Gamma-Tocotrienol Protects the Intestine from Radiation Potentially by Accelerating Mesenchymal Immune Cell Recovery

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Model, GT3 Treatment, Irradiation, Tissue Procurement, and Euthanasia
2.2. TUNEL Assay
2.3. Assessment of Villus Height and Crypt Depth
2.4. In Vivo Intestinal Permeability Assay
2.5. Immunoblotting
2.6. Intestinal Crypt Isolation and Organoid Culture in Matrigel
2.7. Immunohistochemistry
2.8. RNA Extraction, cDNA Preparation, and Quantitative Reverse-Transcription PCR (qRT-PCR)
2.9. Statistical Analysis
3. Results
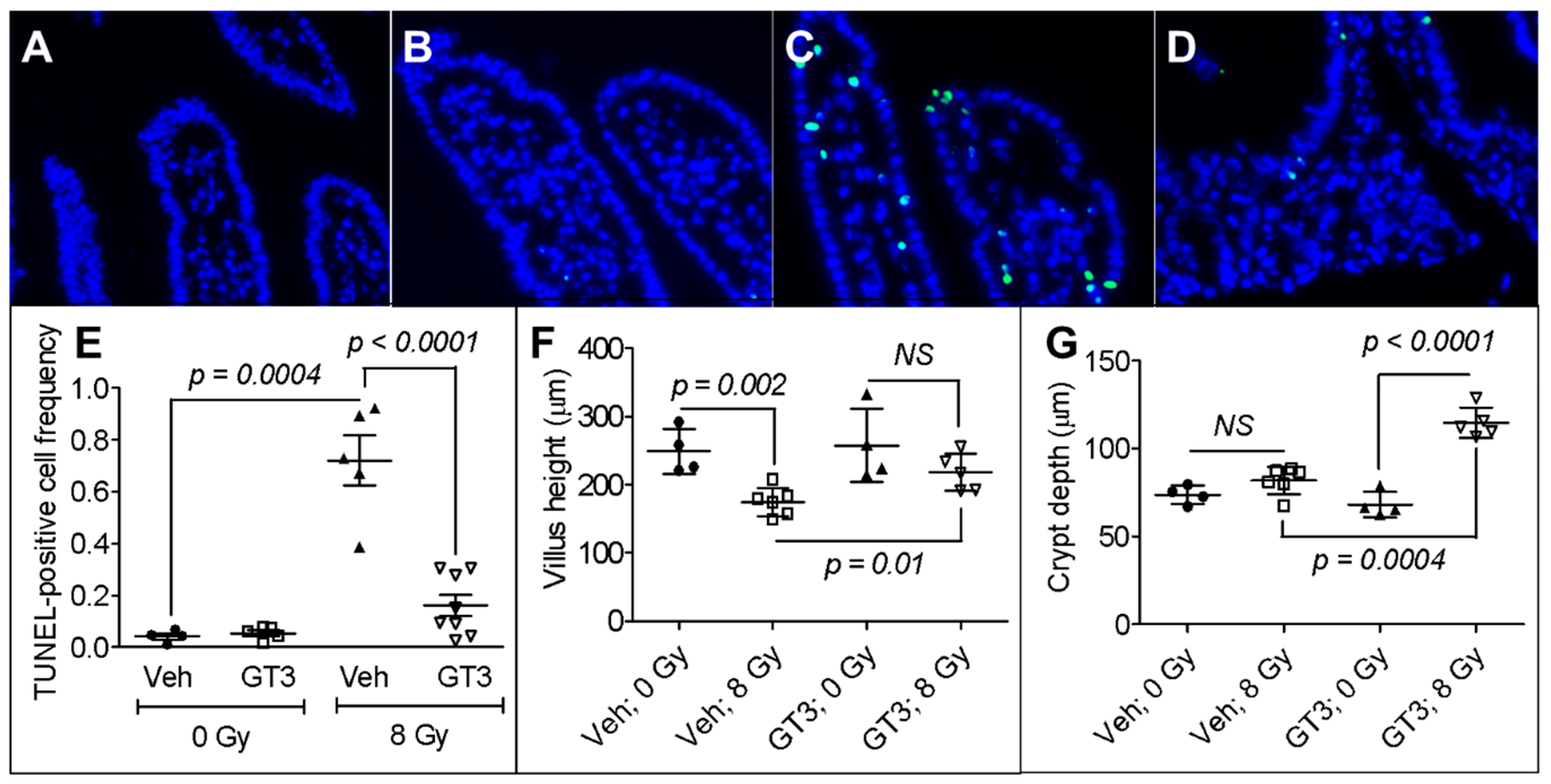
3.1. GT3 Pretreatment Attenuated Intestinal Cell Death, Maintained Villus Height, and Enhanced Crypt Depth in Irradiated Mice
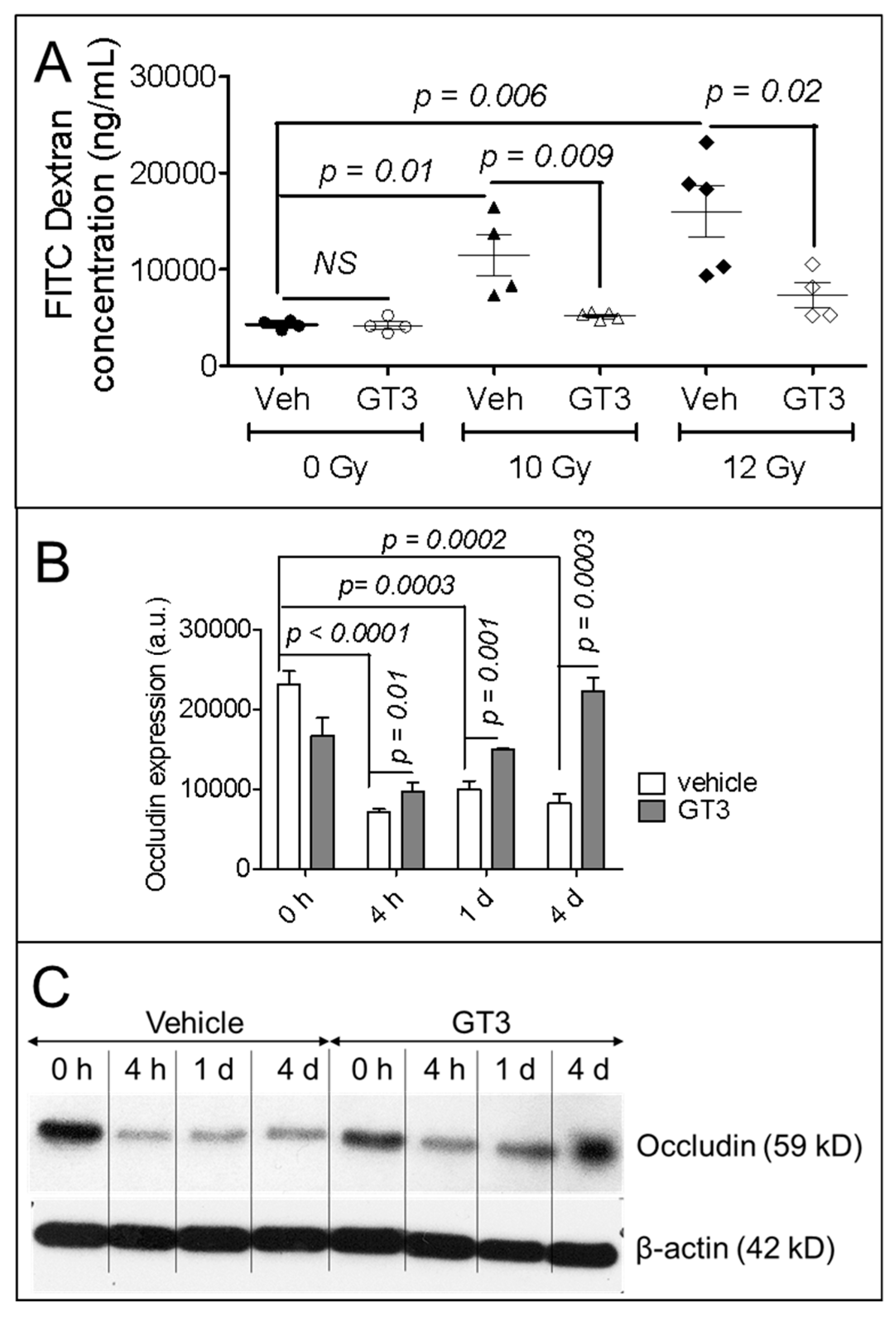
3.2. GT3 Pretreatment Restored the Intestinal Barrier and Upregulated the Level of Occludin Protein in Irradiated Intestinal Tissue
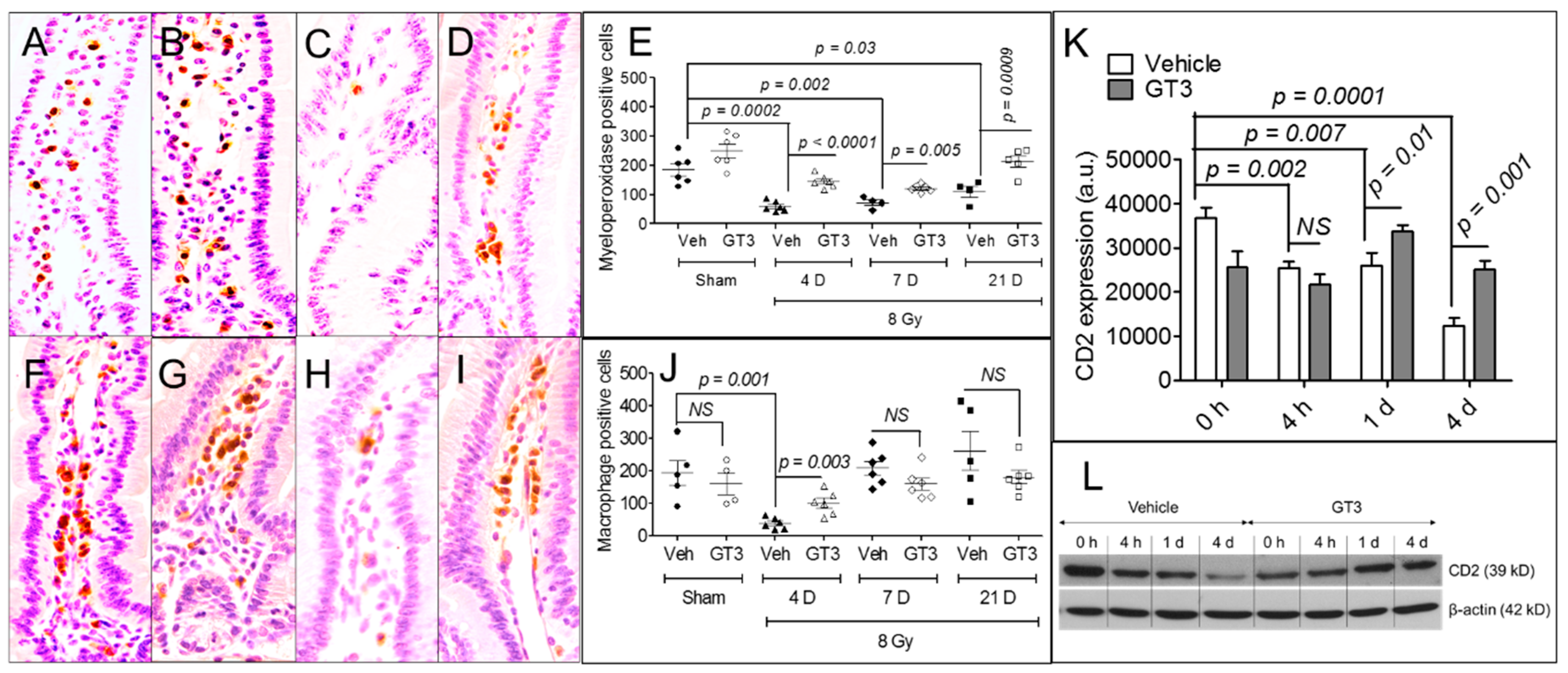
3.3. GT3 Pretreatment Rescued Populations of Intestinal Neutrophils, Macrophages, and Lymphocytes
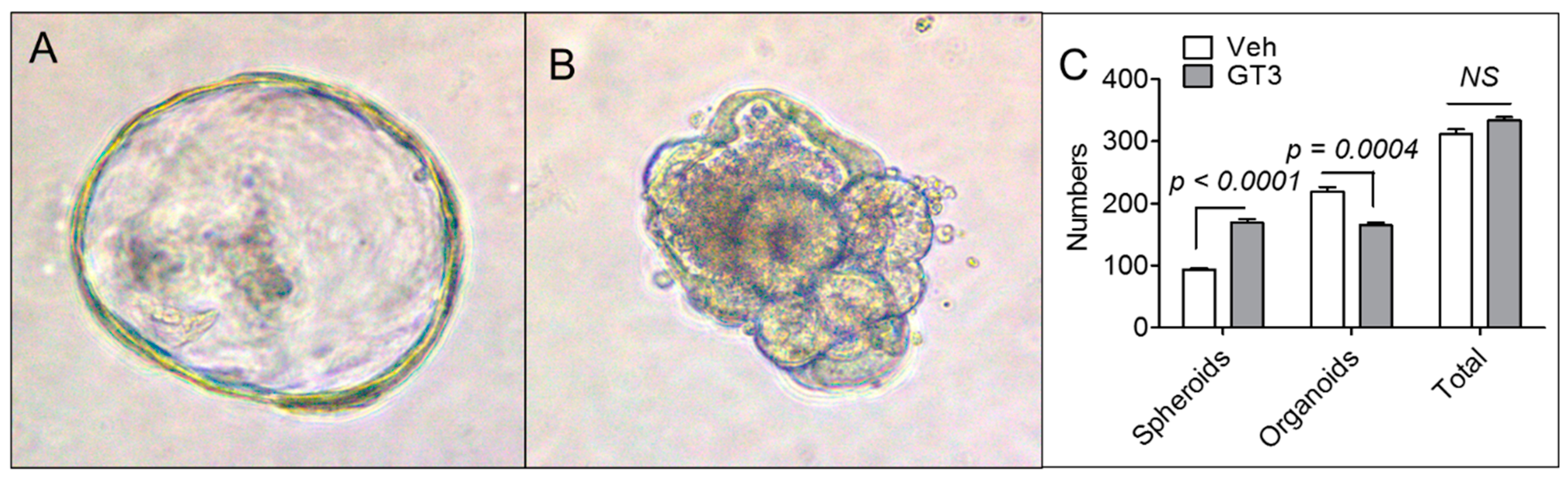
3.4. GT3 Pretreatment Failed to Promote Ex Vivo Intestinal Organoid Formation after TBI
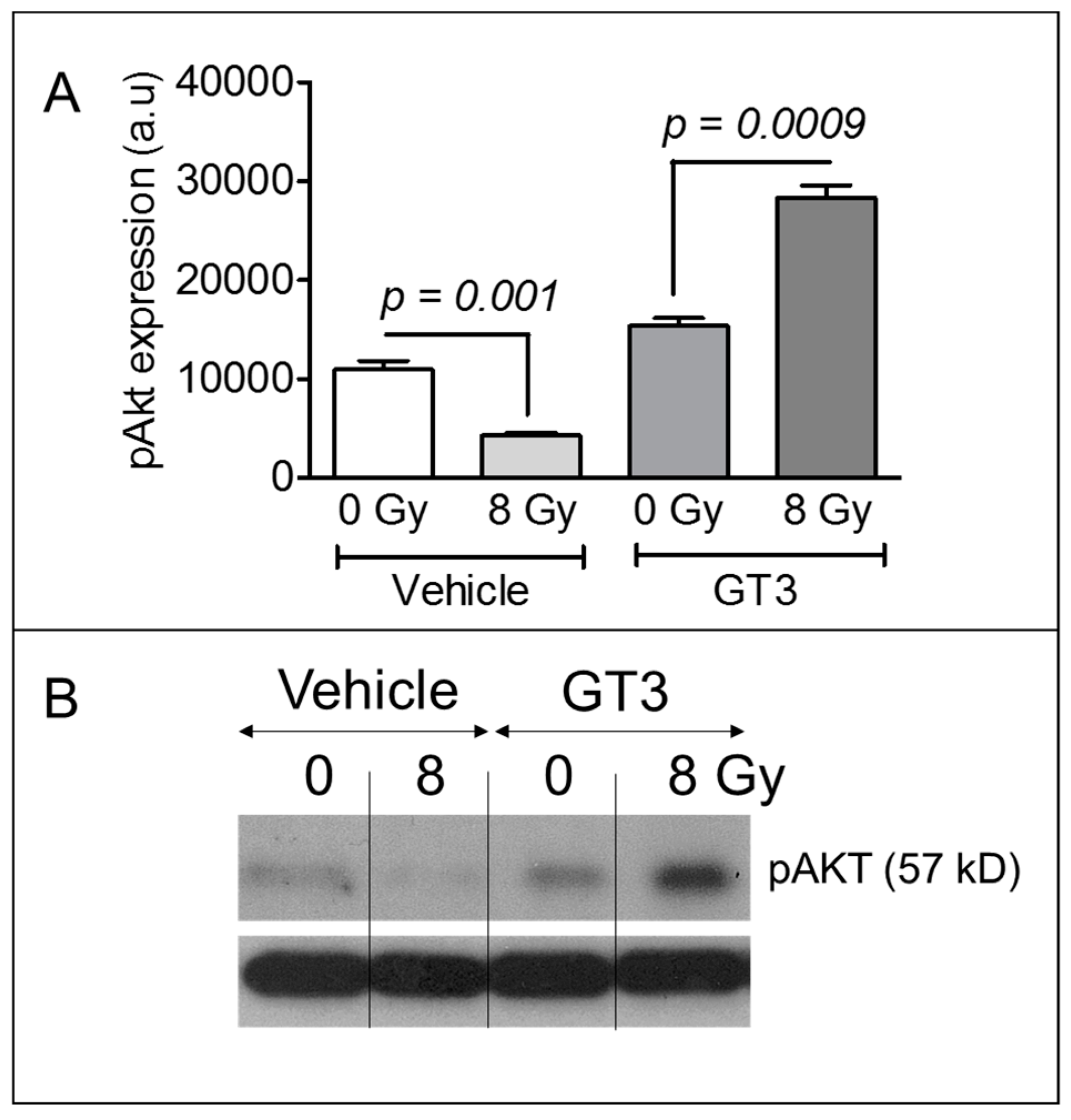
3.5. GT3 Pretreatment Enhanced AKT Phosphorylation on Day 4 after Irradiation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gehart, H.; Clevers, H. Tales from the crypt: New insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Shadad, A.K.; Sullivan, F.J.; Martin, J.D.; Egan, L.J. Gastrointestinal radiation injury: symptoms, risk factors and mechanisms. World J. Gastroenterol. 2013, 19, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Inagaki-Ohara, K.; Takamura, N.; Yada, S.; Alnadjim, Z.; Liu, E.; Yu, X.; Yoshida, H.; Lin, T. Radiation-induced crypt intestinal epithelial cell apoptosis in vivo involves both caspase-3-dependent and -independent pathways. Dig. Dis. Sci. 2002, 47, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Zheng, J.; Wang, J.; Authier, S.; Pouliot, M.; Hauer-Jensen, M. Segmental Differences in Radiation-Induced Alterations of Tight Junction-Related Proteins in Non-Human Primate Jejunum, Ileum and Colon. Radiat. Res. 2016, 185, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.-Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Pull, S.L.; Doherty, J.M.; Mills, J.C.; Gordon, J.I.; Stappenbeck, T.S. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc. Natl. Acad. Sci. USA 2005, 102, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Aranda, E.; Hayakawa, Y.; Bhanja, P.; Atay, S.; Brodin, N.P.; Li, J.; Asfaha, S.; Liu, L.; Tailor, Y.; et al. Macrophage-derived extracellular vesicle-packaged WNTs rescue intestinal stem cells and enhance survival after radiation injury. Nat. Commun. 2016, 7, 13096. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Lindemans, C.A.; Calafiore, M.; Mertelsmann, A.M.; O’Connor, M.H.; Dudakov, J.A.; Jenq, R.R.; Velardi, E.; Young, L.F.; Smith, O.M.; Lawrence, G.; et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 2015, 528, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Yang, W.; Huang, X.; Cao, A.T.; Bilotta, A.J.; Xiao, Y.; Sun, M.; Chen, L.; Ma, C.; Liu, X.; et al. Neutrophils Promote Amphiregulin Production in Intestinal Epithelial Cells through TGF-β and Contribute to Intestinal Homeostasis. J. Immunol. 2018, 201, 2492–2501. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Boerma, M.; Wang, J.; Fu, Q.; Loose, D.S.; Kumar, K.S.; Hauer-Jensen, M. Influence of sublethal total-body irradiation on immune cell populations in the intestinal mucosa. Radiat. Res. 2010, 173, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Hauer-Jensen, M.; Denham, J.W.; Andreyev, H.J.N. Radiation enteropathy--pathogenesis, treatment and prevention. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.P.; Kulkarni, S.; Hieber, K.; Toles, R.; Romanyukha, L.; Kao, T.-C.; Hauer-Jensen, M.; Kumar, K.S. Gamma-tocotrienol, a tocol antioxidant as a potent radioprotector. Int. J. Radiat. Biol. 2009, 85, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Ghosh, S.P.; Satyamitra, M.; Mog, S.; Hieber, K.; Romanyukha, L.; Gambles, K.; Toles, R.; Kao, T.-C.; Hauer-Jensen, M.; et al. Gamma-tocotrienol protects hematopoietic stem and progenitor cells in mice after total-body irradiation. Radiat. Res. 2010, 173, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Berbée, M.; Fu, Q.; Boerma, M.; Wang, J.; Kumar, K.S.; Hauer-Jensen, M. gamma-Tocotrienol ameliorates intestinal radiation injury and reduces vascular oxidative stress after total-body irradiation by an HMG-CoA reductase-dependent mechanism. Radiat. Res. 2009, 171, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Berbee, M.; Fu, Q.; Boerma, M.; Pathak, R.; Zhou, D.; Kumar, K.S.; Hauer-Jensen, M. Reduction of radiation-induced vascular nitrosative stress by the vitamin E analog γ-tocotrienol: evidence of a role for tetrahydrobiopterin. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; Datta, K.; Chakraborty, K.; Kulkarni, S.S.; Doiron, K.; Fornace, A.J.; Sree Kumar, K.; Hauer-Jensen, M.; Ghosh, S.P. Gamma tocotrienol, a potent radioprotector, preferentially upregulates expression of anti-apoptotic genes to promote intestinal cell survival. Food Chem. Toxicol. 2013, 60, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.; Shao, L.; Ghosh, S.P.; Zhou, D.; Boerma, M.; Weiler, H.; Hauer-Jensen, M. Thrombomodulin contributes to gamma tocotrienol-mediated lethality protection and hematopoietic cell recovery in irradiated mice. PLoS ONE 2015, 10, e0122511. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.; Ghosh, S.P.; Zhou, D.; Hauer-Jensen, M. The Vitamin E Analog Gamma-Tocotrienol (GT3) and Statins Synergistically Up-Regulate Endothelial Thrombomodulin (TM). Int. J. Mol. Sci. 2016, 17, 1937. [Google Scholar] [CrossRef] [PubMed]
- Geiger, H.; Pawar, S.A.; Kerschen, E.J.; Nattamai, K.J.; Hernandez, I.; Liang, H.P.H.; Fernández, J.Á.; Cancelas, J.A.; Ryan, M.A.; Kustikova, O.; et al. Pharmacological targeting of the thrombomodulin-activated protein C pathway mitigates radiation toxicity. Nat. Med. 2012, 18, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.; Wang, J.; Garg, S.; Aykin-Burns, N.; Petersen, K.-U.; Hauer-Jensen, M. Recombinant Thrombomodulin (Solulin) Ameliorates Early Intestinal Radiation Toxicity in a Preclinical Rat Model. Radiat. Res. 2016, 186, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Apostolov, E.O.; Soultanova, I.; Savenka, A.; Bagandov, O.O.; Yin, X.; Stewart, A.G.; Walker, R.B.; Basnakian, A.G. Deoxyribonuclease I is essential for DNA fragmentation induced by gamma radiation in mice. Radiat. Res. 2009, 172, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Alegria-Schaffer, A.; Lodge, A.; Vattem, K. Performing and optimizing Western blots with an emphasis on chemiluminescent detection. Methods Enzymol. 2009, 463, 573–599. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zheng, H.; Sung, C.C.; Richter, K.K.; Hauer-Jensen, M. Cellular sources of transforming growth factor-beta isoforms in early and chronic radiation enteropathy. Am. J. Pathol. 1998, 153, 1531–1540. [Google Scholar] [CrossRef]
- Van Bekkum, D.W. Effectiveness and risks of total body irradiation for conditioning in the treatment of autoimmune disease with autologous bone marrow transplantation. Rheumatology 1999, 38, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Deeg, H.J.; Amylon, I.D.; Harris, R.E.; Collins, R.; Beatty, P.G.; Feig, S.; Ramsay, N.; Territo, M.; Khan, S.P.; Pamphilon, D.; et al. Marrow transplants from unrelated donors for patients with aplastic anemia: Minimum effective dose of total body irradiation. Biol. Blood Marrow Transplant. 2001, 7, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Geller, R.B.; Devine, S.M.; O’Toole, K.; Persons, L.; Keller, J.; Mauer, D.; Holland, H.K.; Dix, S.P.; Piotti, M.; Redei, I.; et al. Allogeneic bone marrow transplantation with matched unrelated donors for patients with hematologic malignancies using a preparative regimen of high-dose cyclophosphamide and fractionated total body irradiation. Bone Marrow Transplant. 1997, 20, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Abdel-Azim, H.; McManus, M.; Nemecek, E.; Sposto, R.; Woolfrey, A.; Frangoul, H. Phase I Study of Clofarabine and 2-Gy Total Body Irradiation as a Nonmyeloablative Preparative Regimen for Hematopoietic Stem Cell Transplantation in Pediatric Patients with Hematologic Malignancies: A Therapeutic Advances in Childhood Leukemia Consortium Study. Biol. Blood Marrow Transplantat. 2017, 23, 1134–1141. [Google Scholar] [CrossRef]
- Wei, C.; Candler, T.; Davis, N.; Elson, R.; Crabtree, N.; Stevens, M.; Crowne, E. Bone Mineral Density Corrected for Size in Childhood Leukaemia Survivors Treated with Haematopoietic Stem Cell Transplantation and Total Body Irradiation. Horm. Res. Paediatr. 2018, 89, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Thomas, O.; Mahé, M.; Campion, L.; Bourdin, S.; Milpied, N.; Brunet, G.; Lisbona, A.; Le Mevel, A.; Moreau, P.; Harousseau, J.; et al. Long-term complications of total body irradiation in adults. Int. J. Radiat. Oncol. Biol. Phys. 2001, 49, 125–131. [Google Scholar] [CrossRef]
- Gopal, R.; Ha, C.S.; Tucker, S.L.; Khouri, I.F.; Giralt, S.A.; Gajewski, J.L.; Andersson, B.S.; Cox, J.D.; Champlin, R.E. Comparison of two total body irradiation fractionation regimens with respect to acute and late pulmonary toxicity. Cancer 2001, 92, 1949–1958. [Google Scholar] [CrossRef]
- Lutgens, L.C.H.W.; Blijlevens, N.M.A.; Deutz, N.E.P.; Donnelly, J.P.; Lambin, P.; de Pauw, B.E. Monitoring myeloablative therapy-induced small bowel toxicity by serum citrulline concentration: A comparison with sugar permeability tests. Cancer 2005, 103, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Wu, W.; Yang, S.; Huang, L.; Chen, J.; Gong, C.; Fu, Z.; Lin, R.; Tan, J. Treatment of radiation-induced acute intestinal injury with bone marrow-derived mesenchymal stem cells. Expe. Ther. Med. 2016, 11, 2425–2431. [Google Scholar] [CrossRef] [PubMed]
- Pawar, S.A.; Shao, L.; Chang, J.; Wang, W.; Pathak, R.; Zhu, X.; Wang, J.; Hendrickson, H.; Boerma, M.; Sterneck, E.; et al. C/EBPδ deficiency sensitizes mice to ionizing radiation-induced hematopoietic and intestinal injury. PLoS ONE 2014, 9, e94967. [Google Scholar] [CrossRef] [PubMed]
- Son, T.G.; Gong, E.J.; Bae, M.J.; Kim, S.D.; Heo, K.; Moon, C.; Yang, K.; Kim, J.S. Protective effect of genistein on radiation-induced intestinal injury in tumor bearing mice. BMC Complementary Alternative Med. 2013, 13, 103. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tian, H.; Jiang, J.; Yang, Y.; Tan, S.; Lin, X.; Liu, H.; Wu, B. β-Arrestin-2 modulates radiation-induced intestinal crypt progenitor/stem cell injury. Cell Death Differ. 2016, 23, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.K.; Gangwar, R.; Manda, B.; Meena, A.S.; Yadav, N.; Szabo, E.; Balogh, A.; Lee, S.C.; Tigyi, G.; Rao, R. Rapid disruption of intestinal epithelial tight junction and barrier dysfunction by ionizing radiation in mouse colon in vivo: protection by N-acetyl-l-cysteine. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G705-15. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Leibowitz, B.J.; Epperly, M.; Bi, C.; Li, A.; Steinman, J.; Wipf, P.; Li, S.; Zhang, L.; Greenberger, J.; et al. The GS-nitroxide JP4-039 improves intestinal barrier and stem cell recovery in irradiated mice. Sci. Rep. 2018, 8, 2072. [Google Scholar] [CrossRef] [PubMed]
- Mir, H.; Meena, A.S.; Chaudhry, K.K.; Shukla, P.K.; Gangwar, R.; Manda, B.; Padala, M.K.; Shen, L.; Turner, J.R.; Dietrich, P.; et al. Occludin deficiency promotes ethanol-induced disruption of colonic epithelial junctions, gut barrier dysfunction and liver damage in mice. Biochim. Biophys Acta 2016, 1860, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Bhanja, P.; Kabarriti, R.; Liu, L.; Alfieri, A.A.; Guha, C. Bone marrow stromal cell transplantation mitigates radiation-induced gastrointestinal syndrome in mice. PLoS ONE 2011, 6, e24072. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Sumagin, R.; Brazil, J.C.; Nava, P.; Nishio, H.; Alam, A.; Luissint, A.C.; Weber, D.A.; Neish, A.S.; Nusrat, A.; Parkos, C.A. Neutrophil interactions with epithelial-expressed ICAM-1 enhances intestinal mucosal wound healing. Mucosal Immunol. 2016, 9, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Dahan, S.; Roda, G.; Pinn, D.; Roth-Walter, F.; Kamalu, O.; Martin, A.P.; Mayer, L. Epithelial: Lamina propria lymphocyte interactions promote epithelial cell differentiation. Gastroenterology 2008, 134, 192–203. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garg, S.; Sadhukhan, R.; Banerjee, S.; Savenka, A.V.; Basnakian, A.G.; McHargue, V.; Wang, J.; Pawar, S.A.; Ghosh, S.P.; Ware, J.; et al. Gamma-Tocotrienol Protects the Intestine from Radiation Potentially by Accelerating Mesenchymal Immune Cell Recovery. Antioxidants 2019, 8, 57. https://doi.org/10.3390/antiox8030057
Garg S, Sadhukhan R, Banerjee S, Savenka AV, Basnakian AG, McHargue V, Wang J, Pawar SA, Ghosh SP, Ware J, et al. Gamma-Tocotrienol Protects the Intestine from Radiation Potentially by Accelerating Mesenchymal Immune Cell Recovery. Antioxidants. 2019; 8(3):57. https://doi.org/10.3390/antiox8030057
Chicago/Turabian StyleGarg, Sarita, Ratan Sadhukhan, Sudip Banerjee, Alena V. Savenka, Alexei G. Basnakian, Victoria McHargue, Junru Wang, Snehalata A. Pawar, Sanchita P. Ghosh, Jerry Ware, and et al. 2019. "Gamma-Tocotrienol Protects the Intestine from Radiation Potentially by Accelerating Mesenchymal Immune Cell Recovery" Antioxidants 8, no. 3: 57. https://doi.org/10.3390/antiox8030057
APA StyleGarg, S., Sadhukhan, R., Banerjee, S., Savenka, A. V., Basnakian, A. G., McHargue, V., Wang, J., Pawar, S. A., Ghosh, S. P., Ware, J., Hauer-Jensen, M., & Pathak, R. (2019). Gamma-Tocotrienol Protects the Intestine from Radiation Potentially by Accelerating Mesenchymal Immune Cell Recovery. Antioxidants, 8(3), 57. https://doi.org/10.3390/antiox8030057