Genome–Scale Metabolic Networks Shed Light on the Carotenoid Biosynthesis Pathway in the Brown Algae Saccharina japonica and Cladosiphon okamuranus

 , , , , ,
, , , , ,  ,
,
Abstract

1. Introduction
2. Materials and Methods
2.1. Data Sources and Cleaning
2.2. Reconstruction of Genome–Scale Metabolic Networks
2.3. Exploration and Assessment of Carotenoid Biosynthesis Pathways in Brown Algae
3. Results
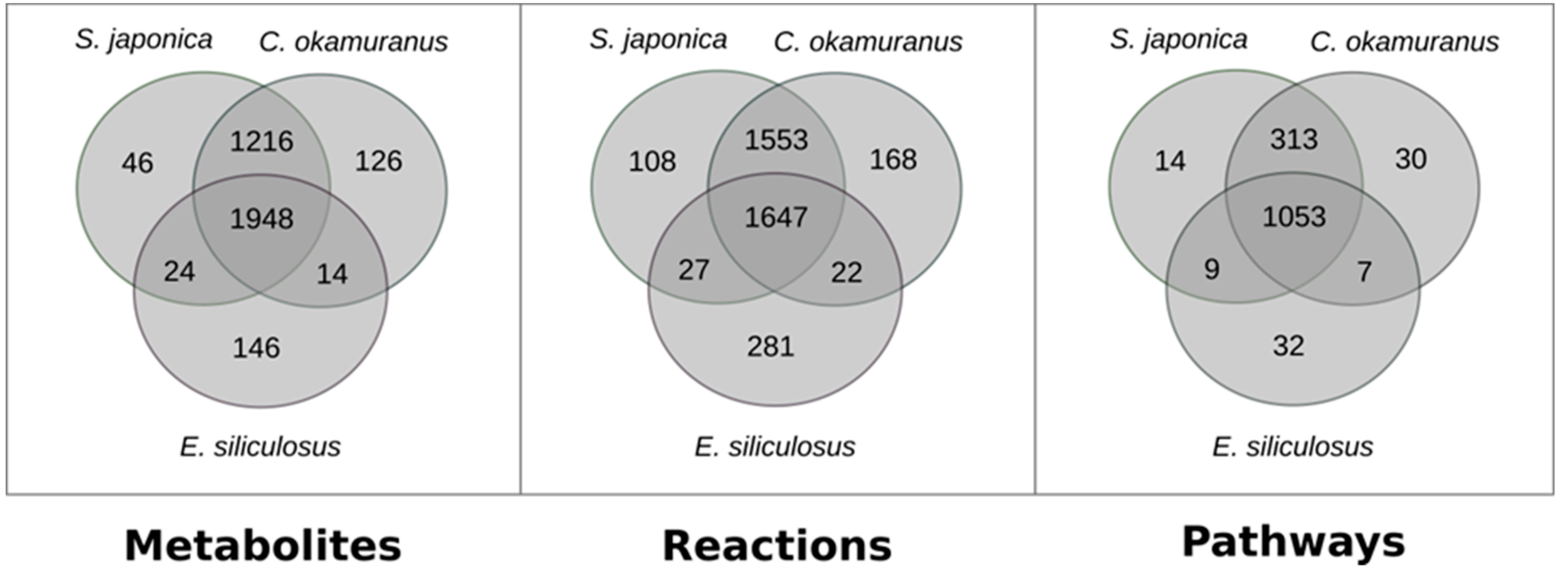
3.1. Genome–Scale Metabolic Network Reconstructions
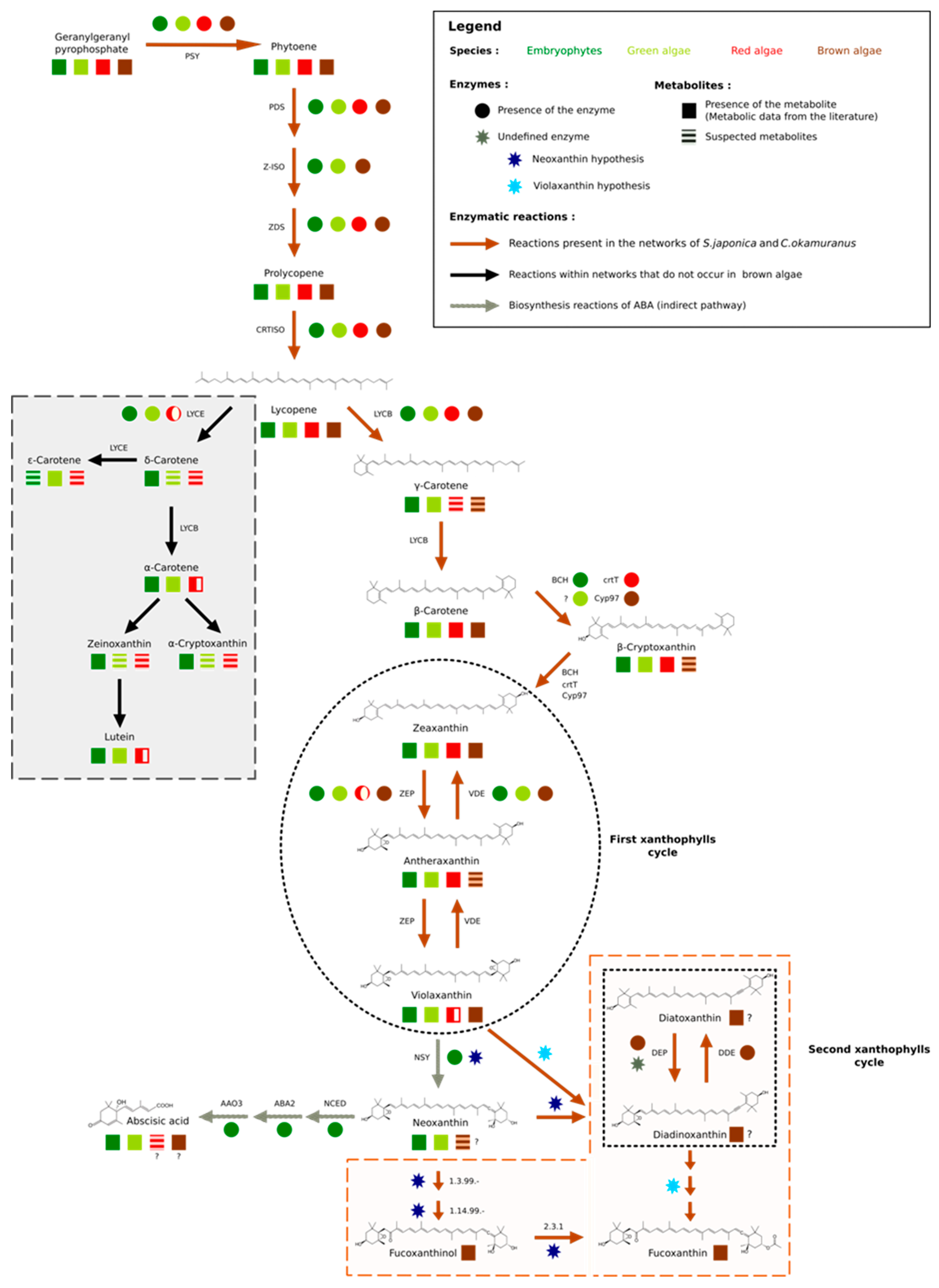
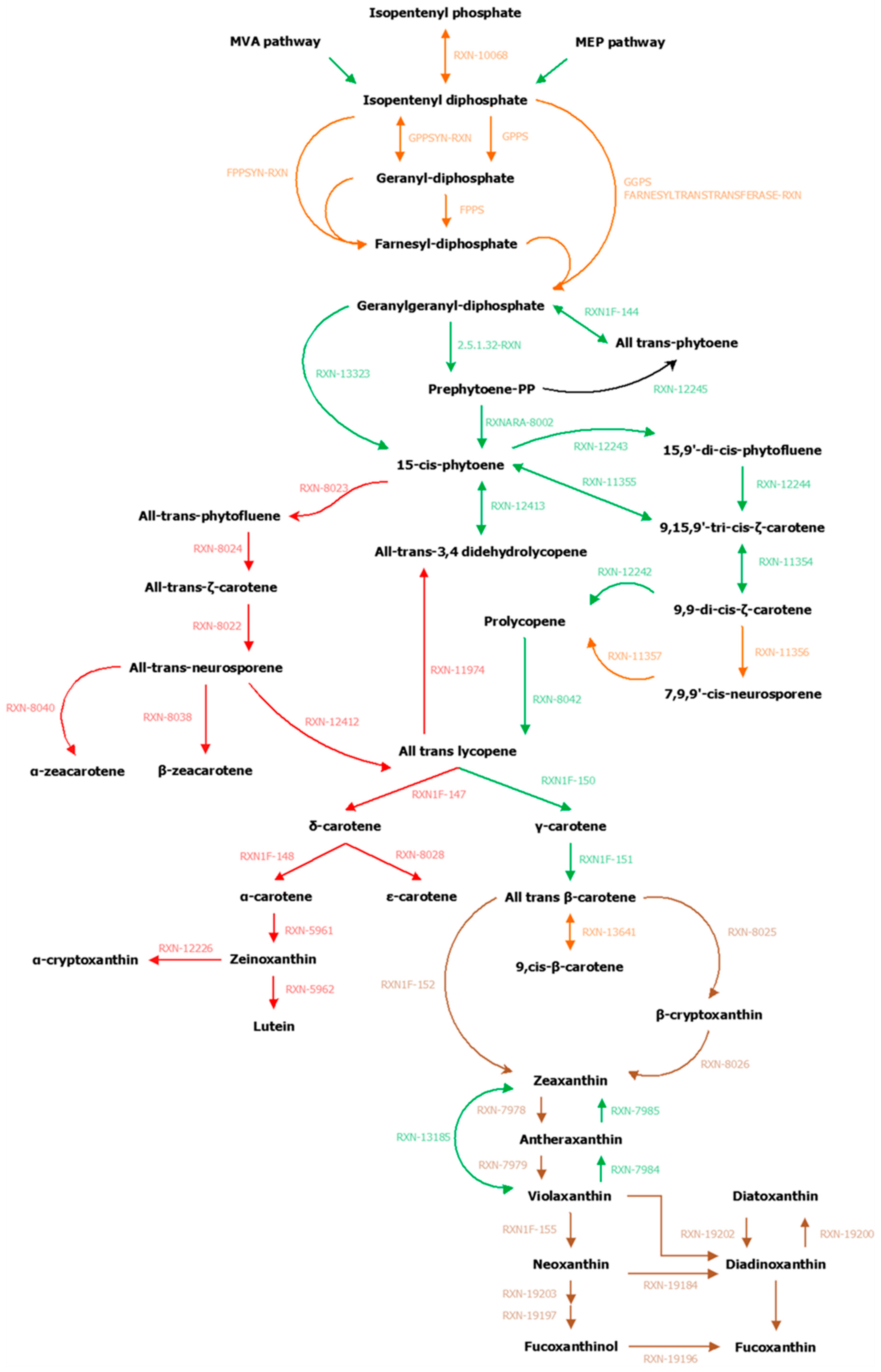
3.2. Focused Exploration of GSMNs Regarding the Carotenoid Biosynthesis Pathway, Generalities, and Specificities
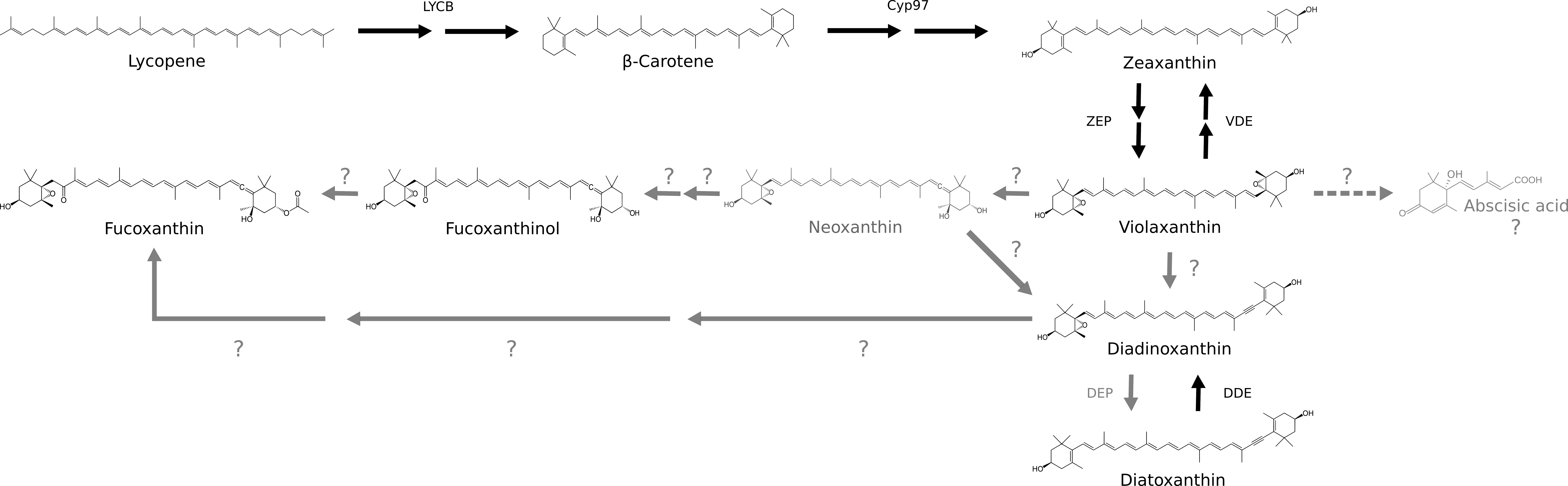
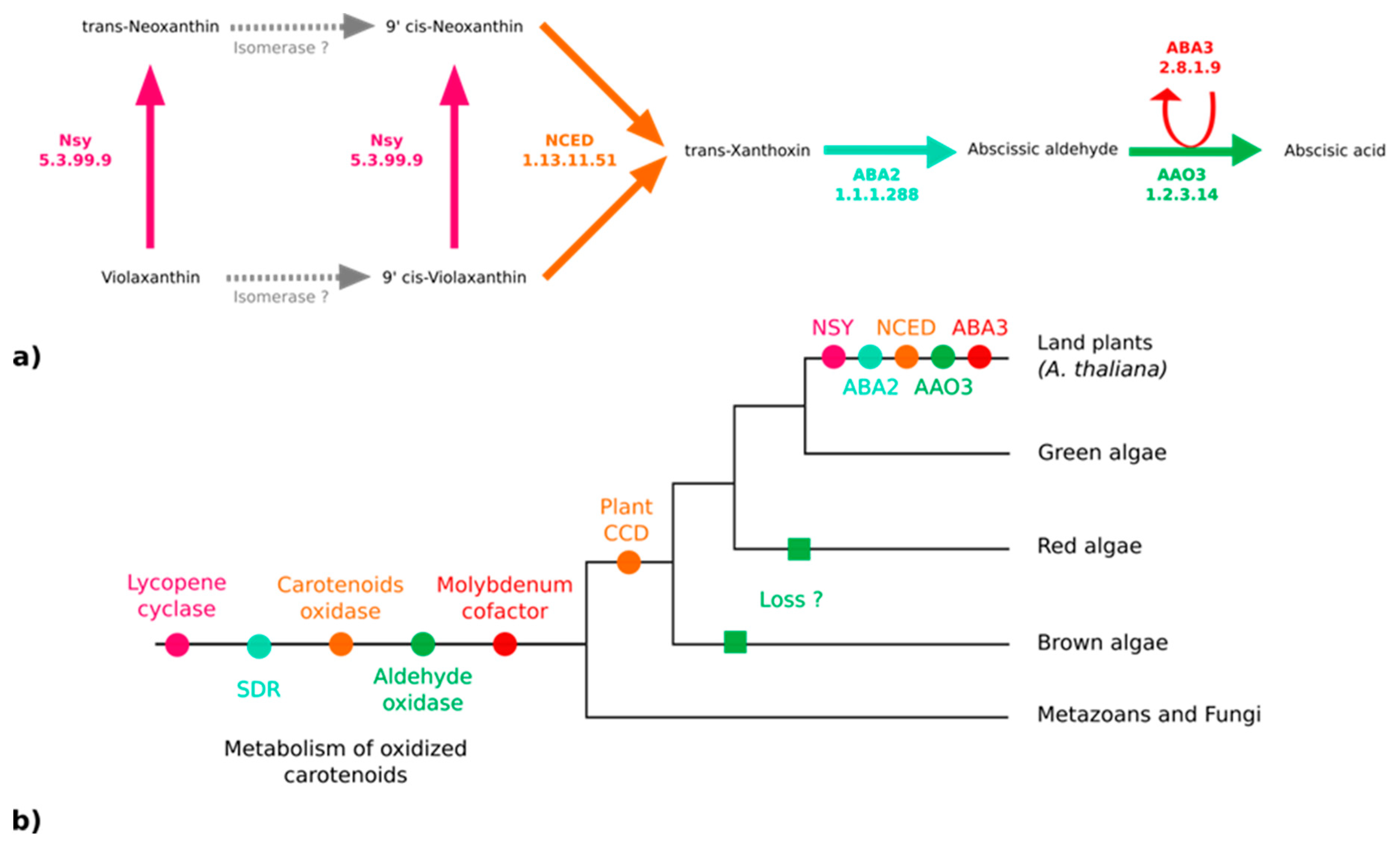
3.3. No Plant-Like Abscisic Acid Synthesis Pathway in Brown Algae
3.4. Extending the Fucoxanthin Biosynthesis Models from Diatoms to Brown Algae
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
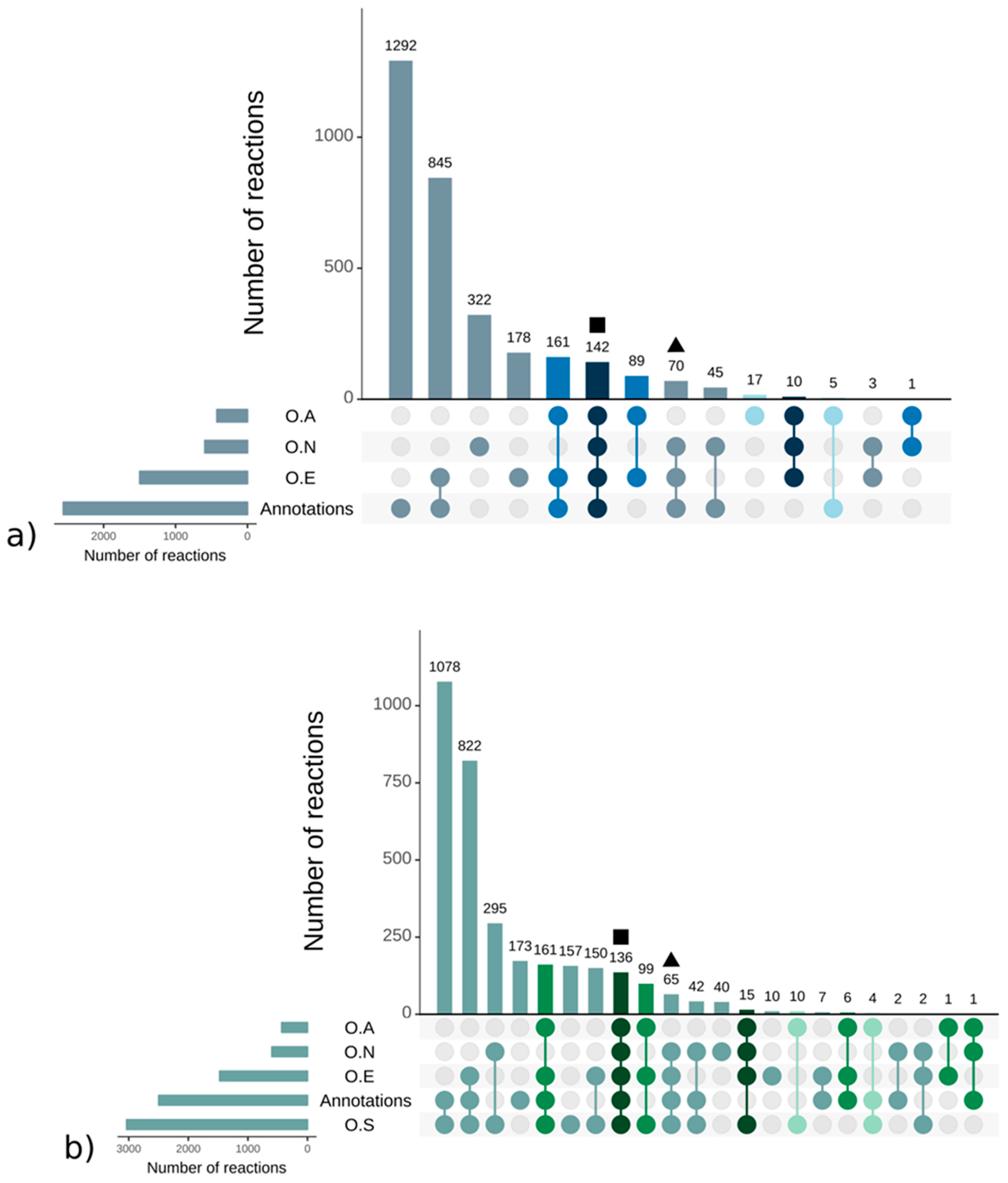
A.1. Qualitative Analysis of Genome–Scale Metabolic Networks and Gap-Filling
A.2. Manual Curation Made to the S. japonica and C. okamuranus Genome–Scale Metabolic Networks

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | ID Reactions (MetaCyc) | Publication | S. japonica (Associated Genes) | C. okamuranus (Associated Genes) |
|---|---|---|---|---|
| Carotenoid biosynthesis and first xanthophyll cycle, well–known reactions whose genes are characterized in brown algae | ||||
| PSY (phytoene synthase) | RXN–13323 | [11,53,55,61,62,104] | SJ02885 | g11610 |
| 2.5.1.32–RXN | ||||
| RXNARA–8002 | ||||
| PDS (phytoene desaturase) | RXN–12243 | [11,53,55,61,62,104] | SJ08891 | g10852 |
| RXN–12244 | ||||
| RXN–11355 | ||||
| Z–iso (ζ–carotene isomerase) | RXN–11354 | [55,104] | SJ04715 | g9721 |
| ZDS (ζ–carotene desaturase) | RXN–12242 | [11,53,55,61,62,104] | SJ05680 SJ05681 | g16199 |
| RXN–11356 | ||||
| RXN–11357 | ||||
| crtISO (prolycopene isomerase) | RXN–8042 | [55,56,61,62,104] | SJ05083 | g8850 |
| LycB (lycopene β–cyclase) | RXN1F–150 | [11,53,55,56,57,61,62,63,104] | SJ04962 | g10898 |
| RXN1F–151 | ||||
| Cyp97B | RXN–8025 | [12,58,62,64,104] | SJ07227 SJ07228 | g4983 g4984 |
| RXN–8026 | ||||
| RXN1F–152 | ||||
| ZEP (zeaxanthin epoxidase) | RXN–7979 | [11,18,53,55,56,62,74,104] | SJ19373 | g5910 |
| RXN–7978 | ||||
| VDE (violaxanthin de–epoxidase) | RXN–7984 | [11,18,53,56] | SJ03764 SJ20456 VDL (VDE–like ~ DDE) SJ19927 VDR (VDE–related) | g11316 g16187 VDL (VDE–like ~ DDE) g4586 VDR (VDE–related) |
| RXN–7985 | ||||
| Reactions removed | ||||
| phytoene desaturase (fungi – al–1 or crtI) | RXN–12413 | SJ18358 (ANNOTATION) | g18971.t1 (ORTHOLOGY) | |
| RXN–11974 | ||||
| RXN–8023 | ||||
| RXN–8024 | ||||
| RXN–8022 | ||||
| RXN–12412 | ||||
| LycE (lycopene ε–cyclase) | RXN–8040 | 7 sequences (ORTHOLOGY x2) | 18 sequences (ORTHOLOGY x3) | |
| LycB (lycopene β–cyclase) | RXN–8038 | |||
| LycE (lycopene ε–cyclase) | RXN1F–147 | |||
| RXN–8028 | ||||
| LycB (lycopene β–cyclase) | RXN1F–148 | |||
| Carotene epsilon monooxygenase | RXN–5961 | 5 sequences (ORTHOLOGY) | 7 sequences (ORTHOLOGY x2) | |
| RXN–5962 | 6 sequences (ORTHOLOGY) | 8 sequences (ORTHOLOGY x2) | ||
| RXN–12226 | — | g13263.t1 (ANNOTATION) | ||
| Reactions added manually | ||||
| DDE (diadinoxanthin de–epoxidase) | RXN–19200 | [12,59,61] | SJ20456 (VDE–like ~ DDE) | g16187 (VDE–like ~ DDE) |
| DEP (diatoxanthin–epoxidase) | RXN–19202 | — | — | |
| 1.3.99. – | RXN–19203 | [5,18,60] | — | — |
| 1.14.99.M8 | RXN–19197 | — | — | |
| 2.3.1. – | RXN–19196 | — | — | |
| — | RXN–19184 | [60] | — | — |
| — | RXN1F–155 | — | — | |
| — | ConversionViolaxanthinToDiadinoxanthin | [18] | — | — |
| — | ConversionDiadinoxanthinToFucoxanthin | [18] | — | — |

References
- Bartsch, I.; Wiencke, C.; Bischof, K.; Buchholz, C.M.; Buck, B.H.; Eggert, A.; Feuerpfeil, P.; Hanelt, D.; Jacobsen, S.; Karez, R.; et al. The genus Laminaria sensu lato: Recent insights and developments. Eur. J. Phycol. 2008, 43, 1–86. [Google Scholar] [CrossRef]
- Cheng, K.-C.; Kuo, P.-C.; Hung, H.-Y.; Yu, K.-H.; Hwang, T.-L.; Shieh, P.-C.; Chang, J.-S.; Wu, T.-S. Four new compounds from edible algae Cladosiphon okamuranus and Chlorella sorokiniana and their bioactivities. Phytochem. Lett. 2016, 18, 113–116. [Google Scholar] [CrossRef]
- Nishitsuji, K.; Arimoto, A.; Iwai, K.; Sudo, Y.; Hisata, K.; Fujie, M.; Arakaki, N.; Kushiro, T.; Konishi, T.; Shinzato, C.; et al. A draft genome of the brown alga, Cladosiphon okamuranus, S-strain: A platform for future studies of ‘mozuku’ biology. DNA Res. 2016, 23, 561–570. [Google Scholar] [CrossRef]
- Bleakley, S.; Hayes, M. Algal Proteins: Extraction, Application, and Challenges Concerning Production. Foods 2017, 6, 33. [Google Scholar] [CrossRef]
- Mikami, K.; Hosokawa, M. Biosynthetic Pathway and Health Benefits of Fucoxanthin, an Algae-Specific Xanthophyll in Brown Seaweeds. Int. J. Mol. Sci. 2013, 14, 13763–13781. [Google Scholar] [CrossRef]
- Christaki, E.; Bonos, E.; Giannenas, I.; Florou-Paneri, P. Functional properties of carotenoids originating from algae. J. Sci. Food Agric. 2013, 93, 5–11. [Google Scholar] [CrossRef]
- Álvarez, R.; Vaz, B.; Gronemeyer, H.; de Lera, Á.R. Functions, Therapeutic Applications, and Synthesis of Retinoids and Carotenoids. Chem. Rev. 2014, 114, 1–125. [Google Scholar] [CrossRef] [PubMed]
- Bohn, T. Carotenoids and Markers of Oxidative Stress in Human Observational Studies and Intervention Trials: Implications for Chronic Diseases. Antioxidants 2019, 8, 179. [Google Scholar] [CrossRef] [PubMed]
- Amengual, J. Bioactive Properties of Carotenoids in Human Health. Nutrients 2019, 11, 2388. [Google Scholar] [CrossRef] [PubMed]
- Mounien, L.; Tourniaire, F.; Landrier, J.-F. Anti-Obesity Effect of Carotenoids: Direct Impact on Adipose Tissue and Adipose Tissue-Driven Indirect Effects. Nutrients 2019, 11, 1562. [Google Scholar] [CrossRef]
- Esteban, R.; Moran, J.F.; Becerril, J.M.; García-Plazaola, J.I. Versatility of carotenoids: An integrated view on diversity, evolution, functional roles and environmental interactions. Environ. Exp. Bot. 2015, 119, 63–75. [Google Scholar] [CrossRef]
- Rodriguez-Concepcion, M.; Avalos, J.; Bonet, M.L.; Boronat, A.; Gomez-Gomez, L.; Hornero-Mendez, D.; Limon, M.C.; Meléndez-Martínez, A.J.; Olmedilla-Alonso, B.; Palou, A.; et al. A global perspective on carotenoids: Metabolism, biotechnology, and benefits for nutrition and health. Prog. Lipid Res. 2018, 70, 62–93. [Google Scholar] [CrossRef] [PubMed]
- Sandmann, G. Antioxidant Protection from UV- and Light-Stress Related to Carotenoid Structures. Antioxidants 2019, 8, 219. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Kiser, P.D.; von Lintig, J.; Palczewski, K. Structural basis of carotenoid cleavage: From bacteria to mammals. Arch. Biochem. Biophys. 2013, 539, 203–213. [Google Scholar] [CrossRef]
- Firn, R.D.; Jones, C.G. A Darwinian view of metabolism: Molecular properties determine fitness. J. Exp. Bot. 2009, 60, 719–726. [Google Scholar] [CrossRef]
- Mise, T.; Ueda, M.; Yasumoto, T. Production of Fucoxanthin-Rich Powder from Cladosiphon okamuranus. Adv. J. Food Sci. Technol. 2011, 3, 73–76. [Google Scholar]
- Kanazawa, K.; Ozaki, Y.; Hashimoto, T.; Das, S.K.; Matsushita, S.; Hirano, M.; Okada, T.; Komoto, A.; Mori, N.; Nakatsuka, M. Commercial-Scale Preparation of Biofunctional Fucoxanthin from Waste Parts of Brown Sea Algae Laminaria japonica. FSTR 2008, 14, 573–582. [Google Scholar] [CrossRef]
- Kuczynska, P.; Jemiola-Rzeminska, M.; Strzalka, K. Photosynthetic Pigments in Diatoms. Mar. Drugs 2015, 13, 5847–5881. [Google Scholar] [CrossRef]
- Schaffelke, B. Abscisic Acid in Sporophytes of Three Laminaria Species (Phaeophyta). J. Plant Physiol. 1995, 146, 453–458. [Google Scholar] [CrossRef]
- Ebrahim, A.; Almaas, E.; Bauer, E.; Bordbar, A.; Burgard, A.P.; Chang, R.L.; Dräger, A.; Famili, I.; Feist, A.M.; Fleming, R.M.; et al. Do genome-scale models need exact solvers or clearer standards? Mol. Syst. Biol. 2015, 11, 831. [Google Scholar] [CrossRef]
- Aite, M.; Chevallier, M.; Frioux, C.; Trottier, C.; Got, J.; Cortés, M.P.; Mendoza, S.N.; Carrier, G.; Dameron, O.; Guillaudeux, N.; et al. Traceability, reproducibility and wiki-exploration for “à-la-carte” reconstructions of genome-scale metabolic models. PLoS Comput. Biol. 2018, 14, e1006146. [Google Scholar] [CrossRef] [PubMed]
- Frainay, C.; Schymanski, E.; Neumann, S.; Merlet, B.; Salek, R.; Jourdan, F.; Yanes, O. Mind the Gap: Mapping Mass Spectral Databases in Genome-Scale Metabolic Networks Reveals Poorly Covered Areas. Metabolites 2018, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Palsson, B.Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [PubMed]
- Prigent, S.; Collet, G.; Dittami, S.M.; Delage, L.; Ethis de Corny, F.; Dameron, O.; Eveillard, D.; Thiele, S.; Cambefort, J.; Boyen, C.; et al. The genome-scale metabolic network of Ectocarpus siliculosus (EctoGEM): A resource to study brown algal physiology and beyond. Plant J. 2014, 80, 367–381. [Google Scholar] [CrossRef]
- Belcour, A.; Girard, J.; Aite, M.; Delage, L.; Trottier, C.; Marteau, C.; Leroux, C.; Dittami, S.M.; Sauleau, P.; Corre, E.; et al. Inferring biochemical reactions and metabolite structures to cope with metabolic pathway drift. bioRxiv 2018, 462556. [Google Scholar] [CrossRef]
- Ye, N.; Zhang, X.; Miao, M.; Fan, X.; Zheng, Y.; Xu, D.; Wang, J.; Zhou, L.; Wang, D.; Gao, Y.; et al. Saccharina genomes provide novel insight into kelp biology. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef]
- Dittami, S.M.; Corre, E. Detection of bacterial contaminants and hybrid sequences in the genome of the kelp Saccharina japonica using Taxoblast. PeerJ 2017, 5, e4073. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.-H.; Davis, F.G.; et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Karp, P.D.; Paley, S.; Romero, P. The Pathway Tools Software. Bioinformatics 2002, 18, S225–S232. [Google Scholar] [CrossRef]
- De Oliveira Dal’Molin, C.G.; Quek, L.-E.; Palfreyman, R.W.; Brumbley, S.M.; Nielsen, L.K. AraGEM, a Genome-Scale Reconstruction of the Primary Metabolic Network in Arabidopsis. Plant Physiol. 2010, 152, 579–589. [Google Scholar] [CrossRef]
- Loira, N.; Mendoza, S.; Paz Cortés, M.; Rojas, N.; Travisany, D.; Genova, A.D.; Gajardo, N.; Ehrenfeld, N.; Maass, A. Reconstruction of the microalga Nannochloropsis salina genome-scale metabolic model with applications to lipid production. BMC Syst. Biol. 2017, 11, 66. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- O’Brien, K.P.; Remm, M.; Sonnhammer, E.L.L. Inparanoid: A comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 2005, 33, D476–D480. [Google Scholar] [CrossRef]
- King, Z.A.; Lu, J.; Dräger, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016, 44, D515–D522. [Google Scholar] [CrossRef]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tissier, C.; et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2008, 36, D623–D631. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Dreher, K.; Karp, P.D. The challenge of constructing, classifying and representing metabolic pathways. FEMS Microbiol. Lett. 2013, 345, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Martin, O.; Van Du Tran, T.; Bridge, A.; Morgat, A.; Pagni, M. MetaNetX/MNXref—Reconciliation of metabolites and biochemical reactions to bring together genome-scale metabolic networks. Nucleic Acids Res. 2016, 44, D523–D526. [Google Scholar] [CrossRef] [PubMed]
- Prigent, S.; Frioux, C.; Dittami, S.M.; Thiele, S.; Larhlimi, A.; Collet, G.; Gutknecht, F.; Got, J.; Eveillard, D.; Bourdon, J.; et al. Meneco, a Topology-Based Gap-Filling Tool Applicable to Degraded Genome-Wide Metabolic Networks. PLoS Comput. Biol. 2017, 13, e1005276. [Google Scholar] [CrossRef]
- Ebenhöh, O.; Handorf, T.; Heinrich, R. Structural analysis of expanding metabolic networks. Genome Inform. 2004, 15, 35–45. [Google Scholar]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74. [Google Scholar] [CrossRef]
- Frioux, C.; Schaub, T.; Schellhorn, S.; Siegel, A.; Wanko, P. Hybrid metabolic network completion. Theory Pract. Log. Program. 2019, 19, 83–108. [Google Scholar] [CrossRef]
- Sterck, L.; Billiau, K.; Abeel, T.; Rouzé, P.; Van de Peer, Y. ORCAE: Online resource for community annotation of eukaryotes. Nat. Methods 2012, 9, 1041. [Google Scholar] [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Cock, J.M.; Sterck, L.; Rouzé, P.; Scornet, D.; Allen, A.E.; Amoutzias, G.; Anthouard, V.; Artiguenave, F.; Aury, J.-M.; Badger, J.H.; et al. The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature 2010, 465, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Hirschberg, J. Carotenoid biosynthesis in flowering plants. Curr. Opin. Plant Biol. 2001, 4, 210–218. [Google Scholar] [CrossRef]
- Zhao, L.; Chang, W.; Xiao, Y.; Liu, H.; Liu, P. Methylerythritol Phosphate Pathway of Isoprenoid Biosynthesis. Annu. Rev. Biochem. 2013, 82, 497–530. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, L.; Chi, S.; Wang, G.; Wang, X.; Liu, T.; Tang, X. Phylogenetic analyses of the genes involved in carotenoid biosynthesis in algae. Acta Oceanol. Sin. 2018, 37, 89–101. [Google Scholar] [CrossRef]
- Takaichi, S. Carotenoids in Algae: Distributions, Biosyntheses and Functions. Mar. Drugs 2011, 9, 1101–1118. [Google Scholar] [CrossRef]
- Cui, H.; Wang, Y.; Qin, S. Molecular Evolution of Lycopene Cyclases Involved in the Formation of Carotenoids in Eukaryotic Algae. Plant Mol. Biol. Rep. 2011, 29, 1013–1020. [Google Scholar] [CrossRef]
- Teng, L.; Fan, X.; Nelson, D.R.; Han, W.; Zhang, X.; Xu, D.; Renault, H.; Markov, G.V.; Ye, N. Diversity and evolution of cytochromes P450 in stramenopiles. Planta 2019, 249, 647–661. [Google Scholar] [CrossRef]
- Lohr, M.; Wilhelm, C. Algae displaying the diadinoxanthin cycle also possess the violaxanthin cycle. Proc. Natl. Acad. Sci. USA 1999, 96, 8784–8789. [Google Scholar] [CrossRef]
- Dambek, M.; Eilers, U.; Breitenbach, J.; Steiger, S.; Büchel, C.; Sandmann, G. Biosynthesis of fucoxanthin and diadinoxanthin and function of initial pathway genes in Phaeodactylum tricornutum. J. Exp. Bot. 2012, 63, 5607–5612. [Google Scholar] [CrossRef]
- Coesel, S.; Oborník, M.; Varela, J.; Falciatore, A.; Bowler, C. Evolutionary Origins and Functions of the Carotenoid Biosynthetic Pathway in Marine Diatoms. PLoS ONE 2008, 3, e2896. [Google Scholar] [CrossRef] [PubMed]
- Frommolt, R.; Werner, S.; Paulsen, H.; Goss, R.; Wilhelm, C.; Zauner, S.; Maier, U.G.; Grossman, A.R.; Bhattacharya, D.; Lohr, M. Ancient Recruitment by Chromists of Green Algal Genes Encoding Enzymes for Carotenoid Biosynthesis. Mol. Biol. Evol. 2008, 25, 2653–2667. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.X.; Pogson, B.; Sun, Z.; McDonald, K.A.; DellaPenna, D.; Gantt, E. Functional analysis of the beta and epsilon lycopene cyclase enzymes of Arabidopsis reveals a mechanism for control of cyclic carotenoid formation. Plant Cell 1996, 8, 1613–1626. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Yu, X.; Wang, Y.; Cui, Y.; Li, X.; Liu, Z.; Qin, S. Evolutionary origins, molecular cloning and expression of carotenoid hydroxylases in eukaryotic photosynthetic algae. BMC Genom. 2013, 14, 457. [Google Scholar] [CrossRef]
- Nimura, K.; Mizuta, H. Inducible effects of abscisic acid on sporophyte discs from Laminaria japonica Areschoug (Laminariales, Phaeophyceae). J. Appl. Phycol. 2002, 14, 159–163. [Google Scholar] [CrossRef]
- Shimizu, K.; Uji, T.; Yasui, H.; Mizuta, H. Control of elicitor-induced oxidative burst by abscisic acid associated with growth of Saccharina japonica (Phaeophyta, Laminariales) sporophytes. J. Appl. Phycol. 2018, 30, 1371–1379. [Google Scholar] [CrossRef]
- Nambara, E.; Marion-Poll, A. Abscisic acid biosynthesis and catabolism. Annu. Rev. Plant Biol. 2005, 56, 165–185. [Google Scholar] [CrossRef]
- Xiong, L.; Zhu, J.-K. Regulation of Abscisic Acid Biosynthesis. Plant Physiol. 2003, 133, 29–36. [Google Scholar] [CrossRef]
- Seo, M.; Koshiba, T. Complex regulation of ABA biosynthesis in plants. Trends Plant Sci. 2002, 7, 41–48. [Google Scholar] [CrossRef]
- Finkelstein, R.R.; Rock, C.D. Abscisic Acid Biosynthesis and Response. Arab. Book 2002, 1, e0058. [Google Scholar] [CrossRef]
- Lee, K.H.; Piao, H.L.; Kim, H.-Y.; Choi, S.M.; Jiang, F.; Hartung, W.; Hwang, I.; Kwak, J.M.; Lee, I.-J.; Hwang, I. Activation of Glucosidase via Stress-Induced Polymerization Rapidly Increases Active Pools of Abscisic Acid. Cell 2006, 126, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Hauser, F.; Waadt, R.; Schroeder, J.I. Evolution of Abscisic Acid Synthesis and Signaling Mechanisms. Curr. Biol. 2011, 21, R346–R355. [Google Scholar] [CrossRef] [PubMed]
- Mackie, A.; Keseler, I.M.; Nolan, L.; Karp, P.D.; Paulsen, I.T. Dead End Metabolites—Defining the Known Unknowns of the E. coli Metabolic Network. PLoS ONE 2013, 8, e75210. [Google Scholar] [CrossRef] [PubMed]
- Sajilata, M.G.; Singhal, R.S.; Kamat, M.Y. The Carotenoid Pigment Zeaxanthin—A Review. Compr. Rev. Food Sci. Food Saf. 2008, 7, 29–49. [Google Scholar] [CrossRef]
- Jahns, P.; Latowski, D.; Strzalka, K. Mechanism and regulation of the violaxanthin cycle: The role of antenna proteins and membrane lipids. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1787, 3–14. [Google Scholar] [CrossRef]
- Haugan, J.A. Algal carotenoids 54. Carotenoids of brown algae (Phaeophyceae). Biochem. Syst. Ecol. 1994, 22, 31–41. [Google Scholar] [CrossRef]
- Bouvier, F.; D’Harlingue, A.; Backhaus, R.A.; Kumagai, M.H.; Camara, B. Identification of neoxanthin synthase as a carotenoid cyclase paralog: Plastid neoxanthin synthase. Eur. J. Biochem. 2000, 267, 6346–6352. [Google Scholar] [CrossRef]
- Pan, S.; Reed, J.L. Advances in gap-filling genome-scale metabolic models and model-driven experiments lead to novel metabolic discoveries. Curr. Opin. Biotechnol. 2018, 51, 103–108. [Google Scholar] [CrossRef]
- Cunningham, F.X.; Gantt, E. One ring or two? Determination of ring number in carotenoids by lycopene epsilon-cyclases. Proc. Natl. Acad. Sci. USA 2001, 98, 2905–2910. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, P.; Liu, X.; Chen, J.; Xu, J.; Chen, H.; Yan, X. Quantitative profiling method for phytohormones and betaines in algae by liquid chromatography electrospray ionization tandem mass spectrometry: Determination of phytohormones and betaines in algae by LC-MS/MS. Biomed. Chromatogr. 2014, 28, 275–280. [Google Scholar] [CrossRef]
- Schwartz, S.H.; Qin, X.; Zeevaart, J.A. Elucidation of the Indirect Pathway of Abscisic Acid Biosynthesis by Mutants, Genes, and Enzymes. Plant Physiol. 2003, 131, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Bittner, F.; Oreb, M.; Mendel, R.R. ABA3 Is a Molybdenum Cofactor Sulfurase Required for Activation of Aldehyde Oxidase and Xanthine Dehydrogenase in Arabidopsis thaliana. J. Biol. Chem. 2001, 276, 40381–40384. [Google Scholar] [CrossRef] [PubMed]
- Kaufholdt, D.; Baillie, C.-K.; Meinen, R.; Mendel, R.R.; Hänsch, R. The Molybdenum Cofactor Biosynthesis Network: In Vivo Protein-Protein Interactions of an Actin Associated Multi-Protein Complex. Front. Plant Sci. 2017, 8, 1946. [Google Scholar] [CrossRef] [PubMed]
- Salt, S.D.; Tuzun, S.; Kuć, J. Effects of β-ionone and abscisic acid on the growth of tobacco and resistance to blue mold. Mimicry of effects of stem infection by Peronospora tabacina Adam. Physiol. Mol. Plant Pathol. 1986, 28, 287–297. [Google Scholar] [CrossRef]
- Havaux, M. Carotenoid oxidation products as stress signals in plants. Plant J. 2014, 79, 597–606. [Google Scholar] [CrossRef]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef]
- Chi, S.; Liu, T.; Wang, X.; Wang, R.; Wang, S.; Wang, G.; Shan, G.; Liu, C. Functional genomics analysis reveals the biosynthesis pathways of important cellular components (alginate and fucoidan) of Saccharina. Curr. Genet. 2018, 64, 259–273. [Google Scholar] [CrossRef]
- Park, J.-N.; Ali-Nehari, A.; Woo, H.-C.; Chun, B.-S. Thermal stabilities of polyphenols and fatty acids in Laminaria japonica hydrolysates produced using subcritical water. Korean J. Chem. Eng. 2012, 29, 1604–1609. [Google Scholar] [CrossRef]
- Getachew, P.; Kang, J.-Y.; Choi, J.-S.; Hong, Y.-K. Does bryozoan colonization alter the biochemical composition of Saccharina japonica affecting food safety and quality? Bot. Mar. 2015, 58, 267–274. [Google Scholar] [CrossRef]
- Patterson, G.W. Sterols of Laminaria. Comp. Biochem. Physiol. 1968, 24, 501–505. [Google Scholar] [CrossRef]
- Zhang, P.; Shao, Z.; Jin, W.; Duan, D. Comparative Characterization of Two GDP-Mannose Dehydrogenase Genes from Saccharina japonica (Laminariales, Phaeophyceae). BMC Plant Biol. 2016, 16, 62. Available online: https://bmcplantbiol.biomedcentral.com/articles/10.1186/s12870-016-0750-3 (accessed on 9 January 2018). [CrossRef] [PubMed]
- Duan, D.; Liu, X.; Pan, F.; Liu, H.; Chen, N.; Fei, X. Extraction and Identification of Cytokinin from Laminaria japonica Aresch. Bot. Mar. 1995, 38, 409–412. [Google Scholar] [CrossRef]
- Honya, M.; Kinoshita, T.; Ishikawa, M.; Mori, H.; Nisizawa, K. Seasonal variation in the lipid content of cultured Laminaria japonica: Fatty acids, sterols, β-carotene and tocopherol. J. Appl. Phycol. 1994, 6, 25–29. [Google Scholar] [CrossRef]
- Hwang, J.-H.; Kim, N.-G.; Woo, H.-C.; Rha, S.-J.; Kim, S.-J.; Shin, T.-S. Variation in the chemical composition of Saccharina japonica with harvest area and culture period. J. Aquac. Res. Dev. 2014, 5, 286. [Google Scholar]
- Groisillier, A.; Shao, Z.; Michel, G.; Goulitquer, S.; Bonin, P.; Krahulec, S.; Nidetzky, B.; Duan, D.; Boyen, C.; Tonon, T. Mannitol metabolism in brown algae involves a new phosphatase family. J. Exp. Bot. 2014, 65, 559–570. [Google Scholar] [CrossRef]
- Saito, H.; Xue, C.; Yamashiro, R.; Moromizato, S.; Itabashi, Y. High Polyunsaturated Fatty Acid Levels in Two Subtropical Macroalgae, Cladosiphon Okamuranus and Caulerpa Lentillifera. J. Phycol. 2010, 46, 665–673. [Google Scholar] [CrossRef]
- Tako, M.; Yoza, E.; Tohma, S. Chemical Characterization of Acetyl Fucoidan and Alginate from Commercially Cultured Cladosiphon okamuranus. Bot. Mar. 2005, 43, 393–398. [Google Scholar] [CrossRef]
- Lim, S.J.; Wan Aida, W.M.; Schiehser, S.; Rosenau, T.; Böhmdorfer, S. Structural elucidation of fucoidan from Cladosiphon okamuranus (Okinawa mozuku). Food Chem. 2019, 272, 222–226. [Google Scholar] [CrossRef]
- Kakisawa, H.; Asari, F.; Kusumi, T.; Toma, T.; Sakurai, T.; Oohusa, T.; Hara, Y.; Chiharai, M. An allelopathic fatty acid from the brown alga Cladosiphon okamuranus. Phytochemistry 1988, 27, 731–735. [Google Scholar] [CrossRef]
- Al-Babili, S.; Hugueney, P.; Schledz, M.; Welsch, R.; Frohnmeyer, H.; Laule, O.; Beyer, P. Identification of a novel gene coding for neoxanthin synthase from Solanum tuberosum. FEBS Lett. 2000, 485, 168–172. [Google Scholar] [CrossRef]
- Tan, B.-C.; Joseph, L.M.; Deng, W.-T.; Liu, L.; Li, Q.-B.; Cline, K.; McCarty, D.R. Molecular characterization of the Arabidopsis 9-cis epoxycarotenoid dioxygenase gene family. Plant J. 2003, 35, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.H.; Strack, D. Carotenoids and their cleavage products: Biosynthesis and functions. Nat. Prod. Rep. 2011, 28, 663–692. [Google Scholar] [CrossRef] [PubMed]
- Priya, R.; Siva, R. Phylogenetic analysis and evolutionary studies of plant carotenoid cleavage dioxygenase gene. Gene 2014, 548, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Ahrazem, O.; Gómez-Gómez, L.; Rodrigo, M.J.; Avalos, J.; Limón, M.C. Carotenoid Cleavage Oxygenases from Microbes and Photosynthetic Organisms: Features and Functions. Int. J. Mol. Sci. 2016, 17, 1781. [Google Scholar] [CrossRef]
- Harrison, P.J.; Bugg, T.D.H. Enzymology of the carotenoid cleavage dioxygenases: Reaction mechanisms, inhibition and biochemical roles. Arch. Biochem. Biophys. 2014, 544, 105–111. [Google Scholar] [CrossRef]
- Seo, M.; Aoki, H.; Koiwai, H.; Kamiya, Y.; Nambara, E.; Koshiba, T. Comparative Studies on the Arabidopsis Aldehyde Oxidase (AAO) Gene Family Revealed a Major Role of AAO3 in ABA Biosynthesis in Seeds. Plant Cell Physiol. 2004, 45, 1694–1703. [Google Scholar] [CrossRef]
- Rodríguez-Trelles, F.; Tarrío, R.; Ayala, F.J. Convergent neofunctionalization by positive Darwinian selection after ancient recurrent duplications of the xanthine dehydrogenase gene. Proc. Natl. Acad. Sci. USA 2003, 100, 13413–13417. [Google Scholar] [CrossRef]
- Moummou, H.; Kallberg, Y.; Tonfack, L.B.; Persson, B.; van der Rest, B. The Plant Short-Chain Dehydrogenase (SDR) superfamily: Genome-wide inventory and diversification patterns. BMC Plant Biol. 2012, 12, 219. [Google Scholar] [CrossRef]
- Peng, T.; Xu, Y.; Zhang, Y. Comparative genomics of molybdenum utilization in prokaryotes and eukaryotes. BMC Genom. 2018, 19, 691. [Google Scholar] [CrossRef]
- Filiz, E.; Distelfeld, A.; Fahima, T.; Metin, Ö.K.; Nevo, E.; Weining, S.; Uncuoğlu, A.A. Barley molybdenum cofactor sulfurase (MCSU): Sequencing, modeling, and its comparison to other higher plants. Turk. J. Agric. For. 2015, 39, 786–796. [Google Scholar] [CrossRef]
- Mendel, R.R. The Molybdenum Cofactor. J. Biol. Chem. 2013, 288, 13165–13172. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Nishino, T.; Bittner, F. Molybdenum enzymes in higher organisms. Coord. Chem. Rev. 2011, 255, 1179–1205. [Google Scholar] [CrossRef] [PubMed]
- Mendel, R.R.; Hänsch, R. Molybdoenzymes and molybdenum cofactor in plants. J. Exp. Bot. 2002, 53, 1689–1698. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nègre, D.; Aite, M.; Belcour, A.; Frioux, C.; Brillet-Guéguen, L.; Liu, X.; Bordron, P.; Godfroy, O.; Lipinska, A.P.; Leblanc, C.; et al. Genome–Scale Metabolic Networks Shed Light on the Carotenoid Biosynthesis Pathway in the Brown Algae Saccharina japonica and Cladosiphon okamuranus. Antioxidants 2019, 8, 564. https://doi.org/10.3390/antiox8110564
Nègre D, Aite M, Belcour A, Frioux C, Brillet-Guéguen L, Liu X, Bordron P, Godfroy O, Lipinska AP, Leblanc C, et al. Genome–Scale Metabolic Networks Shed Light on the Carotenoid Biosynthesis Pathway in the Brown Algae Saccharina japonica and Cladosiphon okamuranus. Antioxidants. 2019; 8(11):564. https://doi.org/10.3390/antiox8110564
Chicago/Turabian StyleNègre, Delphine, Méziane Aite, Arnaud Belcour, Clémence Frioux, Loraine Brillet-Guéguen, Xi Liu, Philippe Bordron, Olivier Godfroy, Agnieszka P. Lipinska, Catherine Leblanc, and et al. 2019. "Genome–Scale Metabolic Networks Shed Light on the Carotenoid Biosynthesis Pathway in the Brown Algae Saccharina japonica and Cladosiphon okamuranus" Antioxidants 8, no. 11: 564. https://doi.org/10.3390/antiox8110564
APA StyleNègre, D., Aite, M., Belcour, A., Frioux, C., Brillet-Guéguen, L., Liu, X., Bordron, P., Godfroy, O., Lipinska, A. P., Leblanc, C., Siegel, A., Dittami, S. M., Corre, E., & Markov, G. V. (2019). Genome–Scale Metabolic Networks Shed Light on the Carotenoid Biosynthesis Pathway in the Brown Algae Saccharina japonica and Cladosiphon okamuranus. Antioxidants, 8(11), 564. https://doi.org/10.3390/antiox8110564