneous mtDNA deletions appear to accumulate with time, and may increase the likelihood that tissue function will be impaired with time [7].
Above all, mtDNA variants (mutations and deletions) have a broad range of impact, from the ones that are causal of monogenic disorders to those that are a risky allele for complex diseases, in which clinical penetrance also depend on environmental factors. For example, de novo mtDNA monogenic mutations m.3243A > G and m.8344A > G trigger MELAS and MERRF, respectively, and can arise spontaneously, because of replication errors. Another high impact variant is a large 4,977-bp deletion of mtDNA, the most common cause of Kearns–Sayre syndrome. These mutations usually occur de novo within two or three human generations and, for deletions, tend to affect only one generation [104]. In all cases, the specific mutation, the level of heteroplasmy, the threshold effect that determines the level of phenotypic penetrance and which tissues are affected, directly determine the inheritance of the disease and the onset and progression of these disorders.
2.2. Mitochondrial Respiratory Chain (MRC)
The MRC contains five multimeric enzymatic complexes. Complexes I–IV oxidize and transfer their electrons, in the form of hydrogen ions, to the next complex. Once in complex IV, electrons are transferred to molecular oxygen, producing water [7,95]. Complex V generates ATP from adenosine diphosphate (ADP) using the transmembrane electrical potential generated by proton translocation along the MRC [14]. This production of ATP from the reduction of oxygen, known as mitochondrial coupling, is what generates the energy needed for cellular function [110]. In parallel, MRC generates ROS as a subproduct of oxygen metabolism, especially in case of disease. Mutations in mtDNA can trigger the impairment of MRC, affecting energy production and cellular dysfunction [111], observed in some NMDs and mitochondrial diseases [112]. mtDNA mutates more than ten times faster than nuclear DNA, and this high mutation rate in mtDNA is based on the proximity of mtDNA to the inner mitochondrial membrane, where ROS are generated as by-products of MRC chain, coupled with a lack of histones and efficient DNA repair mechanisms. In addition, mtDNA has low number of noncoding regions between genes, leading to random mutations that impact coding sequences of genes responsible of MRC.
In particular, most of the muscular dystrophies (MD) have some MRC complexes with impaired or decreased function (Table 1): complex I in Becker MD (BMD), complexes III and IV in DMD, and complexes I and V in oculopharyngeal (OPMD) and in distal MD (DD). Other NMDs with MRC alterations are myofibrillar myopathies (complexes I and IV) (Table 2), ALS (CIV subunit I) (Table 3), SMA (complexes I and IV) (Table 3), GAN (complexes I and IV) (Table 4), and sIBM (complex IV) [108]. Impaired MRC function promotes mitochondrial dysfunction, eventually leading to progressive weakness and muscle wasting.
To understand the extent of the how MRC dysfunction affects the progression of a NMDs or vice versa, here we present the case of hypothyroid myopathy (endocrine myopathy, Table 2) and ALS (motor neuron diseases, Table 3).
In hypothyroid myopathy, there is an abnormal activity of the thyroid gland, leading to reduced hormone levels. Thyroid hormones affect skeletal muscle through T3 receptors, located on the mitochondrial membrane of skeletal muscle fibers [113,114]. This MRC-thyroid hormones connection is mediated by several molecular mechanisms that include both direct and indirect effects on mitochondrial structure, function, and biogenesis. One effect in hypothyroid myopathy is a reduced mitochondrial oxidative metabolism, due to decreased hormonal levels and a reduced amount and activity of complex IV (cytochrome c oxidase negative fibers (or COX-)). In addition, T3 receptors, together with the thyroid hormone receptor, increase the expression of some nuclear-encoded respiratory genes, such as cytochrome c1 and b-F1- ATPase subunit genes [38].
Regarding ALS, ALS-associated P525L mutant interacts with ATP synthase complex subunit TP5B, disrupting the formation of ATP synthase complex, thus inhibiting the production of ATP [115]. Both are examples of the complex and intricated pathways governing NMDs in which mitochondria, and herein explained, MRC, play a role.
2.3. Mitochondrial Quality Control
2.3.1. Mitochondrial Biogenesis
Mitochondrial biogenesis increases the number of mitochondria, what can lead to a higher energy supply and survival of cells; or if mutated mtDNA expands to higher levels, low energy supply, and cell death. Both outcomes depend on random clonal expansion of mitochondria [103]. Although mitochondrial biogenesis is energy-consuming, it helps to increase energy production and often restores mitochondrial function. Mitochondrial biogenesis can react to many external stimuli, such as exercise, hormones, and possibly dietary restriction, in addition to mitochondrial dysfunction [92]. In the case of mitochondrial dysfunction, coordination between mitochondrial synthesis (mitochondrial biogenesis) and degradation (mitophagy) regulates mitochondrial content, quality, and function.
To initiate mitochondrial biogenesis, synthesis of more MRC complexes is required. MRC components are both encoded in nuclear and mtDNA, although their transcripts are not synthesized simultaneously. Cytosolic translation unidirectionally controls mitochondrial translation, both orchestrated by the nuclear genome. The nuclear genes coding for the MRC are rapidly induced under nutrient shift, whereas mitochondrial genes coding for the MRC are induced more slowly. Mitochondrial MRC’s slow kinetics may underlie the lack of environment-responsive mitochondrial transcription factors. Instead of transcription factors, mitochondria contain mRNA-specific translational activators, involved in initiation, elongation, and feedback control of MRC complexes assembly [102].
If mitochondrial biogenesis is disrupted, it may result in mitochondrial dysfunction and loss of respiratory capacity [103], observed in patients with mitochondrial diseases. However, excessive mitochondrial biogenesis can also be a sign of disease. In fact, in the muscle of patients with mitochondrial disease, mitochondrial biogenesis generates the so-called ragged-red fibers (RRF), an increase in the number of mitochondria surrounding muscle fibers aimed to compensate mitochondrial dysfunction (Figure 2). RRF typically loss COX activity (partially encoded in the mitochondrial genome) and increase complex II function (succinate dehydrogenase, SDH, entirely encoded in the nuclear genome). Thus, RRF mainly accumulate abnormal proliferated mitochondria [94].
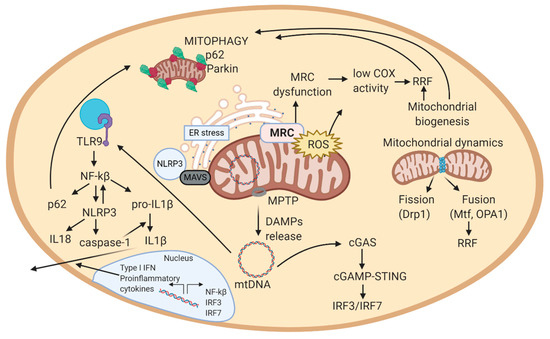
Figure 2.
Schematic view of the mechanistic model of mitochondrial-related pathways in a cell. 1. MRC generates ATP in the inner mitochondrial membrane. In this process, also ROS are produced. 2. In case of excessive ROS levels, mitochondrial membrane potential decreases, allowing MPTP to open and release DAMPs (mtDNA, cardiolipin, ceramides) to the cytosol. These DAMPs can trigger an inflammatory response. In this scheme, the inflammatory cascade represented is the one activated by mtDNA. mtDNA activates cGAS in cytosol, that eventually induces the expression of interferon-related genes (IRF3/IRF7). In parallel, mtDNA activates TLR9, which activates NF-kB, and thus, NLRP3 inflammasome, expression of pro-inflammatory cytokines, and among them, IL-1 response. 3. Increased ROS levels can result from MRC dysfunction. To compensate dysfunctional mitochondria, mitochondrial biogenesis increases the number of mitochondria in a cell. Excessive mitochondria around muscle cells form RRF. 4. Excessive ROS, RRF, inflammation, and ER stress damage mitochondria, which alterations are sensed by autophagy and mitophagy. Autophagy and mitophagy remove damaged mitochondria, and consequently stop the inflammatory response, supporting repair mechanisms in the cell. If autophagy and mitophagy are altered, mitochondria cannot be recycled, leading to apoptosis. Abbreviations: MRC: mitochondrial respiratory chain; ROS: reactive oxygen species; MPTP: mitochondrial permeability transition pore; DAMPs: damage-associated molecular patterns; mtDNA: mitochondrial DNA; cGAS: cyclic GMP-AMP synthase; TLR9: toll-like receptor 9; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3: NOD-like receptor protein 3; RRF: ragged-red fibers; ER: endoplasmic reticulum; MAVS: mitochondrial antiviral signaling protein. Figure created with BioRender.com.
Alterations in some pathways may trigger accumulation of mitochondria in RRF (e.g., changes in mitochondrial protein synthesis). In addition, RRF usually contain a higher amount of mutant mtDNA genomes than non-RRF. Overall, RRF are a tag of defective oxidative phosphorylation in the affected muscle fibers [7]. In fact, RRF are found in many NMDs like limb-girdle MD (LGMD), sIBM, myofibrillar myopathies, myasthenia gravis, ALS, and mitochondrial myopathies (Table 1, Table 2, Table 3 and Table 4).
2.3.2. Mitochondrial Dynamics
Mitochondria are highly dynamic organelles that continuously fuse, divide, and move, and mitochondrial function is controlled and maintained by these morphologic changes. Mitochondrial fission is mainly mediated by dynamin-related guanosine triphosphatase (GTPase) protein 1 (Drp1); while in mitochondrial fusion, the outer and inner mitochondrial membranes primarily fuse mediated by dynamin-related GTPases mitofusin (Mfn 1 and 2) and optic atrophy 1 (OPA1), respectively [103]. The most direct consequence of mitochondrial division and fusion is the change in size of the mitochondria [105]. Interestingly, these processes promote the exchange of genetic and structural material among participant mitochondria, thus enabling the renewal of damaged components, or its expansion throughout the mitochondrial net (Figure 2). However, mitochondria can also increase in number when there are the specific requirements.
When mitochondria are damaged, they undergo changes in mitochondrial membrane potential (MMP) causing depolarization. Altered MMP can also impact the mitochondrial fission/fusion machinery and thereby mitochondrial morphology. Mitochondrial fission generates fragmented mitochondria with increased ROS production [106], which may help in the separation of dysfunctional mitochondria from healthy mitochondria and is a prerequisite for mitochondrial degradation (in a process called mitophagy) [107]. The detrimental effect of mitochondrial fission is the release of cytochrome c from mitochondria, which could activate apoptosis signaling through activation of Bax/Bak pore [103]. Apoptotic cell death may be an important mediator of nervous system injury in the mitochondrial encephalomyopathies [7]. Other NMD with increased apoptosis are OPMD, spinal-bulbar muscular atrophy (SBMA), and ion channel diseases. Defects in ion channel diseases may directly reduce MMP and delay repolarization, affecting the modulation of cell survival and increasing cell apoptosis.
Mitochondrial dynamics is also implicated in the pathogenesis of NMDs like inherited peripheral neuropathy, GAN [76], infantile-onset encephalopathy [116], autosomal dominant optic atrophy [101], and CMT2A [74]. The last three are characterized by mutations involving mitochondrial fusion regulatory genes, OPA1 and MFN2, respectively [117]. Some mutations in mitochondrial dynamics genes can trigger severe effects or even death. An example is a lethal dominant negative allele of DRP1 that affects mitochondrial and peroxisomes fission in newborns and triggers abnormalities in brain development and optic atrophy [118].
Mitochondrial dynamics have also been thoroughly studied in ALS. As mutations in several genes are responsible of ALS, depending on the gene mutated, fusion or fission of mitochondria are enhanced. For example, TDP-43 mutations enhance Mitochondrial fission 1 protein (Fis 1) expression, which increases DRP1 recruitment, leading to higher mitochondrial fission, and a fragmented morphology of mitochondria. In ALS caused by SOD1, increased DRP1 and reduced Mfn1/Opa1 lead to higher level of mitochondrial fission and a fragmented mitochondrial network; while in ALS related to C9ORF72 mutation, higher Mfn1 levels triggered increased mitochondrial fusion and an elongated morphology [119]. Thus, the deregulation of mitochondrial dynamics is frequently associated to NMDs and its proper function protects and counteracts the progression of some NMDs.
2.3.3. Autophagy and Mitophagy
Lysosomes were discovered in 1955 [109], and soon was observed that cytoplasmic components could be located in lysosomes [120]. This was the basis for De Duve (1963) to coin the term autophagy [121]. Autophagy is a lysosomal degradation process characterized by the formation of double-membrane autophagosomes from cytoplasmic material and damaged organelles. Autophagosomes fuse with lysosomes to degrade their content by acidic hydrolases. Autophagic degradation generates amino acids and fatty acids for protein synthesis or oxidation in the MRC to produce ATP for cell survival under starvation conditions. Other functions of autophagy include removal of damaged organelles and protein aggregates, control of cellular biomass and elimination of intracellular pathogens [92]. There are three subtypes of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy, divided according to the mechanism used to send intracellular material to be degraded in the lysosomes and the kind of cargo for degradation.
Briefly, mitophagy—the specific autophagy of mitochondria—initiates with the recognition of damaged mitochondria by the E3-ubiquitin ligase Parkin, which decorates the outer mitochondrial membrane proteins with poly-Ub chains [122]. p62 binds polyubiquitinated proteins and damaged organelles and target them to autophagosomal clearance via its ubiquitin association domain (UBA) and LC3 binding motif (LIR), respectively [123]. Absence of specific mitophagy proteins can block autophagosomal or lysosomal clearance leading to an accumulation of damaged proteins and dysfunctional organelles in autophagosomes [124,125].
Mitophagy can occur under several circumstances: metabolic alterations (starvation, particularly), differentiation of some cell types (requiring mitochondrial clearance), and selective targeting and removal of dysfunctional mitochondria [126]. Mitophagy of dysfunctional mitochondria is also called PTEN-induced putative kinase 1 (PINK1)-Parkin-mediated mitochondrial quality-control system [92], since both PTEN and PINK1 are among the main effectors of mitophagy.
The extent of mitophagy varies in different tissues. For instance, heart, skeletal muscle, nervous system, hepatic and renal tissue have higher mitophagy than thymus and spleen [127]. Defects in mitophagy can lead to accumulation of dysfunctional mitochondria and increased levels of mitochondrial ROS and damage-associated molecular pattern (DAMPs), which are self-molecules that resemble pathogenic ones and can trigger an inflammatory response (Figure 2). Both mitochondrial ROS and DAMPs can initiate an inflammatory response [128]. In addition, impaired mitophagy has been linked with aging, metabolic disorders, cancer, senescence, inflammation, and genomic instability [107]. Regarding NMDs, impaired autophagy has been revealed in congenital MD (CMD), endocrine myopathies, inflammatory myopathies, myofibrillar myopathy and congenital myasthenic syndromes, among others.
In CMD linked to collagen VI deficiency, impaired autophagy is responsible for spontaneous apoptosis, dysfunctional mitochondria and myofibers degeneration. Defective autophagy causes accumulation of abnormal organelles and apoptotic degeneration of muscle fibers. Accumulation of defective mitochondria increases oxidative stress and ROS production, which also contribute to increase apoptosis. The cause of defective autophagy in CMD is a reduced protein amount of beclin-1 and Bnip3, what leads to a defective activation of the autophagy process [15].
As mitophagy protects from the accumulation of defective mitochondria and ROS overproduction, this process is frequently impaired in NMDs affecting both nerve and muscle function.
2.4. Mitochondrial ROS and ER Stress
ROS comprises superoxide anion (O2•−), hydrogen peroxide (H2O2), hydroxyl radical (OH•), peroxyl radical (ROO•), and nitric oxide (NO•). Although mitochondrial oxidative metabolism is a major source of ROS in many cell types, outside mitochondria there are other enzymes associated with ROS production, like NADPH oxidase, neural nitric oxide synthase, and monoamine oxidase [129]. A balance between oxidative species and antioxidant defense mechanisms is required for cell homeostasis [130]. Briefly, the antioxidant response is mediated by enzymes responsible for metabolizing or neutralizing ROS, such as catalase, glutathione peroxidase, or superoxide dismutase (SODs) [131].
The high proportion of ROS generation in MRC is due to the transfer of electrons in the inner mitochondrial membrane until they encounter oxygen, when electrons are converted in water. Under hypoxia or pathologic conditions this process is not completed, resulting in an increase of superoxide anions [131]. For example, mutation in SOD2, an antioxidant enzyme encoded in the mitochondrial genome, triggers the development of CMT, a NMD [132]. Mutations in SOD1 gene are found in ALS. SOD1 encodes the Cu-Zn superoxide dismutase, one of three proteins involved in the conversion of free superoxide radicals to molecular oxygen and hydrogen peroxide. Mutations in the SOD1 gene are found in 10–20% of familial ALS cases and 1–5% of sporadic ALS cases globally [133]; so far, more than 170 mutations of the SOD1 gene are known in ALS [119].
Other NMDs in which high ROS levels are not compensated and cells hold excessive oxidative stress are BMD, DMD, FSHD, ALS, SBMA, mitochondrial myopathies, and FA.
When overproduction of ROS cannot be compensated by antioxidant defenses or mitophagy, ER receives ROS signals through MAMs, which may eventually activate cell stress responses to compensate and overcome this adverse situation [134].
ER is the main organelle in charge of protein biosynthesis and folding and one of the main cell sensors to stress insults. Alterations of the ER redox status can negatively affect protein folding and result in ER stress. ER stress occurs when the rate at which new protein entering the ER exceeds its folding capacity, which can lead to the activation of three transmembrane proteins: inositol-requiring enzyme 1α (IRE1α), PKR-like ER kinase (PERK), and the activation transcription factor 6 (ATF6), which altogether trigger a UPR [134]. The UPR is comprised of a series of transcriptional, translational, and post-translational processes that can lower the rate of protein synthesis and increase the protein folding capacity of the ER machinery. This results in an increase of misfolded protein elimination, and escalating the size of the ER compartment thereby reducing the ER stress [98]. Usually, transient ER stress can be overcome by UPR, but if the damaging stimulus persists, inflammatory response genes are activated [131].
ER-mitochondria crosstalk is involved in several pathways including cell proliferation, apoptosis, autophagy, lipid metabolism, Ca2+ signaling, UPR, inflammation, and bioenergetics. Alterations in this ER-mitochondria crosstalk are associated with multiple diseases, like motor neuron diseases, myotonic dystrophy, OPMD, endocrine myopathies, myofibrillar myopathies, SBMA, CMT, and mitochondrial myopathies. In ALS, over expression of both wild-type and mutated TDP-43 leads to a decrease in ER-mitochondria physical and functional coupling, affecting Ca2+ regulation. Ca2+ dysregulation is considered to be a primary cause of motor neuron death in ALS [135]. Ca2+ is responsible for the link between metabolic impairment and MAMs interaction. This is seen in cancer cells, which are able to remodel their intracellular Ca2+ signaling, enhancing their survival and proliferation. Modulation of ER-mitochondrial Ca2+ crosstalk favors resistance to apoptosis [136].
In addition to triggering ER stress, if ROS damage overwhelms these repair mechanisms, MMP may decrease, thus potentially leading to the increase in the permeability of mitochondrial membranes. Higher permeability in mitochondrial membrane can activate the mitochondrial permeability transition pore (MPTP) and thus, the release of DAMPs to the cytosol (mtDNA), ceramides, formaldehyde) [137,138]. MPTP and ROS can also promote the activation of mitochondrial fission and mitophagy to eliminate damaged mitochondria (Figure 2). In mitochondrial fission, MAMs signal the ER tubules to encircle mitochondria and tag the sites for mitochondrial division [139]. Accordingly to mitochondrial implication in NMDs, dysregulation of MPTP opening and defective autophagy have been associated with muscular dystrophies, like collagen VI myopathies (CMD), BMD, DMD, LGMD, and DD [14,15] and, remarkably, most of NMDs show some level of deregulation in most of these pathways.
2.5. Mitochondrially Induced Inflammatory Response
Inflammatory responses recognize pathogen-associated molecular patterns (PAMPs), derived from infection, through a variety of receptors. However, because of the bacterial origin of mitochondria, their proteins are structurally similar to those in bacteria and enable their recognition by the same receptors of the immune system, reinforcing the notion of mitochondria as hubs of immunity [140]. Thus, mitochondrial DAMPs can also trigger an inflammatory response [128]. DAMPs include proteins and peptides, such as N-formyl peptides and TFAM, as well as lipids, and metabolites such as cardiolipin, succinate and ATP, and mtDNA [139].
2.5.1. Implication of mtDNA in Inflammation
In particular, mtDNA is recognized by multiple innate immune receptors: cytosolic cyclic GMP-AMP synthase (cGAS), endosomal Toll-like receptor 9 (TLR9), and NOD, LRR, and Pyrin domain-containing protein 3 (NLRP3) [140]. Here we briefly remark the main effects of these innate immune responses.
Cytosolic mtDNA enhances cytosolic antiviral signaling and expression of interferon-stimulated genes (IRF3/IRF7), which results in the activation of cGAS DNA sensor and STING-IRF3-dependent signaling [141]. These pathways activate transcription factors NF- kB and IRF3 through the kinases IKK and TBK1, respectively. MtDNA stress triggered by TFAM deficiency has been reported to release mtDNA to the cytosol [101].
TLR9 is a nucleic acid sensor that binds to unmethylated CpG DNA, like mtDNA. In basal conditions, TLR9 is located in the ER, but when it is stimulated, TLR9 translocates to the membrane of endosomes or lysosomes, where it binds to ligands and triggers cell inflammation [140].
TLR9 interaction with mtDNA occurs in the endolysosomal compartment and activates the myeloid differentiation primary response protein 88 (MyD88), which induces a number of kinases and transcriptional factors, namely mitogen-activated protein kinases (MAPK) [101]. MAPK leads to nuclear factor-kB (NF-κB) and IRF7 activation to enhance pro-inflammatory and interferon type I (IFN-1) responses, respectively [89]. NF-kB signaling increases the expression of other pro-inflammatory cytokines such as tumor necrosis factor-a (TNF-a), interleukin (IL)-6, IL-1b [128]. Furthermore, TLR9 senses incomplete digestion in lysosomes of damaged mitochondria, because of impaired mitophagy, without the mtDNA release to cytosol.
The activation of the inflammasome NLRP3 involves two sequential signals. The inflammasome priming and assembly signal is induced by NF-κB, which acts downstream of TLRs and other immune receptors [142]. NF-kB activates the expression of inflammasome components and inactive forms of the cytokines, including pro-IL-1b. When active, NLRP3 inflammasome activates caspase-1, which processes pro-IL-1β and pro-IL-18 [100,143]. DAMPs directly activate NLRP3 inflammasome as well. Mitochondria are suggested to regulate the activity of the NLRP3 inflammasome complex, by: activating mitophagy (reduces inflammation by clearing mitochondrial-bound NLRP3 complexes) and releasing mitochondrial ROS (amplifies inflammasome immunogenic signal) [140].
In normal conditions, NLRP3 protein is located on the ER. Under oxidative stress and in response to inflammation, NLRP3 can be translocated to MAMs. MAMs transmit danger signals through physical interaction, promoting coordinated responses to oxidative stress, such as triggering mitophagy to remove damaged mitochondria [131].
Altogether, mitochondria-induced inflammation usually is initiated by mitochondrial ROS, which can cause the activation of MAPKs by inhibiting MAPK phosphatase. Activated MAPK may aid in the production of IL-6 and TNF. Mitochondrial ROS accumulation can also activate NLRP3 inflammasome, which promotes the maturation of IL-1β and IL-18. mtDNA accumulation in the cytosol can interact with TLR-9 (in lysosomes) to induce inflammatory responses (e.g., in autosomal dominant optic atrophy [101]. This pathologic feature has been described in sIBM and in LMNA-related NMDs (CMD, Emery-Dreifuss MD, LGMD, CMT, etc.)) [144]. Further, cytosolic mtDNA can activate cGAS-STING to induce IFN-1 or the NLRP3 inflammasome which can induce the maturation/secretion of pro-inflammatory cytokines [101]. Thus, mtDNA can activate major innate immune responses by acting as a DAMP from the mitochondria. Overall, mtDNA is released from mitochondria to the cytosol, where it is recognized by immune receptors, which trigger a signaling cascade that leads to the production of cytokines or transcription of inflammatory genes in the nucleus (Figure 2) [143]. This close association between mitochondria and inflammation triggered from affected cells, explains why NMDs are usually associated with inflammatory effects.
2.5.2. Implication of Mitochondrial ROS in Inflammation
Regarding mitochondrial ROS, they are sensed by mitochondrial antiviral signaling protein (MAVS), key in viral RNA infections. MAVS can activate pathways that regulate NF-kB and IRF3/IRF7 to induce gene expression [145]. It also can interact with the outer mitochondrial membrane, where mitochondrial ROS can trigger MAVS oligomerization, promoting IFN-1 production. In addition, MAVS can associate with NLRP3 and triggers its oligomerization, leading to caspase-1 activation [146].
NLRP3 is activated by mitochondrial ROS but also by cardiolipin, an inner mitochondrial membrane lipid. Cardiolipin translocates to the outer mitochondrial membrane after mitochondrial membrane depolarization, where it can recruit NLRP3. Cardiolipin-NLRP3 interaction suggests the role of mitochondria as a hub for the activation of innate immunity [143].
Similar to MAVS, NLRP3 also responds to mitochondrial ROS and can cause mitochondrial damage that promotes the generation of additional ROS. NLRP3 drives the production of IL-1β, IL-18, and pyroptosis. mtDNA can also activate NLRP3, and is also sensed by TLR9, which leads to immune and inflammatory gene expression. Mitochondria are therefore critical for signaling by three major innate immune pathways: RIG-I/MAVS, NLRP3, and TLR9 [143]. All these pathways rely on mitochondria-inflammatory response and, consequently, are altered in numerous NMDs.
2.5.3. Feedback Regulation of the Mitochondrial Inflammatory Response
NF-kB has a role as a pro-inflammatory and anti-inflammatory transcription factor in mitochondrial-induced inflammation. NF-kB anti-inflammatory functions prevent premature and excessive NLRP3-inflammasome activation, avoiding chronic inflammatory disease and inflammation caused by cell and tissue stress [147]. Self-limiting inflammation is also important for maintaining homeostasis, tissue repair, and regeneration, which restores integrity of epithelial barriers and thereby attenuates PAMP and DAMP availability [100].
Autophagy is the mechanism underlying NF-kB-mediated inhibition of NLRP3 inflammasome. After inducing the expression of pro-inflammatory cytokines and NLRP3, NF-kB is able to switch the initial pro-inflammatory to an anti-inflammatory state to avoid excessive inflammation. Thus, NF-kB induces the expression of p62, which eliminates NLRP3 by mitophagy [138]. Therefore, the “NF-kB-p62/SQSTM1-mitophagy” pathway provides an essential regulatory loop through which NF-kB orchestrates a reparative inflammatory response and prevents excessive collateral damage [100].
Dysregulation of the NLRP3-inflammasome is frequently associated with diverse inflammatory, metabolic, and malignant diseases, including gouty arthritis, Alzheimer’s disease, obesity, type II diabetes, and colorectal cancer [148]. Other NMDs with increased inflammation are ALS (with aberrant activation of NF-kB) [17], muscular dystrophies (like BMD and DMD), and inflammatory myopathies. Therefore, proper control of NLRP3-inflammasome activity and overall inflammatory cascade are critical for preventing disease development.
The aging process itself is linked to elevated low-grade inflammation or para-inflammation, characterized by constitutive production of low amounts of IL-1b, TNF, and IL-6. The NLRP3-inflammasome activation and insufficient mitophagy link accumulation of damaged mitochondria to the para-inflammatory state. Normal, healthy aging may be compromised and greatly enhanced by accumulation of somatically mutated mitochondria, which are more likely to release mitochondrial ROS and fragmented mtDNA that act as direct NLRP3-inflammasome activators [100]. This explains why most of NMDs arise hand by hand to aging processes.
3. Treatment Strategies in NMDs
As stated before, mitochondria play a key role in the early process of nerve and muscle degeneration and affects the progression of NMDs. Although current understanding of mitochondrial dysfunction in NMD is still evolving, there are some potential therapeutic strategies that tackle mitochondrial deregulated pathways.
The number of NMDs for which treatments are available is limited. This scarcity of therapeutic options lays on the rarity and diversity of NMDs, the complexity of their genetic inheritance, the abundance and distribution of muscle tissue, and low treatment efficacy [14]. The rarity of NMDs limits the available patients for recruitment for clinical trials, which highly depends on the quality and reliability of data obtained in cell and animal experiments [149].
Despite there is no cure for most NMDs, some can be effectively managed and treated. Some common interventions include drug therapy, surgery, or patient support groups, to improve physical and psychological wellbeing of NMD patients [6]. Innovative breakthroughs are approaching owing to antisense oligonucleotides (ASO) and gene replacement therapies, currently under development. Here we summarize the main points of different treatment strategies, most of them posing supportive pharmacologic options aimed not to cure but to slow down disease progression.
Drug therapy in NMDs comprises a wide range of treatments. Immunosuppressive drugs are widely used to treat certain muscle, nerve, and NMJ diseases. Anticonvulsants and antidepressants might aid in treating the pain of neuropathy [6]. More specifically, senolytic drugs (e.g., resveratrol) help to remove senescent cells to reduce the overall pro-inflammatory profile, although inflammation differs among tissues and makes it difficult to only target senescent cells [82]. Interestingly, resveratrol has been associated to promote mitochondrial health and renewal through different mechanisms [150].
Other treatment options are being developed to specifically target mitochondria. For example metformin, used as an anticancer drug, increases the activation of AMPK, favoring autophagy of damaged mitochondria (e.g., in mitochondrial diseases and DMD); caloric restriction, which reduces ROS production and thus, DNA damage (in collagen VI myopathies); and mitohormesis, based in inducing mild mitochondrial stress during life to induce an antioxidant response (e.g., by vitamin E). Some dietary supplements boost mitochondrial capacity like niacin (vitamin B3) in mitochondrial myopathy; epicatechin in BMD, or triheptanoin, CoQ10 and tocotrienols in ALS [151].
Moreover, exercise has shown to induce autophagy to overcome sarcopenia (e.g., in metabolic myopathies) [152]. In young and old mice, exercise training has even promoted biogenesis of mitochondria and mitochondrial autophagy to reduce the expression of proinflammatory cytokines by up to 49% [82]. Unfortunately, exercise is not always possible in patients affected by NMDs.
Other mitochondrial pathways that could be tackled therapeutically are dysregulation of MPTP opening, mitochondrial fission, and ER stress. MPTP modifiers (e.g., CsA, cyclophilin D inhibitors) might potentially aid in many NMDs [14]. Targeting mitochondrial fission is based on its direct relation to apoptosis, thus reducing fission might reduce cell death. Treating ER stress could be relevant in many NMD and other proteinopathies [30]. Currently, these therapeutic approaches are under development.
In addition to drug therapy, some NMDs benefit from surgery. Depending on the NMD, patients may receive neurological, thoracic, orthopedic, or other surgeries. Physical, occupational, or rehabilitation therapies may help before or after these interventions [6].
Recently, other approaches more related to personalized medicine are being developed or even in the market, like ASO and gene therapy. ASOs are short, synthetic single-stranded nucleic acids that bind targeted RNAs sequences to promote their degradation or modify splicing by impeding the binding of RNA splicing factors [153]. This approach has been developed in SMA and DMD and approved by FDA. In SMA, an ASO that switches the splicing to include an exon in the pre-mRNA of SMN2 gene has become the first treatment for SMA approved by FDA, commercialized by Biogen (MA, USA) under the name Nusinersen [67].
In DMD, an exon skipping ASO called Eteplirsen is also approved and commercially available. In this case, exon-skipping of damaged dystrophin in DMD generates a shortened but still functional version of dystrophin [154]. Other NMDs like LGMD could benefit from ASO exon skipping, by restoring the disrupted reading frame of the SGCG gene [5,27].
Apart from ASOs, gene replacement therapies are under development. In SMA, there is a gene replacement therapy available for patients younger than 2 years old. In this case, SMN1 gene cDNA is packaged in an adeno-associated virus 9 vector (AAV9), which are non-pathogenic, delivered intravenously and can cross the blood brain barrier to transduce neurons [155]. Once inside neurons, SMN1 cDNA constitutively expresses, with long-term expression in differentiated cells after a single dose, but not in mitotically active cells. This drug has shown efficacy in infantile SMA patients and is commercialized by Avexis under the name Zolgensma [67,156]. Other gene therapies using CRISPR/Cas-mediated approaches in NMD have shown efficacy in models of DMD, CMD, LGMD, DM1, and FSHD [157], becoming promising therapeutic approaches for further transference into clinical settings.
Altogether, currently there are nearly 200 potential therapies under development for NMD (in preclinical and clinical stages), mainly targeting ALS, SMA, and DMD [158]. Around 43% are small molecules (targeting receptor modulation, epigenetic reprogramming, redox metabolism, etc.), 14% gene therapy and 9% antisense oligonucleotides (ASOs) [159]. This shows that personalized medicine is increasingly offering new treatments for rare diseases, like NMDs. Indeed, the number of molecules in clinical trials for NMD has sharply increased in the last 5 years [158]. This proves NMD therapies are becoming a highly dynamic and potentially achieving field [8].
Notably, treatments not only focus in improving physical health of NMD patients, but also patients and families receive education and counselling to check their wellbeing. This may include getting involved in support groups or genetic counsellors (e.g., physical therapists, nutritionists). Patient’s associations are highly interested in creating support groups and analyze the quality of life of NMD patients and carers. One ongoing project, promoted by Share4Rare and released in the beginning of 2020, consists in a study called “Understanding how neuromuscular diseases impact learning and working opportunities for patients and carers.” They claim that patients with rare NMDs are a scattered community, often dispersed, and poorly represented. Even with the increased interest in developing research and potential therapeutic options in clinical trials in the last 10 years, little is known about the impact on quality of life of NMDs. With this study, they focus on understanding the psychosocial impact of NMD on patients, families, and caregivers [160].
4. Discussion
Despite we must acknowledge the impossibility of summarizing all previous knowledge in the current review, the present revision aimed to outline the state-of-the-art in the main etiologies of NMDs related to mitochondrial metabolism. The role of oxidative stress, mitochondrial biogenesis, autophagy, and inflammation related to mitochondria have been thoroughly described and linked to the affected NMDs (Figure 2). Overall, mitochondrial defects play a key role in the early process of neuronal and muscle degeneration and further affects the progression of NMDs.
NMDs can appear as a result of damage in nerve, muscle, or both tissues. In fact, the classification of NMDs include seven groups, two of them with primary muscle affection (MD and myopathies other than dystrophies), three of them with primary nerve dysfunction (NMJ diseases, peripheral nerve diseases and motor neuron diseases) and last two with both tissues affected (mitochondrial diseases and ion channel diseases). More attention should be focused on identifying prognostic and diagnostic biomarkers that target a specific pathway to accurately diagnose every NMD and control its progression.
Despite the number of treatments for NMD is still limited, some experimental treatments are currently under development to tackle mitochondrial deregulated pathways in NMDs. Current studies and clinical trials in many disabling NMDs aim to improve the wellbeing of patients and their families, but deep understanding of mitochondrial dysfunction in NMD will be essential to validate and establish further therapeutic targets.
The preclinical and clinical research advances in NMDs in the last years have been astounding. Better cell and animal models have helped in the characterization of many NMDs. Furthermore, the broad diversity and rarity of these diseases have revealed the importance of carefully studying every NMD history and progression. This more accurate approach is translating to more personalized treatments, as seen with new ASOs and gene therapies recently released to the market, and will eventually lead to a higher success rate in clinical trials for these disabling diseases.
5. Conclusions
- NMDs have a direct or indirect influence of mitochondria in their etiology. Mitochondrial genetic defects directly affect nerves and muscles in NMDs; but when there is an alternative cause of disease, unproper mitochondrial function can limit energetic supply of compensatory mechanisms, thus indirectly conditioning the progression of disease.
- Defects in oxidative metabolism can lead to accumulation of ROS, potentially affecting the ER and triggering inflammation, which eventually may lead to cell death and tissue damage.
- To avoid cell damage and excessive oxidative stress, mitochondrial quality control processes closely monitor changes in mitochondrial metabolism. In case of trouble, different responses arise by the increase in the number of mitochondria (mitochondrial biogenesis), by changing their morphology and size (mitochondrial dynamics) or recycling them (mitophagy). Most of these processes are altered in NMDs, highlighting the relevance of mitochondria in the development and progression of these disorders.
- Understanding nerves, muscles, and mitochondrial defects in NMDs is essential to improve diagnosis and treatments for these major incapacitating diseases.
- Despite growing efforts to clarify the etiology of NMD, the complexity of reality is still far beyond current knowledge and information provided in the present review, challenging new researchers to develop novel approaches.
Author Contributions
Conceptualization: J.C.-S. and G.G.; formal analysis: J.C.-S. and G.G.; investigation: J.C.-S. and G.G.; writing—original draft preparation: J.C.-S.; writing—review and editing: J.C.-S., J.M.G.-J. and G.G.; supervision: J.M.G.-J. and G.G.; project administration: J.M.G.-J. and G.G.; funding acquisition: J.M.G.-J. and G.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Instituto de Salud Carlos III (ISCIII) and Fondos Europeos de Desarrollo Regional (FEDER), ‘una manera de hacer Europa’, FIS PI1800498.
Acknowledgments
We are in dept to all patients and their families, always devoted to collaborate and widen the state-of-the-art in diseases.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Diagnosis—European Reference Network—EURO-NMD. Available online: https://ern-euro-nmd.eu/ (accessed on 22 April 2020).
- Scotton, C.; Passarelli, C.; Neri, M.; Ferlini, A. Biomarkers in rare neuromuscular diseases. Exp. Cell Res. 2013, 325, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Rivier, F.; Hamroun, D. The 2018 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 2017, 27, 1152–1183. [Google Scholar] [CrossRef] [PubMed]
- Deenen, C.W.J.; Horlings, G.C.C.; Verschuuren, J.G.M.J.; Verbeek, L.M.A.; van Engelen, G.M.B. The Epidemiology of Neuromuscular Disorders: A Comprehensive Overview of the Literature. J. Neuromuscul. Dis. 2015, 2, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Van Putten, M.; Hmeljak, J.; Aartsma-Rus, A.; Dowling, J.J. Moving neuromuscular disorders research forward: From novel models to clinical studies. Dis. Model. Mech. 2020, 13, 44370. [Google Scholar] [CrossRef]
- Neurology Neuromuscular Disorders Causes & Symptoms Beaumont Health. Available online: https://www.beaumont.org/conditions/neuromuscular-disorders (accessed on 19 May 2020).
- Nardin, R.A.; Johns, D.R. Mitochondrial dysfunction and neuromuscular disease. Muscle Nerve 2001, 24, 170–191. [Google Scholar] [CrossRef]
- Cowling, B.S.; Thielemans, L. Translational medicine in neuromuscular disorders: From academia to industry. Dis. Model. Mech. 2019, 13, 41434. [Google Scholar] [CrossRef]
- Find A Neuromuscular Disease Muscular Dystrophy Association. Available online: https://www.mda.org/disease/list (accessed on 19 May 2020).
- GeneTable. Available online: http://www.musclegenetable.fr/index.html (accessed on 7 July 2020).
- Hewitt, J.E.; Pollard, A.K.; Lesanpezeshki, L.; Deane, C.S.; Gaffney, C.J.; Etheridge, T.; Szewczyk, N.J.; Vanapalli, S.A. Muscle strength deficiency and mitochondrial dysfunction in a muscular dystrophy model of Caenorhabditis elegans and its functional response to drugs. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Heydemann, A. Skeletal muscle metabolism in duchenne and becker muscular dystrophy—Implications for therapies. Nutrients 2018, 10, 796. [Google Scholar] [CrossRef]
- Mendell, J.R.; Sahenk, Z.; Malik, V.; Gomez, A.M.; Flanigan, K.M.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Meadows, E.; Lewis, S.; et al. A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol. Ther. 2015, 23, 192–201. [Google Scholar] [CrossRef]
- Katsetos, C.D.; Koutzaki, S.; Melvin, J.J. Mitochondrial dysfunction in neuromuscular disorders. Semin. Pediatric Neurol. 2013, 20, 202–215. [Google Scholar] [CrossRef]
- Grumati, P.; Grumati, P.; Coletto, L.; Sabatelli, P.; Cescon, M.; Angelin, A.; Bertaggia, E.; Blaauw, B.; Urciuolo, A.; Tiepolo, T.; et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 2010, 16, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Jongpiputvanich, S.; Sueblinvong, T.; Norapucsunton, T. Mitochondrial respiratory chain dysfunction in various neuromuscular diseases. J. Clin. Neurosci. 2005, 12, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Pleasure, D. Advances in Translational Research in Neuromuscular Diseases. Arch. Neurol. 2011, 68, 429. [Google Scholar] [CrossRef][Green Version]
- Lamar, K.-M.; Mcnally, E.M. Genetic Modifiers for Neuromuscular Diseases. J. Neuromuscul. Dis. 2014, 1, 3–13. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophin-deficient Mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, 115763. [Google Scholar] [CrossRef] [PubMed]
- Godin, R.; Daussin, F.; Matecki, S.; Li, T.; Petrof, B.J.; Burelle, Y. Peroxisome proliferator-activated receptor γ coactivator 1-α gene transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post-necrotic mdx mouse skeletal muscle. J. Physiol. 2012, 590, 5487–5502. [Google Scholar] [CrossRef]
- Pauly, M.; Daussin, F.; Burelle, Y.; Li, T.; Godin, R.; Fauconnier, J.; Koechlin-Ramonatxo, C.; Hugon, G.; Lacampagne, A.; Coisy-Quivy, M.; et al. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am. J. Pathol. 2012, 181, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Zuela, N.; Dorfman, J.; Gruenbaum, Y. Global transcriptional changes caused by an EDMD mutation correlate to tissue specific disease phenotypes in C. elegans. Nucleus 2017, 8, 60–69. [Google Scholar] [CrossRef]
- Puckelwartz, M.; McNally, E.M. Emery-Dreifuss muscular dystrophy. Handb. Clin. Neurol. 2011, 101, 155–166. [Google Scholar] [CrossRef]
- Orphanet. Available online: https://www.orpha.net/consor4.01/www/cgi-bin/?lng=ES (accessed on 22 May 2020).
- Lemmers, R.J.L.F.; Van Der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camaño, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; Van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef]
- Deng, H.-X.; Klein, C.J.; Yan, J.; Shi, Y.; Wu, Y.; Fecto, F.; Yau, H.-J.; Yang, Y.; Zhai, H.; Siddique, N.; et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat. Genet. 2010, 42, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Demonbreun, A.R.; Wyatt, E.J.; Fallon, K.S.; Oosterbaan, C.C.; Page, P.G.; Hadhazy, M.; Quattrocelli, M.; Barefield, D.Y.; McNally, E.M. A gene-edited mouse model of limb-girdle muscular dystrophy 2C for testing exon skipping. DMM Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Lilleker, J.B.; Keh, Y.S.; Roncaroli, F.; Sharma, R.; Roberts, M. Metabolic myopathies: A practical approach. Pract. Neurol. 2018, 18, 14–26. [Google Scholar] [CrossRef] [PubMed]
- CNBP Gene—Genetics Home Reference—NIH. Available online: https://ghr.nlm.nih.gov/gene/CNBP#conditions (accessed on 25 May 2020).
- Malerba, A.; Klein, P.; Bachtarzi, H.; Jarmin, S.A.; Cordova, G.; Ferry, A.; Strings, V.; Espinoza, M.P.; Mamchaoui, K.; Blumen, S.C.; et al. PABPN1 gene therapy for oculopharyngeal muscular dystrophy. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Malerba, A.; Roth, F.; Harish, P.; Dhiab, J.; Lu-Nguyen, N.; Cappellari, O.; Jarmin, S.; Mahoudeau, A.; Ythier, V.; Lainé, J.; et al. Pharmacological modulation of the ER stress response ameliorates oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2019, 28, 1694–1708. [Google Scholar] [CrossRef]
- Chartier, A.; Klein, P.; Pierson, S.; Barbezier, N.; Gidaro, T.; Casas, F.; Carberry, S.; Dowling, P.; Maynadier, L.; Bellec, M.; et al. Mitochondrial Dysfunction Reveals the Role of mRNA Poly(A) Tail Regulation in Oculopharyngeal Muscular Dystrophy Pathogenesis. PLoS Genet. 2015, 11, 1005092. [Google Scholar] [CrossRef]
- Rossi, D.; Palmio, J.; Evilä, A.; Galli, L.; Barone, V.; Caldwell, T.A.; Policke, R.A.; Aldkheil, E.; Berndsen, C.E.; Wright, N.T.; et al. A novel FLNC frameshift and an OBSCN variant in a family with distal muscular dystrophy. PLoS ONE 2017, 12, 186642. [Google Scholar] [CrossRef]
- Vincent, A.E.; Rosa, H.S.; Alston, C.L.; Grady, J.P.; Rygiel, K.A.; Rocha, M.C.; Barresi, R.; Taylor, R.W.; Turnbull, D.M. Dysferlin mutations and mitochondrial dysfunction. Neuromuscul. Disord. 2016, 26, 782–788. [Google Scholar] [CrossRef]
- GeneTable. Available online: http://www.musclegenetable.fr/4DACTION/Blob_groupe11/ (accessed on 6 July 2020).
- Fusto, A.; Moyle, L.A.; Gilbert, P.M.; Pegoraro, E. Cored in the act: The use of models to understand core myopathies. DMM Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef]
- Gilbreath, H.R.; Castro, D.; Iannaccone, S.T. Congenital myopathies and muscular dystrophies. Neurol. Clin. 2014, 32, 689–703. [Google Scholar] [CrossRef]
- Siciliano, G.; Monzani, F.; Manca, M.L.; Tessa, A.; Caraccio, N.; Tozzi, G.; Piemonte, F.; Mancuso, M.; Santorelli, F.M.; Ferrannini, E.; et al. Human Mitochondrial Transcription Factor A Reduction and Mitochondrial Dysfunction in Hashimoto’s Hypothyroid Myopathy. Mol. Med. 2002, 8, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Lloreta, J.; Roquer, J.; Corominas, J.M.; Serrano, S. Hyperthyroid myopathy with mitochondrial paracrystalline rectangular inclusions. Ultrastruct. Pathol. 1996, 20, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Gong, Y.; Dong, M.; Pei, Z.; Ren, J. Role of autophagy in inherited metabolic and endocrine myopathies. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Selva-O’callaghan, A.; Pinal-Fernandez, I.; Trallero-Araguás, E.; Milisenda, J.C.; Grau-Junyent, M.; Mammen, A.L. Classification and management of adult inflammatory myopathies. Lancet Neurol. 2018, 17, 816–828. [Google Scholar] [CrossRef]
- Catalan-Garcia, M.; Garrabou, G.; Moren, C.; Guitart-Mampel, M.; Hernando, A.; Diaz-Ramos, A.; Gonzalez-Casacuberta, I.; Juarez, D.-L.; Bano, M.; Enrich-Bengoa, J.; et al. Mitochondrial DNA disturbances and deregulated expression of oxidative phosphorylation and mitochondrial fusion proteins in sporadic inclusion body myositis. Clin. Sci. 2016, 130, 1741–1751. [Google Scholar] [CrossRef] [PubMed]
- Sharp, L.J.; Haller, R.G. Metabolic and mitochondrial myopathies. Neurol. Clin. 2014, 32, 777–799. [Google Scholar] [CrossRef]
- Selcen, D.; Ohno, K.; Engel, A.G. Myofibrillar myopathy: Clinical, morphological and genetic studies in 63 patients. Brain 2004, 127, 439–451. [Google Scholar] [CrossRef]
- Perrone, L.; Squillaro, T.; Napolitano, F.; Terracciano, C.; Sampaolo, S.; Anna, M.; Melone, B. The Autophagy Signaling Pathway: A Potential Multifunctional Therapeutic Target of Curcumin in Neurological and Neuromuscular Diseases. Nutrients 2019, 11, 1881. [Google Scholar] [CrossRef]
- Vincent, A.E.; Grady, J.P.; Rocha, M.C.; Alston, C.L.; Rygiel, K.A.; Barresi, R.; Taylor, R.W.; Turnbull, D.M. Mitochondrial dysfunction in myofibrillar myopathy. Neuromuscul. Disord. 2016, 26, 691–701. [Google Scholar] [CrossRef]
- Pá, E.; Bedekovics Istvá, T.; Ti, G. Familial Scapuloperoneal Myopathy and Mitochondrial DNA Defect. Eur Neurol. 1999, 42, 211–216. [Google Scholar]
- Zukosky, K.; Meilleur, K.; Traynor, B.J.; Dastgir, J.; Medne, L.; Devoto, M.; Collins, J.; Rooney, J.; Zou, Y.; Yang, M.L.; et al. Association of a novel ACTA1 mutation with a dominant progressive scapuloperoneal myopathy in an extended family. JAMA Neurol. 2015, 72, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.H.; Raskind, W.H.; Parson, W.W.; Sonnen, J.A.; Vu, T.; Zheng, Y.; Matsushita, M.; Wolff, J.; Lipe, H.; Bird, T.D. A novel mutation in FHL1 in a family with X-linked scapuloperoneal myopathy: Phenotypic spectrum and structural study of FHL1 mutations. J. Neurol. Sci. 2010, 296, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Shen, X.M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef]
- Chaouch, A.; Porcelli, V.; Cox, D.; Edvardson, S.; Scarcia, P.; De Grassi, A.; Pierri, C.L.; Cossins, J.; Laval, S.H.; Griffin, H.; et al. Chaouch et al. Mutations in the Mitochondrial Citrate Carrier SLC25A1. J. Neuromuscul. Dis. 2014, 1, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Congenital myasthenic syndromes. Orphanet J. Rare Dis. 2019, 14, 57. [Google Scholar] [CrossRef]
- Bogdanik, L.P.; Burgess, R.W. A valid mouse model of AGRIN-associated congenital myasthenic syndrome. Hum. Mol. Genet. 2011, 20, 4617–4633. [Google Scholar] [CrossRef]
- Kesner, V.G.; Oh, S.J.; Dimachkie, M.M.; Barohn, R.J. Lambert-Eaton Myasthenic Syndrome. Neurol. Clin. 2018, 36, 379–394. [Google Scholar] [CrossRef]
- Hülsbrink, R.; Hashemolhosseini, S. Lambert-Eaton myasthenic syndrome-Diagnosis, pathogenesis and therapy. Clin. Neurophysiol. 2014, 125, 2328–2336. [Google Scholar] [CrossRef]
- Herrmann, D.N.; Horvath, R.; Sowden, J.E.; Gonzales, M.; Sanchez-Mejias, A.; Guan, Z.; Whittaker, R.G.; Almodovar, J.L.; Lane, M.; Bansagi, B.; et al. Synaptotagmin 2 mutations cause an autosomal-dominant form of lambert-eaton myasthenic syndrome and nonprogressive motor neuropathy. Am. J. Hum. Genet. 2014, 95, 332–339. [Google Scholar] [CrossRef]
- Marchiori, P.E.; Levy, J.A.; Carvalho-Alegro, M.S.; Lusvarghi, E.S.; Tsanaclis, A.M.; De Assis, J.L.; Scaff, M. Mitochondrial dysfunction in myasthenia gravis. Report of a case. Arq. Neuropsiquiatr. 1989, 47, 355–358. [Google Scholar] [CrossRef]
- Gilhus, N.E. Myasthenia Gravis. N. Engl. J. Med. 2016, 375, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nat. Lett. 2010, 465. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.; Paul, P.; Chen, H.-J.; Morris, A.; Payling, M.; Falchi, M.; Habgood, J.; Panoutsou, S.; Winkler, S.; Tisato, V.; et al. Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc. Natl. Acad. Sci. USA 2010, 107, 7556–7561. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Van Den Berg, L.H.; Kiernan, M.C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 639–649. [Google Scholar] [CrossRef]
- Chiò, A.; Mora, G.; Lauria, G. Pain in amyotrophic lateral sclerosis. Lancet Neurol. 2017, 16, 144–157. [Google Scholar] [CrossRef]
- Comi, G.P.; Bordoni, A.; Salani, S.; Franceschina, L.; Sciacco, M.; Prelle, A.; Fortunato, F.; Zeviani, M.; Napoli, L.; Bresolin, N.; et al. Cytochrome c oxidase subunit I microdeletion in a patient with motor neuron disease. Ann. Neurol. 1998, 43, 110–116. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Guo, K.; Murdock, B.J.; Paez-Colasante, X.; Bassis, C.M.; Mikhail, K.A.; Raue, K.D.; Evans, M.C.; Taubman, G.F.; McDermott, A.J.; et al. Temporal evolution of the microbiome, immune system and epigenome with disease progression in ALS mice. DMM Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef]
- Lynch, E.; Semrad, T.; Belsito, V.S.; FitzGibbons, C.; Reilly, M.; Hayakawa, K.; Suzuki, M. C9ORF72-related cellular pathology in skeletal myocytes derived from ALS-patient induced pluripotent stem cells. DMM Dis. Model. Mech. 2019, 12, 1–12. [Google Scholar] [CrossRef]
- Ranganathan, S.; Harmison, G.G.; Meyertholen, K.; Pennuto, M.; Burnett, B.G.; Fischbeck, K.H. Mitochondrial abnormalities in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2008, 18, 27–42. [Google Scholar] [CrossRef]
- Ravi, B.; Antonellis, A.; Sumner, C.J.; Lieberman, A.P. Genetic approaches to the treatment of inherited neuromuscular diseases. Hum. Mol. Genet. 2019, 28, 1–55. [Google Scholar] [CrossRef]
- Giorgetti, E.; Lieberman, A.P. Polyglutamine androgen receptor-mediated neuromuscular disease. Cell Mol. Life Sci. 2016, 73, 3991–3999. [Google Scholar] [CrossRef] [PubMed]
- Grunseich, C.; Miller, R.; Swan, T.; Glass, D.J.; El Mouelhi, M.; Fornaro, M.; Petricoul, O.; Vostiar, I.; Roubenoff, R.; Meriggioli, M.N.; et al. Safety, tolerability, and preliminary efficacy of an IGF-1 mimetic in patients with spinal and bulbar muscular atrophy: A randomised, placebo-controlled trial. Lancet Neurol. 2018, 17, 1043–1052. [Google Scholar] [CrossRef]
- Hashizume, A.; Katsuno, M.; Suzuki, K.; Banno, H.; Takeuchi, Y.; Kawashima, M.; Suga, N.; Mano, T.; Araki, A.; Hijikata, Y.; et al. Efficacy and safety of leuprorelin acetate for subjects with spinal and bulbar muscular atrophy: Pooled analyses of two randomized-controlled trials. J. Neurol. 2019, 266, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Passini, M.A.; Bu, J.; Roskelley, E.M.; Richards, A.M.; Sardi, S.P.; O’Riordan, C.R.; Klinger, K.W.; Shihabuddin, L.S.; Cheng, S.H. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Investig. 2010, 120, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Tinelli, E.; Pereira, J.A.; Suter, U. Muscle-specific function of the centronuclear myopathy and Charcot-Marie-Tooth neuropathy-associated dynamin 2 is required for proper lipid metabolism, mitochondria, muscle fibers, neuromuscular junctions and peripheral nerves. Hum. Mol. Genet. 2013, 22, 4417–4429. [Google Scholar] [CrossRef]
- Patel, P.I.; Roa, B.B.; Welcher, A.A.; Schoener-Scott, R.; Trask, B.J.; Pentao, L.; Snipes, G.J.; Garcia, C.A.; Francke, U.; Shooter, E.M.; et al. The gene for the peripheral myelin protein PMP–22 is a candidate for Charcot–Marie–Tooth disease type 1A. Nat. Genet. 1992, 1, 159–165. [Google Scholar] [CrossRef]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Pezeshkpour, G.; Krarup, C.; Buchthal, F.; DiMauro, S.; Bresolin, N.; McBurney, J. Peripheral neuropathy in mitochondrial disease. J. Neurol. Sci. 1987, 77, 285–304. [Google Scholar] [CrossRef]
- Bomont, P.; Cavalier, L.; Blondeau, F.; Hamida, C.B.; Belal, S.; Tazir, M.; Demir, E.; Topaloglu, H.; Korinthenberg, R.; Tüysüz, B.; et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat. Lett. 2000, 26, 370–374. [Google Scholar] [CrossRef]
- Milone, M.; Wong, L.-J. Diagnosis of mitochondrial myopathies. Mol. Genet. Metab. 2013, 110, 35–41. [Google Scholar] [CrossRef]
- Khan, N.A.; Auranen, M.; Paetau, I.; Pirinen, E.; Euro, L.; Forsström, S.; Pasila, L.; Velagapudi, V.; Carroll, C.J.; Auwerx, J.; et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 2014, 6, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.E.; Ng, Y.S.; White, K.; Davey, T.; Mannella, C.; Falkous, G.; Feeney, C.; Schaefer, A.M.; Mcfarland, R.; Gorman, G.S.; et al. The Spectrum of Mitochondrial Ultrastructural Defects in Mitochondrial Myopathy. Sci. Rep. 2016, 6, 30610. [Google Scholar] [CrossRef]
- Cook, A.; Giunti, P. Friedreich’s ataxia: Clinical features, pathogenesis and management. Br. Med. Bull. 2017, 124, 19–30. [Google Scholar] [CrossRef]
- Alfedi, G.; Luffarelli, R.; Condò, I.; Pedini, G.; Mannucci, L.; Massaro, D.S.; Benini, M.; Toschi, N.; Alaimo, G.; Panarello, L.; et al. Drug repositioning screening identifies etravirine as a potential therapeutic for friedreich’s ataxia. Mov. Disord. 2019, 34, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Strickland, M.; Yacoubi-Loueslati, B.; Bouhaouala-Zahar, B.; Pender, S.L.F.; Larbi, A. Relationships between ion channels, mitochondrial functions and inflammation in human aging. Front. Physiol. 2019, 10, 158. [Google Scholar] [CrossRef] [PubMed]
- Jurkat-Rott, K.; Lehmann-Horn, F. State of the art in hereditary muscle channelopathies. Acta Myol. 2010, 29, 343–350. [Google Scholar]
- Pi, Y.; Goldenthal, M.J.; Marín-García, J.; Pi, Y.; Goldenthal, M.J.; Marín-García, J. Mitochondrial channelopathies in aging. J. Mol. Med. 2007, 85, 937–951. [Google Scholar] [CrossRef]
- Vicart, S.; Sternberg, D.; Fontaine, B.; Meola, G. Human skeletal muscle sodium channelopathies. Neurol. Sci. 2005, 26, 194–202. [Google Scholar] [CrossRef]
- Rudolf, R.; Khan, M.M.; Witzemann, V. Motor Endplate—Anatomical, Functional, and Molecular Concepts in the Historical Perspective. Cells 2019, 8, 387. [Google Scholar] [CrossRef]
- Calderón, J.C.; Bolaños, P.; Caputo, C. The excitation-contraction coupling mechanism in skeletal muscle. Biophys. Rev. 2014, 6, 133–160. [Google Scholar] [CrossRef]
- Bach-y-Rita, P.; Ito, F. In vivo studies on fast and slow muscle fibers in cat extraocular muscles. J. Gen. Physiol. 1966, 49, 1177–1198. [Google Scholar] [CrossRef] [PubMed]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcárcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.F.; Feener, C.; Kunkel’tt, L.M. Complete Cloning of the Duchenne Muscular Dystrophy (DMD) cDNA and Preliminary Genomic Organization of the DMD Gene in Normal and Affected Individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Siekevitz, P. Powerhouse of the Cell. Nature 1957, 197, 131–144. [Google Scholar] [CrossRef]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Ran Larsson, N.-G. Cell Metabolism Review Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Suliman, H.B. Transcriptional control of mitochondrial biogenesis and its interface with inflammatory processes. Biochim. Biophys. Acta 2012, 1820, 532–541. [Google Scholar] [CrossRef]
- Kepp, O.; Galluzzi, L.; Kroemer, G. Mitochondrial control of the NLRP3 inflammasome. Nat. Immunol. 2011, 12. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 595–609. [Google Scholar] [CrossRef]
- Manevski, M.; Muthumalage, T.; Devadoss, D.; Sundar, I.K.; Wang, Q.; Singh, K.P.; Unwalla, H.J.; Chand, H.S.; Rahman, I. Cellular stress responses and dysfunctional Mitochondrial–cellular senescence, and therapeutics in chronic respiratory diseases. Redox Biol. 2020, 33, 101443. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, H.; Uematsu, S.; Yang, B.-G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1b production. Nat. Lett. 2008, 456. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Nuevo, A.; Díaz-Ramos, A.; Noguera, E.; Díaz-Sáez, F.; Duran, X.; Muñoz, J.P.; Romero, M.; Plana, N.; Sebastián, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Couvillion, M.T.; Soto, I.C.; Shipkovenska, G.; Stirling, L. Synchronized mitochondrial and cytosolic translation programs. Nature 2016, 533, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Tokuyama, T.; Hirai, A.; Shiiba, I.; Ito, N.; Matsuno, K.; Takeda, K.; Saito, K.; Mii, K.; Matsushita, N.; Fukuda, T.; et al. Mitochondrial Dynamics Regulation in Skin Fibroblasts from Mitochondrial Disease Patients. Biomolecules 2020, 10, 450. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling inflamm-aging through mitochondrial dysfunction: Mechanisms and molecular targets. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 2297–2310. [Google Scholar] [CrossRef]
- Babbar, M.; Basu, S.; Yang, B.; Croteau, D.L.; Bohr, V.A. Mitophagy and DNA damage signaling in human aging. Mech. Ageing Dev. 2020, 186. [Google Scholar] [CrossRef]
- Bhatt, P.S.; Tzoulis, C.; Balafkan, N.; Miletic, H.; Tran, G.T.T.; Sanaker, P.S.; Bindoff, L.A. Mitochondrial DNA depletion in sporadic inclusion body myositis. Neuromuscul. Disord. 2019, 29, 242–246. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.H.; Willems, P.H.G.M.; Smeitink, J.A.M. Monogenic Mitochondrial Diseases. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA mutations in neurodegeneration. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Lightowlers, R.N.; Taylor, R.W.; Turnbull, D.M. Mutations causing mitochondrial disease: What is new and what challenges remain? Science 2015, 349, 1494–1499. [Google Scholar] [CrossRef]
- Argov, Z.; Renshaw, P.F.; Boden, B.; Winokur, A.; Bank, W.J. Effects of thyroid hormones on skeletal muscle bioenergetics. In vivo phosphorus-31 magnetic resonance spectroscopy study of humans and rats. J. Clin. Investig. 1988, 81, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Monzani, F.; Caraccio, N.; Siciliano, G.; Manca, L.; Murri, L.; Ferrannini, E. Clinical and Biochemical Features of Muscle Dysfunction in Subclinical Hypothyroidism. J. Clin. Endocrinol. Metab. 1997, 82, 3315–3318. [Google Scholar] [CrossRef]
- Deng, J.; Wang, P.; Chen, X.; Cheng, H.; Liu, J.; Fushimi, K.; Zhu, L.; Wu, J.Y. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. USA 2018, 115, E9678–E9686. [Google Scholar] [CrossRef]
- Spiegel, R.; Saada, A.; Flannery, P.J.; Burté, F.; Soiferman, D.; Khayat, M.; Eisner, V.; Vladovski, E.; Taylor, R.W.; Bindoff, L.A.; et al. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J. Med. Genet. 2016, 53, 127–131. [Google Scholar] [CrossRef]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.T.; Mooyer, P.A.W.; Wanders, R.J.A.; Leonard, J.V. A Lethal Defect of Mitochondrial and Peroxisomal Fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Kodavati, M.; Wang, H.; Hegde, M.L. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cells 2020, 9, 1065. [Google Scholar] [CrossRef] [PubMed]
- Clark, S. Newborn mice studied with the electron mircroscope. Biophys. Biochem. Cytol. 1957, 3, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Seabright, A.P.; Fine, N.H.F.; Barlow, J.P.; Lord, S.O.; Musa, I.; Gray, A.; Bryant, J.A.; Banzhaf, M.; Lavery, G.G.; Hardie, D.G.; et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J. 2020, 34, 6284–6301. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nat. Lett. 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hu, X.; Shen, Q.; Xing, D. Mitochondria-specific drug release and reactive oxygen species burst induced by polyprodrug nanoreactors can enhance chemotherapy. Nat. Commun. 2019, 10, 1–14. [Google Scholar]
- Lerner, C.A.; Sundar, I.K.; Rahman, I. Mitochondrial redox system, dynamics, and dysfunction in lung inflammaging and COPD. Int. J. Biochem. Cell Biol. 2016, 81, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Domènech, B.E.; Marfany, G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants 2020, 9, 347. [Google Scholar] [CrossRef] [PubMed]
- Peterman, E.M.; Sullivan, C.; Goody, M.F.; Rodriguez-Nunez, I.; Yoder, J.A.; Kim, C.H. Neutralization of mitochondrial superoxide by superoxide dismutase 2 promotes bacterial clearance and regulates phagocyte numbers in zebrafish. Infect. Immun. 2015, 83, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M.; Nilsson, P.; Forsgren, L.; Marklund, S.L. CuZn-Superoxide Dismutase, Extracellular Superoxide Dismutase, and Glutathione Peroxidase in Blood from Individuals Homozygous for Asp90Ala CuZn-Superoxide Dismutase Mutation. J. Neurochem. 2002, 70, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef] [PubMed]
- Naon, D.; Scorrano, L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2184–2194. [Google Scholar] [CrossRef]
- Nadalutti, C.A.; Stefanick, D.F.; Zhao, M.-L.; Horton, J.K.; Prasad, R.; Brooks, A.M.; Griffith, J.D.; Wilson, S.H. Mitochondrial dysfunction and DNA damage accompany enhanced levels of formaldehyde in cultured primary human fibroblasts. Sci. Rep. 2020, 10, 5575. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Pyo Kim, H.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 8, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Treviño, P.; Velásquez, M.; García, N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. BBA Mol. Basis Dis. 2020, 1866, 165761. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Nuevo, A.; Zorzano, A. The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress 2019, 3, 195–207. [Google Scholar] [CrossRef] [PubMed]
- West, A.P. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology 2017, 391, 54–63. [Google Scholar] [CrossRef]
- Baroja-Mazo, A.; Martín-Sánchez, F.; Gomez, A.I.; Martínez, C.M.; Amores-Iniesta, J.; Compan, V.; Barberà-Cremades, M.; Yagüe, J.; Ruiz-Ortiz, E.; Antón, J. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Cappelletti, C.; Salerno, F.; Canioni, E.; Mora, M.; Mantegazza, R.; Bernasconi, P.; Maggi, L. Up-regulation of toll-like receptors 7 and 9 and its potential implications in the pathogenic mechanisms of lmna-related myopathies. Nucleus 2018, 9, 398–409. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.-W.; Alnemri, E.S. The mitochondrial anti-viral protein MAVS associates with NLRP3 and regulates its inflammasome activity 1. J. Immunol. 2013, 191, 4358–4366. [Google Scholar] [CrossRef]
- Deswaerte, V.; Ruwanpura, S.M.; Jenkins, B.J. Transcriptional regulation of inflammasome-associated pattern recognition receptors, and the relevance to disease pathogenesis. Mol. Immunol. 2017, 86, 3–9. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Kornegay, J.N.; Spurney, C.F.; Nghiem, P.P.; Brinkmeyer-Langford, C.L.; Hoffman, E.P.; Nagaraju, K. Pharmacologic Management of Duchenne Muscular Dystrophy: Target Identification and Preclinical Trials. ILAR J. 2014, 55, 119–149. [Google Scholar] [CrossRef] [PubMed]
- Antosh, M.; Whitaker, R.; Kroll, A.; Hosier, S.; Chang, C.; Bauer, J.; Cooper, L.; Neretti, N.; Helfand, S.L. Comparative transcriptional pathway bioinformatic analysis of dietary restriction, Sir2, p53 and resveratrol life span extension in Drosophila. Cell Cycle 2011, 10, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Interventional Studies Neuromuscular Diseases. Available online: https://clinicaltrials.gov/ct2/results?cond=Neuromuscular+Diseases&term=mitochondria&type=Intr&rslt=&age_v=&gndr=&intr=&titles=&outc=&spons=&lead=&id=&cntry=&state=&city=&dist=&locn=&rsub=&strd_s=&strd_e=&prcd_s=&prcd_e=&sfpd_s=&sfpd_e=&rfpd_s=&rfpd_e=&lu (accessed on 29 May 2020).
- Liang, J.; Zeng, Z.; Zhang, Y.; Chen, N. Regulatory role of exercise-induced autophagy for sarcopenia. Exp. Gerontol. 2020, 130, 110789. [Google Scholar] [CrossRef]
- Schoch, K.M.; Miller, T.M. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron 2017, 94, 1056–1070. [Google Scholar] [CrossRef]
- Levin, A.A. Treating disease at the RNA level with oligonucleotides. N. Engl. J. Med. 2019, 380, 57–70. [Google Scholar] [CrossRef]
- Saraiva, J.; Nobre, R.J.; Pereira De Almeida, L. Gene therapy for the CNS using AAVs: The impact of systemic delivery by AAV9. J. Control. Release 2016, 241, 94–109. [Google Scholar] [CrossRef]
- Mendell, J.R. Single-Dose Gene-Replacement Therapy for SMA. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Young, C.S.; Pyle, A.D.; Spencer, M.J. CRISPR for Neuromuscular Disorders: Gene Editing and Beyond. Physiology 2019, 34, 341–353. [Google Scholar] [CrossRef]
- Understanding Neuromuscular Disease Care—IQVIA. Available online: https://www.iqvia.com/insights/the-iqvia-institute/reports/understanding-neuromuscular-disease-care (accessed on 28 May 2020).
- Patridge, E.V.; Gareiss, P.C.; Kinch, M.S.; Hoyer, D.W. An analysis of original research contributions toward FDA-approved drugs. Drug Discov. Today 2015, 20, 1182–1187. [Google Scholar] [CrossRef]
- S4R Registration Share4Rare. Available online: https://www.share4rare.org/registration/understanding-neuromuscular-diseases (accessed on 19 May 2020).


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).