Anthocyanins from Hibiscus syriacus L. Inhibit Oxidative Stress-Mediated Apoptosis by Activating the Nrf2/HO-1 Signaling Pathway





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of PS
2.2. Reagents and Antibodies
2.3. Cell Culture and Relative Cell Viability
2.4. Viable Cell Count, Viability, and Dead Cell Populations
2.5. Annexin V Staining for Apoptosis
2.6. Analysis of Intracellular ROS
2.7. Analysis of mtROS
2.8. Analysis of Mitochondrial Depolarization
2.9. Analysis of Caspase3/7 Activity
2.10. Protein Extraction and Western Blot Analysis
2.11. Nrf2 Immunostaining
2.12. Statistical Analysis
3. Results
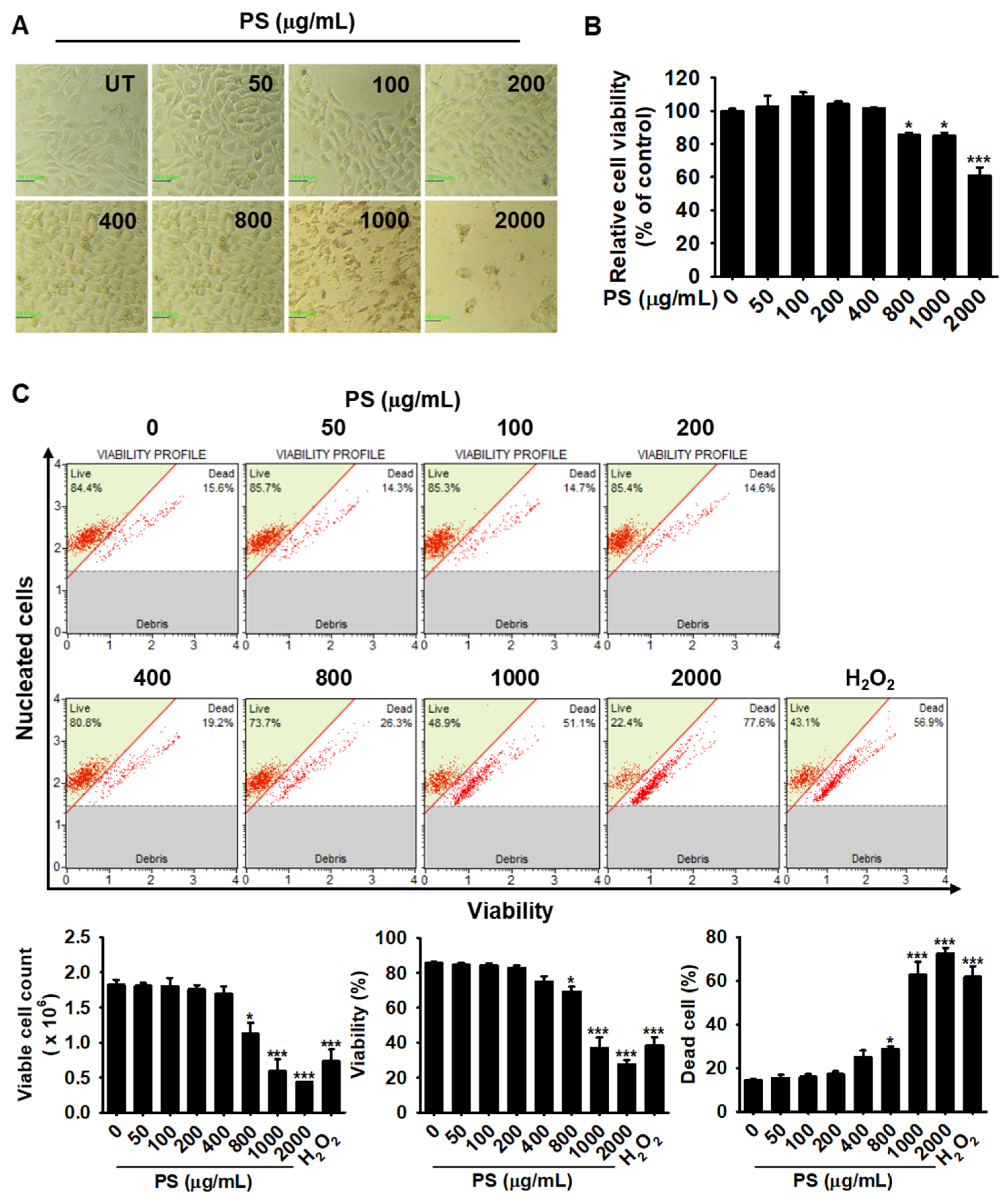
3.1. Low Concentrations of PS Have No Cytotoxic Effect on HaCaT Keratinocytes
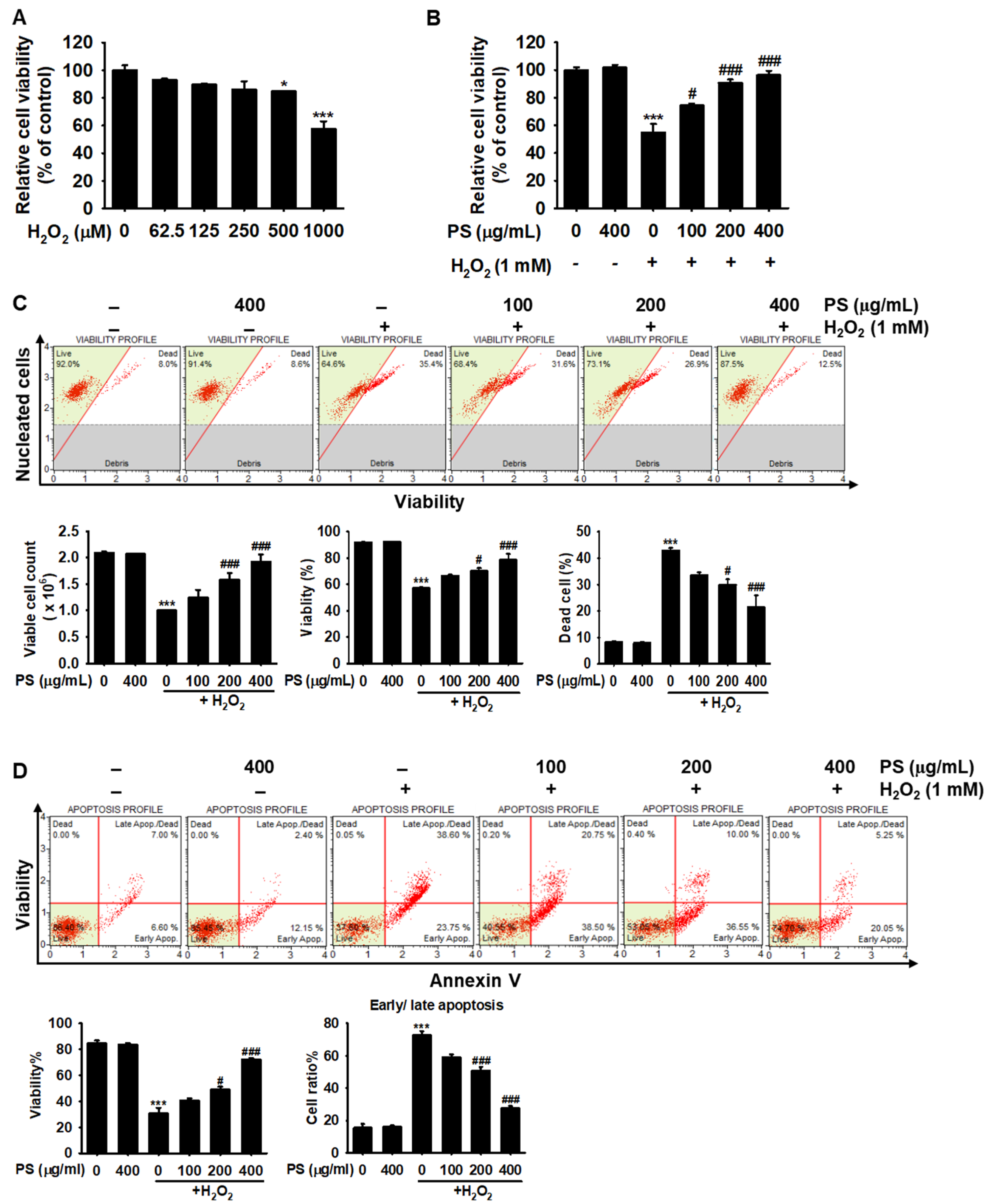
3.2. PS Protects HaCaT Keratinocytes from H2O2-Induced Apoptosis
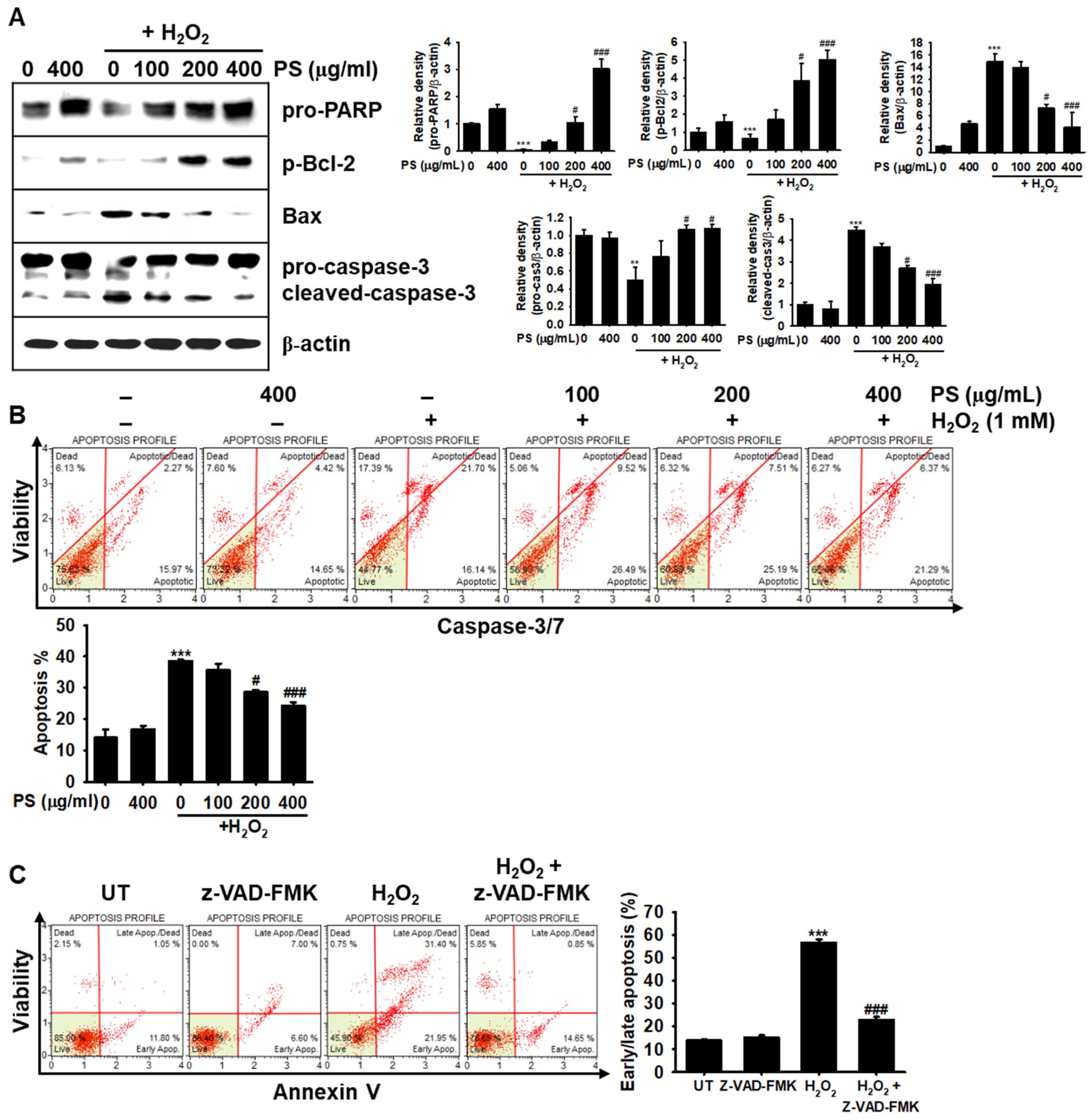
3.3. The Cytoprotective Effect of PS against H2O2-Induced Apoptosis Is Mediated through Modulation of Apoptosis-Related Proteins in HaCaT Keratinocytes
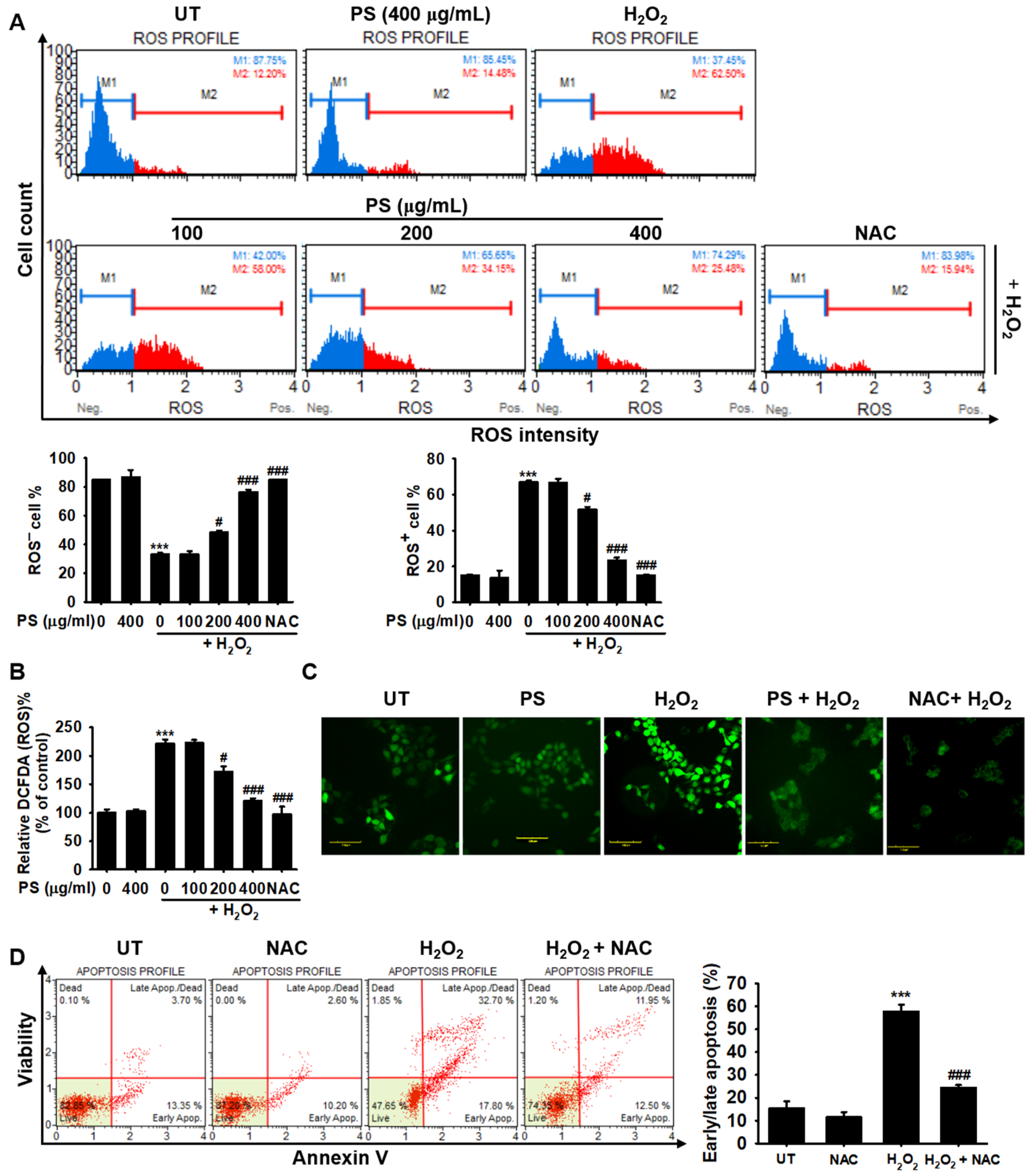
3.4. PS Protects HaCaT Keratinocytes from H2O2-Induced Apoptosis by Reducing Intracellular ROS Generation
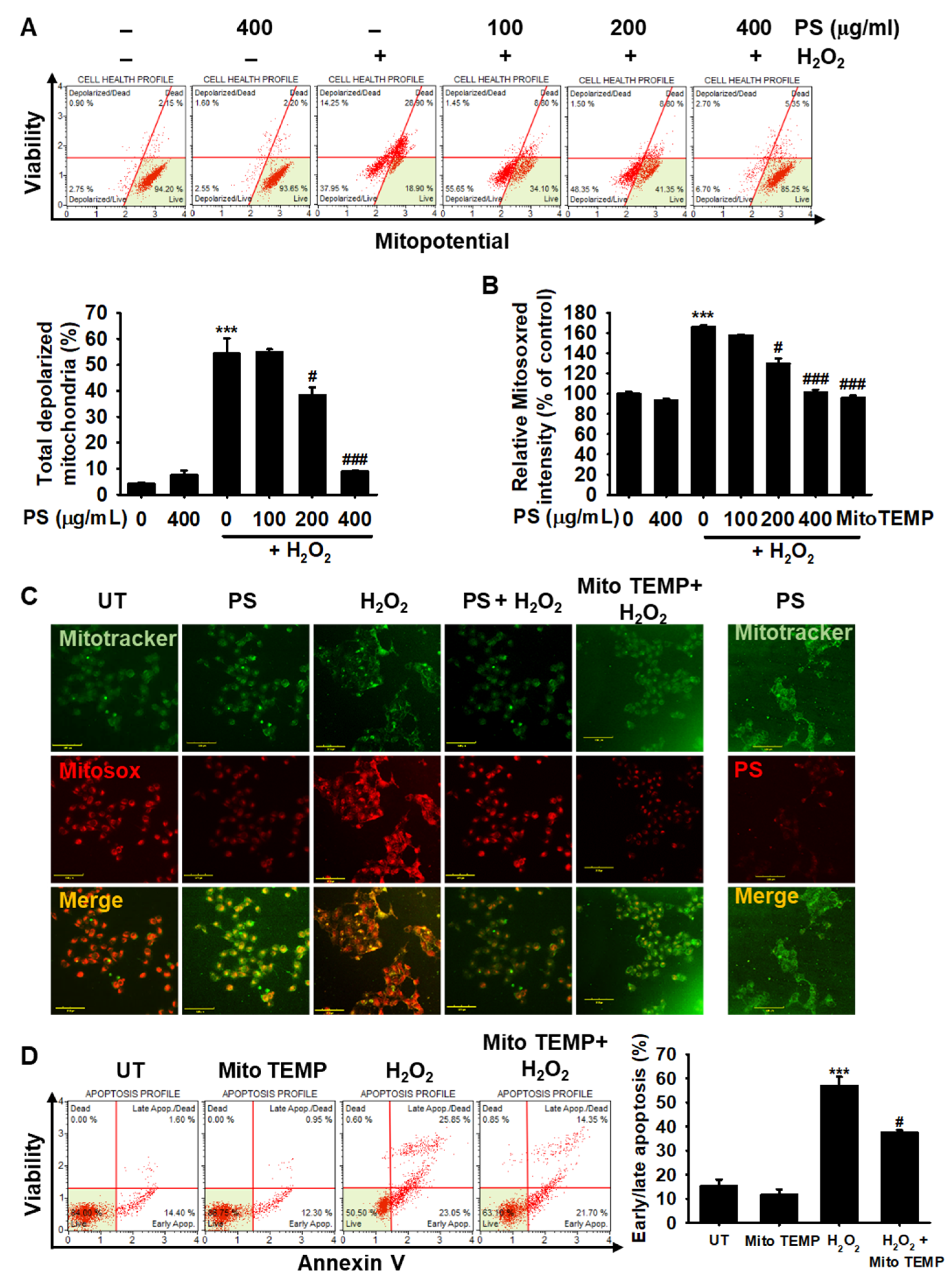
3.5. PS Protects HaCaT Keratinocytes from H2O2-Induced Mitochondrial Depolarization and mtROS Generation
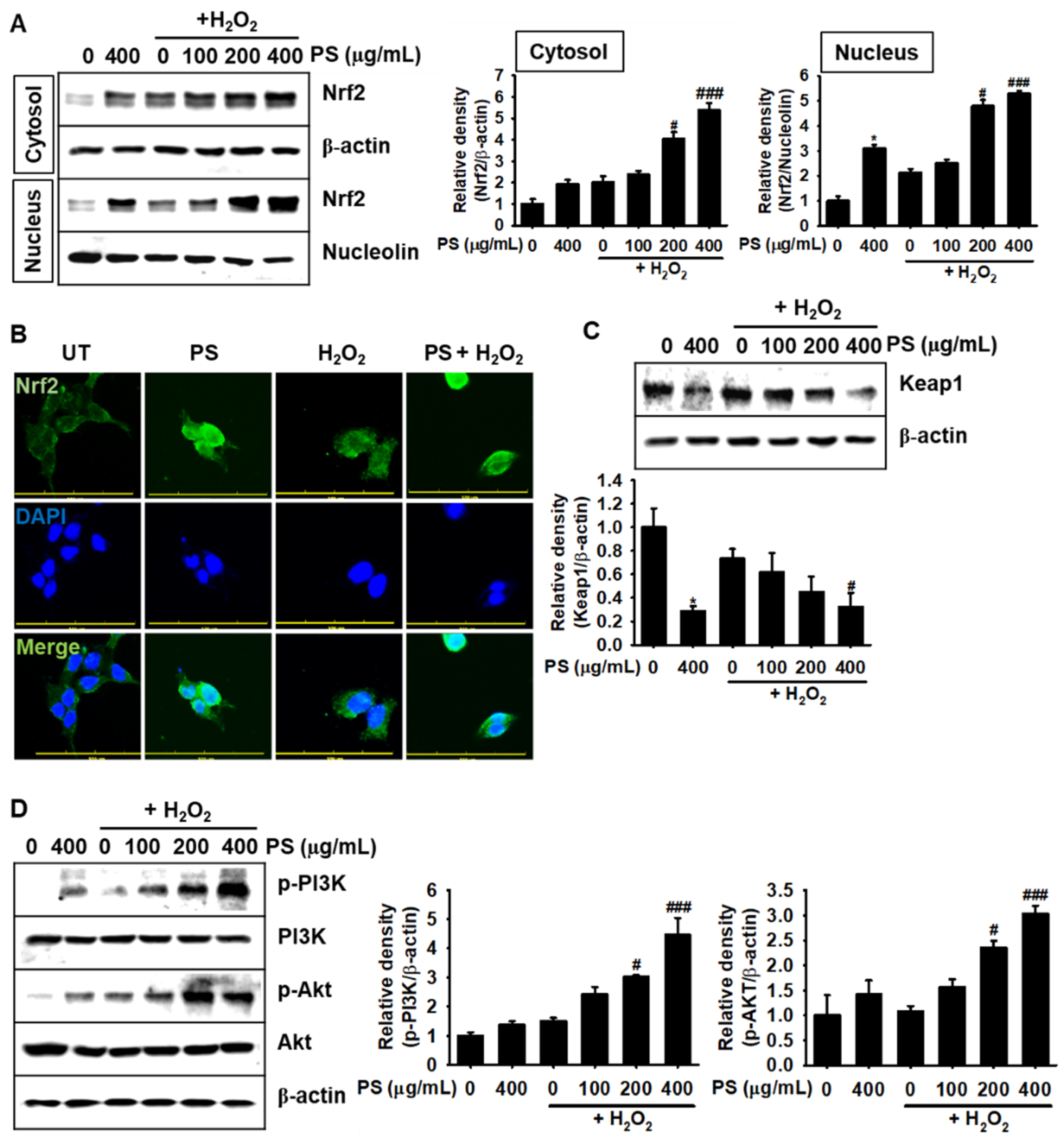
3.6. Under H2O2-Induced Oxidative Stress, PS Stimulates the Nrf2-Mediated Defense System in HaCaT Keratinocytes
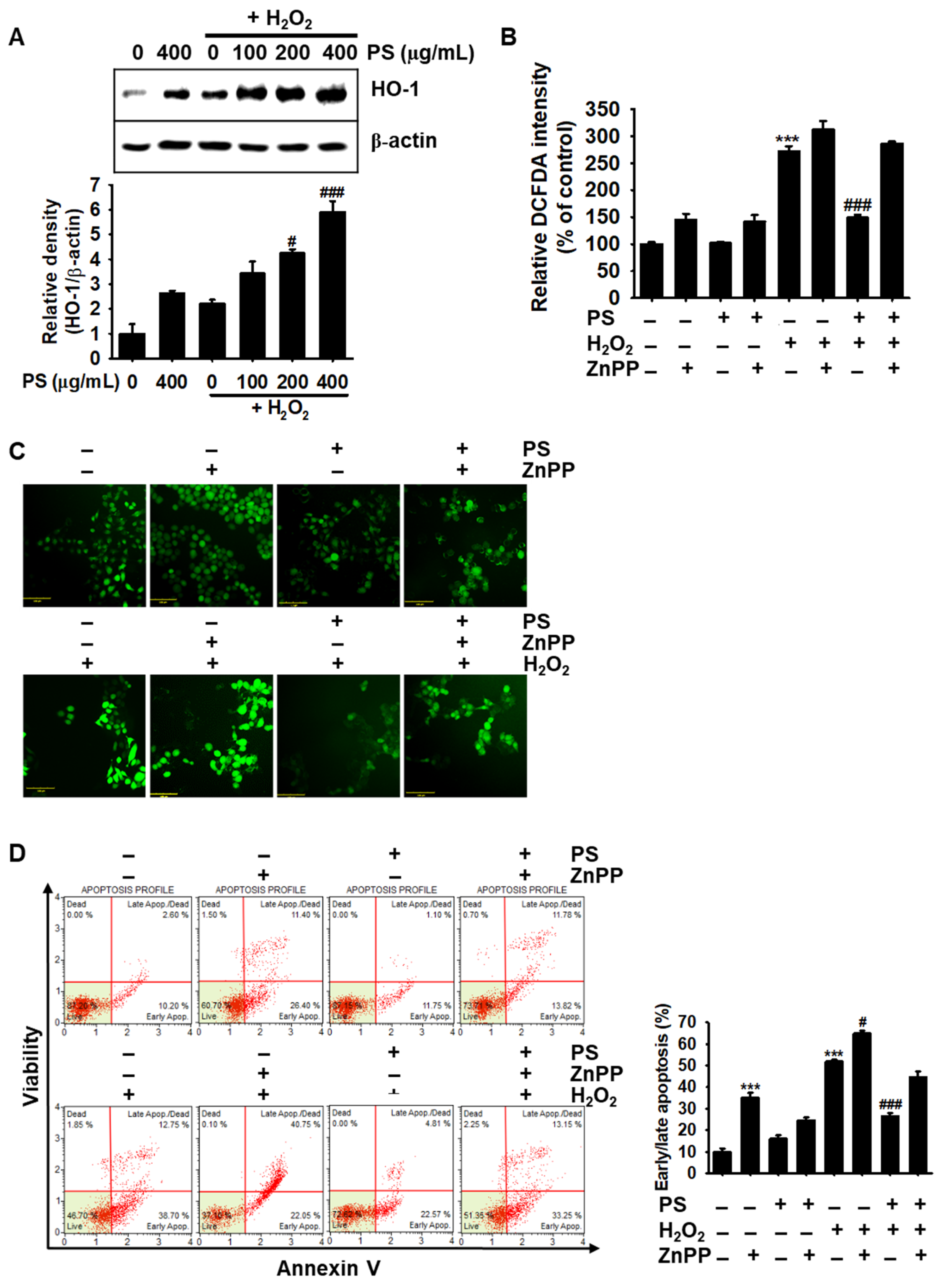
3.7. PS-Mediated Cytoprotection Depends on the Canonical Nrf2/HO-1 Signaling Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Proksch, E.; Brandner, J.M.; Jensen, J.M. The skin: An indispensable barrier. Exp. Dermatol. 2008, 17, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yin, M.; Zhang, L.J. Keratin 6, 16 and 17-critical barrier alarmin molecules in skin wounds and psoriasis. Cells 2019, 8, 807. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Wagener, F.A.; Carels, C.E.; Lundvig, D.M. Targeting the redox balance in inflammatory skin conditions. Int. J. Mol. Sci. 2013, 14, 9126–9167. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Li, X.-K. Oxidative stress in atopic dermatitis. Oxid. Med. Cell. Longev. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Birch-Machin, M.A.; Bowman, A. Oxidative stress and ageing. Br. J. Dermatol. 2016, 175, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, A.; Chrousos, G.P. Stress-related skin disorders. Rev. Endocr. Metab. Disord. 2016, 17, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Yankner, B.A. The aging stress response. Mol. Cell 2010, 40, 333–344. [Google Scholar] [CrossRef]
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting free radicals in oxidative stress-related human diseases. Trends Pharmacol. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef]
- Keum, Y.-S. Regulation of Nrf2-mediated phase II detoxification and anti-oxidant genes. Biomol. Ther. 2012, 20, 144–151. [Google Scholar] [CrossRef]
- Kim, H.J.; Joe, Y.; Surh, Y.-J.; Chung, H.T. Metabolic signaling functions of the heme oxygenase/CO system in metabolic diseases. Cell. Mol. Immunol. 2018, 15, 1085–1087. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Luna, A.; Ávila-Román, J.; Oliveira, H.; Motilva, V.; Talero, E. Fucoxanthin and rosmarinic acid combination has anti-inflammatory effects through regulation of NLRP3 inflammasome in UVB-exposed HaCaT keratinocytes. Mar. Drugs 2019, 17, 451. [Google Scholar] [CrossRef] [PubMed]
- Kahremany, S.; Babaev, I.; Gvirtz, R.; Ogen-Stern, N.; Azoulay-Ginsburg, S.; Senderowitz, H.; Cohen, G.; Gruzman, A. Nrf2 activation by SK-119 attenuates oxidative stress, UVB, and LPS-induced damage. Skin Pharmacol. Physiol. 2019, 32, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhu, M.-L.; Li, D.-H.; Zeng, R.; Han, B.-N. N-Me-trichodermamide B isolated from Penicillium janthinellum, with antioxidant properties through Nrf2-mediated signaling pathway. Bioorg. Med. Chem. 2017, 25, 6614–6622. [Google Scholar] [CrossRef] [PubMed]
- Trouba, K.J.; Hamadeh, H.K.; Amin, R.P.; Germolec, D.R. Oxidative stress and its role in skin disease. Antioxid. Redox Signal. 2002, 4, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Witzig, T.E.; Adjei, A.A. Targeting apoptosis pathways in cancer therapy. CA Cancer J. Clin. 2005, 55, 178–194. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Fleury, C.; Mignotte, B.; Vayssiere, J.L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002, 84, 131–141. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmstrom, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef]
- Kim, Y.; Oh, Y.J.; Han, K.Y.; Kim, G.H.; Ko, J.; Park, J. The complete chloroplast genome sequence of Hibiscus syriacus L. ‘Mamonde’ (Malvaceae). Mitochondrial DNA Part B 2019, 4, 558–559. [Google Scholar] [CrossRef]
- Geng, M.; Ren, M.; Liu, Z.; Shang, X. Free radical scavenging activities of pigment extract from Hibiscus syriacus L. petals in vitro. Afr. J. Biotechnol. 2012, 11, 429–435. [Google Scholar]
- Karunarathne, W.; Molagoda, I.M.N.; Park, S.R.; Kim, J.W.; Lee, O.K.; Kwon, H.Y.; Oren, M.; Choi, Y.H.; Ryu, H.W.; Oh, S.R.; et al. Anthocyanins from Hibiscus syriacus L. inhibit melanogenesis by activating the ERK signaling pathway. Biomolecules 2019, 9, 645. [Google Scholar] [CrossRef] [PubMed]
- Ndisang, J.F. Synergistic interaction between heme oxygenase (HO) and nuclear-factor E2-related factor-2 (Nrf2) against oxidative stress in cardiovascular related diseases. Curr. Pharm. Des. 2017, 23, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Wu, S.B.; Hong, C.H.; Yu, H.S.; Wei, Y.H. Molecular mechanisms of UV-induced apoptosis and its effects on skin residential cells: The implication in UV-based phototherapy. Int. J. Mol. Sci. 2013, 14, 6414–6435. [Google Scholar] [CrossRef] [PubMed]
- Salucci, S.; Burattini, S.; Buontempo, F.; Martelli, A.M.; Falcieri, E.; Battistelli, M. Protective effect of different antioxidant agents in UVB-irradiated keratinocytes. Eur. J. Histochem. 2017, 61, 2784. [Google Scholar] [CrossRef]
- Forman, H.J. Use and abuse of exogenous H2O2 in studies of signal transduction. Free Radic. Biol. Med. 2007, 42, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xie, H.; Chen, X.; Shi, W.; Xiao, X.; Lei, D.; Li, J. Differential response of normal human epidermal keratinocytes and HaCaT cells to hydrogen peroxide-induced oxidative stress. Clin. Exp. Dermatol. 2012, 37, 772–780. [Google Scholar] [CrossRef]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochim. Biophys. Acta 2006, 1757, 639–647. [Google Scholar] [CrossRef]
- He, X.; Kan, H.; Cai, L.; Ma, Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J. Mol. Cell. Cardiol. 2009, 46, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Li, K.; Liu, L.; Zhang, Y.; Zhou, Z.; Li, C.; Gao, T. Heme oxygenase-1 protects human melanocytes from H2O2-induced oxidative stress via the Nrf2-ARE pathway. J. Investig. Dermatol. 2011, 131, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Nishi, S.; Sakai, M. Transcription factor Nrf2/MafK regulates rat placental glutathione S-transferase gene during hepatocarcinogenesis. Biochem. J. 2004, 380, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, B.; Tong, K.I.; Ohta, T.; Nakamura, Y.; Scharlock, M.; Ohtsuji, M.; Kang, M.-I.; Kobayashi, A.; Yokoyama, S.; Yamamoto, M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell 2006, 21, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, Y.; Kijima, A.; Ishii, Y.; Takasu, S.; Tsuchiya, T.; Umemura, T. Effects of Nrf2 silencing on oxidative stress-associated intestinal carcinogenesis in mice. Cancer Med. 2016, 5, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jia, Z.; Trush, M.A.; Li, Y.R. Nrf2 deficiency promotes melanoma growth and lung metastasis. React. Oxyg. Species 2016, 2, 308. [Google Scholar] [CrossRef][Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molagoda, I.M.N.; Lee, K.T.; Choi, Y.H.; Kim, G.-Y. Anthocyanins from Hibiscus syriacus L. Inhibit Oxidative Stress-Mediated Apoptosis by Activating the Nrf2/HO-1 Signaling Pathway. Antioxidants 2020, 9, 42. https://doi.org/10.3390/antiox9010042
Molagoda IMN, Lee KT, Choi YH, Kim G-Y. Anthocyanins from Hibiscus syriacus L. Inhibit Oxidative Stress-Mediated Apoptosis by Activating the Nrf2/HO-1 Signaling Pathway. Antioxidants. 2020; 9(1):42. https://doi.org/10.3390/antiox9010042
Chicago/Turabian StyleMolagoda, Ilandarage Menu Neelaka, Kyoung Tae Lee, Yung Hyun Choi, and Gi-Young Kim. 2020. "Anthocyanins from Hibiscus syriacus L. Inhibit Oxidative Stress-Mediated Apoptosis by Activating the Nrf2/HO-1 Signaling Pathway" Antioxidants 9, no. 1: 42. https://doi.org/10.3390/antiox9010042
APA StyleMolagoda, I. M. N., Lee, K. T., Choi, Y. H., & Kim, G.-Y. (2020). Anthocyanins from Hibiscus syriacus L. Inhibit Oxidative Stress-Mediated Apoptosis by Activating the Nrf2/HO-1 Signaling Pathway. Antioxidants, 9(1), 42. https://doi.org/10.3390/antiox9010042