Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
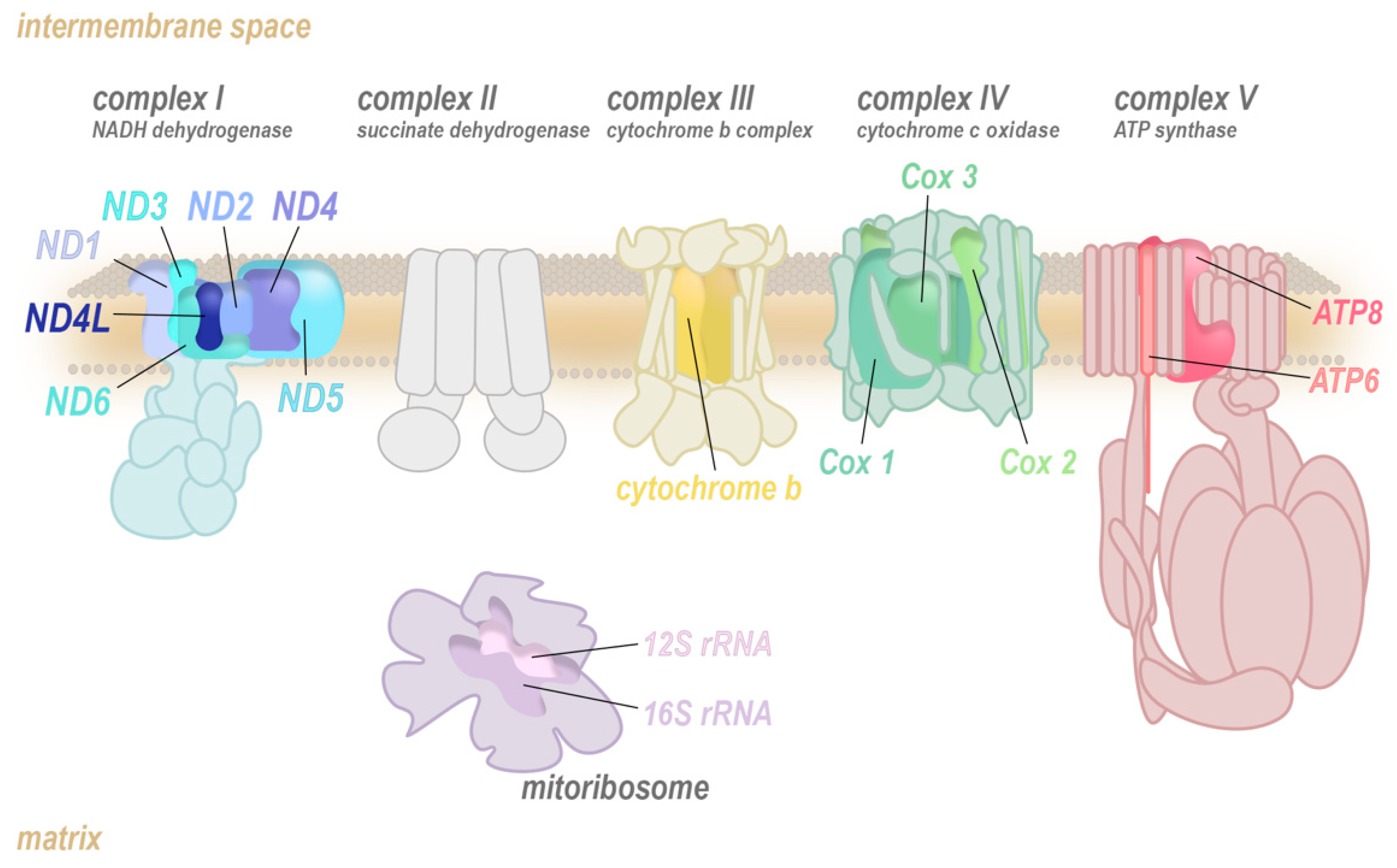
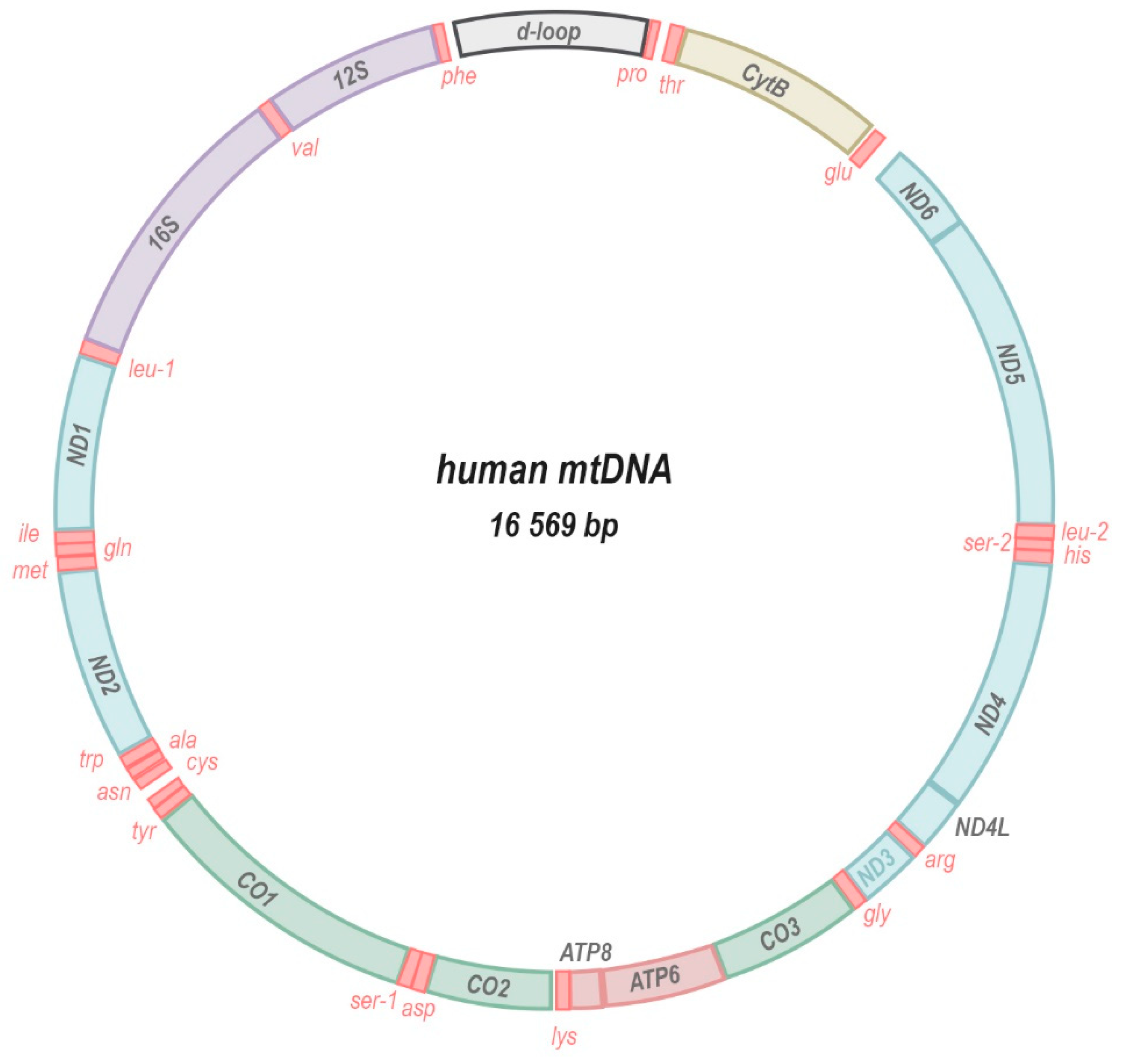
1. The Mitochondrial Genome
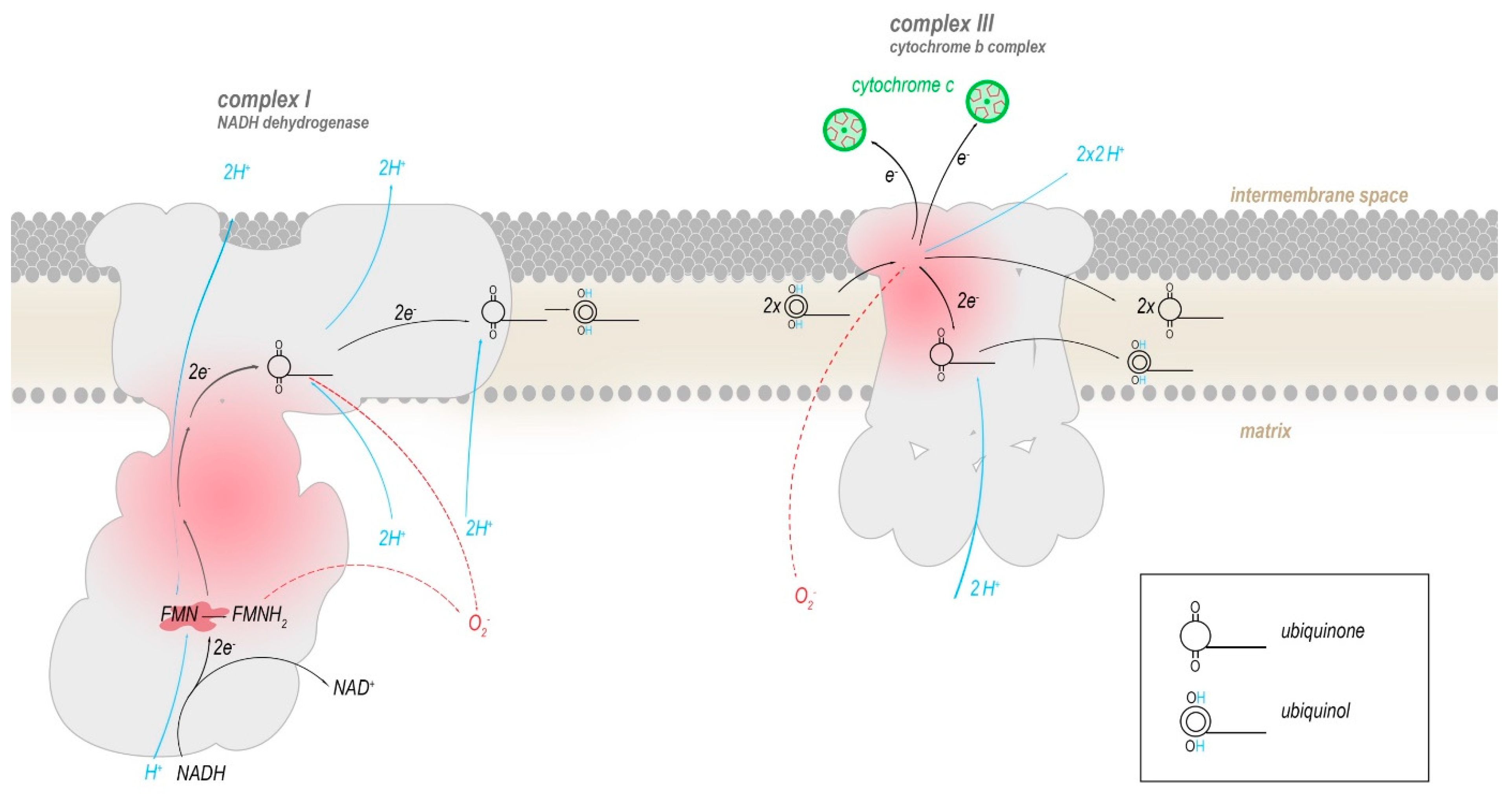
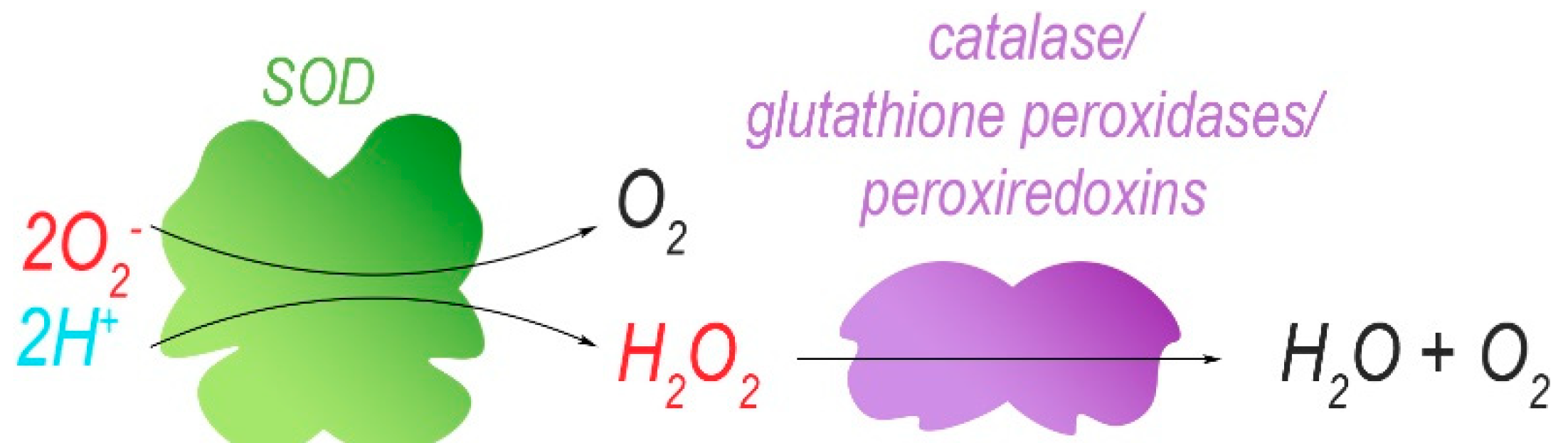
2. Mitochondrial-Encoded Complex I (NADH Dehydrogenase) Mutations
3. Mitochondrial-Encoded Complex III (Coenzyme Q-Cytochrome C Reductase) Mutations
4. Mitochondrial-Encoded Complex IV (Cytochrome C Oxidase) Mutations
5. Mitochondrial-Encoded Complex V (ATP Synthase) Mutations
6. Mitochondrial-Encoded tRNA and rRNA Mutations
7. Mutations in Polymerase γ (Nuclear-Encoded)
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kolesnikov, A.A.; Gerasimov, E.S. Diversity of mitochondrial genome organization. Biochemistry 2012, 77, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Alverson, A.J.; Rice, D.W.; Dickinson, S.; Barry, K.; Palmer, J.D. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant. Cell 2011, 23, 2499–2513. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Zuryn, S. The Cellular Mitochondrial Genome Landscape in Disease. Trends Cell Biol. 2019, 29, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Soong, N.W.; Hinton, D.R.; Cortopassi, G.; Arnheim, N. Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat. Genet. 1992, 2, 318–323. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515. [Google Scholar] [CrossRef]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518. [Google Scholar] [CrossRef]
- Ahier, A.; Dai, C.-Y.; Tweedie, A.; Bezawork-Geleta, A.; Kirmes, I.; Zuryn, S. Affinity purification of cell-specific mitochondria from whole animals resolves patterns of genetic mosaicism. Nat. Cell Biol. 2018, 20, 352–360. [Google Scholar] [CrossRef]
- Ahier, A.; Cummins, N.; Dai, C.-Y.; Götz, J.; Zuryn, S. PINK1-parkin-mediated mitophagy generates stereotyped somatic mosaicism of the mitochondrial genome. BioRxiv 2019. [Google Scholar] [CrossRef]
- Barrell, B.G.; Bankier, A.T.; Drouin, J. A different genetic code in human mitochondria. Nature 1979, 282, 189. [Google Scholar] [CrossRef]
- Macino, G.; Coruzzi, G.; Nobrega, F.G.; Li, M.; Tzagoloff, A. Use of the UGA terminator as a tryptophan codon in yeast mitochondria. Proc. Natl. Acad. Sci. USA 1979, 76, 3784–3785. [Google Scholar] [CrossRef] [PubMed]
- Haag, S.; Sloan, K.E.; Ranjan, N.; Warda, A.S.; Kretschmer, J.; Blessing, C.; Hübner, B.; Seikowski, J.; Dennerlein, S.; Rehling, P.; et al. NSUN3 and ABH1 modify the wobble position of mt-tRNAMet to expand codon recognition in mitochondrial translation. EMBO J. 2016, 35, 2104–2119. [Google Scholar] [CrossRef] [PubMed]
- Osawa, S.; Jukes, T.H.; Watanabe, K.; Muto, A. Recent evidence for evolution of the genetic code. Microbiol. Rev. 1992, 56, 229–264. [Google Scholar] [PubMed]
- Bender, A.; Hajieva, P.; Moosmann, B. Adaptive antioxidant methionine accumulation in respiratory chain complexes explains the use of a deviant genetic code in mitochondria. Proc. Natl. Acad. ASci. USA 2008, 105, 16496–16501. [Google Scholar] [CrossRef] [PubMed]
- Garey, J.R.; Wolstenholme, D.R. Platyhelminth mitochondrial DNA: Evidence for early evolutionary origin of a tRNAserAGN that contains a dihydrouridine arm replacement loop, and of serine-specifying AGA and AGG codons. J. Mol. Evol. 1989, 28, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.A. Replication and Transcription of Vertebrate Mitochondrial DNA. Annu. Rev. Cell Biol. 1991, 7, 453–478. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Clayton, D.A. Mitochondrial transcription initiation. Variation and conservation. J. Biol. Chem. 1993, 268, 16083–16086. [Google Scholar] [PubMed]
- Fernandez-Silva, P.; Enriquez, J.A.; Montoya, J. Replication and transcription of mammalian mitochondrial DNA. Exp. Physiol. 2003, 88, 41–56. [Google Scholar] [CrossRef]
- Kasamatsu, H.; Robberson, D.L.; Vinograd, J. A Novel Closed-Circuit Mitochondrial DNA with Properties of a Replicating Intermediate. Proc. Natl. Acad. Sci. USA 1971, 68, 2252–2257. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. Endogenous oxidative damage of mtDNA. Mutation Res. Fundam. Mol. Mech. Mutagen. 1999, 424, 51–58. [Google Scholar] [CrossRef]
- Haag-Liautard, C.; Coffey, N.; Houle, D.; Lynch, M.; Charlesworth, B.; Keightley, P.D. Direct Estimation of the Mitochondrial DNA Mutation Rate in Drosophila melanogaster. PLoS Biol. 2008, 6, e204. [Google Scholar] [CrossRef] [PubMed]
- Radzvilavicius, A.L.; Hadjivasiliou, Z.; Pomiankowski, A.; Lane, N. Selection for Mitochondrial Quality Drives Evolution of the Germline. PloS Biol. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Elliott, H.R.; Samuels, D.C.; Eden, J.A.; Relton, C.L.; Chinnery, P.F. Pathogenic Mitochondrial DNA Mutations Are Common in the General Population. Am. J. Hum. Genet. 2008, 83, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of Reactive Oxygen Species by Mitochondria: Central Role of Complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of Superoxide Production from Different Sites in the Mitochondrial Electron Transport Chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Utsumi, H.; Kang, D.; Hattori, N.; Uchida, K.; Arimura, K.-i.; Egashira, K.; Takeshita, A. Mitochondrial Electron Transport Complex I Is a Potential Source of Oxygen Free Radicals in the Failing Myocardium. Circ. Res. 1999, 85, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metabol. 2005, 1, 401–408. [Google Scholar] [CrossRef]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial Complex III ROS Regulate Adipocyte Differentiation. Cell Metabol. 2011, 14, 537–544. [Google Scholar] [CrossRef]
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum. Mol. Genet. 2009, 18, 1578–1589. [Google Scholar] [CrossRef]
- Sun, W.; Zhou, S.; Chang, S.S.; McFate, T.; Verma, A.; Califano, J.A. Mitochondrial mutations contribute to HIF1alpha accumulation via increased reactive oxygen species and up-regulated pyruvate dehydrogenease kinase 2 in head and neck squamous cell carcinoma. Clinic. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Bonora, E.; Porcelli, A.M.; Gasparre, G.; Biondi, A.; Ghelli, A.; Carelli, V.; Baracca, A.; Tallini, G.; Martinuzzi, A.; Lenaz, G.; et al. Defective Oxidative Phosphorylation in Thyroid Oncocytic Carcinoma Is Associated with Pathogenic Mitochondrial DNA Mutations Affecting Complexes I and III. Cancer Res. 2006, 66, 6087–6096. [Google Scholar] [CrossRef] [PubMed]
- Koshikawa, N.; Hayashi, J.; Nakagawara, A.; Takenaga, K. Reactive oxygen species-generating mitochondrial DNA mutation up-regulates hypoxia-inducible factor-1alpha gene transcription via phosphatidylinositol 3-kinase-Akt/protein kinase C/histone deacetylase pathway. J. Biol. Chem. 2009, 284, 33185–33194. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.-I. ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, W.; Li, H.; Yu, Y.; Tao, J.; Huang, S.; Zeng, Z. Nonsense and missense mutation of mitochondrial ND6 gene promotes cell migration and invasion in human lung adenocarcinoma. BMC Cancer 2015, 15, 346. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Sharpley, M.S.; Fan, W.; Waymire, K.G.; Sadun, A.A.; Carelli, V.; Ross-Cisneros, F.N.; Baciu, P.; Sung, E.; McManus, M.J.; et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 20065–20070. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Bermudez, A.; Vallejo, C.G.; Vicente-Blanco, R.J.; Gallardo, M.E.; Fernandez-Moreno, M.A.; Quintanilla, M.; Garesse, R. Enhanced tumorigenicity by mitochondrial DNA mild mutations. Oncotarget 2015, 6, 13628–13643. [Google Scholar] [CrossRef]
- Beretta, S.; Mattavelli, L.; Sala, G.; Tremolizzo, L.; Schapira, A.H.V.; Martinuzzi, A.; Carelli, V.; Ferrarese, C. Leber hereditary optic neuropathy mtDNA mutations disrupt glutamate transport in cybrid cell lines. Brain 2004, 127, 2183–2192. [Google Scholar] [CrossRef]
- Perier, C.; Tieu, K.; Guegan, C.; Caspersen, C.; Jackson-Lewis, V.; Carelli, V.; Martinuzzi, A.; Hirano, M.; Przedborski, S.; Vila, M. Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc. Natl. Acad. Sci. USA 2005, 102, 19126–19131. [Google Scholar] [CrossRef]
- Wong, A.; Cavelier, L.; Collins-Schramm, H.E.; Seldin, M.F.; McGrogan, M.; Savontaus, M.L.; Cortopassi, G.A. Differentiation-specific effects of LHON mutations introduced into neuronal NT2 cells. Hum. Mol. Genet. 2002, 11, 431–438. [Google Scholar] [CrossRef]
- Gonzalo, R.; Garcia-Arumi, E.; Llige, D.; Marti, R.; Solano, A.; Montoya, J.; Arenas, J.; Andreu, A.L. Free radicals-mediated damage in transmitochondrial cells harboring the T14487C mutation in the ND6 gene of mtDNA. FEBS Lett. 2005, 579, 6909–6913. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Srivastava, A.; Kalaiarasan, P.; Manvati, S.; Chopra, R.; Bamezai, R.N.K. mtDNA germ line variation mediated ROS generates retrograde signaling and induces pro-cancerous metabolic features. Sci. Rep. 2014, 4, 6571. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, C.; Roolf, C.; Timmer, K.; Sekora, A.; Knübel, G.; Escobar, H.M.; Fuellen, G.; Ibrahim, S.M.; Tiedge, M.; Baltrusch, S.; et al. Polymorphisms of the murine mitochondrial ND4, CYTB and COX3 genes impact hematopoiesis during aging. Oncotarget 2016, 7, 74460–74472. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Cui, J.; Liu, F.; Gao, L.; Luo, Y.; Li, P.; Guan, L.; Gao, Y. Mitochondrial DNA 10609T Promotes Hypoxia-Induced Increase of Intracellular ROS and Is a Risk Factor of High Altitude Polycythemia. PLoS ONE 2014, 9, e87775. [Google Scholar] [CrossRef] [PubMed]
- Bilal, E.; Rabadan, R.; Alexe, G.; Fuku, N.; Ueno, H.; Nishigaki, Y.; Fujita, Y.; Ito, M.; Arai, Y.; Hirose, N.; et al. Mitochondrial DNA Haplogroup D4a Is a Marker for Extreme Longevity in Japan. PLoS ONE 2008, 3, e2421. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.-G.; Kong, Q.-P.; Bandelt, H.-J.; Kivisild, T.; Zhang, Y.-P. Phylogeographic Differentiation of Mitochondrial DNA in Han Chinese. Am. J. Hum. Genet. 2002, 70, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.; Coo, I.D.; Diaz, F.; Smeets, H.; Moraes, C.T. An out-of-frame cytochrome b gene deletion from a patient with parkinsonism is associated with impaired complex III assembly and an increase in free radical production. Ann. Neurol. 2000, 48, 774–781. [Google Scholar] [CrossRef]
- Fliss, M.S.; Usadel, H.; Caballero, O.L.; Wu, L.; Buta, M.R.; Eleff, S.M.; Jen, J.; Sidransky, D. Facile Detection of Mitochondrial DNA Mutations in Tumors and Bodily Fluids. Science 2000, 287, 2017–2019. [Google Scholar] [CrossRef]
- Dasgupta, S.; Hoque, M.O.; Upadhyay, S.; Sidransky, D. Mitochondrial Cytochrome B Gene Mutation Promotes Tumor Growth in Bladder Cancer. Cancer Res. 2008, 68, 700–706. [Google Scholar] [CrossRef]
- Schuelke, M.; Krude, H.; Finckh, B.; Mayatepek, E.; Janssen, A.; Schmelz, M.; Trefz, F.; Trijbels, F.; Smeitink, J. Septo-optic dysplasia associated with a new mitochondrial cytochrome b mutation. Ann. Neurol. 2002, 51, 388–392. [Google Scholar] [CrossRef]
- Arnold, R.S.; Sun, Q.; Sun, C.Q.; Richards, J.C.; O’Hearn, S.; Osunkoya, A.O.; Wallace, D.C.; Petros, J.A. An inherited heteroplasmic mutation in mitochondrial gene COI in a patient with prostate cancer alters reactive oxygen, reactive nitrogen and proliferation. BioMed Res. Int. 2013, 2013, 239257. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Waymire, K.G.; Narula, N.; Li, P.; Rocher, C.; Coskun, P.E.; Vannan, M.A.; Narula, J.; MacGregor, G.R.; Wallace, D.C. A Mouse Model of Mitochondrial Disease Reveals Germline Selection Against Severe mtDNA Mutations. Science 2008, 319, 958–962. [Google Scholar] [CrossRef] [PubMed]
- D’Aurelio, M.; Pallotti, F.; Barrientos, A.; Gajewski, C.D.; Kwong, J.Q.; Bruno, C.; Beal, M.F.; Manfredi, G. In Vivo Regulation of Oxidative Phosphorylation in Cells Harboring a Stop-codon Mutation in Mitochondrial DNA-encoded Cytochrome c Oxidase Subunit I. J. Biol. Chem. 2001, 276, 46925–46932. [Google Scholar] [CrossRef] [PubMed]
- Bruno, C.; Martinuzzi, A.; Tang, Y.; Andreu, A.L.; Pallotti, F.; Bonilla, E.; Shanske, S.; Fu, J.; Sue, C.M.; Angelini, C.; et al. A Stop-Codon Mutation in the Human mtDNA Cytochrome c Oxidase I Gene Disrupts the Functional Structure of Complex IV. Am. J. Hum. Genet. 1999, 65, 611–620. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vives-Bauza, C.; Gonzalo, R.; Manfredi, G.; Garcia-Arumi, E.; Andreu, A.L. Enhanced ROS production and antioxidant defenses in cybrids harbouring mutations in mtDNA. Neurosci. Lett. 2006, 391, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Schüll, S.; Günther, S.D.; Brodesser, S.; Seeger, J.M.; Tosetti, B.; Wiegmann, K.; Pongratz, C.; Diaz, F.; Witt, A.; Andree, M.; et al. Cytochrome c oxidase deficiency accelerates mitochondrial apoptosis by activating ceramide synthase 6. Cell Death Amp. Dis. 2015, 6, e1691. [Google Scholar] [CrossRef]
- Campos, Y.; García-Redondo, A.; Fernández-Moreno, M.A.; Martínez-Pardo, M.; Goda, G.; Rubio, J.C.; Martín, M.A.; Hoyo, P.D.; Cabello, A.; Bornstein, B.; et al. Early onset multisystem mitochondrial disorder caused by a nonsense mutation in the mitochondrial DNA Cytochrome C oxidase II gene. Ann. Neurol. 2001, 50, 409–413. [Google Scholar] [CrossRef]
- Moreno-Lastres, D.; Fontanesi, F.; García-Consuegra, I.; Martín, M.A.; Arenas, J.; Barrientos, A.; Ugalde, C. Mitochondrial Complex I Plays an Essential Role in Human Respirasome Assembly. Cell Metabol. 2012, 15, 324–335. [Google Scholar] [CrossRef]
- Rahman, S.; Taanman, J.-W.; Cooper, J.M.; Nelson, I.; Hargreaves, I.; Meunier, B.; Hanna, M.G.; García, J.J.; Capaldi, R.A.; Lake, B.D.; et al. A Missense Mutation of Cytochrome Oxidase Subunit II Causes Defective Assembly and Myopathy. Am. J. Hum. Genet. 1999, 65, 1030–1039. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Goto, Y.-i. Respiratory Chain Complex Disorganization Impairs Mitochondrial and Cellular Integrity: Phenotypic Variation in Cytochrome c Oxidase Deficiency. Am. J. Hum. Pathol. 2017, 187, 110–121. [Google Scholar] [CrossRef]
- Lenaz, G.; Baracca, A.; Carelli, V.; D’Aurelio, M.; Sgarbi, G.; Solaini, G. Bioenergetics of mitochondrial diseases associated with mtDNA mutations. Biochim. Biophys. Acta Bioenerg. 2004, 1658, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Degoul, F.; Diry, M.; Rodriguez, D.; Robain, O.; Francois, D.; Ponsot, G.; Marsac, C.; Desguerre, I. Clinical, biochemical, and molecular analysis of a maternally inherited case of Leight syndrome (MILS) associated with the mtDNA T8993G point mutation. J. Inherit. Metab. Dis. 1995, 18, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Mattiasson, G. Flow cytometric analysis of isolated liver mitochondria to detect changes relevant to cell death. Cytom. Part. A 2004, 60a, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, M.; Vijayvergiya, C.; Gajewski, C.D.; DeVivo, D.C.; Lenaz, G.; Wiedmann, M.; Manfredi, G. The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum. Mol. Genet. 2004, 13, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Geromel, V.; Kadhom, N.; Cebalos-Picot, I.; Ouari, O.; Polidori, A.; Munnich, A.; Rötig, A.; Rustin, P. Superoxide-induced massive apoptosis in cultured skin fibroblasts harboring the neurogenic ataxia retinitis pigmentosa (NARP) mutation in the ATPase-6 gene of the mitochondrial DNA. Hum. Mol. Genet. 2001, 10, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Baracca, A.; Sgarbi, G.; Mattiazzi, M.; Casalena, G.; Pagnotta, E.; Valentino, M.L.; Moggio, M.; Lenaz, G.; Carelli, V.; Solaini, G. Biochemical phenotypes associated with the mitochondrial ATP6 gene mutations at nt8993. Biochim. Biophys. Acta Bioenerg. 2007, 1767, 913–919. [Google Scholar] [CrossRef]
- Auré, K.; Dubourg, O.; Jardel, C.; Clarysse, L.; Sternberg, D.; Fournier, E.; Laforêt, P.; Streichenberger, N.; Petiot, P.; Gervais-Bernard, H.; et al. Episodic weakness due to mitochondrial DNA MT-ATP6/8 mutations. Neurology 2013, 81, 1810–1818. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.; Wester-Rosenloef, L.; Koch, C.; Koch, F.; Baltrusch, S.; Tiedge, M.; Ibrahim, S. The Mitochondrial Atp8 Mutation Induces Mitochondrial ROS Generation, Secretory Dysfunction, and β-Cell Mass Adaptation in Conplastic B6-mtFVB Mice. Endocrinology 2012, 153, 4666–4676. [Google Scholar] [CrossRef]
- Yu, X.; Wester-Rosenlöf, L.; Gimsa, U.; Holzhueter, S.-A.; Marques, A.; Jonas, L.; Hagenow, K.; Kunz, M.; Nizze, H.; Tiedge, M.; et al. The mtDNA nt7778 G/T polymorphism affects autoimmune diseases and reproductive performance in the mouse. Hum. Mol. Genet. 2009, 18, 4689–4698. [Google Scholar] [CrossRef]
- Wu, S.-B.; Ma, Y.-S.; Wu, Y.-T.; Chen, Y.-C.; Wei, Y.-H. Mitochondrial DNA Mutation-Elicited Oxidative Stress, Oxidative Damage, and Altered Gene Expression in Cultured Cells of Patients with MERRF Syndrome. Mol. Neurobiol. 2010, 41, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Wang, M.; Li, H.; Wang, H.; Jiang, F.; Hou, L.; Geng, J.; Lin, Z.; Peng, Y.; Zhou, H.; et al. Mitochondrial tRNA mutations in 2070 Chinese Han subjects with hypertension. Mitochondrion 2016, 30, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Kulawiec, M.; Owens, K.M.; Singh, K.K. Cancer cell mitochondria confer apoptosis resistance and promote metastasis. Cancer Biol. Ther. 2009, 8, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Loshuertos, R.; Acín-Pérez, R.; Fernández-Silva, P.; Movilla, N.; Pérez-Martos, A.; de Cordoba, S.R.; Gallardo, M.E.; Enríquez, J.A. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat. Genet. 2006, 38, 1261. [Google Scholar] [CrossRef] [PubMed]
- Niemann, J.; Johne, C.; Schröder, S.; Koch, F.; Ibrahim, S.M.; Schultz, J.; Tiedge, M.; Baltrusch, S. An mtDNA mutation accelerates liver aging by interfering with the ROS response and mitochondrial life cycle. Free Radic. Biol. Med. 2017, 102, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wen, C.; Li, W.; Wang, H.; Guan, X.; Zhang, W.; Ye, W.; Lu, J. The tRNAGly T10003C mutation in mitochondrial haplogroup M11b in a Chinese family with diabetes decreases the steady-state level of tRNAGly, increases aberrant reactive oxygen species production, and reduces mitochondrial membrane potential. Mol. Cell. Biochem. 2015, 408, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Zhu, C.; Tian, L.; Guan, M.; Chen, Y. Mitochondrial biogenesis dysfunction and metabolic dysfunction from a novel mitochondrial tRNAMet 4467 C>A mutation in a Han Chinese family with maternally inherited hypertension. Sci. Rep. 2017, 7, 3034. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Peng, Y.; Jiang, P.; Wang, M.; Fan, M.; Wang, X.; Zhou, H.; Li, H.; Yan, Q.; Huang, T.; et al. A deafness-associated tRNAHis mutation alters the mitochondrial function, ROS production and membrane potential. Nucleic Acids Res. 2014, 42, 8039–8048. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Katayama, A.; Komaki, H.; Nishino, I.; Goto, Y.-i. Molecular pathomechanisms and cell-type-specific disease phenotypes of MELAS caused by mutant mitochondrial tRNATrp. Acta Neuropathol. Commun. 2015, 3, 52. [Google Scholar] [CrossRef]
- Horváth, R.; Bender, A.; Abicht, A.; Holinski-Feder, E.; Czermin, B.; Trips, T.; Schneiderat, P.; Lochmüller, H.; Klopstock, T. Heteroplasmic mutation in the anticodon-stem of mitochondrial tRNAVal causing MNGIE-like gastrointestinal dysmotility and cachexia. J. Neurol. 2009, 256, 810–815. [Google Scholar] [CrossRef]
- Chauhan, A.; Chauhan, V.; Brown, W.T.; Cohen, I. Oxidative stress in autism: Increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin—The antioxidant proteins. Life Sci. 2004, 75, 2539–2549. [Google Scholar] [CrossRef] [PubMed]
- Saigo, K.; Takenokuchi, M.; Hiramatsu, Y.; Tada, H.; Hishita, T.; Takata, M.; Misawa, M.; Imoto, S.; Imashuku, S. Oxidative Stress Levels in Myelodysplastic Syndrome Patients: Their Relationship to Serum Ferritin and Haemoglobin Values. J. Int. Med. Res. 2011, 39, 1941–1945. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, R.; Fettermann, A.; Li, Z.; Qian, Y.; Liu, Y.; Wang, X.; Zhou, A.; Mo, J.Q.; Yang, L.; et al. Maternally Inherited Essential Hypertension Is Associated With the Novel 4263A>G Mutation in the Mitochondrial tRNAIle Gene in a Large Han Chinese Family. Circ. Res. 2011, 108, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Geng, J.; Yu, H.; Tang, X.; Yang, X.; Xue, L. Mitochondrial tRNAThr 15909A>G mutation associated with hypertension in a Chinese Han pedigree. Biochem. Biophys. Res. Commun. 2018, 495, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xue, L.; Chen, Y.; Li, H.; He, Q.; Wang, B.; Meng, F.; Wang, M.; Guan, M.-X. A hypertension-associated mitochondrial DNA mutation introduces an m1G37 modification into tRNAMet, altering its structure and function. J. Biol. Chem. 2018, 293, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Ballana, E.; Morales, E.; Rabionet, R.; Montserrat, B.; Ventayol, M.; Bravo, O.; Gasparini, P.; Estivill, X. Mitochondrial 12S rRNA gene mutations affect RNA secondary structure and lead to variable penetrance in hearing impairment. Biochem. Biophys. Res. Commun. 2006, 341, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.-X. Mitochondrial 12S rRNA mutations associated with aminoglycoside ototoxicity. Mitochondrion 2011, 11, 237–245. [Google Scholar] [CrossRef]
- Yu, J.; Zheng, J.; Zhao, X.; Liu, J.; Mao, Z.; Ling, Y.; Chen, D.; Chen, C.; Hui, L.; Cui, L.; et al. Aminoglycoside Stress Together with the 12S rRNA 1494C>T Mutation Leads to Mitophagy. PLoS ONE 2014, 9, e114650. [Google Scholar] [CrossRef] [PubMed]
- Raimundo, N.; Song, L.; Shutt, T.E.; McKay, S.E.; Cotney, J.; Guan, M.-X.; Gilliland, T.C.; Hohuan, D.; Santos-Sacchi, J.; Shadel, G.S. Mitochondrial Stress Engages E2F1 Apoptotic Signaling to Cause Deafness. Cell 2012, 148, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Satoshi, M.; Yasuyuki, M.; Yujiro, S.; Sadamitsu, A.; Akira, T.; Takashi, T.; Shigeo, O. Mutations in the mitochondrial genome confer resistance of cancer cells to anticancer drugs. Cancer Sci. 2009, 100, 1680–1687. [Google Scholar] [CrossRef]
- Liu, Z.; Song, Y.; Li, D.; He, X.; Li, S.; Wu, B.; Wang, W.; Gu, S.; Zhu, X.; Wang, X.; et al. The novel mitochondrial 16S rRNA 2336T>C mutation is associated with hypertrophic cardiomyopathy. J. Med. Genet. 2014, 51, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Michikawa, Y.; Mazzucchelli, F.; Bresolin, N.; Scarlato, G.; Attardi, G. Aging-Dependent Large Accumulation of Point Mutations in the Human mtDNA Control Region for Replication. Science 1999, 286, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Michikawa, Y.; Mallidis, C.; Bai, Y.; Woodhouse, L.; Yarasheski, K.E.; Miller, C.A.; Askanas, V.; Engel, W.K.; Bhasin, S.; et al. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. Proc. Natl. Acad. Sci. USA 2001, 98, 4022–4027. [Google Scholar] [CrossRef] [PubMed]
- Bua, E.; Johnson, J.; Herbst, A.; Delong, B.; McKenzie, D.; Salamat, S.; Aiken, J.M. Mitochondrial DNA–Deletion Mutations Accumulate Intracellularly to Detrimental Levels in Aged Human Skeletal Muscle Fibers. Am. J. Hum. Genet. 2006, 79, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Arnheim, N.; Cortopassi, G. Deleterious mitochondrial DNA mutations accumulate in aging human tissues. Mutation Res. DNAging 1992, 275, 157–167. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Arnheim, N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990, 18, 6927–6933. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly-Y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417. [Google Scholar] [CrossRef]
- Logan, A.; Shabalina, I.G.; Prime, T.A.; Rogatti, S.; Kalinovich, A.V.; Hartley, R.C.; Budd, R.C.; Cannon, B.; Murphy, M.P. In vivo levels of mitochondrial hydrogen peroxide increase with age in mtDNA mutator mice. Aging Cell 2014, 13, 765–768. [Google Scholar] [CrossRef]
- Macao, B.; Uhler, J.P.; Siibak, T.; Zhu, X.; Shi, Y.; Sheng, W.; Olsson, M.; Stewart, J.B.; Gustafsson, C.M.; Falkenberg, M. The exonuclease activity of DNA polymerase γ is required for ligation during mitochondrial DNA replication. Nat. Commun. 2015, 6, 7303. [Google Scholar] [CrossRef]
- Ponamarev, M.V.; Longley, M.J.; Nguyen, D.; Kunkel, T.A.; Copeland, W.C. Active site mutation in DNA polymerase gamma associated with progressive external ophthalmoplegia causes error-prone DNA synthesis. J. Biol..Chem. 2002, 277, 15225–15228. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.; Day, B.J.; Kohler, J.J.; Hosseini, S.H.; Chan, S.S.; Green, E.C.; Haase, C.P.; Keebaugh, E.S.; Long, R.; Ludaway, T.; et al. Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase gamma. Lab. Investig. J. Tech. Methods Pathol. 2007, 87, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hahn, A.; Zuryn, S. Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants 2019, 8, 392. https://doi.org/10.3390/antiox8090392
Hahn A, Zuryn S. Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants. 2019; 8(9):392. https://doi.org/10.3390/antiox8090392
Chicago/Turabian StyleHahn, Anne, and Steven Zuryn. 2019. "Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species" Antioxidants 8, no. 9: 392. https://doi.org/10.3390/antiox8090392
APA StyleHahn, A., & Zuryn, S. (2019). Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants, 8(9), 392. https://doi.org/10.3390/antiox8090392