Neural Underpinnings of Obesity: The Role of Oxidative Stress and Inflammation in the Brain

 , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Mitochondrial Dysfunction in the CNS
2.1. Hypothalamus
2.2. Extra-Hypothalamic Areas
3. Endoplasmic Reticulum (ER) Stress in the CNS
3.1. Hypothalamus
3.2. Extra-Hypothalamic Areas
4. Role of Antioxidants/Anti-Inflammatory Agents in the CNS
4.1. Quercetin
4.2. Curcumin
4.3. Resveratrol
4.4. Celastrol
4.5. Epigallocatechin-3-gallate (EGCG)
4.6. Grape Seed and Skin Extract (GSSE)
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Finucane, M.M.; Stevens, G.A.; Cowan, M.J.; Danaei, G.; Lin, J.K.; Paciorek, C.J.; Singh, G.M.; Gutierrez, H.R.; Lu, Y.; Bahalim, A.N.; et al. National, regional, and global trends in body-mass index since 1980: Systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet 2011, 377, 557–567. [Google Scholar] [CrossRef]
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of obesity among adults and youth: US, 2015–2016. NCHS Data Brief 2017, 288, 1–8. [Google Scholar]
- Heron, M. Deaths: Leading causes for 2016. Natl. Vital Stat. Rep. 2018, 67, 1–77. [Google Scholar] [PubMed]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Maury, E.; Brichard, S.M. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol. Cell Endocrinol. 2010, 314, 1–16. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, R.W. Inflammation in obesity-related diseases. Surgery 2009, 145, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondria and cell signalling. J. Cell Sci. 2012, 125, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol. Life. Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, N.; Asada, R.; Saito, A.; Kanemoto, S.; Imaizumi, K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci. Rep. 2012, 2, 799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef]
- Posey, K.A.; Clegg, D.J.; Printz, R.L.; Byun, J.; Morton, G.J.; Vivekanandan-Giri, A.; Pennathur, S.; Baskin, D.G.; Heinecke, J.W.; Woods, S.C.; et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1003–E1012. [Google Scholar] [CrossRef]
- Cazettes, F.; Cohen, J.I.; Yau, P.L.; Talbot, H.; Convit, A. Obesity-mediated inflammation may damage the brain circuit that regulates food intake. Brain Res. 2011, 1373, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J.; Broadwell, R.D. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation 1995, 2, 241–248. [Google Scholar] [CrossRef]
- Kleinridders, A.; Schenten, D.; Konner, A.C.; Belgardt, B.F.; Mauer, J.; Okamura, T.; Wunderlich, F.T.; Medzhitov, R.; Bruning, J.C. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009, 10, 249–259. [Google Scholar] [CrossRef]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef]
- Konner, A.C.; Bruning, J.C. Toll-like receptors: Linking inflammation to metabolism. Trends Endocrinol. Metab. 2011, 22, 16–23. [Google Scholar] [CrossRef]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef]
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.; Velloso, L.A. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 2005, 146, 4192–4199. [Google Scholar] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [PubMed]
- Douglass, J.D.; Dorfman, M.D.; Fasnacht, R.; Shaffer, L.D.; Thaler, J.P. Astrocyte IKKbeta/NF-kappaB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol. Metab. 2017, 6, 366–373. [Google Scholar] [PubMed]
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nature 2006, 443, 289–295. [Google Scholar]
- Shin, A.C.; Zheng, H.; Berthoud, H.R. An expanded view of energy homeostasis: Neural integration of metabolic, cognitive, and emotional drives to eat. Physiol. Behav. 2009, 97, 572–580. [Google Scholar]
- Goldenthal, M.J.; Marin-Garcia, J. Mitochondrial signaling pathways: A receiver/integrator organelle. Mol. Cell Biochem. 2004, 262, 1–16. [Google Scholar]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 330. [Google Scholar]
- Carraro, R.S.; Souza, G.F.; Solon, C.; Razolli, D.S.; Chausse, B.; Barbizan, R.; Victorio, S.C.; Velloso, L.A. Hypothalamic mitochondrial abnormalities occur downstream of inflammation in diet-induced obesity. Mol. Cell Endocrinol. 2018, 460, 238–245. [Google Scholar]
- Schneeberger, M.; Dietrich, M.O.; Sebastian, D.; Imbernon, M.; Castano, C.; Garcia, A.; Esteban, Y.; Gonzalez-Franquesa, A.; Rodriguez, I.C.; Bortolozzi, A.; et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 2013, 155, 172–187. [Google Scholar]
- Kim, J.D.; Leyva, S.; Diano, S. Hormonal regulation of the hypothalamic melanocortin system. Front. Physiol. 2014, 5, 480. [Google Scholar]
- Dietrich, M.O.; Liu, Z.W.; Horvath, T.L. Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell 2013, 155, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Colombani, A.L.; Carneiro, L.; Benani, A.; Galinier, A.; Jaillard, T.; Duparc, T.; Offer, G.; Lorsignol, A.; Magnan, C.; Casteilla, L.; et al. Enhanced hypothalamic glucose sensing in obesity: Alteration of redox signaling. Diabetes 2009, 58, 2189–2197. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, S.; Nestorov, J.; Matic, G.; Elakovic, I. Chronic stress combined with a fructose diet reduces hypothalamic insulin signaling and antioxidative defense in female rats. Neuroendocrinology 2019, 108, 278–290. [Google Scholar] [PubMed]
- De Bona Schraiber, R.; De Mello, A.H.; Garcez, M.L.; De Bem Silveira, G.; Zacaron, R.P.; De Souza Goldim, M.P.; Budni, J.; Silveira, P.C.L.; Petronilho, F.; Ferreira, G.K.; et al. Diet-induced obesity causes hypothalamic neurochemistry alterations in Swiss mice. Metab. Brain Dis. 2019, 34, 565–573. [Google Scholar] [CrossRef]
- De Mello, A.H.; Schraiber, R.B.; Goldim, M.P.S.; Garcez, M.L.; Gomes, M.L.; de Bem Silveira, G.; Zaccaron, R.P.; Schuck, P.F.; Budni, J.; Silveira, P.C.L.; et al. Omega-3 fatty acids attenuate brain alterations in high-fat diet-induced obesity model. Mol. Neurobiol. 2019, 56, 513–524. [Google Scholar] [CrossRef]
- Doll, D.N.; Rellick, S.L.; Barr, T.L.; Ren, X.; Simpkins, J.W. Rapid mitochondrial dysfunction mediates TNF-alpha-induced neurotoxicity. J. Neurochem. 2015, 132, 443–451. [Google Scholar] [CrossRef]
- Kastl, L.; Sauer, S.W.; Ruppert, T.; Beissbarth, T.; Becker, M.S.; Suss, D.; Krammer, P.H.; Gulow, K. TNF-alpha mediates mitochondrial uncoupling and enhances ROS-dependent cell migration via NF-kappaB activation in liver cells. FEBS Lett. 2014, 588, 175–183. [Google Scholar]
- Taylor, D.J.; Faragher, E.B.; Evanson, J.M. Inflammatory cytokines stimulate glucose uptake and glycolysis but reduce glucose oxidation in human dermal fibroblasts in vitro. Circ. Shock 1992, 37, 105–110. [Google Scholar]
- Zell, R.; Geck, P.; Werdan, K.; Boekstegers, P. TNF-alpha and IL-1 alpha inhibit both pyruvate dehydrogenase activity and mitochondrial function in cardiomyocytes: Evidence for primary impairment of mitochondrial function. Mol. Cell Biochem. 1997, 177, 61–67. [Google Scholar]
- Clemmensen, C.; Muller, T.D.; Woods, S.C.; Berthoud, H.R.; Seeley, R.J.; Tschop, M.H. Gut-brain cross-talk in metabolic control. Cell 2017, 168, 758–774. [Google Scholar] [CrossRef]
- Batterink, L.; Yokum, S.; Stice, E. Body mass correlates inversely with inhibitory control in response to food among adolescent girls: An fMRI study. Neuroimage 2010, 52, 1696–1703. [Google Scholar] [CrossRef] [PubMed]
- Kishinevsky, F.I.; Cox, J.E.; Murdaugh, D.L.; Stoeckel, L.E.; Cook, E.W., III; Weller, R.E. fMRI reactivity on a delay discounting task predicts weight gain in obese women. Appetite 2012, 58, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, G.; Trinchese, G.; Penna, E.; Cimmino, F.; Pirozzi, C.; Lama, A.; Annunziata, C.; Catapano, A.; Mattace Raso, G.; Meli, R.; et al. High-fat diet induces neuroinflammation and mitochondrial impairment in mice cerebral cortex and synaptic fraction. Front. Cell Neurosci. 2019, 13, 509. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, D.; Galizzi, G.; Amato, A.; Terzo, S.; Picone, P.; Cristaldi, L.; Mule, F.; Di Carlo, M. Regular intake of pistachio mitigates the deleterious effects of a high fat-diet in the brain of obese mice. Antioxidants 2020, 9, 317. [Google Scholar]
- Leffa, D.D.; Rezin, G.T.; Daumann, F.; Longaretti, L.M.; Dajori, A.L.; Gomes, L.M.; Silva, M.C.; Streck, E.L.; De Andrade, V.M. Effects of acerola (Malpighia emarginata DC.) juice intake on brain energy metabolism of mice fed a cafeteria diet. Mol. Neurobiol. 2017, 54, 954–963. [Google Scholar] [CrossRef]
- De Farias, B.X.; Costa, A.B.; Engel, N.A.; De Souza Goldim, M.P.; Da Rosa Turatti, C.; Cargnin-Cavalho, A.; Fortunato, J.J.; Petronilho, F.; Jeremias, I.C.; Rezin, G.T.; et al. Donepezil prevents inhibition of cerebral energetic metabolism without altering behavioral parameters in animal model of obesity. Neurochem. Res. 2020, 45, 2487–2498. [Google Scholar] [CrossRef]
- Davidson, T.L.; Jones, S.; Roy, M.; Stevenson, R.J. The cognitive control of eating and body weight: It’s more than what you “think”. Front. Psychol. 2019, 10, 62. [Google Scholar] [CrossRef]
- Parcet, M.A.; Adrian-Ventura, J.; Costumero, V.; Avila, C. Individual differences in hippocampal volume as a function of BMI and reward sensitivity. Front. Behav. Neurosci. 2020, 14, 53. [Google Scholar]
- Mestre, Z.L.; Bischoff-Grethe, A.; Eichen, D.M.; Wierenga, C.E.; Strong, D.; Boutelle, K.N. Hippocampal atrophy and altered brain responses to pleasant tastes among obese compared with healthy weight children. Int. J. Obes. 2017, 41, 1496–1502. [Google Scholar] [CrossRef]
- Cheke, L.G.; Bonnici, H.M.; Clayton, N.S.; Simons, J.S. Obesity and insulin resistance are associated with reduced activity in core memory regions of the brain. Neuropsychologia 2017, 96, 137–149. [Google Scholar] [CrossRef]
- Zanini, P.; Arbo, B.D.; Niches, G.; Czarnabay, D.; Benetti, F.; Ribeiro, M.F.; Cecconello, A.L. Diet-induced obesity alters memory consolidation in female rats. Physiol. Behav. 2017, 180, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Woodie, L.; Blythe, S. The differential effects of high-fat and high-fructose diets on physiology and behavior in male rats. Nutr. Neurosci. 2018, 21, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Gandia, M.C.; Minarro, J.; Rodriguez-Arias, M. Behavioral profile of intermittent vs continuous access to a high fat diet during adolescence. Behav. Brain. Res. 2019, 368, 111891. [Google Scholar] [CrossRef] [PubMed]
- Hargrave, S.L.; Davidson, T.L.; Zheng, W.; Kinzig, K.P. Western diets induce blood-brain barrier leakage and alter spatial strategies in rats. Behav. Neurosci. 2016, 130, 123–135. [Google Scholar] [CrossRef]
- Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Mitschelen, M.; Koller, A.; Szalai, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A.; et al. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: Effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Carnell, S.; Gibson, C.; Benson, L.; Ochner, C.N.; Geliebter, A. Neuroimaging and obesity: Current knowledge and future directions. Obes. Rev. 2012, 13, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kroemer, N.B.; Oehme, L.; Beuthien-Baumann, B.; Goschke, T.; Smolka, M.N. Lower dopamine tone in the striatum is associated with higher body mass index. Eur. Neuropsychopharmacol. 2018, 28, 719–731. [Google Scholar] [CrossRef]
- Michaelides, M.; Miller, M.L.; Egervari, G.; Primeaux, S.D.; Gomez, J.L.; Ellis, R.J.; Landry, J.A.; Szutorisz, H.; Hoffman, A.F.; Lupica, C.R.; et al. Striatal Rgs4 regulates feeding and susceptibility to diet-induced obesity. Mol. Psychiatry 2020, 25, 2058–2069. [Google Scholar] [CrossRef]
- Nummenmaa, L.; Saanijoki, T.; Tuominen, L.; Hirvonen, J.; Tuulari, J.J.; Nuutila, P.; Kalliokoski, K. Mu-opioid receptor system mediates reward processing in humans. Nat. Commun. 2018, 9, 1500. [Google Scholar]
- Letra, L.; Pereira, D.; Castelo-Branco, M. Functional Neuroimaging in Obesity Research. Adv. Neurobiol. 2017, 19, 239–248. [Google Scholar] [CrossRef]
- Ansari, A.; Rahman, M.S.; Saha, S.K.; Saikot, F.K.; Deep, A.; Kim, K.H. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell 2017, 16, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Nerurkar, P.V.; Johns, L.M.; Buesa, L.M.; Kipyakwai, G.; Volper, E.; Sato, R.; Shah, P.; Feher, D.; Williams, P.G.; Nerurkar, V.R. Momordica charantia (bitter melon) attenuates high-fat diet-associated oxidative stress and neuroinflammation. J. Neuroinflamm. 2011, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Nguyen, C.U.; Chong, T.; Michel, C.R.; Fritz, K.S.; Reisdorph, N.; Knaub, L.; Reusch, J.E.B.; Pugazhenthi, S. SIRT3 deficiency-induced mitochondrial dysfunction and inflammasome formation in the brain. Sci. Rep. 2018, 8, 17547. [Google Scholar] [CrossRef] [PubMed]
- Porter, G.A., Jr.; Beutner, G. Cyclophilin D, Somehow a master regulator of mitochondrial function. Biomolecules 2018, 8, 176. [Google Scholar] [CrossRef]
- Devalaraja-Narashimha, K.; Diener, A.M.; Padanilam, B.J. Cyclophilin D deficiency prevents diet-induced obesity in mice. FEBS Lett. 2011, 585, 677–682. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Foufelle, F.; Velloso, L.A. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 2012, 18, 59–68. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, J.; Shi, Y.C.; Zhang, Y.; Lin, S. The role of inflammation and endoplasmic reticulum stress in obesity-related cognitive impairment. Life Sci. 2019, 233, 116707. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Nakatani, Y.; Kaneto, H.; Kawamori, D.; Yoshiuchi, K.; Hatazaki, M.; Matsuoka, T.A.; Ozawa, K.; Ogawa, S.; Hori, M.; Yamasaki, Y.; et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J. Biol. Chem. 2005, 280, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S.; et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G., Jr.; Ozcan, U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Cakir, I.; Cyr, N.E.; Perello, M.; Litvinov, B.P.; Romero, A.; Stuart, R.C.; Nillni, E.A. Obesity induces hypothalamic endoplasmic reticulum stress and impairs proopiomelanocortin (POMC) post-translational processing. J. Biol. Chem. 2013, 288, 17675–17688. [Google Scholar] [CrossRef] [PubMed]
- Contreras, C.; Gonzalez-Garcia, I.; Martinez-Sanchez, N.; Seoane-Collazo, P.; Jacas, J.; Morgan, D.A.; Serra, D.; Gallego, R.; Gonzalez, F.; Casals, N.; et al. Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell Rep. 2014, 9, 366–377. [Google Scholar] [CrossRef]
- Diaz, B.; Fuentes-Mera, L.; Tovar, A.; Montiel, T.; Massieu, L.; Martinez-Rodriguez, H.G.; Camacho, A. Saturated lipids decrease mitofusin 2 leading to endoplasmic reticulum stress activation and insulin resistance in hypothalamic cells. Brain Res. 2015, 1627, 80–89. [Google Scholar] [CrossRef]
- Timper, K.; Bruning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis. Models Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef]
- Won, J.C.; Jang, P.G.; Namkoong, C.; Koh, E.H.; Kim, S.K.; Park, J.Y.; Lee, K.U.; Kim, M.S. Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity 2009, 17, 1861–1865. [Google Scholar] [CrossRef]
- Contreras, C.; Gonzalez-Garcia, I.; Seoane-Collazo, P.; Martinez-Sanchez, N.; Linares-Pose, L.; Rial-Pensado, E.; Ferno, J.; Tena-Sempere, M.; Casals, N.; Dieguez, C.; et al. Reduction of hypothalamic endoplasmic reticulum stress activates browning of white fat and ameliorates obesity. Diabetes 2017, 66, 87–99. [Google Scholar] [CrossRef]
- Halperin, L.; Jung, J.; Michalak, M. The many functions of the endoplasmic reticulum chaperones and folding enzymes. IUBMB Life 2014, 66, 318–326. [Google Scholar] [CrossRef]
- Williams, K.W.; Liu, T.; Kong, X.; Fukuda, M.; Deng, Y.; Berglund, E.D.; Deng, Z.; Gao, Y.; Sohn, J.W.; Jia, L.; et al. Xbp1s in POMC neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab. 2014, 20, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Chowen, J.A.; Argente-Arizon, P.; Freire-Regatillo, A.; Frago, L.M.; Horvath, T.L.; Argente, J. The role of astrocytes in the hypothalamic response and adaptation to metabolic signals. Prog. Neurobiol. 2016, 144, 68–87. [Google Scholar] [CrossRef] [PubMed]
- Mendes, N.F.; Kim, Y.B.; Velloso, L.A.; Araujo, E.P. Hypothalamic microglial activation in obesity: A mini-review. Front. Neurosci. 2018, 12, 846. [Google Scholar] [CrossRef]
- Baufeld, C.; Osterloh, A.; Prokop, S.; Miller, K.R.; Heppner, F.L. High-fat diet-induced brain region-specific phenotypic spectrum of CNS resident microglia. Acta Neuropathol. 2016, 132, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Valdearcos, M.; Douglass, J.D.; Robblee, M.M.; Dorfman, M.D.; Stifler, D.R.; Bennett, M.L.; Gerritse, I.; Fasnacht, R.; Barres, B.A.; Thaler, J.P.; et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab. 2017, 26, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Valdearcos, M.; Robblee, M.M.; Benjamin, D.I.; Nomura, D.K.; Xu, A.W.; Koliwad, S.K. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014, 9, 2124–2138. [Google Scholar] [CrossRef]
- Lu, J.; Wu, D.M.; Zheng, Y.L.; Hu, B.; Cheng, W.; Zhang, Z.F.; Shan, Q. Ursolic acid improves high fat diet-induced cognitive impairments by blocking endoplasmic reticulum stress and IkappaB kinase beta/nuclear factor-kappaB-mediated inflammatory pathways in mice. Brain Behav. Immun. 2011, 25, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Sims-Robinson, C.; Zhao, S.; Hur, J.; Feldman, E.L. Central nervous system endoplasmic reticulum stress in a murine model of type 2 diabetes. Diabetologia 2012, 55, 2276–2284. [Google Scholar] [CrossRef]
- Sims-Robinson, C.; Bakeman, A.; Glasser, R.; Boggs, J.; Pacut, C.; Feldman, E.L. The role of endoplasmic reticulum stress in hippocampal insulin resistance. Exp. Neurol. 2016, 277, 261–267. [Google Scholar] [CrossRef]
- Lloyd, D.J.; McCormick, J.; Helmering, J.; Kim, K.W.; Wang, M.; Fordstrom, P.; Kaufman, S.A.; Lindberg, R.A.; Veniant, M.M. Generation and characterization of two novel mouse models exhibiting the phenotypes of the metabolic syndrome: Apob48−/-Lepob/ob mice devoid of ApoE or Ldlr. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E496–E505. [Google Scholar] [CrossRef][Green Version]
- Liang, L.; Chen, J.; Zhan, L.; Lu, X.; Sun, X.; Sui, H.; Zheng, L.; Xiang, H.; Zhang, F. Endoplasmic reticulum stress impairs insulin receptor signaling in the brains of obese rats. PLoS ONE 2015, 10, e0126384. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Wang, H.; Li, J.J.; Zhang, Y.L.; Xin, L.; Li, F.; Lou, S.J. The signaling mechanisms of hippocampal endoplasmic reticulum stress affecting neuronal plasticity-related protein levels in high fat diet-induced obese rats and the regulation of aerobic exercise. Brain Behav. Immun. 2016, 57, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, P.; Yang, Y. Neural circuit mechanisms underlying emotional regulation of homeostatic feeding. Trends Endocrinol. Metab. 2017, 28, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Geha, P.; Cecchi, G.; Todd Constable, R.; Abdallah, C.; Small, D.M. Reorganization of brain connectivity in obesity. Hum. Brain Mapp. 2017, 38, 1403–1420. [Google Scholar] [CrossRef] [PubMed]
- Castro, G.; MF, C.A.; Weissmann, L.; Quaresma, P.G.; Katashima, C.K.; Saad, M.J.; Prada, P.O. Diet-induced obesity induces endoplasmic reticulum stress and insulin resistance in the amygdala of rats. FEBS Open Bio. 2013, 3, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.S.; Shruthi, K.; Reddy, V.S.; Raghu, G.; Suryanarayana, P.; Giridharan, N.V.; Reddy, G.B. Altered ubiquitin-proteasome system leads to neuronal cell death in a spontaneous obese rat model. Biochim. et Biophys. Acta Gen. Subj. 2014, 1840, 2924–2934. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, B.B.; Cai, M.; Li, J.J.; Lou, S.J. Excessive endoplasmic reticulum stress and decreased neuroplasticity-associated proteins in prefrontal cortex of obese rats and the regulatory effects of aerobic exercise. Brain Res. Bull. 2018, 140, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Horwath, J.A.; Hurr, C.; Butler, S.D.; Guruju, M.; Cassell, M.D.; Mark, A.L.; Davisson, R.L.; Young, C.N. Obesity-induced hepatic steatosis is mediated by endoplasmic reticulum stress in the subfornical organ of the brain. JCI Insight 2017, 2, e90170. [Google Scholar] [CrossRef] [PubMed]
- Mimee, A.; Smith, P.M.; Ferguson, A.V. Circumventricular organs: Targets for integration of circulating fluid and energy balance signals? Physiol. Behav. 2013, 121, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Tun, S.; Spainhower, C.J.; Cottrill, C.L.; Lakhani, H.V.; Pillai, S.S.; Dilip, A.; Chaudhry, H.; Shapiro, J.I.; Sodhi, K. Therapeutic efficacy of antioxidants in ameliorating obesity phenotype and associated comorbidities. Front. Pharmacol. 2020, 11, 1234. [Google Scholar] [CrossRef]
- Khan, H.; Ullah, H.; Martorell, M.; Valdes, S.E.; Belwal, T.; Tejada, S.; Sureda, A.; Kamal, M.A. Flavonoids nanoparticles in cancer: Treatment, prevention and clinical prospects. Semin. Cancer Biol. 2019, 19, 30182-8. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Mukai, Y. Modulation of chronic inflammation by quercetin: The beneficial effects on obesity. J. Inflamm. Res. 2020, 13, 421–431. [Google Scholar] [CrossRef]
- Kobori, M.; Masumoto, S.; Akimoto, Y.; Oike, H. Chronic dietary intake of quercetin alleviates hepatic fat accumulation associated with consumption of a Western-style diet in C57/BL6J mice. Mol. Nutr. Food Res. 2011, 55, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Oest, M.E.; Prater, M.R. Intrauterine exposure to high saturated fat diet elevates risk of adult-onset chronic diseases in C57BL/6 mice. Birth Defects Res. B Dev. Reprod. Toxicol. 2009, 86, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Ohkoshi, E.; Miyazaki, H.; Shindo, K.; Watanabe, H.; Yoshida, A.; Yajima, H. Constituents from the leaves of Nelumbo nucifera stimulate lipolysis in the white adipose tissue of mice. Planta Med. 2007, 73, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kim, C.S.; Tu, T.H.; Kim, M.S.; Goto, T.; Kawada, T.; Choi, M.S.; Park, T.; Sung, M.K.; Yun, J.W.; et al. Quercetin protects obesity-induced hypothalamic inflammation by reducing microglia-mediated inflammatory responses via HO-1 induction. Nutrients 2017, 9, 650. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.F.; Xie, Z.X.; Qiao, Y.; Li, L.R.; Cheng, X.R.; Tang, X.; Shi, Y.H.; Le, G.W. Differential effects of quercetin on hippocampus-dependent learning and memory in mice fed with different diets related with oxidative stress. Physiol. Behav. 2015, 138, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.M.; Huang, H.I.; Fenton, M.R.; Fong, D. In vivo inhibition of nitric oxide synthase gene expression by curcumin, a cancer preventive natural product with anti-inflammatory properties. Biochem. Pharmacol. 1998, 55, 1955–1962. [Google Scholar] [CrossRef]
- Gonzales, A.M.; Orlando, R.A. Curcumin and resveratrol inhibit nuclear factor-kappaB-mediated cytokine expression in adipocytes. Nutr. Metab. 2008, 5, 17. [Google Scholar] [CrossRef]
- Kuo, J.J.; Chang, H.H.; Tsai, T.H.; Lee, T.Y. Positive effect of curcumin on inflammation and mitochondrial dysfunction in obese mice with liver steatosis. Int. J. Mol. Med. 2012, 30, 673–679. [Google Scholar] [CrossRef]
- Pendurthi, U.R.; Williams, J.T.; Rao, L.V. Inhibition of tissue factor gene activation in cultured endothelial cells by curcumin. Suppression of activation of transcription factors Egr-1, AP-1, and NF-kappa B. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3406–3413. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.L.; Li, Y.; Wen, Y.; Chen, Y.F.; Na, L.X.; Li, S.T.; Sun, C.H. Curcumin, a potential inhibitor of up-regulation of TNF-alpha and IL-6 induced by palmitate in 3T3-L1 adipocytes through NF-kappaB and JNK pathway. Biomed. Environ. Sci. 2009, 22, 32–39. [Google Scholar] [CrossRef]
- Weber, W.M.; Hunsaker, L.A.; Roybal, C.N.; Bobrovnikova-Marjon, E.V.; Abcouwer, S.F.; Royer, R.E.; Deck, L.M.; Vander Jagt, D.L. Activation of NFkappaB is inhibited by curcumin and related enones. Bioorg. Med. Chem. 2006, 14, 2450–2461. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, Q.; Wang, X.; Yu, S.; Li, L.; Wu, X.; Chen, Y.; Zhao, J.; Zhao, Y. Neuroprotection by curcumin in ischemic brain injury involves the Akt/Nrf2 pathway. PLoS ONE 2013, 8, e59843. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Zeng, Q.; Mitchell, E.S.; Xiu, J.; Duan, Y.; Li, C.; Tiwari, J.K.; Hu, Y.; Cao, X.; Zhao, Z. Curcumin enhances neurogenesis and cognition in aged rats: Implications for transcriptional interactions related to growth and synaptic plasticity. PLoS ONE 2012, 7, e31211. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lin, D.; Li, S.; Li, G.; Shyamala, S.G.; Barish, P.A.; Vernon, M.M.; Pan, J.; Ogle, W.O. Curcumin reverses impaired cognition and neuronal plasticity induced by chronic stress. Neuropharmacology 2009, 57, 463–471. [Google Scholar] [CrossRef]
- Yu, S.Y.; Gao, R.; Zhang, L.; Luo, J.; Jiang, H.; Wang, S. Curcumin ameliorates ethanol-induced memory deficits and enhanced brain nitric oxide synthase activity in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 44, 210–216. [Google Scholar] [CrossRef]
- Reeta, K.H.; Mehla, J.; Gupta, Y.K. Curcumin ameliorates cognitive dysfunction and oxidative damage in phenobarbitone and carbamazepine administered rats. Eur. J. Pharmacol. 2010, 644, 106–112. [Google Scholar] [CrossRef]
- Jaques, J.A.; Rezer, J.F.; Carvalho, F.B.; da Rosa, M.M.; Gutierres, J.M.; Goncalves, J.F.; Schmatz, R.; de Bairros, A.V.; Mazzanti, C.M.; Rubin, M.A.; et al. Curcumin protects against cigarette smoke-induced cognitive impairment and increased acetylcholinesterase activity in rats. Physiol. Behav. 2012, 106, 664–669. [Google Scholar] [CrossRef]
- Franco-Robles, E.; Campos-Cervantes, A.; Murillo-Ortiz, B.O.; Segovia, J.; Lopez-Briones, S.; Vergara, P.; Perez-Vazquez, V.; Solis-Ortiz, M.S.; Ramirez-Emiliano, J. Effects of curcumin on brain-derived neurotrophic factor levels and oxidative damage in obesity and diabetes. Appl. Physiol. Nutr. Metab. 2014, 39, 211–218. [Google Scholar] [CrossRef]
- Xu, M.X.; Yu, R.; Shao, L.F.; Zhang, Y.X.; Ge, C.X.; Liu, X.M.; Wu, W.Y.; Li, J.M.; Kong, L.D. Up-regulated fractalkine (FKN) and its receptor CX3CR1 are involved in fructose-induced neuroinflammation: Suppression by curcumin. Brain Behav. Immun. 2016, 58, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, J.; Shi, J.S. Anti-inflammatory activities of resveratrol in the brain: Role of resveratrol in microglial activation. Eur. J. Pharmacol. 2010, 636, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Das, D.K. Anti-inflammatory responses of resveratrol. Inflamm. Allergy Drug Targets 2007, 6, 168–173. [Google Scholar] [CrossRef]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Kashyap, M.P.; Kumar, V.; Al-Khedhairy, A.A.; Musarrat, J.; Pant, A.B. Protective potential of trans-resveratrol against 4-hydroxynonenal induced damage in PC12 cells. Toxicol. In Vitro 2010, 24, 1592–1598. [Google Scholar] [CrossRef]
- Rege, S.D.; Kumar, S.; Wilson, D.N.; Tamura, L.; Geetha, T.; Mathews, S.T.; Huggins, K.W.; Broderick, T.L.; Babu, J.R. Resveratrol protects the brain of obese mice from oxidative damage. Oxid. Med. Cell Longev. 2013, 2013, 419092. [Google Scholar] [CrossRef] [PubMed]
- Jeon, B.T.; Jeong, E.A.; Shin, H.J.; Lee, Y.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S.; et al. Resveratrol attenuates obesity-associated peripheral and central inflammation and improves memory deficit in mice fed a high-fat diet. Diabetes 2012, 61, 1444–1454. [Google Scholar] [CrossRef]
- Jimenez-Gomez, Y.; Mattison, J.A.; Pearson, K.J.; Martin-Montalvo, A.; Palacios, H.H.; Sossong, A.M.; Ward, T.M.; Younts, C.M.; Lewis, K.; Allard, J.S.; et al. Resveratrol improves adipose insulin signaling and reduces the inflammatory response in adipose tissue of rhesus monkeys on high-fat, high-sugar diet. Cell Metab. 2013, 18, 533–545. [Google Scholar] [CrossRef]
- Ramadori, G.; Gautron, L.; Fujikawa, T.; Vianna, C.R.; Elmquist, J.K.; Coppari, R. Central administration of resveratrol improves diet-induced diabetes. Endocrinology 2009, 150, 5326–5333. [Google Scholar] [CrossRef]
- Cascao, R.; Fonseca, J.E.; Moita, L.F. Celastrol: A spectrum of treatment opportunities in chronic diseases. Front. Med. 2017, 4, 69. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, W.; Wang, D.; Ma, X. Chinese medicine in the battle against obesity and metabolic diseases. Front. Physiol. 2018, 9, 850. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Davis, K.C.; Morgan, D.A.; Toth, B.A.; Jiang, J.; Singh, U.; Berglund, E.D.; Grobe, J.L.; Rahmouni, K.; Cui, H.; et al. Celastrol reduces obesity in MC4R deficiency and stimulates sympathetic nerve activity affecting metabolic and cardiovascular functions. Diabetes 2019, 68, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lee, J.; Salazar Hernandez, M.A.; Mazitschek, R.; Ozcan, U. Treatment of obesity with celastrol. Cell 2015, 161, 999–1011. [Google Scholar] [CrossRef]
- Kyriakou, E.; Schmidt, S.; Dodd, G.T.; Pfuhlmann, K.; Simonds, S.E.; Lenhart, D.; Geerlof, A.; Schriever, S.C.; De Angelis, M.; Schramm, K.W.; et al. Celastrol promotes weight loss in diet-induced obesity by inhibiting the protein tyrosine phosphatases PTP1B and TCPTP in the hypothalamus. J. Med. Chem. 2018, 61, 11144–11157. [Google Scholar] [CrossRef]
- Potenza, M.A.; Iacobazzi, D.; Sgarra, L.; Montagnani, M. The intrinsic virtues of EGCG, an extremely good cell guardian, on prevention and treatment of diabesity complications. Molecules 2020, 25, 3061. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Gao, C.; Yan, P.; Zhang, M.; Wang, Y.; Hu, Y.; Wu, X.; Wang, X.; Sheng, J. EGCG reduces obesity and white adipose tissue gain partly through AMPK activation in mice. Front. Pharmacol. 2018, 9, 1366. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Shin, Y.; Jung, S.; Kim, Y. Effects of epigallocatechin-3-gallate on thermogenesis and mitochondrial biogenesis in brown adipose tissues of diet-induced obese mice. Food Nutr. Res. 2017, 61, 1325307. [Google Scholar] [CrossRef]
- Mi, Y.; Qi, G.; Fan, R.; Qiao, Q.; Sun, Y.; Gao, Y.; Liu, X. EGCG ameliorates high-fat- and high-fructose-induced cognitive defects by regulating the IRS/AKT and ERK/CREB/BDNF signaling pathways in the CNS. FASEB J. 2017, 31, 4998–5011. [Google Scholar] [CrossRef] [PubMed]
- Okuda, M.H.; Zemdegs, J.C.; De Santana, A.A.; Santamarina, A.B.; Moreno, M.F.; Hachul, A.C.; Dos Santos, B.; Do Nascimento, C.M.; Ribeiro, E.B.; Oyama, L.M.; et al. Green tea extract improves high fat diet-induced hypothalamic inflammation, without affecting the serotoninergic system. J. Nutr. Biochem. 2014, 25, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Hochstetter, D.; Yao, L.; Zhao, Y.; Zhou, J.; Wang, Y.; Xu, P. Green tea polyphenol (-)-epigallocatechin gallate (EGCG) attenuates neuroinflammation in palmitic acid-stimulated BV-2 microglia and high-fat diet-induced obese mice. Int. J. Mol. Sci. 2019, 20, 5081. [Google Scholar] [CrossRef]
- Akaberi, M.; Hosseinzadeh, H. Grapes (Vitis vinifera) as a potential candidate for the therapy of the metabolic syndrome. Phytother. Res. 2016, 30, 540–556. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yun, P.; Hu, Y.; Yang, J.; Khadka, R.B.; Peng, X. Effects of grape seed proanthocyanidin extract on obesity. Obes. Facts 2020, 13, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Perez, C.; Garcia-Villanova, B.; Guerra-Hernandez, E.; Verardo, V. Grape seeds proanthocyanidins: An overview of in vivo bioactivity in animal models. Nutrients 2019, 11, 2435. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, D.; Bagchi, M.; Stohs, S.J.; Das, D.K.; Ray, S.D.; Kuszynski, C.A.; Joshi, S.S.; Pruess, H.G. Free radicals and grape seed proanthocyanidin extract: Importance in human health and disease prevention. Toxicology 2000, 148, 187–197. [Google Scholar] [CrossRef]
- Charradi, K.; Elkahoui, S.; Karkouch, I.; Limam, F.; Hassine, F.B.; Aouani, E. Grape seed and skin extract prevents high-fat diet-induced brain lipotoxicity in rat. Neurochem. Res. 2012, 37, 2004–2013. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Charradi, K.; Limam, F.; Aouani, E. Grape seed and skin extract as an adjunct to xenical therapy reduces obesity, brain lipotoxicity and oxidative stress in high fat diet fed rats. Obes. Res. Clin. Pract. 2018, 12, 115–126. [Google Scholar] [CrossRef]
- Langley, M.R.; Yoon, H.; Kim, H.N.; Choi, C.I.; Simon, W.; Kleppe, L.; Lanza, I.R.; LeBrasseur, N.K.; Matveyenko, A.; Scarisbrick, I.A. High fat diet consumption results in mitochondrial dysfunction, oxidative stress, and oligodendrocyte loss in the central nervous system. Biochim. et Biophys. Acta Mol. Basis Dis. 2020, 1866, 165630. [Google Scholar] [CrossRef]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef]


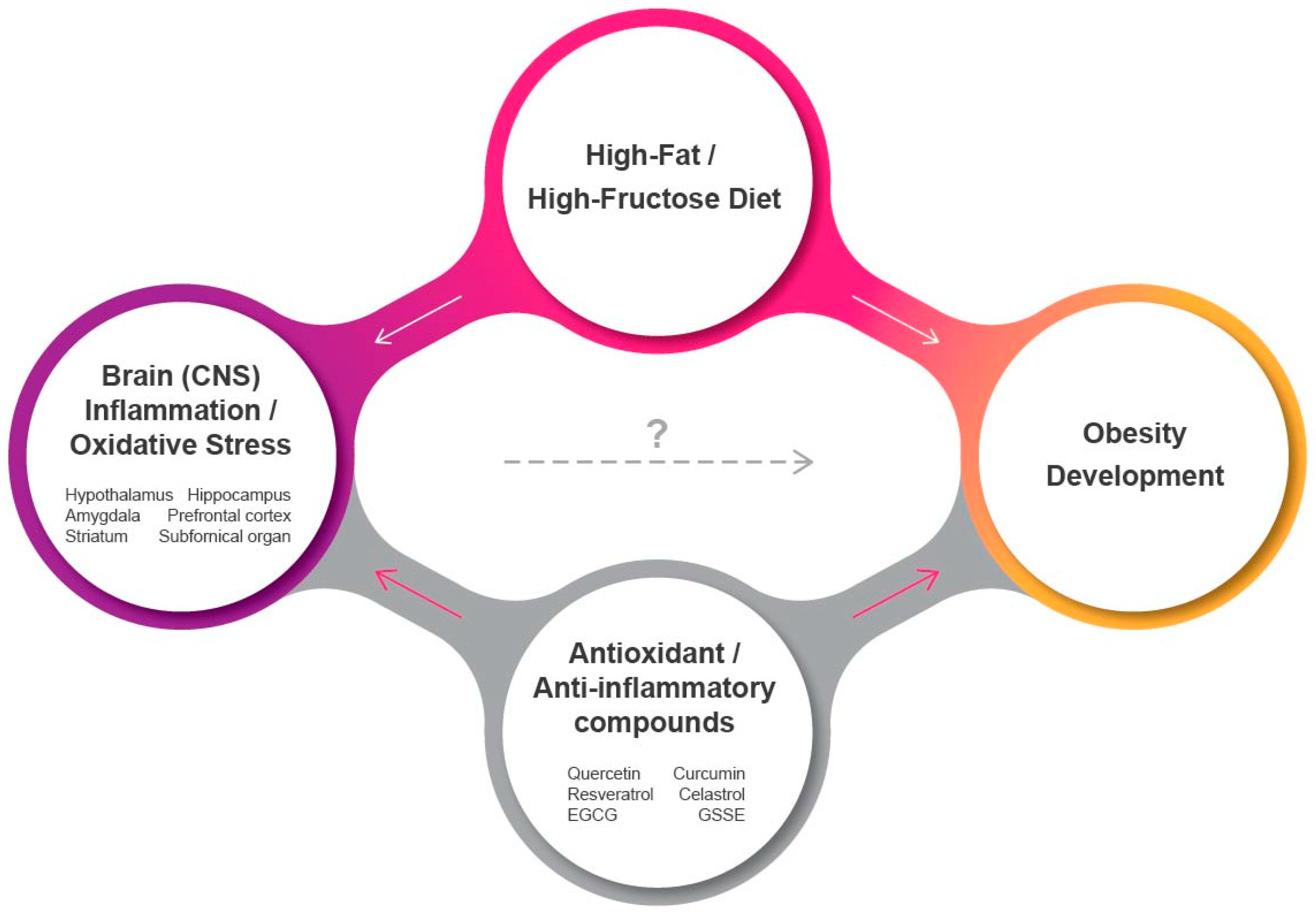{kind=link}
| Natural Compounds | Obese Model | Body Weight | Brain Area | Outcomes | Ref. |
|---|---|---|---|---|---|
| Quercetin | HFD | NR | Hypothalamus | ↓Inflammation | [106] |
| HFD | NR | Hippocampus | ↓Oxidative stress | [107] | |
| Curcumin | db/db | ↓ | Hippocampus | ↓Oxidative stress | [120] |
| 30% Fructose | Resisted gain | Hippocampus Hypothalamus | ↓Inflammation ↓Microglia activity | [121] | |
| Resveratrol | ob/ob | − | Whole brain | ↓Lipid peroxidation ↓Antioxidant activity | [126] |
| HFD | − | Hippocampus | ↓Inflammation ↓Lipid peroxidation | [127] | |
| HFD | − | Hypothalamus | ↓Inflammation | [129] | |
| Celastrol | MC4R KO | ↓ | Hypothalamus | −Inflammation −ER stress | [132] |
| HFD | ↓ | Hypothalamus | ↓ER stress | [133] | |
| ob/ob db/db | − | ||||
| Epigallocatechin-3-gallate (EGCG) | HFD+ 10% fructose | ↓ | Whole Brain | ↓Inflammation | [138] |
| HFD | − | Hypothalamus | ↓Inflammation | [139] | |
| Green tea extract (>90% EGCG) | HFD | Resisted gain | Hypothalamus | ↓Inflammation | [140] |
| Grape seed and skin extract (GSSE) | HFD | Resisted gain | Whole brain | ↓Lipotoxicityv ↓Antioxidant activity | [145] |
| HFD | Resisted gain | Whole brain | ↓Lipid peroxidation ↓Oxidative stress ↑Antioxidant activity | [146] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mullins, C.A.; Gannaban, R.B.; Khan, M.S.; Shah, H.; Siddik, M.A.B.; Hegde, V.K.; Reddy, P.H.; Shin, A.C. Neural Underpinnings of Obesity: The Role of Oxidative Stress and Inflammation in the Brain. Antioxidants 2020, 9, 1018. https://doi.org/10.3390/antiox9101018
Mullins CA, Gannaban RB, Khan MS, Shah H, Siddik MAB, Hegde VK, Reddy PH, Shin AC. Neural Underpinnings of Obesity: The Role of Oxidative Stress and Inflammation in the Brain. Antioxidants. 2020; 9(10):1018. https://doi.org/10.3390/antiox9101018
Chicago/Turabian StyleMullins, Caitlyn A., Ritchel B. Gannaban, Md Shahjalal Khan, Harsh Shah, Md Abu B. Siddik, Vijay K. Hegde, P. Hemachandra Reddy, and Andrew C. Shin. 2020. "Neural Underpinnings of Obesity: The Role of Oxidative Stress and Inflammation in the Brain" Antioxidants 9, no. 10: 1018. https://doi.org/10.3390/antiox9101018
APA StyleMullins, C. A., Gannaban, R. B., Khan, M. S., Shah, H., Siddik, M. A. B., Hegde, V. K., Reddy, P. H., & Shin, A. C. (2020). Neural Underpinnings of Obesity: The Role of Oxidative Stress and Inflammation in the Brain. Antioxidants, 9(10), 1018. https://doi.org/10.3390/antiox9101018