Traumatic Brain Injury: Oxidative Stress and Novel Anti-Oxidants Such as Mitoquinone and Edaravone

 , , ,
, , ,  ,
,  ,
,  ,
,  ,
, 
 and
and
Abstract
1. Traumatic Brain Injury: Definition and Pathogenesis
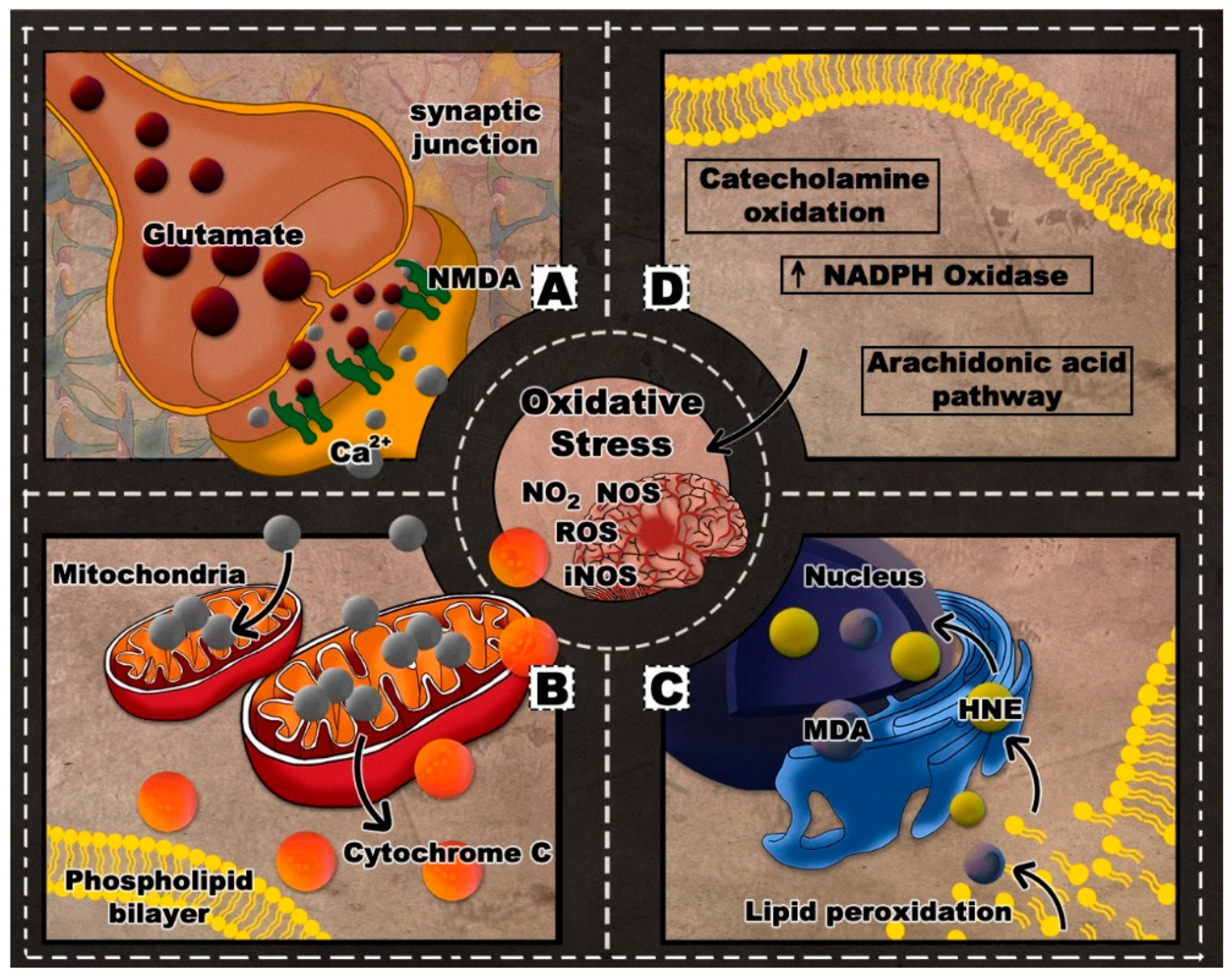
2. Oxidative Stress in TBI
3. Therapeutic Options Targeting Oxidative Stress in TBI
3.1. A Brief History of Multiple Antioxidants Utilized in TBI
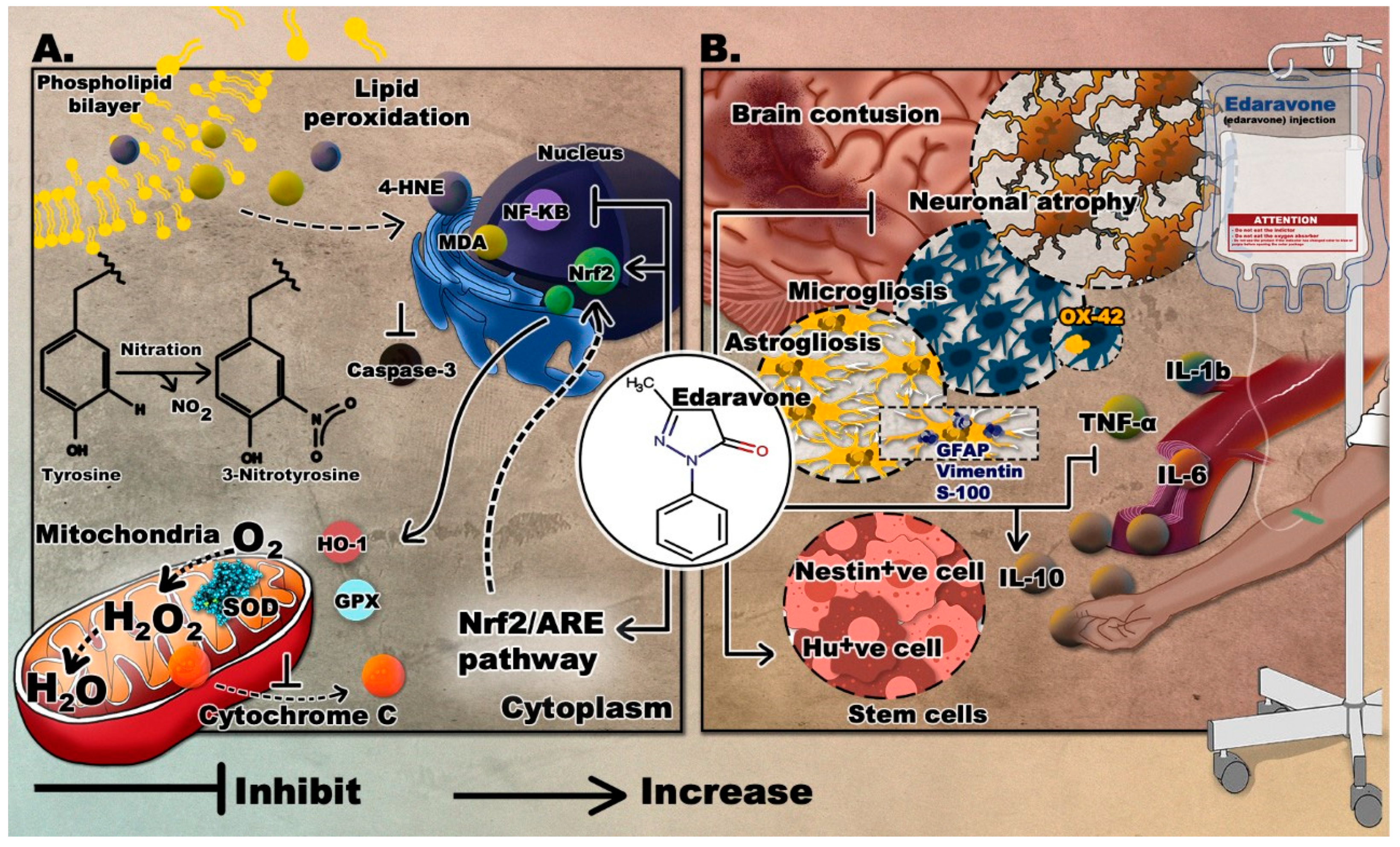
3.2. Edaravone
3.2.1. Edaravone in ALS
3.2.2. Edaravone in TBI
Dose, Therapeutic Window, and Time Frame
Providing Neuroprotection
Antagonizing Oxidative Stress
Attenuating the Immune Response and Managing Cerebral Edema
Enhancing Stem Cell Production
Modifying Behavior
Use of Edaravone in Patients with TBI
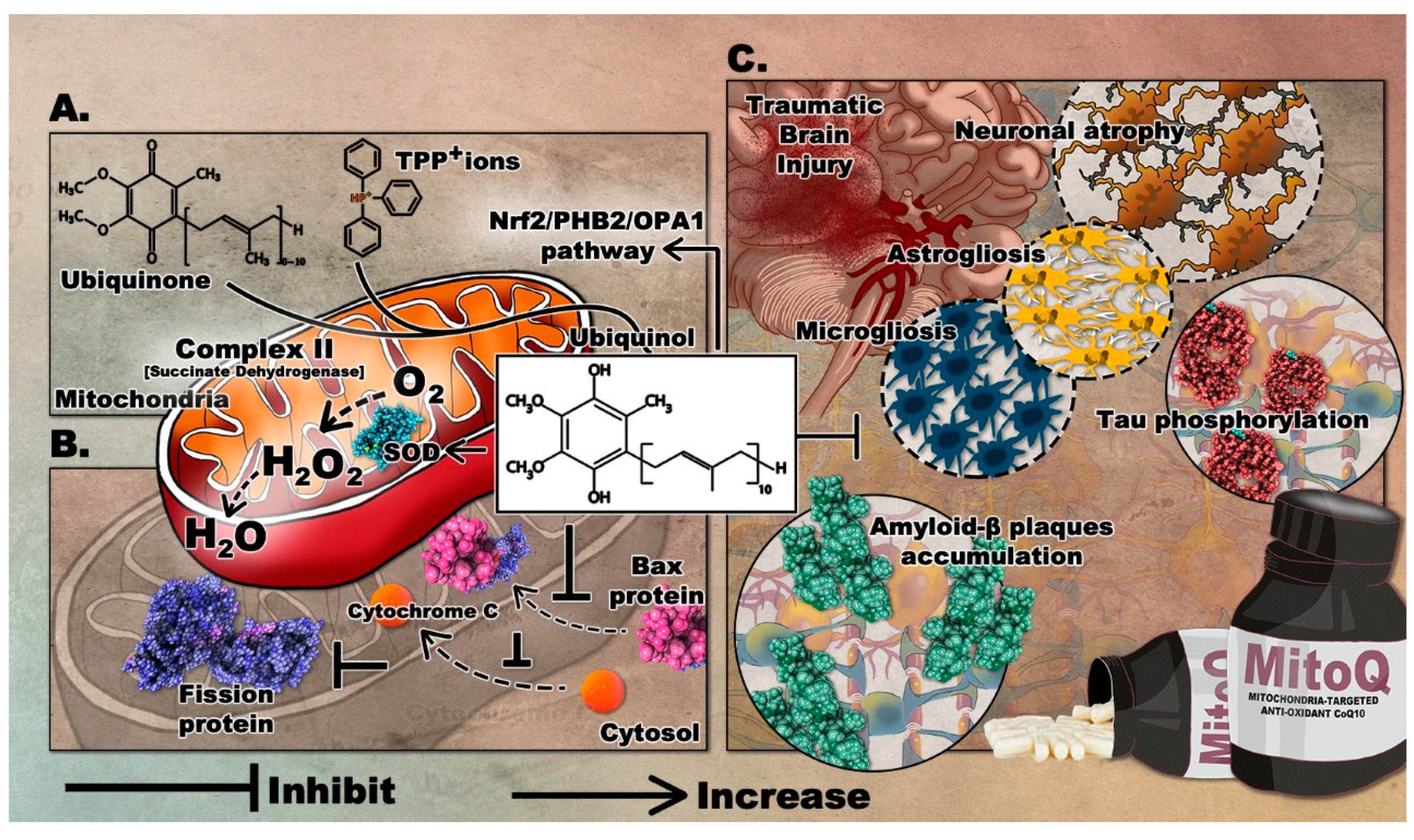
3.3. Mitoquinone (MitoQ)
3.3.1. Effects of MitoQ in Preclinical Studies of Neurodegenerative Diseases
3.3.2. Effects of MitoQ in Preclinical Models of TBI
3.3.3. Safety of MitoQ in Clinical Trials in Neurodegenerative Disorders
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Menon, D.K.; Schwab, K.; Wright, D.W.; Maas, A.I. Position statement: Definition of traumatic brain injury. Arch. Phys. Med. Rehabil. 2010, 91, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Peeters, W.; van den Brande, R.; Polinder, S.; Brazinova, A.; Steyerberg, E.W.; Lingsma, H.F.; Maas, A.I. Epidemiology of traumatic brain injury in Europe. Acta Neurochir. 2015, 157, 1683–1696. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.; Woessner, D. Sports-related traumatic brain injury. Prim. Care 2015, 42, 243–248. [Google Scholar] [CrossRef]
- Wojcik, B.E.; Stein, C.R.; Bagg, K.; Humphrey, R.J.; Orosco, J. Traumatic brain injury hospitalizations of U.S. army soldiers deployed to Afghanistan and Iraq. Am. J. Prev. Med. 2010, 38, S108–S116. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.D.; Griswold, D.P. Traumatic brain injury: A global challenge. Lancet Neurol. 2017, 16, 949–950. [Google Scholar] [CrossRef]
- El-Menyar, A.; Mekkodathil, A.; Al-Thani, H.; Consunji, R.; Latifi, R. Incidence, demographics, and outcome of traumatic brain injury in the Middle East: A systematic review. World Neurosurg. 2017, 107, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Buki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef]
- Teasdale, G.; Jennett, B. Assessment of coma and impaired consciousness. A practical scale. Lancet (Lond. Engl.) 1974, 2, 81–84. [Google Scholar] [CrossRef]
- McCrea, M.; Guskiewicz, K.M.; Marshall, S.W.; Barr, W.; Randolph, C.; Cantu, R.C.; Onate, J.A.; Yang, J.; Kelly, J.P. Acute effects and recovery time following concussion in collegiate football players: The NCAA Concussion Study. JAMA 2003, 290, 2556–2563. [Google Scholar] [CrossRef]
- Yang, Z.; Lin, F.; Weissman, A.S.; Jaalouk, E.; Xue, Q.S.; Wang, K.K. A repetitive concussive head injury model in mice. J. Vis. Exp. 2016, 116, 54530. [Google Scholar] [CrossRef] [PubMed]
- Pervez, M.; Kitagawa, R.S.; Chang, T.R. Definition of traumatic brain injury, neurosurgery, trauma orthopedics, neuroimaging, psychology, and psychiatry in mild traumatic brain injury. Neuroimaging Clin. N. Am. 2018, 28, 1–13. [Google Scholar] [CrossRef]
- Galgano, M.; Toshkezi, G.; Qiu, X.; Russell, T.; Chin, L.; Zhao, L.-R. Traumatic brain injury: Current treatment strategies and future endeavors. Cell Transplant. 2017, 26, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Fehily, B.; Fitzgerald, M. Repeated mild traumatic brain injury: Potential mechanisms of damage. Cell Transplant. 2017, 26, 1131–1155. [Google Scholar] [CrossRef]
- Albensi, B.C. Models of brain injury and alterations in synaptic plasticity. J. Neurosci. Res. 2001, 65, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Zohar, O.; Schreiber, S.; Getslev, V.; Schwartz, J.P.; Mullins, P.G.; Pick, C.G. Closed-head minimal traumatic brain injury produces long-term cognitive deficits in mice. Neuroscience 2003, 118, 949–955. [Google Scholar] [CrossRef]
- Barkhoudarian, G.; Hovda, D.A.; Giza, C.C. The molecular pathophysiology of concussive brain injury—An update. Phys. Med. Rehabil. Clin. N. Am. 2016, 27, 373–393. [Google Scholar] [CrossRef]
- Hayes, R.L.; Jenkins, L.W.; Lyeth, B.G. Neurotransmitter-mediated mechanisms of traumatic brain injury: Acetylcholine and excitatory amino acids. J. Neurotrauma 1992, 9 (Suppl. S1), S173–S187. [Google Scholar]
- Strecker, G.J.; Jackson, M.B.; Dudek, F.E. Blockade of NMDA-activated channels by magnesium in the immature rat hippocampus. J. Neurophysiol. 1994, 72, 1538–1548. [Google Scholar] [CrossRef]
- Starkov, A.A.; Chinopoulos, C.; Fiskum, G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium 2004, 36, 257–264. [Google Scholar] [CrossRef]
- Lewen, A.; Matz, P.; Chan, P.H. Free radical pathways in CNS injury. J. Neurotrauma 2000, 17, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.C.; Beart, P.M.; Shin, Y.S.; Chen, M.J.; Cheung, N.S.; Nagley, P. Oxidative stress: Emerging mitochondrial and cellular themes and variations in neuronal injury. J. Alzheimers Dis. 2010, 20 (Suppl. S2), S453–S473. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting free radicals in oxidative Stress-related human diseases. Trends Pharmacol. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, B.; Tann, A.W.; Mitra, S. Age- and tissue-specific changes in mitochondrial and nuclear DNA base excision repair activity in mice: Susceptibility of skeletal muscles to oxidative injury. Mech. Ageing Dev. 2010, 131, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Shohami, E.; Beit-Yannai, E.; Horowitz, M.; Kohen, R. Oxidative stress in closed-head injury: Brain antioxidant capacity as an indicator of functional outcome. J. Cereb. Blood Flow Metab. 1997, 17, 1007–1019. [Google Scholar] [CrossRef]
- Cernak, I.; Savic, V.J.; Kotur, J.; Prokic, V.; Veljovic, M.; Grbovic, D. Characterization of plasma magnesium concentration and oxidative stress following graded traumatic brain injury in humans. J. Neurotrauma 2000, 17, 53–68. [Google Scholar] [CrossRef]
- Hall, E.D.; Yonkers, P.A.; Andrus, P.K.; Cox, J.W.; Anderson, D.K. Biochemistry and pharmacology of lipid antioxidants in acute brain and spinal cord injury. J. Neurotrauma 1992, 9 (Suppl. S2), S425–S442. [Google Scholar] [PubMed]
- Marklund, N.; Clausen, F.; Lewander, T.; Hillered, L. Monitoring of reactive oxygen species production after traumatic brain injury in rats with microdialysis and the 4-hydroxybenzoic acid trapping method. J. Neurotrauma 2001, 18, 1217–1227. [Google Scholar] [CrossRef]
- Choi, B.Y.; Jang, B.G.; Kim, J.H.; Lee, B.E.; Sohn, M.; Song, H.K.; Suh, S.W. Prevention of traumatic brain injury-induced neuronal death by inhibition of NADPH oxidase activation. Brain Res. 2012, 1481, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.G.; Laird, M.D.; Han, D.; Nguyen, K.; Scott, E.; Dong, Y.; Dhandapani, K.M.; Brann, D.W. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS ONE 2012, 7, e34504. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, C.; Prata, C.; Dalla Sega, F.V.; Piperno, R.; Hrelia, S. Traumatic brain injury and NADPH oxidase: A deep relationship. Oxidative Med. Cell. Longev. 2015, 2015, 370312. [Google Scholar] [CrossRef] [PubMed]
- Tavazzi, B.; Signoretti, S.; Lazzarino, G.; Amorini, A.M.; Delfini, R.; Cimatti, M.; Marmarou, A.; Vagnozzi, R. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery 2005, 56, 582–589. [Google Scholar] [CrossRef]
- Hall, E.D.; Vaishnav, R.A.; Mustafa, A.G. Antioxidant therapies for traumatic brain injury. Neurotherapeutics 2010, 7, 51–61. [Google Scholar] [CrossRef]
- Kim, J.S.; He, L.; Lemasters, J.J. Mitochondrial permeability transition: A common pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 463–470. [Google Scholar] [CrossRef]
- Tavazzi, B.; Vagnozzi, R.; Signoretti, S.; Amorini, A.M.; Finocchiaro, A.; Cimatti, M.; Delfini, R.; Di Pietro, V.; Belli, A.; Lazzarino, G. Temporal window of metabolic brain vulnerability to concussions: Oxidative and nitrosative stresses—Part II. Neurosurgery 2007, 61, 390–396. [Google Scholar] [CrossRef]
- Hummel, S.G.; Fischer, A.J.; Martin, S.M.; Schafer, F.Q.; Buettner, G.R. Nitric oxide as a cellular antioxidant: A little goes a long way. Free Radic. Biol. Med. 2006, 40, 501–506. [Google Scholar] [CrossRef]
- Bains, M.; Hall, E.D. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim. Biophys. Acta 2012, 1822, 675–684. [Google Scholar] [CrossRef]
- Di Pietro, V.; Yakoub, K.M.; Caruso, G.; Lazzarino, G.; Signoretti, S.; Barbey, A.K.; Tavazzi, B.; Lazzarino, G.; Belli, A.; Amorini, A.M. Antioxidant Therapies in traumatic brain injury. Antioxidants 2020, 9, 260. [Google Scholar] [CrossRef]
- Hall, E.D. Beneficial effects of acute intravenous ibuprofen on neurologic recovery of head-injured mice: Comparison of cyclooxygenase inhibition with inhibition of thromboxane A2 synthetase or 5-lipoxygenase. Cent. Nerv. Syst. Trauma 1985, 2, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Muizelaar, J.P.; Kupiec, J.W.; Rapp, L.A. PEG-SOD after head injury. J. Neurosurg. 1995, 83, 942. [Google Scholar] [CrossRef] [PubMed]
- Carroll, R.T.; Galatsis, P.; Borosky, S.; Kopec, K.K.; Kumar, V.; Althaus, J.S.; Hall, E.D. 4-Hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (Tempol) inhibits peroxynitrite-mediated phenol nitration. Chem. Res. Toxicol. 2000, 13, 294–300. [Google Scholar] [CrossRef]
- Hall, E.D.; Yonkers, P.A.; McCall, J.M.; Braughler, J.M. Effects of the 21-aminosteroid U74006F on experimental head injury in mice. J. Neurosurg. 1988, 68, 456–461. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, T.K.; Thomas, M.; Smith, D.; Banbury, M. The novel 21-aminosteroid U74006F attenuates cerebral edema and improves survival after brain injury in the rat. J. Neurotrauma 1992, 9, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Dimlich, R.V.; Tornheim, P.A.; Kindel, R.M.; Hall, E.D.; Braughler, J.M.; McCall, J.M. Effects of a 21-aminosteroid (U-74006F) on cerebral metabolites and edema after severe experimental head trauma. Adv. Neurol. 1990, 52, 365–375. [Google Scholar]
- Marshall, L.F.; Maas, A.I.; Marshall, S.B.; Bricolo, A.; Fearnside, M.; Iannotti, F.; Klauber, M.R.; Lagarrigue, J.; Lobato, R.; Persson, L. A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. J. Neurosurg. 1998, 89, 519–525. [Google Scholar] [CrossRef]
- Rizvi, S.; Raza, S.T.; Ahmed, F.; Ahmad, A.; Abbas, S.; Mahdi, F. The role of vitamin E in human health and some diseases. Sultan Qaboos Univ. Med. J. 2014, 14, e157–e165. [Google Scholar]
- Sharma, S.; Zhuang, Y.; Ying, Z.; Wu, A.; Gomez-Pinilla, F. Dietary curcumin supplementation counteracts reduction in levels of molecules involved in energy homeostasis after brain trauma. Neuroscience 2009, 161, 1037–1044. [Google Scholar] [CrossRef]
- Ozdemir, D.; Uysal, N.; Gonenc, S.; Acikgoz, O.; Sonmez, A.; Topcu, A.; Ozdemir, N.; Duman, M.; Semin, I.; Ozkan, H. Effect of melatonin on brain oxidative damage induced by traumatic brain injury in immature rats. Physiol. Res. 2005, 54, 631–637. [Google Scholar]
- Toklu, H.Z.; Hakan, T.; Biber, N.; Solakoglu, S.; Ogunc, A.V.; Sener, G. The protective effect of alpha lipoic acid against traumatic brain injury in rats. Free Radic. Res. 2009, 43, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.D.; Braughler, J.M.; Yonkers, P.A.; Smith, S.L.; Linseman, K.L.; Means, E.D.; Scherch, H.M.; Von Voigtlander, P.F.; Lahti, R.A.; Jacobsen, E.J. U-78517F: A potent inhibitor of lipid peroxidation with activity in experimental brain injury and ischemia. J. Pharmacol. Exp. Ther. 1991, 258, 688–694. [Google Scholar] [PubMed]
- Long, D.A.; Ghosh, K.; Moore, A.N.; Dixon, C.E.; Dash, P.K. Deferoxamine improves spatial memory performance following experimental brain injury in rats. Brain Res. 1996, 717, 109–117. [Google Scholar] [CrossRef]
- Gu, Y.; Hua, Y.; Keep, R.F.; Morgenstern, L.B.; Xi, G. Deferoxamine reduces intracerebral hematoma-induced iron accumulation and neuronal death in piglets. Stroke 2009, 40, 2241–2243. [Google Scholar] [CrossRef] [PubMed]
- Panter, S.S.; Braughler, J.M.; Hall, E.D. Dextran-coupled deferoxamine improves outcome in a murine model of head injury. J. Neurotrauma 1992, 9, 47–53. [Google Scholar] [CrossRef]
- Kikuchi, K.; Kawahara, K.-I.; Uchikado, H.; Miyagi, N.; Kuramoto, T.; Miyagi, T.; Morimoto, Y.; Ito, T.; Tancharoen, S.; Miura, N.; et al. Potential of edaravone for neuroprotection in neurologic diseases that do not involve cerebral infarction. Exp. Ther. Med. 2011, 2, 771–775. [Google Scholar] [CrossRef]
- Yoshino, H. Edaravone for the treatment of amyotrophic lateral sclerosis. Expert Rev. Neurother. 2019, 19, 185–193. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Ito, H.; Wate, R.; Zhang, J.; Ohnishi, S.; Kaneko, S.; Ito, H.; Nakano, S.; Kusaka, H. Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp. Neurol. 2008, 213, 448–455. [Google Scholar] [CrossRef]
- Aoki, M.; Warita, H.; Mizuno, H.; Suzuki, N.; Yuki, S.; Itoyama, Y. Feasibility study for functional test battery of SOD transgenic rat (H46R) and evaluation of edaravone, a free radical scavenger. Brain Res. 2011, 1382, 321–325. [Google Scholar] [CrossRef]
- Ikeda, K.; Iwasaki, Y. Edaravone, a free radical scavenger, delayed symptomatic and pathological progression of motor neuron disease in the wobbler mouse. PLoS ONE 2015, 10, e0140316. [Google Scholar] [CrossRef] [PubMed]
- Shou, L.; Bei, Y.; Song, Y.; Wang, L.; Ai, L.; Yan, Q.; He, W. Nrf2 mediates the protective effect of edaravone after chlorpyrifos-induced nervous system toxicity. Environ. Toxicol. 2019, 34, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Guo, Y.; Wang, H.; Zhao, L.; Ma, Z.; Li, T.; Liu, J.; Sun, M.; Jian, Y.; Yao, L.; et al. Edaravone reduces Abeta-induced oxidative damage in SH-SY5Y cells by activating the Nrf2/ARE signaling pathway. Life Sci. 2019, 221, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yu, D.; Xu, Z. Edaravone prevents neurotoxicity of mutant L166P DJ-1 in Parkinson’s disease. J. Mol. Neurosci. 2013, 51, 539–549. [Google Scholar] [CrossRef]
- Miyamoto, K.; Ohtaki, H.; Dohi, K.; Tsumuraya, T.; Song, D.; Kiriyama, K.; Satoh, K.; Shimizu, A.; Aruga, T.; Shioda, S. Therapeutic time window for edaravone treatment of traumatic brain injury in mice. Biomed. Res. Int. 2013, 2013, 379206. [Google Scholar] [CrossRef]
- Wang, G.H.; Jiang, Z.L.; Li, Y.C.; Li, X.; Shi, H.; Gao, Y.Q.; Vosler, P.S.; Chen, J. Free-radical scavenger edaravone treatment confers neuroprotection against traumatic brain injury in rats. J. Neurotrauma 2011, 28, 2123–2134. [Google Scholar] [CrossRef]
- Itoh, T.; Satou, T.; Nishida, S.; Tsubaki, M.; Hashimoto, S.; Ito, H. The novel free radical scavenger, edaravone, increases neural stem cell number around the area of damage following rat traumatic brain injury. Neurotox. Res. 2009, 16, 378–389. [Google Scholar] [CrossRef]
- Itoh, T.; Satou, T.; Nishida, S.; Tsubaki, M.; Imano, M.; Hashimoto, S.; Ito, H. Edaravone protects against apoptotic neuronal cell death and improves cerebral function after traumatic brain injury in rats. Neurochem. Res. 2010, 35, 348–355. [Google Scholar] [CrossRef]
- Higashi, Y.; Hoshijima, M.; Yawata, T.; Nobumoto, A.; Tsuda, M.; Shimizu, T.; Saito, M.; Ueba, T. Suppression of oxidative stress and 5-lipoxygenase activation by edaravone improves depressive-like behavior after concussion. J. Neurotrauma 2014, 31, 1689–1699. [Google Scholar] [CrossRef]
- Zhang, M.; Teng, C.H.; Wu, F.F.; Ge, L.Y.; Xiao, J.; Zhang, H.Y.; Chen, D.Q. Edaravone attenuates traumatic brain injury through anti-inflammatory and anti-oxidative modulation. Exp. Ther. Med. 2019, 18, 467–474. [Google Scholar] [CrossRef]
- Tomasello, G.; Armenia, I.; Molla, G. The protein imager: A full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics 2020, 36, 2909–2911. [Google Scholar] [CrossRef]
- Yoshino, H.; Kimura, A. Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph. Lateral Scler. 2006, 7, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Itoyama, Y.; Sobue, G.; Tsuji, S.; Aoki, M.; Doyu, M.; Hamada, C.; Kondo, K.; Yoneoka, T.; Akimoto, M.; et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Front. Degener 2014, 15, 610–617. [Google Scholar] [CrossRef]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Watanabe, T.; Tahara, M.; Todo, S. The novel antioxidant edaravone: From bench to bedside. Cardiovasc. Ther. 2008, 26, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Lapchak, P.A. A critical assessment of edaravone acute ischemic stroke efficacy trials: Is edaravone an effective neuroprotective therapy? Expert Opin. Pharmacother. 2010, 11, 1753–1763. [Google Scholar] [CrossRef]
- Kikuchi, K.; Miura, N.; Kawahara, K.I.; Murai, Y.; Morioka, M.; Lapchak, P.A.; Tanaka, E. Edaravone (Radicut), a free radical scavenger, is a potentially useful addition to thrombolytic therapy in patients with acute ischemic stroke. Biomed. Rep. 2013, 1, 7–12. [Google Scholar] [CrossRef]
- Dohi, K.; Satoh, K.; Mihara, Y.; Nakamura, S.; Miyake, Y.; Ohtaki, H.; Nakamachi, T.; Yoshikawa, T.; Shioda, S.; Aruga, T. Alkoxyl radical-scavenging activity of edaravone in patients with traumatic brain injury. J. Neurotrauma 2006, 23, 1591–1599. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Ann. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef]
- James, A.M.; Cocheme, H.M.; Smith, R.A.; Murphy, M.P. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 2005, 280, 21295–21312. [Google Scholar] [CrossRef]
- Doughan, A.K.; Dikalov, S.I. Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxid. Redox Signal. 2007, 9, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.A.; Klein, S.R.; Bonar, S.J.; Zielonka, J.; Mizuno, N.; Dickey, J.S.; Keller, P.W.; Joseph, J.; Kalyanaraman, B.; Shacter, E. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J. Biol. Chem. 2010, 285, 34447–34459. [Google Scholar] [CrossRef]
- Solesio, M.E.; Prime, T.A.; Logan, A.; Murphy, M.P.; Jiménez, M.D.M.A.; Jordan, J.; Galindo, M.F. The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochim. Biophys. Acta 2013, 1832, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Chandran, K.; Kalivendi, S.V.; Joseph, J.; Antholine, W.E.; Hillard, C.J.; Kanthasamy, A.; Kanthasamy, A.; Kalyanaraman, B. Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free Radic. Biol. Med. 2010, 49, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Ünal, İ.; Çalışkan-Ak, E.; Üstündağ, Ü.V.; Ateş, P.S.; Alturfan, A.A.; Altinoz, M.A.; Elmaci, I.; Emekli-Alturfan, E. Neuroprotective effects of mitoquinone and oleandrin on Parkinson’s disease model in zebrafish. Int. J. Neurosci. 2019, 130, 1–9. [Google Scholar] [CrossRef]
- Pinho, B.R.; Duarte, A.I.; Canas, P.M.; Moreira, P.I.; Murphy, M.P.; Oliveira, J.M.A. The interplay between redox signalling and proteostasis in neurodegeneration: In vivo effects of a mitochondria-targeted antioxidant in Huntington’s disease mice. Free Radic. Biol. Med. 2020, 146, 372–382. [Google Scholar] [CrossRef]
- Chen, W.; Guo, C.; Jia, Z.; Wang, J.; Xia, M.; Li, C.; Li, M.; Yin, Y.; Tang, X.; Chen, T.; et al. Inhibition of mitochondrial ROS by MitoQ alleviates white matter injury and improves outcomes after intracerebral haemorrhage in mice. Oxidative Med. Cell. Longev. 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- Miquel, E.; Cassina, A.; Martinez-Palma, L.; Souza, J.M.; Bolatto, C.; Rodriguez-Bottero, S.; Logan, A.; Smith, R.A.; Murphy, M.P.; Barbeito, L.; et al. Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2014, 70, 204–213. [Google Scholar] [CrossRef]
- Young, M.L.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Mol. Cell. Neurosci. 2019, 101, 103409. [Google Scholar] [CrossRef]
- Zhang, T.; Xu, S.; Wu, P.; Zhou, K.; Wu, L.; Xie, Z.; Xu, W.; Luo, X.; Li, P.; Ocak, U.; et al. Mitoquinone attenuates blood-brain barrier disruption through Nrf2/PHB2/OPA1 pathway after subarachnoid hemorrhage in rats. Exp. Neurol. 2019, 317, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, H.; Shen, R.; Fang, J.; Yang, Y.; Dai, W.; Zhu, Y.; Zhou, M. Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. Am. J. Transl. Res. 2018, 10, 1887–1899. [Google Scholar] [PubMed]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.L.; Scafidi, S.; McKenna, M.C.; Fiskum, G. Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp. Neurol. 2009, 218, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H. Mitochondria-targeted peptide antioxidants: Novel neuroprotective agents. AAPS J. 2006, 8, E521–E531. [Google Scholar] [CrossRef] [PubMed]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 1670–1674. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Animal Species | TBI Model/Device | Drug Administration | Behavioral Outcomes Following Edaravone Administration | Molecular Outcomes Following Edaravone Administration | Ref |
|---|---|---|---|---|---|
| C57BL/6 mice | Electromagnetic CCI | Injection of 3 mg/kg (100–150 µL volume) into jugular vein | - |
| [65] |
| Male Sprague-Dawley rats | Feeney’s weight-drop model | Injection of 1.5 mg/kg via vena caudalis at 2 h and 10 h after TBI | - |
| [66] |
| 10-week old male Wistar rats | Pneumatic-controlled injury device | Intravenous injection of 3 mg/mL edaravone at 3 mg/kg directly after TBI | Decrease in the arrival time to platform (MWM) |
| [67,68] |
| dMale C57BL/6 mice (12–16 weeks old) | Concussive head trauma device (using a vertical metal guide tube) | Intravenous injection of 3 mg/mL edaravone at 3 mg/kg, directly after TBI | Decrease in immobility time in FST |
| [69] |
| C57BL/6 mice | Pneumatic controlled cortical impact | Intraperitoneal injection of 3 mg/kg of edaravone, 1 h post-TBI | Decrease in neurological deficits (NSS) |
| [70] |
| Model | Experimental Procedure | Behavioral Outcomes | Molecular Outcomes | Biomarkers | Ref |
|---|---|---|---|---|---|
| Huntington’s disease |
|
|
|
| [87] |
| TBI |
| Lowered NSS scoring at day 1 and 3 post-TBI |
|
| [92] |
| ALS |
|
|
|
| [89] |
| Alzheimer’s disease |
| Enhanced learning and spatial memory retention by MWM |
|
| [93] |
| Parkinson’s Disease |
| Improved locomotor activities in open field and Rotarod |
|
| [85] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismail, H.; Shakkour, Z.; Tabet, M.; Abdelhady, S.; Kobaisi, A.; Abedi, R.; Nasrallah, L.; Pintus, G.; Al-Dhaheri, Y.; Mondello, S.; et al. Traumatic Brain Injury: Oxidative Stress and Novel Anti-Oxidants Such as Mitoquinone and Edaravone. Antioxidants 2020, 9, 943. https://doi.org/10.3390/antiox9100943
Ismail H, Shakkour Z, Tabet M, Abdelhady S, Kobaisi A, Abedi R, Nasrallah L, Pintus G, Al-Dhaheri Y, Mondello S, et al. Traumatic Brain Injury: Oxidative Stress and Novel Anti-Oxidants Such as Mitoquinone and Edaravone. Antioxidants. 2020; 9(10):943. https://doi.org/10.3390/antiox9100943
Chicago/Turabian StyleIsmail, Helene, Zaynab Shakkour, Maha Tabet, Samar Abdelhady, Abir Kobaisi, Reem Abedi, Leila Nasrallah, Gianfranco Pintus, Yusra Al-Dhaheri, Stefania Mondello, and et al. 2020. "Traumatic Brain Injury: Oxidative Stress and Novel Anti-Oxidants Such as Mitoquinone and Edaravone" Antioxidants 9, no. 10: 943. https://doi.org/10.3390/antiox9100943
APA StyleIsmail, H., Shakkour, Z., Tabet, M., Abdelhady, S., Kobaisi, A., Abedi, R., Nasrallah, L., Pintus, G., Al-Dhaheri, Y., Mondello, S., El-Khoury, R., Eid, A. H., Kobeissy, F., & Salameh, J. (2020). Traumatic Brain Injury: Oxidative Stress and Novel Anti-Oxidants Such as Mitoquinone and Edaravone. Antioxidants, 9(10), 943. https://doi.org/10.3390/antiox9100943