Peroxiredoxin II Maintains the Mitochondrial Membrane Potential against Alcohol-Induced Apoptosis in HT22 Cells

 ,
,  and
and 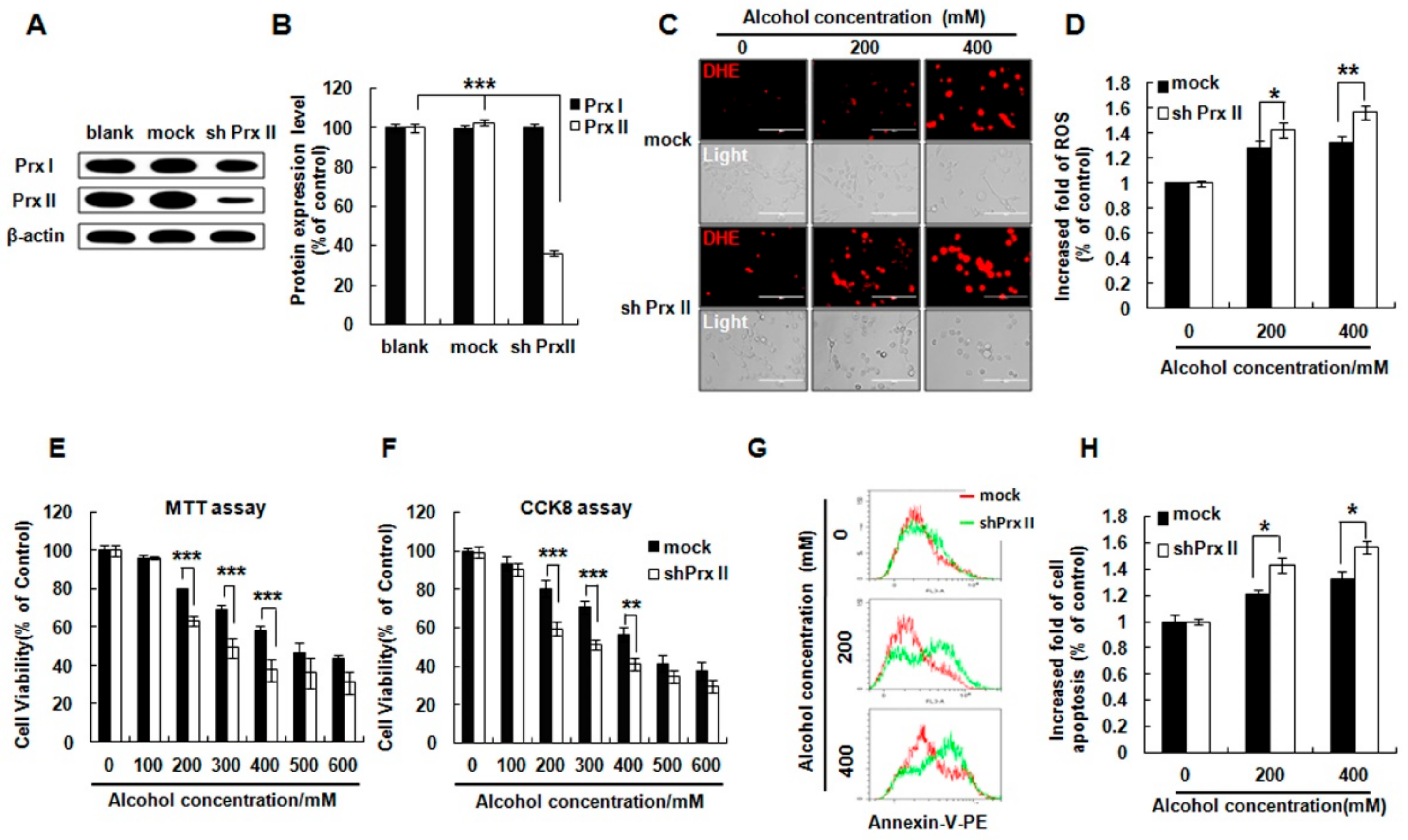{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Transduction of HT22 Cells with Lentiviral-Delivered Prx II Wild Type and Prx II siRNA
2.4. Cell Viability Assay
2.5. Detection of ROS and Superoxide Anion
2.6. Flow Cytometric Detection of Mitochondrial Membrane Potential
2.7. Detection of Apoptosis
2.8. Western Blot
2.9. Electron Microscopy Analysis
2.10. Statistical Analysis
3. Results
3.1. Prx II Knockdown Elevated Alcohol Induced Apoptosis and ROS Accumulation in HT22 Cells
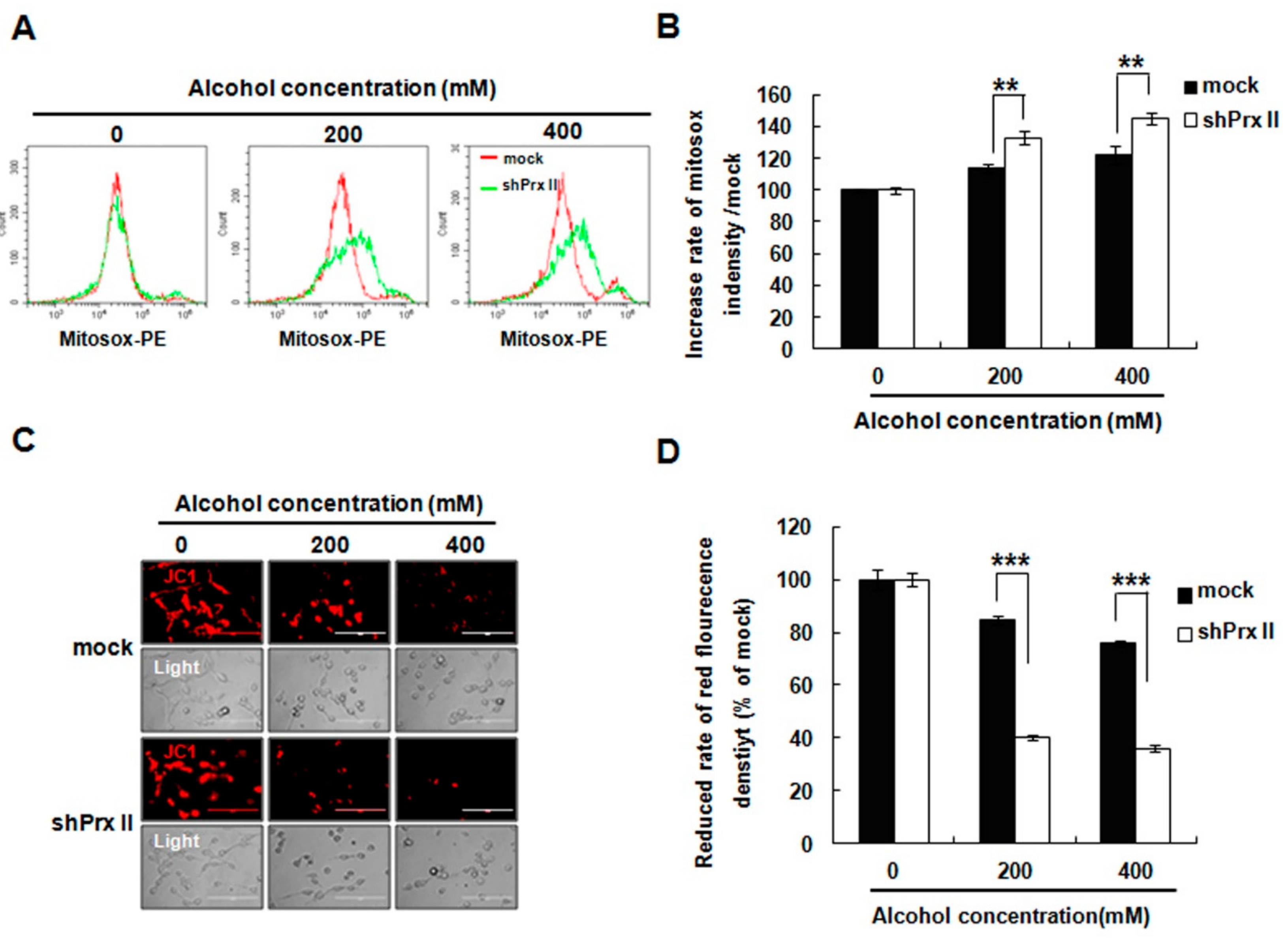
3.2. Knockdown of Prx II Elevated the Alcohol-Induced Mitochondria ROS and Membrane Permeability in HT22 Cells
3.3. Knockdown of Prx II Results in Up-Regulating the Mitochondria Dependent Cellular Apoptosis
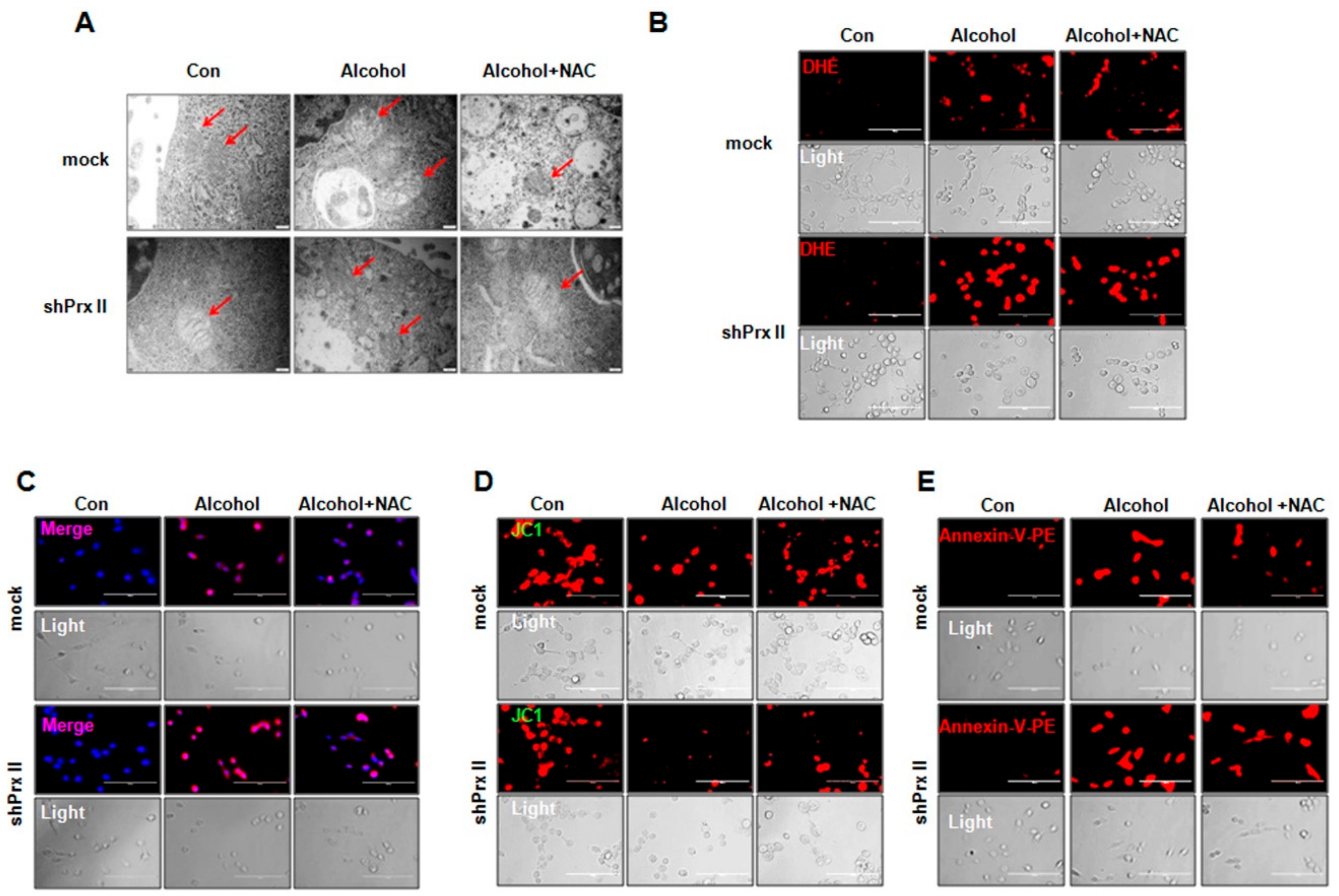
3.4. Regulatory Function of Prx II on Alcohol-Induced Mitochondria Dysfunctions is Dependent on Cellular ROS Levels
3.5. NAC Treatment Restores the Alcohol-Induced Up-Regulating of the Apoptosis-Related Protein Expressions in shPrx II HT22 Cells
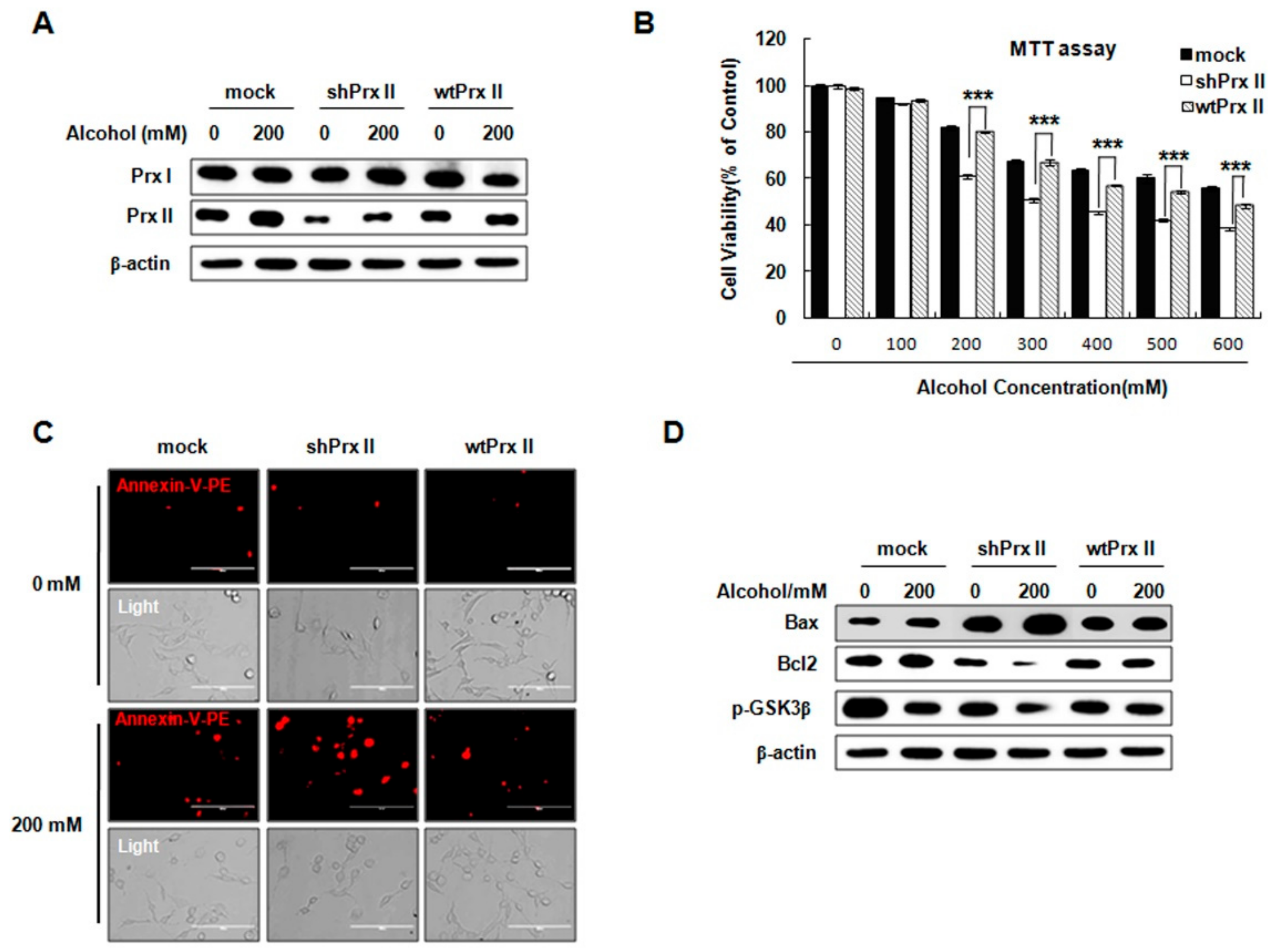
3.6. Prx II Inhibits the Cellular Apoptosis by Down-Regulating the Mitochondria-Dependent Apoptotic Pathway
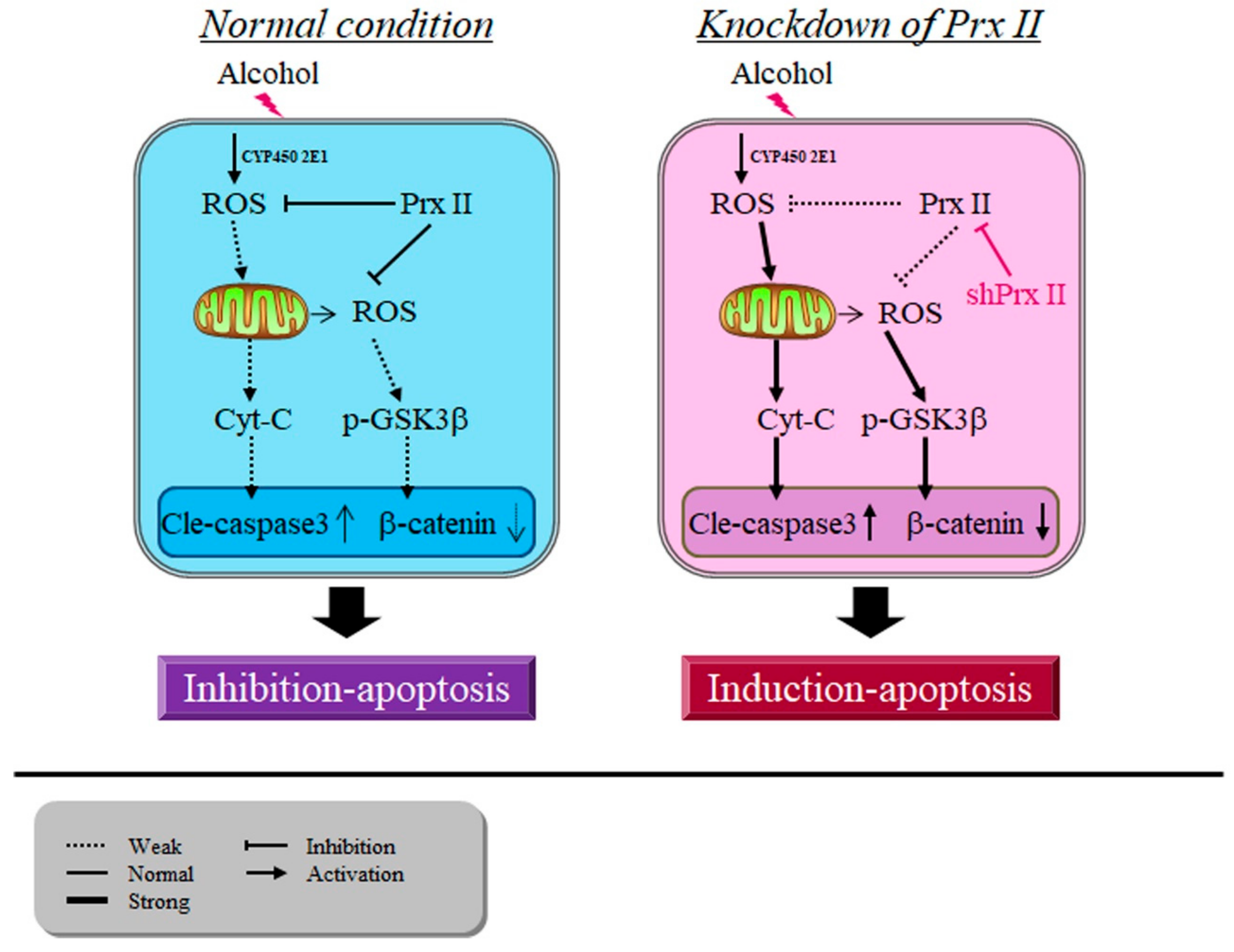
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gonzalez-Reimers, E.; Camino, M.; Fernández-Rodríguez, M.; Martín-González, C.; Hernández-Betancor, I.; Abreu-González, P.; de la Vega-Prieto, M.J.; Elvira-Cabrera, O.; Santolaria-Fernández, F. Antioxidant vitamins and brain dysfunction in alcoholics. Alcohol Alcohol. 2014, 49, 45–50. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rice, J.; Gu, C. Function and Mechanism of Myelin Regulation in Alcohol Abuse and Alcoholism. Bioessays 2019, 41, e1800255. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Allen, J.W.; Kimelberg, H.K.; LoPachin, R.M.; Streit, W.J. Glial cells in neurotoxicity development. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 151–173. [Google Scholar] [CrossRef] [PubMed]
- Corso, T.D.; Mostafa, H.M.; Collins, M.A.; Neafsey, E.J. Brain neuronal degeneration caused by episodic alcohol intoxication in rats: Effects of nimodipine, 6,7-dinitro-quinoxaline-2,3-dione, and MK-801. Alcohol. Clin. Exp. Res. 1998, 22, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Rojas, C.; Mira, R.G.; Torres, A.K.; Jara, C.; Pérez, M.J.; Vergara, E.H.; Cerpa, W.; Quintanilla, R.A. Alcohol consumption during adolescence: A link between mitochondrial damage and ethanol brain intoxication. Birth Defects Res. 2017, 109, 1623–1639. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Collins, M.A.; Dlugos, C.; Littleton, J.; Wilkins, L.; Neafsey, E.J.; Pentney, R.; Snell, L.D.; Tabakoff, B.; Zou, J. Alcohol-induced neurodegeneration: when, where and why? Alcohol. Clin. Exp. Res. 2004, 28, 350–364. [Google Scholar] [CrossRef]
- Switzer, R.C. Application of silver degeneration stains for neurotoxicity testing. Toxicol. Pathol. 2000, 28, 70–83. [Google Scholar] [CrossRef]
- Khan, M.; Shah, S.A.; Kim, M.O. 17beta-Estradiol via SIRT1/Acetyl-p53/NF-kB Signaling Pathway Rescued Postnatal Rat Brain Against Acute Ethanol Intoxication. Mol. Neurobiol. 2018, 55, 3067–3078. [Google Scholar] [CrossRef]
- Eskay, R.L.; Chautard, T.; Torda, T.; Daoud, R.I.; Hamelink, C. Alcohol, corticosteroids, energy utilization, and hippocampal endangerment. Ann. N. Y. Acad. Sci. 1995, 771, 105–114. [Google Scholar] [CrossRef]
- Obernier, J.A.; White, A.M.; Swartzwelder, H.S.; Crews, F.T. Cognitive deficits and CNS damage after a 4-day binge ethanol exposure in rats. Pharmacol. Biochem. Behav. 2002, 72, 521–532. [Google Scholar] [CrossRef]
- Wilson, S.; Bair, J.L.; Thomas, K.M.; Iacono, W.G. Problematic alcohol use and reduced hippocampal volume: A meta-analytic review. Psychol. Med. 2017, 47, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Herrera, D.G.; Yagüe, A.G.; Johnsen-Soriano, S.; Bosch-Morell, F.; Collado-Morente, L.; Muriach, M.; Romero, F.J.; García-Verdugo, J.M. Selective impairment of hippocampal neurogenesis by chronic alcoholism: protective effects of an antioxidant. Proc. Natl. Acad. Sci. USA 2003, 100, 7919–7924. [Google Scholar] [CrossRef] [PubMed]
- Obernier, J.A.; Bouldin, T.W.; Crews, F.T. Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol. Clin. Exp. Res. 2002, 26, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Christian, K.M.; Song, H.; Ming, G.L. Functions and dysfunctions of adult hippocampal neurogenesis. Annu. Rev. Neurosci. 2014, 37, 243–262. [Google Scholar] [CrossRef]
- Ali, T.; Rehman, S.U.; Shah, F.A.; Kim, M.O. Acute dose of melatonin via Nrf2 dependently prevents acute ethanol-induced neurotoxicity in the developing rodent brain. J. Neuroinflamm. 2018, 15, 119. [Google Scholar] [CrossRef]
- Wang, X.; Yu, H.; You, J.; Wang, C.; Feng, C.; Liu, Z.; Li, Y.; Wei, R.; Xu, S.; Zhao, R.; et al. Memantine can improve chronic ethanol exposure-induced spatial memory impairment in male C57BL/6 mice by reducing hippocampal apoptosis. Toxicology 2018, 406, 21–32. [Google Scholar] [CrossRef]
- Kocyigit, A.; Guler, E.M. Curcumin induce DNA damage and apoptosis through generation of reactive oxygen species and reducing mitochondrial membrane potential in melanoma cancer cells. Cell Mol. Biol. 2017, 63, 97–105. [Google Scholar] [CrossRef]
- Li, H.; Zhu, C.; Wang, B.; Zhu, W.; Feng, Y.; Du, F.; Wang, S.; Hu, C.; Ma, J.; Yu, X. 17beta-Estradiol Protects the Retinal Nerve Cells Suppressing TLR2 Mediated Immune-Inflammation and Apoptosis from Oxidative Stress Insult Independent of PI3K. J. Mol. Neurosci. 2016, 60, 195–204. [Google Scholar] [CrossRef]
- Liu, J.Y.; Guo, F.; Wu, H.L.; Wang, Y.; Liu, J.S. Midazolam anesthesia protects neuronal cells from oxidative stress-induced death via activation of the JNK-ERK pathway. Mol. Med. Rep. 2017, 15, 169–179. [Google Scholar] [CrossRef]
- Wu, D.; Cederbaum, A.I. Oxidative stress mediated toxicity exerted by ethanol-inducible CYP2E1. Toxicol. Appl. Pharmacol. 2005, 207, 70–76. [Google Scholar] [CrossRef]
- Kong, L.Z.; Chandimali, N.; Han, Y.H.; Lee, D.H.; Kim, J.S.; Kim, S.U.; Kim, T.D.; Jeong, D.K.; Sun, H.N.; Lee, D.S.; et al. Pathogenesis, Early Diagnosis, and Therapeutic Management of Alcoholic Liver Disease. Int. J. Mol. Sci. 2019, 20, 2712. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Demeilliers, C.; Amsellem, S.; Pessayre, D.; Fromenty, B. Acute ethanol administration oxidatively damages and depletes mitochondrial dna in mouse liver, brain, heart, and skeletal muscles: Protective effects of antioxidants. J. Pharmacol. Exp. Ther. 2001, 298, 737–743. [Google Scholar] [PubMed]
- Boyadjieva, N.I.; Sarkar, D.K. Microglia play a role in ethanol-induced oxidative stress and apoptosis in developing hypothalamic neurons. Alcohol. Clin. Exp. Res. 2013, 37, 252–262. [Google Scholar] [CrossRef] [PubMed]
- McDonough, K.H. Antioxidant nutrients and alcohol. Toxicology 2003, 189, 89–97. [Google Scholar] [CrossRef]
- Soleimani, E.; Goudarzi, I.; Abrari, K.; Lashkarbolouki, T. Maternal administration of melatonin prevents spatial learning and memory deficits induced by developmental ethanol and lead co-exposure. Physiol. Behav. 2017, 173, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Akbar, M.; Essa, M.M.; Daradkeh, G.; Abdelmegeed, M.A.; Choi, Y.; Mahmood, L.; Song, B.J. Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res. 2016, 1637, 34–55. [Google Scholar] [CrossRef]
- Bhola, P.D.; Letai, A. Mitochondria-Judges and Executioners of Cell Death Sentences. Mol. Cell 2016, 61, 695–704. [Google Scholar] [CrossRef]
- Hampton, M.B.; O’Connor, K.M. Peroxiredoxins and the Regulation of Cell Death. Mol. Cells 2016, 39, 72–76. [Google Scholar]
- Chandimali, N.; Zhang, J.J.; Lee, J.C.; Yu, D.Y.; Jeong, D.K.; Kwon, T. MicroRNA-122 negatively associates with peroxiredoxin-II expression in human gefitinib-resistant lung cancer stem cells. Cancer Gene Ther. 2018, 26, 292–304. [Google Scholar] [CrossRef]
- Chandimali, N.; Jeong, D.K.; Kwon, T. Peroxiredoxin II Regulates Cancer Stem Cells and Stemness-Associated Properties of Cancers. Cancers 2018, 10, 305. [Google Scholar] [CrossRef]
- Jin, M.H.; Lee, Y.H.; Kim, J.M.; Sun, H.N.; Moon, E.Y.; Shong, M.H.; Kim, S.U.; Lee, S.H.; Lee, T.H.; Yu, D.Y.; et al. Characterization of neural cell types expressing peroxiredoxins in mouse brain. Neurosci. Lett. 2005, 381, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.U.; Jin, M.H.; Kim, Y.S.; Lee, S.H.; Cho, Y.S.; Cho, K.J.; Lee, K.S.; Kim, Y.I.; Kim, G.W.; Kim, J.M.; et al. Peroxiredoxin II preserves cognitive function against age-linked hippocampal oxidative damage. Neurobiol. Aging 2011, 32, 1054–1068. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Weng, Z.; Chu, C.T.; Zhang, L.; Cao, G.; Gao, Y.; Signore, A.; Zhu, J.; Hastings, T.; Greenamyre, J.T.; et al. Peroxiredoxin-2 protects against 6-hydroxydopamine-induced dopaminergic neurodegeneration via attenuation of the apoptosis signal-regulating kinase (ASK1) signaling cascade. J. Neurosci. 2011, 31, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Xie, Y.; Meng, Y.; Ma, W.; Tong, Z.; Yang, X.; Lai, S.; Zhou, Y.; He, M.; Liao, Z. Resveratrol protects cardiomyocytes against anoxia/reoxygenation via dephosphorylation of VDAC1 by Akt-GSK3 beta pathway. Eur. J. Pharmacol. 2019, 843, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; Zou, J.Y.; Neafsey, E.J. Brain damage due to episodic alcohol exposure in vivo and in vitro: Furosemide neuroprotection implicates edema-based mechanism. FASEB J. 1998, 12, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Majchrowicz, E. Induction of physical dependence upon ethanol and the associated behavioral changes in rats. Psychopharmacologia 1975, 43, 245–254. [Google Scholar] [CrossRef]
- Haorah, J.; Knipe, B.; Leibhart, J.; Ghorpade, A.; Persidsky, Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J. Leukoc. Biol. 2005, 78, 1223–1232. [Google Scholar] [CrossRef]
- Katz, G.G.; Shear, N.H.; Malkiewicz, I.M.; Valentino, K.; Neuman, M.G. Signaling for ethanol-induced apoptosis and repair in vitro. Clin. Biochem. 2001, 34, 219–227. [Google Scholar] [CrossRef]
- Alfonso-Loeches, S.; Guerri, C. Molecular and behavioral aspects of the actions of alcohol on the adult and developing brain. Crit. Rev. Clin. Lab. Sci. 2011, 48, 19–47. [Google Scholar] [CrossRef]
- White, A.M. What happened? Alcohol, memory blackouts, and the brain. Alcohol. Res. Health 2003, 27, 186–196. [Google Scholar]
- Crews, F.; Nixon, K.; Kim, D.; Joseph, J.; Shukitt-Hale, B.; Qin, L.; Zou, J. BHT blocks NF-kappaB activation and ethanol-induced brain damage. Alcohol. Clin. Exp. Res. 2006, 30, 1938–1949. [Google Scholar] [CrossRef] [PubMed]
- Alijan-Pour, J.; Abrari, K.; Ghorbanian, M.T.; Goudarzi, I.; Salmani, M.E.; Mirshekar, M. Acute ethanol administration affects memory reactivation: A look at the neuronal density and apoptosis in the rat hippocampus. Pharmacol. Biochem. Behav. 2012, 102, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Ali Shah, S.; Ullah, I.; Lee, H.Y.; Kim, M.O. Anthocyanins protect against ethanol-induced neuronal apoptosis via GABAB1 receptors intracellular signaling in prenatal rat hippocampal neurons. Mol. Neurobiol. 2013, 48, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.M.; Atkinson, R.M. Alcoholism and dementia. Int. J. Addict. 1995, 30, 1843–1869. [Google Scholar] [CrossRef] [PubMed]
- Sripathirathan, K.; Brown III, J.; Neafsey, E.J.; Collins, M.A. Linking binge alcohol-induced neurodamage to brain edema and potential aquaporin-4 upregulation: Evidence in rat organotypic brain slice cultures and in vivo. J. Neurotrauma 2009, 26, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Huang, Y.; Qin, J.; Wang, Y.; Ke, B.; Yang, Y. Inhibition of MAPKs Signaling Pathways Prevents Acrolein-Induced Neurotoxicity in HT22 Mouse Hippocampal Cells. Biol. Pharm. Bull. 2019, 42, 617–622. [Google Scholar] [CrossRef]
- Hampson, A.J.; Grimaldi, M.; Lolic, M.; Wink, D.; Rosenthal, R.; Axelrod, J. Neuroprotective antioxidants from marijuana. Ann. NY Acad. Sci. 2000, 899, 274–282. [Google Scholar] [CrossRef]
- Tabakoff, B.; Hoffman, P.L. Alcohol interactions with brain opiate receptors. Life Sci. 1983, 32, 197–204. [Google Scholar] [CrossRef]
- Anderson, M.L.; Nokia, M.S.; Govindaraju, K.P.; Shors, T.J. Moderate drinking? Alcohol consumption significantly decreases neurogenesis in the adult hippocampus. Neuroscience 2012, 224, 202–209. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.; Lee, S.; Kang, S.W. 2-cys peroxiredoxins: emerging hubs determining redox dependency of Mammalian signaling networks. Int. J. Cell Biol. 2014, 2014, 715867. [Google Scholar] [CrossRef]
- Wang, Y.; Ge, C.; Chen, J.; Tang, K.; Liu, J. GSK-3beta inhibition confers cardioprotection associated with the restoration of mitochondrial function and suppression of endoplasmic reticulum stress in sevoflurane preconditioned rats following ischemia/reperfusion injury. Perfusion 2018, 33, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Boveris, A.; Cadenas, E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid. Redox Signal. 2014, 20, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sun, T.; Xin, F.; Cui, W.; Guo, J.; Hu, J. Nerve Growth Factor Protects Against Alcohol-Induced Neurotoxicity in PC12 Cells via PI3K/Akt/mTOR Pathway. Alcohol Alcohol. 2017, 52, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Jope, R.S. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 2006, 79, 173–189. [Google Scholar] [CrossRef]
- Mazor, M.; Kawano, Y.; Zhu, H.; Waxman, J.; Kypta, R.M. Inhibition of glycogen synthase kinase-3 represses androgen receptor activity and prostate cancer cell growth. Oncogene 2004, 23, 7882–7892. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, M.-H.; Yu, J.-B.; Sun, H.-N.; Jin, Y.-H.; Shen, G.-N.; Jin, C.-H.; Cui, Y.-D.; Lee, D.-S.; Kim, S.-U.; Kim, J.-S.; et al. Peroxiredoxin II Maintains the Mitochondrial Membrane Potential against Alcohol-Induced Apoptosis in HT22 Cells. Antioxidants 2020, 9, 1. https://doi.org/10.3390/antiox9010001
Jin M-H, Yu J-B, Sun H-N, Jin Y-H, Shen G-N, Jin C-H, Cui Y-D, Lee D-S, Kim S-U, Kim J-S, et al. Peroxiredoxin II Maintains the Mitochondrial Membrane Potential against Alcohol-Induced Apoptosis in HT22 Cells. Antioxidants. 2020; 9(1):1. https://doi.org/10.3390/antiox9010001
Chicago/Turabian StyleJin, Mei-Hua, Jia-Bin Yu, Hu-Nan Sun, Ying-Hua Jin, Gui-Nan Shen, Cheng-Hao Jin, Yu-Dong Cui, Dong-Seok Lee, Sun-Uk Kim, Ji-Su Kim, and et al. 2020. "Peroxiredoxin II Maintains the Mitochondrial Membrane Potential against Alcohol-Induced Apoptosis in HT22 Cells" Antioxidants 9, no. 1: 1. https://doi.org/10.3390/antiox9010001
APA StyleJin, M.-H., Yu, J.-B., Sun, H.-N., Jin, Y.-H., Shen, G.-N., Jin, C.-H., Cui, Y.-D., Lee, D.-S., Kim, S.-U., Kim, J.-S., Kwon, T., & Han, Y.-H. (2020). Peroxiredoxin II Maintains the Mitochondrial Membrane Potential against Alcohol-Induced Apoptosis in HT22 Cells. Antioxidants, 9(1), 1. https://doi.org/10.3390/antiox9010001