1. Introduction
A noise exposure-induced reduction in hearing ability is referred to as noise-induced hearing loss (NIHL). Temporary NIHL is referred to as temporary threshold shift of hearing, and it is characterized by a reduced sensitivity to sound over a wide frequency range that is restored gradually to its original level within a short period of time. However, if the noise is intense or the duration of exposure is long enough, hearing loss might be irreversible, referred to as permanent threshold shift of hearing. The pathological mechanisms behind NIHL can be (1) direct mechanical destruction of the hair cell membranes and supporting structures of the organ of Corti, and (2) intense metabolic activities that lead to increased free radical (reactive oxygen and nitrogen species, ROS/RNS) generation in the inner ear tissue mitochondria. Excess generation of free radicals during and after noise exposure suppresses cochlear blood flow that promotes ischemia with further increase in free radical production and a decrease in blood flow. This positive feedback loop eventually induces both necrotic and apoptotic cell death in the organ of Corti [
1,
2].
Preconditioning is a non-damaging or minimal damage stress condition that potentiates the protective capability against a later and more injurious damage. Various preconditioning strategies such as hyperthermia and noise itself have been applied to protect the cochlea from potential hearing deficits that might be caused by later noise exposure [
3,
4,
5]. In particular, hypoxic preconditioning (8% O
2 for 4 h) confers significant protection against broadband noise administered 24–48 h after preconditioning in CBA mice. This inner ear resistance to noise injury is associated with increased expression of hypoxia-inducible factor-1α (HIF-1α) within the organ of Corti [
6]. It was reported previously that pretreatment with CoCl
2 under normoxic conditions mimics hypoxic preconditioning and upregulates HIF-1 expression that subsequently confers tolerance to severe hypoxic injury in chick embryos [
7]. Additionally, it has been reported that in vivo administration of CoCl
2 preconditions the mouse heart against global ischemia-reperfusion injury through the activation of HIF-1α, activator protein-1, and inducible nitric oxide synthase [
8]. Induction of HIF-1α in inner ears of CoCl
2-preconditioned mice inhibits noise-induced damage to the inner ear and leads to better recovery of hearing after noise exposure compared to its counterparts [
9]. However, the downstream pathway of HIF-1α–mediated preconditioning effect on inner ear remains to be elucidated.
Peroxiredoxins (Prdxs) belong to a superfamily of nonseleno and thiol-dependent peroxidases, catalyzing the reduction of H
2O
2, short-chain hydroperoxides, and peroxinitrite. They are widely distributed throughout all kingdoms and are classified as 1/2-cys Prdxs according to the number of conserved catalytic cysteine residues [
10]. Of the six mammalian Prdxs, the only 1-cys enzyme is Prdx6 that exhibits a bifunctional enzyme with both peroxidase and Ca
+2-independent phospholipase A
2 (aiPLA
2) activities. Prdx6 has unique characteristics that distinguish it from the other Prdx family members. Notably, it utilizes glutathione (GSH) instead of thioredoxin as a physiological reductant, forms heterodimerization with glutathione S-transferase π (GSTπ) to complete its catalytic cycle, and reduces/hydrolyzes phospholipid hydroperoxides [
11]. Prdx6 plays an important role in oxidative stress response as its overexpression in lung carcinoma cells (NCI-H441) reduced cellular OH
- levels, and attenuated membrane phospholipid peroxidation and apoptosis induced by Cu
2+-ascorbate treatment [
12]. Meanwhile, antisense-mediated decrease in Prdx6 expression resulted in the accumulation of lipid peroxidation products in the plasma membrane with subsequent apoptotic cell death [
13]. The antioxidant protective function of Prdx6 has been further supported by mouse model phenotypes. For example, transgenic Prdx6-overexpressing mice exhibited increased resistance to hyperoxia-induced lung injury [
14], while Prdx6-null mice were more susceptible to lung damage with paraquat administration, resulting in increased mortality [
15]. Multiple analyses of the Prdx6 promoter region have shown binding sites for various putative regulatory elements [
16], Pax5 [
17], and several redox-active transcription factors [
18,
19,
20], suggesting that interior and exterior factors such as a change in redox milieu contribute to its transcriptional expression. We have reported recently that retinoic acid-induced Prdx6 expression is involved in rapid hearing recovery after temporary noise exposure. Its transactivation is mediated by a retinoic acid response element on the Prdx6 promoter [
21].
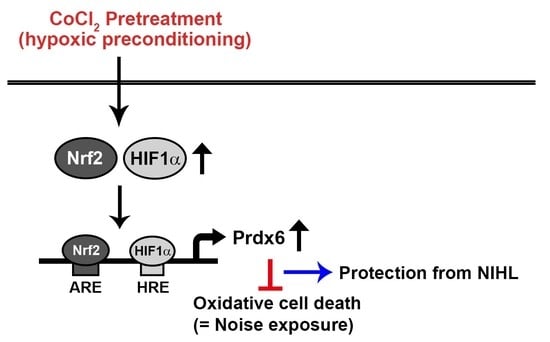
In the present study, we examined the protective mechanism behind hypoxia-mediated preconditioning against oxidative burst. Pretreatment of inner ear sensory hair cells with CoCl2 resulted in the activation of HIF-1α and Nrf-2 that function as positive transcription factors of Prdx6 gene expression. Additionally, we aimed to determine the immunolocalization of HIF-1α, Nrf-2, and Prdx6 proteins in the cochlear tissues of CoCl2-administrated mice.
2. Materials and Methods
2.1. Materials
All cell culture medium components were purchased from Life Technologies (Gaithersburg, MD, USA) unless otherwise stated. The primary antibodies used in the present study are polyclonal rabbit-anti-Nrf-2 (sc-722), goat-anti-Keap1 (sc-15246), and monoclonal mouse anti-Lamin B (sc-374015) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), polyclonal rabbit anti-HIF-1α, (ab2185, Abcam, Cambridge, MA, USA), polyclonal rabbit anti-Prdx6 (LF-PA0011) and rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH, LF-PA0018), AbFrontier Co., Seoul, Korea). Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA, USA). An immortalized auditory cell line (HEI-OC1) derived from the organ of Corti of Immortomouse transgenic mice was kindly provided by Dr. Federico Kalinec (Dept. of Cell and Molecular Biology, House Ear Institute, Los Angeles, CA, USA). All other chemicals (biotechnology grade) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
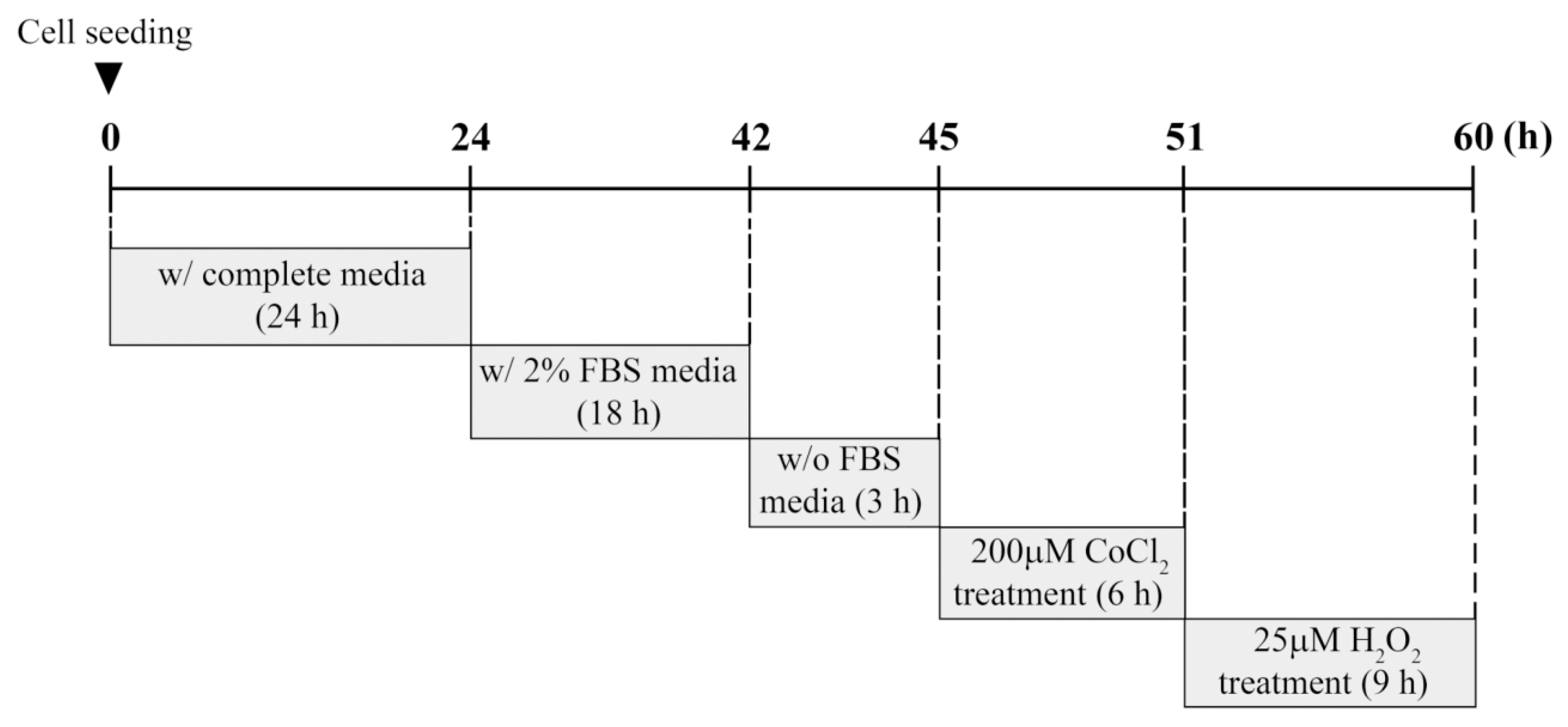
2.2. Cell Culture and CoCl2/H2O2 Treatment
The establishment and characterization of the immortalized HEI-OC1 auditory cells were performed as described previously [
22]. HEI-OC1 cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS) and penicillin (100 U/mL)/streptomycin (100 μg/mL) at 33 °C with 10% CO
2 atmosphere in a humidified incubator. For experiments involving CoCl
2 or H
2O
2 exposure, cells were seeded at ~70% confluence on 60-mm culture dishes and cultured for 24 h under standard conditions. Cells were deprived gradually of serum by incubation in 2% FBS overnight, followed by incubation in serum-free medium for 3 h. These serum-starved cells were exposed to different concentrations of CoCl
2 or H
2O
2 at the indicated times. In preconditioning studies, cells were preincubated with 200 μM CoCl
2 for 6 h. Next, the culture medium containing CoCl
2 was changed, and cells were treated with 25 μM H
2O
2 for 9 h. This experimental procedure including time schedule is summarized in
Figure 1.
2.3. Cytotoxicity Assay
Cell viability was measured by a Cell Counting Kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instructions. The cells were seeded onto 96-well plates at a density of 5 × 103 cells/well. Following serum starvation, cells were treated with CoCl2, H2O2, or both for 24 h as described above. The amount of dark blue formazan product was determined by measuring absorbance at 450 nm using a microplate spectrophotometer (Molecular Devices Corp., Sunnyvale, CA, USA). The absorbance value in untreated control cells was taken as 100% of viability.
2.4. Detection of Intracellular Oxygen Radicals
Intracellular ROS level was measured using a fluorescent dye, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetylester (CM-H2DCFDA, Molecular Probes, Inc., Eugene, OR, USA). Serum-starved cells grown on 96-well plates (4 × 103 cells/well) were treated with 200 μM CoCl2 for 6 h, 25 μM H2O2 for 9 h, or both as described above. Cells were then washed twice with Hank’s balanced salt solution (HBSS) and incubated with 5 μM CM-H2DCFDA at 33 °C for 20 min in the dark. After washing with HBSS, the levels of DCF fluorescence were immediately measured using a luminescence spectrofluorometer (VICTOR 3; Perkin-Elmer, Waltham, MA, USA) with excitation and emission wavelengths of 485 and 535 nm, respectively. The values were converted to folds for comparison with the untreated control.
2.5. Construction of Murine NRF-2 or HIF-1α Gene Knockdown (KD) Plasmids
The shRNA expression vectors targeting the murine Nrf-2 and HIF-1α genes were generated by designing target DNA oligonucleotides (
Table 1). The expression vectors were subcloned into pLKO.1-puro lentiviral vector (Addgene, Cambridge, MA, USA) double-digested with Age I and EcoR I (pLKO-mNrf-2 or pLKO-mHIF-1α). Subsequently, each recombinant plasmid was transformed into competent DH5α
E. coli cells. Correct short hairpin (sh) sequence formation was confirmed by DNA sequencing.
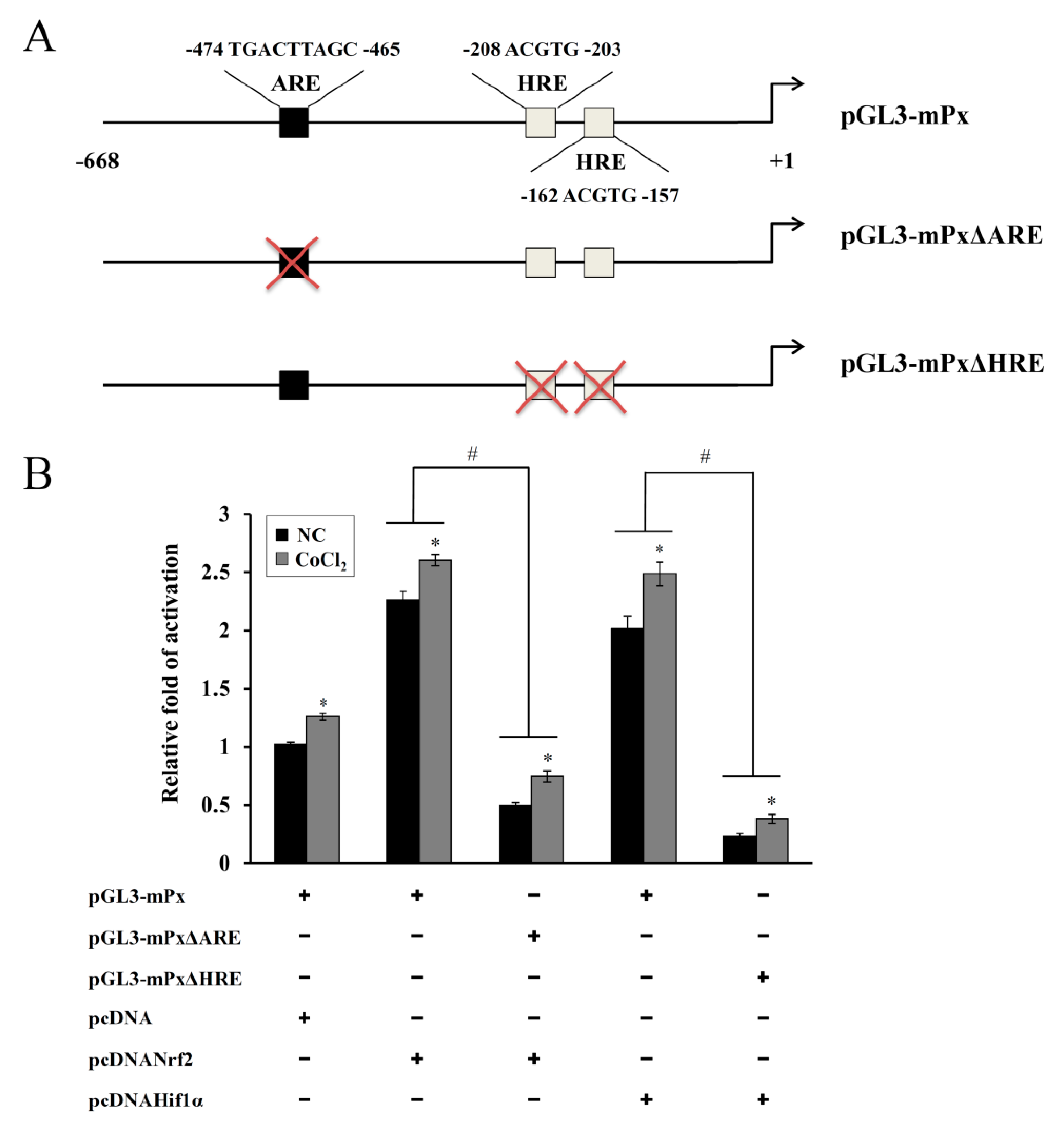
2.6. Construction of ARE- and HRE-Deleted Reporter Plasmids (pGL3-mPxΔARE or pGL3-mPxΔHRE)
A luciferase reporter plasmid (pGL3-mPx) containing the proximal 5′-flanking region of the murine Prdx6 promoter (668bp) has been described elsewhere [
17,
21]. In each mutant-reporter plasmid, the deletion of the HRE or antioxidant response element (ARE) consensus sequences within the Prdx6 promoter was done with the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) using pGL3-mPx as the template and appropriate sets of primers. DNA sequencing was used to verify the mutant constructs with the deletions (pGL3-mPxΔARE or pGL3-mPxΔHRE).
2.7. Transfection of NRF-2 or HIF-1α KD Plasmids into HEI-OC1 Cells
HEI-OC1 cells that reached ~70% confluence, were transfected with the reporter plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). HEI-OC1 cells were transfected with the same amount of mock vector as an internal control. Forty-eight hours after transfection, cells were harvested for immunoblot analysis of Nrf-2 or HIF-1α expression. To evaluate the cell viability, each transfectant was treated with CoCl2, H2O2, or both as described above.
2.8. Luciferase Reporter Assay
HEI-OC1 cells were seeded in 24-well culture plates at a density of 1 × 10
5 cells/well and cotransfected with pGL3-mPx and each shRNA expression vector (pLKO-mNrf-2 or pLKO-mHIF-1α). Additionally, cells were cotransfected with wild- or mutant-reporter plasmids (pGL3-mPxΔARE or pGL3-mPxΔHRE) and Nrf-2 or HIF-1α overexpression plasmids (pcDNA-Nrf-2 or HA-HIF-1α-pcDNA3) as described elsewhere [
20]. For the normalization of transfection efficiency, the pCMV-β-gal plasmid was simultaneously transfected into the cells. About 48 h after transfection, cells were treated with CoCl
2 (200 μM) for 6 h. Luciferase and β-galactosidase activities from total cell lysate were measured according to respective Bright-Glo Luciferase and Beta-Glo Assay kits (Promega, Madison, WI, USA) using a microplate luminometer (Perkin-Elmer). The luciferase activities of individual reporter plasmids were normalized to those of β-galactosidase.
2.9. Immunoblot Analysis
Cells were washed with ice-cold PBS and lysed with RIPA buffer (Sigma-Aldrich) supplemented with complete protease inhibitor cocktail and centrifuged at 13,000× g at 4 °C for 20 min. Nuclear proteins were isolated using the NE-PER Cytoplasmic and Nuclear Protein extraction kit (Pierce Biotechnology, Rockford, IL, USA) according to the manufacturer’s instructions. Protein concentration was determined with the BCA Protein Assay kit (Pierce Biotechnology). Next, 30 μg of total soluble proteins or 5 μg of nuclear proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes (Merck-Millipore, Billerica, MA, USA). The membranes were probed with primary antibodies (1:1000 dilution) described in Materials, followed by incubation with appropriate secondary antibodies (1:5000 dilution). The immunoreactive bands were visualized with a West-Q chemiluminescent Substrate kit (GenDEPOT, Barker, TX, USA) and the band intensities on films were analyzed by densitometry to quantify protein expression using a FluorS MultiImager (Bio-Rad, Hercules, CA, USA). The membranes then were washed with Restore Western Blot Stripping Buffer (Thermo Scientitis, Waltham, MA, USA) and reprobed with 1:1000 diluted anti-GAPDH polyclonal or anti-Lamin B polyclonal antibodies to normalize for cytosolic or nuclear protein loading, respectively.
2.10. Administration of Mice with CoCl2
All experimental procedures were performed in compliance with the guidelines of the National Institutes of Health and the Declaration of Helsinki. The Committee on the Use and Care of Animals of the University of Ulsan approved protocols. Animal care was performed under the supervision of the Laboratory Animal Unit of the Asan Institute for Life Sciences (IACUC No. 2016-13-271; date of approval, November 14, 2016). Male CBA mice at 5–6 weeks (~20 g) of age (Oriental Charles River Technology, Seoul, Korea) were intraperitoneally administered with vehicle (saline) or CoCl2 (60 mg/kg body weight). After 6 h, both cochleae were removed, fixed with 4% formaldehyde/1% glutaraldehyde in 0.1 M sodium phosphate buffer, decalcified in 5.5% EDTA, and embedded in paraffin.
2.11. Immunohistochemistry
Deparaffinized and rehydrated cochlear paraffin sections (5-μm thick) were heated in a microwave oven in 10 mM sodium citrate buffer (pH 6.0) for antigen retrieval and then pretreated with 3% H2O2 in 0.1 M Tris-buffered saline (TBS) (pH 7.4) to quench endogenous peroxidase activity. The sections were incubated with TBS containing 5% normal goat serum and the primary antibodies (1:500 dilution for HIF-1α and Nrf-2, 1:7500 dilution for Prdx6) overnight at 4 °C, followed by goat anti-rabbit polyclonal HRP-conjugated secondary antibody (1:100 dilution; DakoNorth America, Inc., Carpinteria, CA, USA). Immunostaining of each protein was visualized with an ImmPACT™ DAB Peroxidase Substrate kit (Vector Laboratories, Burlingame, CA, USA). The sections were counterstained with Mayer’s hematoxylin, dehydrated, cleared in xylene, and mounted in Permount. Images of sections were recorded with an upright microscope (Nikon Eclipse Ci, Tokyo, Japan).
2.12. Statistical Analysis
Data were expressed as means ± standard deviation (SD) of three or more independent experiments. Differences between groups were evaluated using the Student t-test or two-way ANOVA with Tukey’s post-hoc test, as appropriate. Differences between mean values were considered statistically significant at p < 0.05.
4. Discussion
It is well established that a noise exposure induced elevation in free radical generation in the cochlea causes oxidative stress-induced sensory hair cell damage with subsequent acoustic trauma such as NIHL. Since the regeneration of injured auditory neurons and hair cells is difficult, there is no effective treatment after the development of a permanent threshold shift. In this respect, the paradigm of preconditioning is one of the most effective approaches for intrinsic protection of the cochlea against noise-induced damage. We reported previously the protective effect of intraperitoneal CoCl
2 preinjection on sensory hair cell loss and hearing impairment induced by white band noise exposure in mice [
9]. In the present study, we found that CoCl
2 pretreatment protected auditory hair cells (HEI-OC1) from H
2O
2-mediated cytotoxicity via the activation of redox-sensitive transcription factors HIF-1α and Nrf-2 and their target gene Prdx6, demonstrating an in vitro hypoxic preconditioning mechanism for noise-induced cochlear cell injury.
Preconditioning with mild hypoxia can protect various tissues against subsequent lethal hypoxic/ischemic injuries. In the cochlea, sublethal ischemia prevented lethal ischemia-induced hair cell degeneration and ameliorated hearing impairment, suggesting ischemic tolerance [
24]. Systemic hypoxia in mice resulted in smaller permanent threshold shifts at all tested frequencies, compared to the air-exposed control [
6]. An appealing mechanism for hypoxia-mediated protection is the elevated antioxidative capacity regulation of later excessive free radical formation within the cochlea and other inner ear structures during and after noise exposure. Hypoxic preconditioning can be chemically mimicked using CoCl
2, where ionized cobalt interferes with intracellular oxygen homeostasis. Cobalt is an essential element found in the form of cyanocobalamin (vitamin B
12) and it is critical for animals due to its involvement in red blood cell production and nervous system function. Although exposure to a high dose or prolonged exposure to a low dose of CoCl
2 induces apoptosis and necrosis with inflammatory responses, its beneficial effects include stimulation of erythropoiesis and angiogenesis, promotion of tissue adaptation to hypoxia, and improvement of ischemic/hypoxic tolerance [
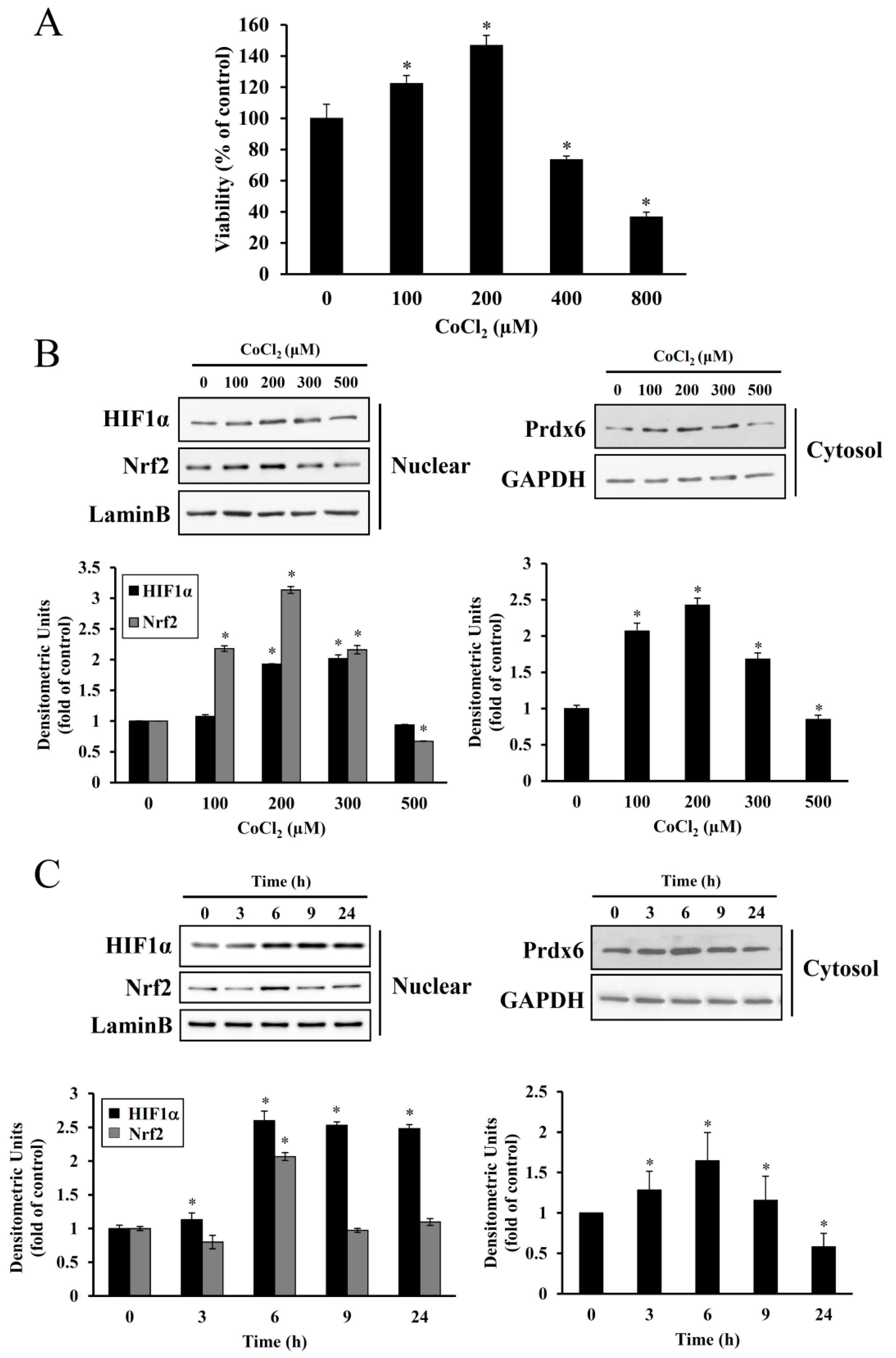
25]. In the present study, treatment with 100–200 μM CoCl
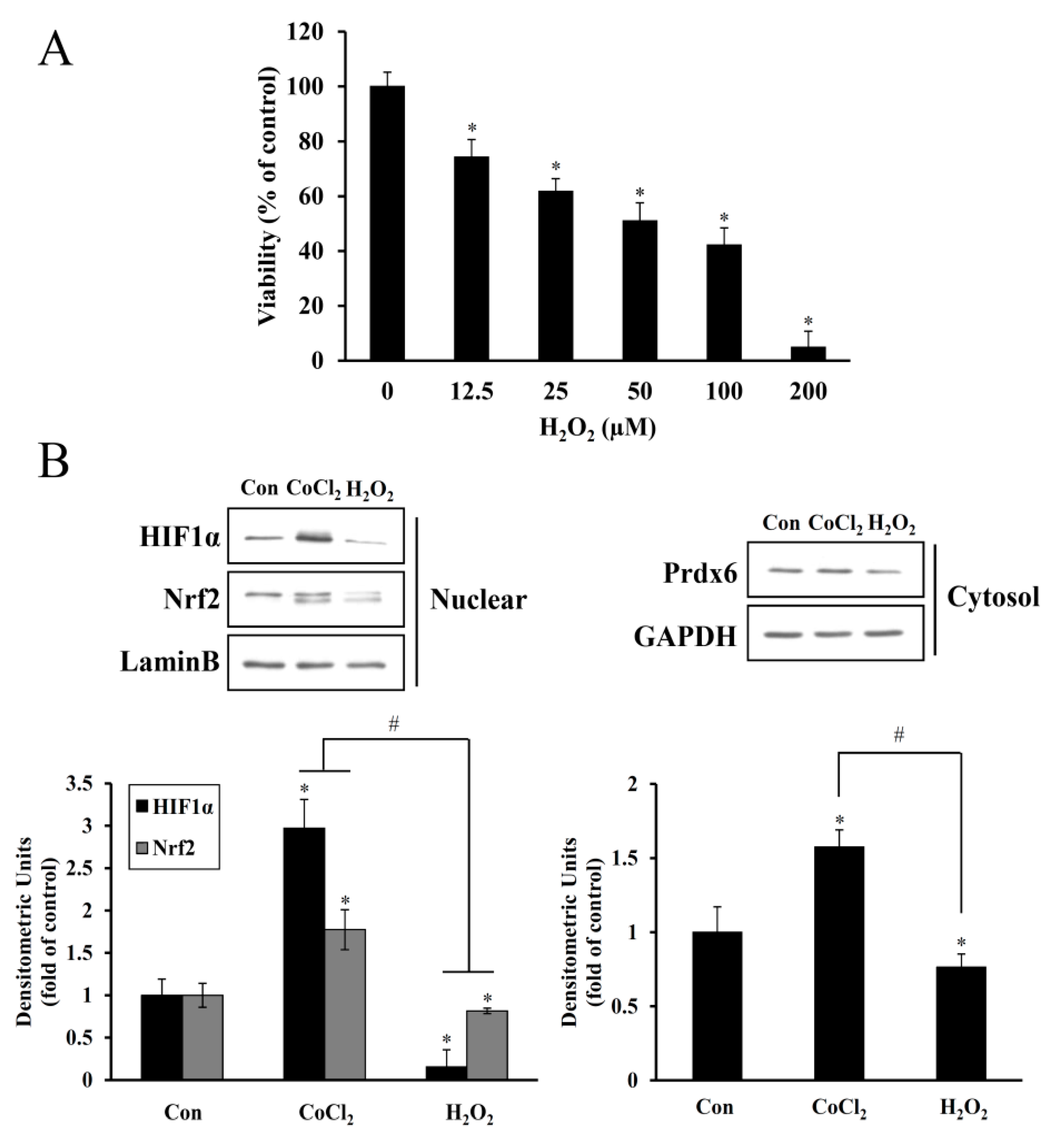
2 for 6 h resulted in the proliferation of HEI-OC1 cells. However, at higher dose ranges, this proliferative effect was reduced (
Figure 2), suggesting a dose and exposure time-dependent optimal preconditioning effect. The same CoCl
2 concentration range has been reported to promote the proliferation and migration of rhesus choroid endothelial cells [
26].
Cobalt triggers free radical generation via a Fenton-like reaction. We observed intracellular ROS generation slightly increased in HEI-OC1 cells exposed to CoCl
2 for 6 h, as assessed by a peroxide-sensitive fluorescent probe, CM-H
2DCFDA (
Figure 4C). In contrast to the detrimental effects of an abrupt alternation of redox status probably due to massive ROS production, moderate amounts of ROS act as second messengers in signal transduction and gene regulation in various cell types, thus protecting against lethal oxidative stress. HIF-1α and Nrf-2 are well known redox-sensitive transcription factors that activate the expression of genes involved in the adaptation of cells and tissues to hypoxia and detoxification/antioxidant defense, respectively [
27]. In the present study, maximal accumulation of HIF-1α and Nrf-2 was detected in the nuclei of HEI-OC1 cells treated with CoCl
2 for 6 h, and a concurrent maximal elevation was observed in the cytosolic levels of their target, Prdx6 (
Figure 2). This exposure time coincided with the highest proliferation rate mediated by CoCl
2, indicating that ROS generation under these conditions promotes the induction of redox response genes and cellular proliferation.
Excessive free radical generation induced by noise exposure causes the accumulation of lipid peroxidation products, oxidized proteins, and oxidative DNA adducts in the cochlea and other inner ear cells, promoting the death of the hair cells and nerve endings with subsequent hearing loss. Exposure of mice to intense broadband noise resulted in the significant elevation of cochlear ROS levels within 1–2 h following the exposure, along with anatomical abnormalities of the OC and a permanent threshold shift [
28]. Additionally, intense noise increased the expression of lipid peroxidation products such as 8-isoprostane in the SV, SGC, and the OC of guinea pigs. In the OC, the heavily immunoreactive outer hair cells than inner hair cells were well correlated with permanent hair cell damage [
29]. The accumulation of ROS and RNS markers appeared rapidly and transiently in the inner ear during and following high-intensity noise exposure, while hair cell loss progressively increased over time, stabilizing two or more weeks after a single insult. Therefore, oxidative/nitrative stress triggered by noise begins early and persists for an extended time even after termination of noise exposure [
30]. In all, these findings imply that noise-induced immediate hair cell injury might be attributable to mechanical irritation plus transiently intensive free radical generation, whereas its continuous generation contributes to long-term hair cell loss and eventually, permanent hearing loss. In the present study, noise-induced oxidative stress in hair cells was mimicked by direct H
2O
2 exposure, causing a dose-dependent increase in cell death and suppression of HIF-1α, NRF-2, and Prdx6 expression to below the expression levels observed in the untreated control (
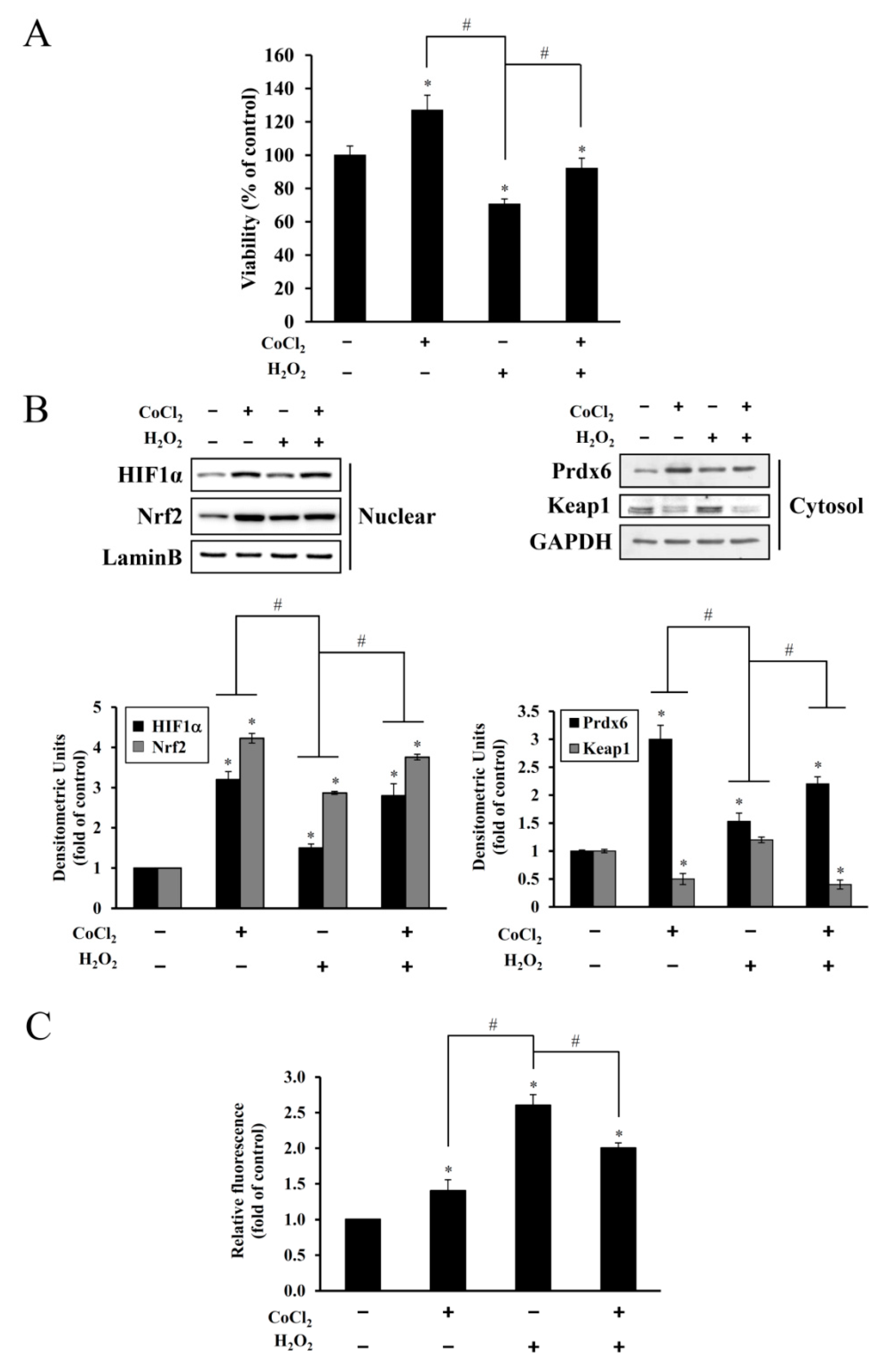
Figure 3). This oxidative stress-mediated cytotoxicity, protein degradation, and intracellular ROS accumulation were attenuated by CoCl
2 pretreatment (
Figure 4). The protective effect of CoCl
2 on oxidative injury has been also reported in HepG2 tumor cells treated with tert-butyl hydroperoxide/serum deprivation [
31], neonatal piglets that underwent hypothermic circulatory arrest [
32], and in rats exposed to hypobaric hypoxia-induced oxidative stress [
33], where the upregulation of HIF-1α and its target proteins play crucial roles for benefiting from CoCl
2 preconditioning.
In addition to HIF-1α [
6,
9], a protective role has been reported for Nrf-2 in NIHL animal models. The Nrf-2/heme oxygenase-1 (HO-1) signaling pathway was activated in the OC of rats intraperitoneally administered with rosmarinic acid, resulting in the reduction of the noise-induced oxidative stress triggered a generation of superoxide and lipid peroxidation [
34]. Nrf-2-null mice exhibited more severe impairment of hearing than its WT counterparts at day 7 after noise exposure, whereas treatment with Nrf-2-activating drugs before noise exposure preserved the integrity of hair cells and improved post-exposure hearing levels in only the WT mice. Moreover, a single nucleotide polymorphism in the human NRF-2 promoter was associated with sensory neuronal hearing loss [
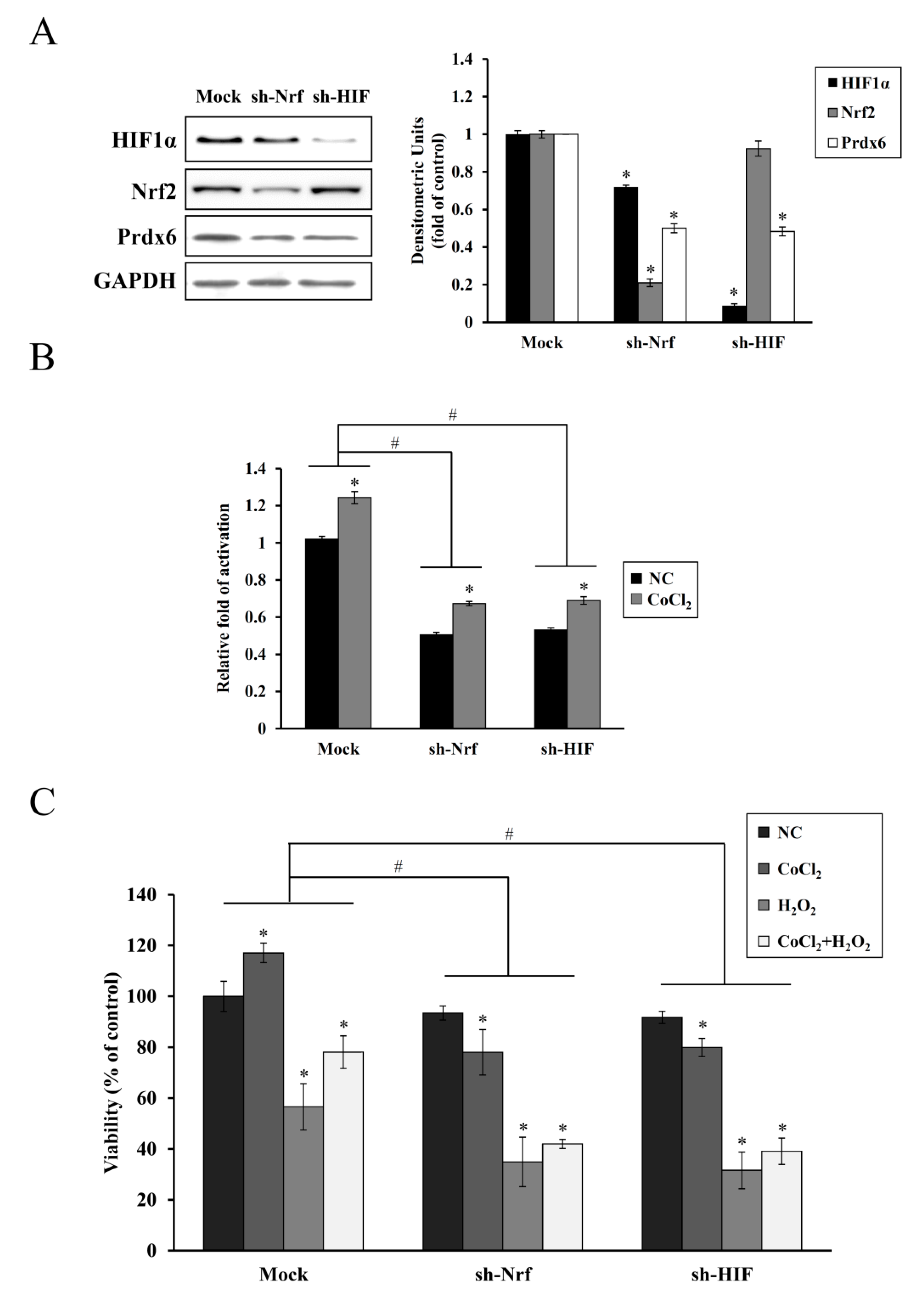
35]. In the present study, shRNA KD of HIF-1α or NRF-2 abolished the endogenous upregulation of HIF-1α or NRF-2 along with Prdx6 expression and cell proliferation induced by CoCl
2 pretreatment, thus, failing subsequent cytoprotection against oxidative insults (
Figure 5). A similar correlation between reduced HIF-1α and Nrf-2 expression and increased apoptotic cell death has been previously reported in chronic hypoxia-mediated neurodegeneration [
36]. In particular, we found that Nrf-2 KD attenuated the induction of HIF-1α expression, suggesting its role as a transcriptional regulator of
HIF-1α expression. This is consistent with a previous finding, where downregulation of
NRF-2 expression via shRNA interference impaired the hypoxia-mediated induction of HIF-1α and its target gene expression in glioblastoma cells [
37]. Nrf-2 is also required for the arsenite-mediated upregulation of HIF-1α in HepG2 hepatoma cells [
38]. A direct regulatory connection between Nrf-2 and HIF-1α via the Nrf-2 binding site (ARE) in the enhancer region upstream of HIF-1α gene has been recently reported, indicating that Nrf-2 plays a role in HIF-1α expression [
39].
Elevated ROS generation or oxidative stress actively induces Prdx6 expression. Prdx6 functions as an antioxidant through its peroxidase activity, reducing various oxidants including H
2O
2, short-chain hydroperoxides, oxidized fatty acids, and phospholipid hydroperoxides [
11]. Transcriptional regulation of
Prdx6 gene is mediated by multiple redox-active transcription factors to its promoter [
20]. In the present study, one ARE and two HRE consensus sequences were observed in the murine Prdx6 promoter and both Nrf-2 and HIF-1α contributed to the CoCl
2-induced expression of Prdx6, as confirmed by the KD and mutant promoter/reporter assays (
Figure 5 and
Figure 6). ChIP analyses using primer sets covering the ARE and HRE regions and a respective antibody revealed that their binding abilities were substantially increased by CoCl
2 treatment (data not shown), confirming that both sequences are functionally responsible for the binding of Nrf-2 and HIF-1α, respectively. These results indicate that Nrf-2 and HIF-1α recognize their respective ARE and HRE sequences within the murine Prdx6 promoter and function as transactivators for the Prdx6 gene during the CoCl
2 preconditioning period. Consistent with the findings from the present study, it has been shown in other studies that the transcriptional upregulation of other antioxidants/phase II-detoxifying enzymes such as HO-1 in Kupffer cells from ethanol-fed rats is mediated by the binding of Nrf-2 and HIF-1α to their respective sequences within the HO-1 promoter, thus protecting the liver from alcohol-induced injury [
40]. The Keap1/Nrf-2/ARE pathway-mediated upregulation of
Prdx6 mRNA expression has been reported in human lung carcinoma cells (A549) and primary rat alveolar type II cells treated with H
2O
2 or tert-butylhydroquinone [
19]. Additionally, Prdx6 expression has been associated with cell viability in retinal ganglion cells treated with CoCl
2, where the prolonged exposure triggered reduction in Prdx6 expression was related to an increase in hypoxia-induced cell death [
41]. In all, these findings implicate that an Nrf-2/HIF-1α pathway regulated induction of Prdx6 expression plays protective roles in oxidative stress-induced cell death.
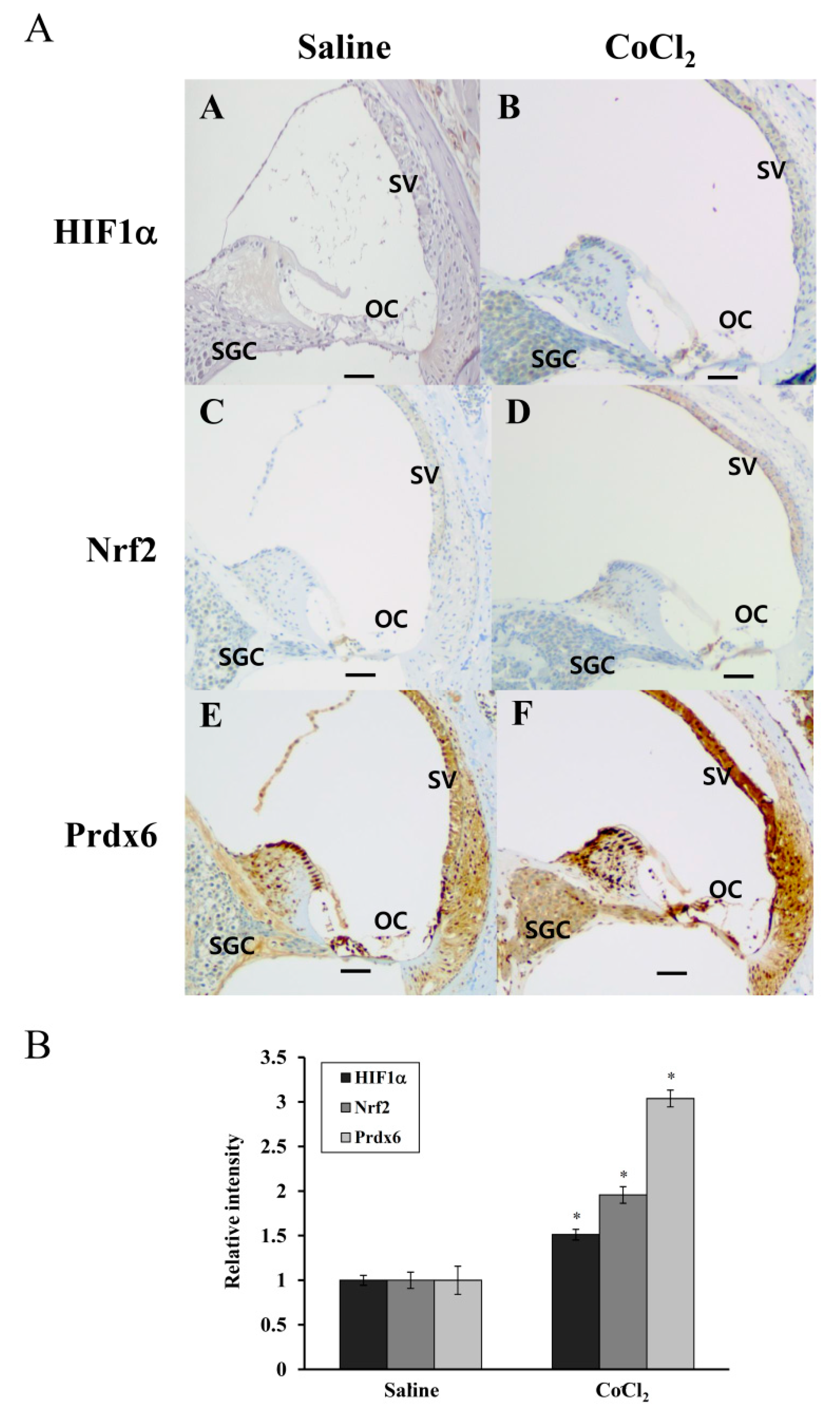
Accumulation of HIF-1α was reported previously in the noise-exposed cochlea of mice, where its expression was elevated in OC, SV, and SGC after a 3h exposure to white band noise with 120dB peak equivalent sound pressure level [
42]. Furthermore, the induction of HIF-1α expression by CoCl
2 preinjection minimized sensory hair cell loss caused by subsequent noise exposure in these regions, compared with the saline preinjected controls [
9]. In the human cochlea, Nrf-2 immunoreactivity was detected in the cytoplasm and nuclei of hair and supporting cells of OC and the vestibular sensory epithelia, and its reactivity significantly decreased with age [
43]. Consistent with these findings, the present study showed a more intense immunoreactivity for HIF-1α, Nrf-2, and Prdx6 in the OC, SV, and SGC regions of CoCl
2-injected cochleae compared to that observed in the same regions of saline-injected controls (
Figure 7). Considering the accumulation of free radicals in these regions during and after noise exposure and their antioxidant capacities, it is likely that the induction of HIF-1α and Nrf-2 by CoCl
2 injection leads to the transcriptional upregulation of Prdx6, in turn protecting the cochlea from noise-triggered oxidative damage. Prdx6 is widely distributed throughout all major mammalian organs with high contents in the lung, brain, liver, kidney, and testis. Within tissues, Prdx6 expression is the highest in epithelium, including apical regions of olfactory and respiratory epithelium and skin epidermis, where it functions in vivo as an antioxidant enzyme [
11]. Since noise-induced cochlear damage is caused by direct mechanical destruction, excessive free radical generation, and reduction of cochlear blood flow, elevated protein expression of Prdx6 in OC, SV, and SGC postulates that it might be involved in repairing noise-mediated mechanical wounds, antioxidative defense, as well as restoration of redox homeostasis. This is supported by findings from previous studies showing endogenous Prdx6 overexpression in the hyperproliferative epidermis of mouse skin wounds [
44] and actively proliferating epithelia and anterior stroma of wounded rat corneas after photorefractive keratectomy [
45]. Moreover, Prdx6 has been reported to participate in the protection of the epidermis from UV-induced damage and the formation of new blood vessels in inflamed tissues caused by excision injury, suggesting its requirement for blood vessel integrity and viability in the wounded tissue [
46]. To our knowledge, this is the first report of Prdx6 immunolocalization to mouse cochlear tissues.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}