Syntaxin-17-Dependent Mitochondrial Dynamics Is Essential for Protection against Oxidative-Stress-Induced Apoptosis

Abstract
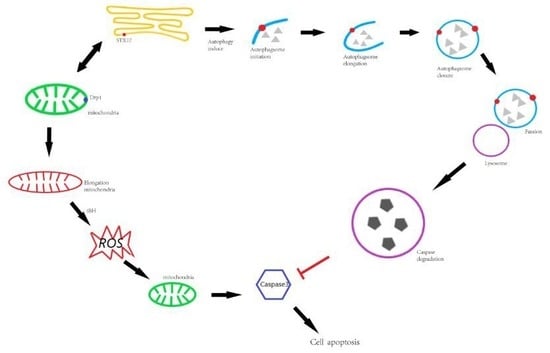
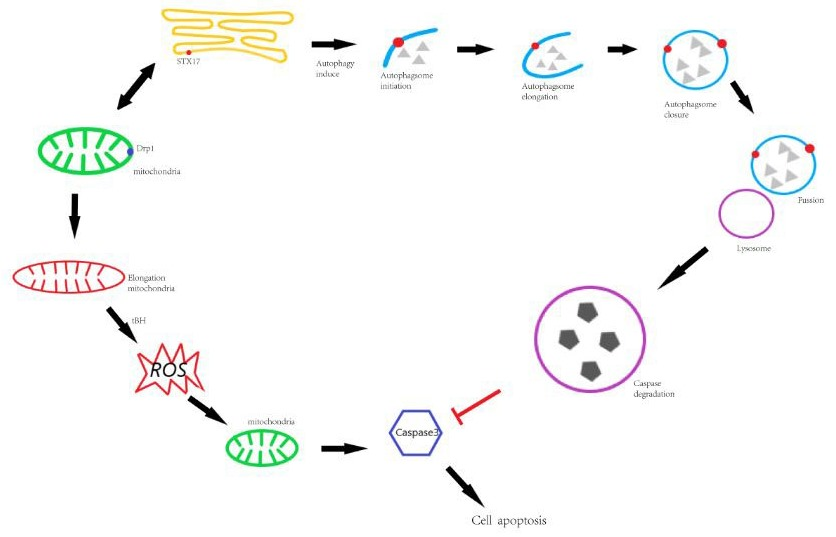{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture, Cell Transfection and Cell Lysate Preparation
2.3. Immunofluorescence Staining
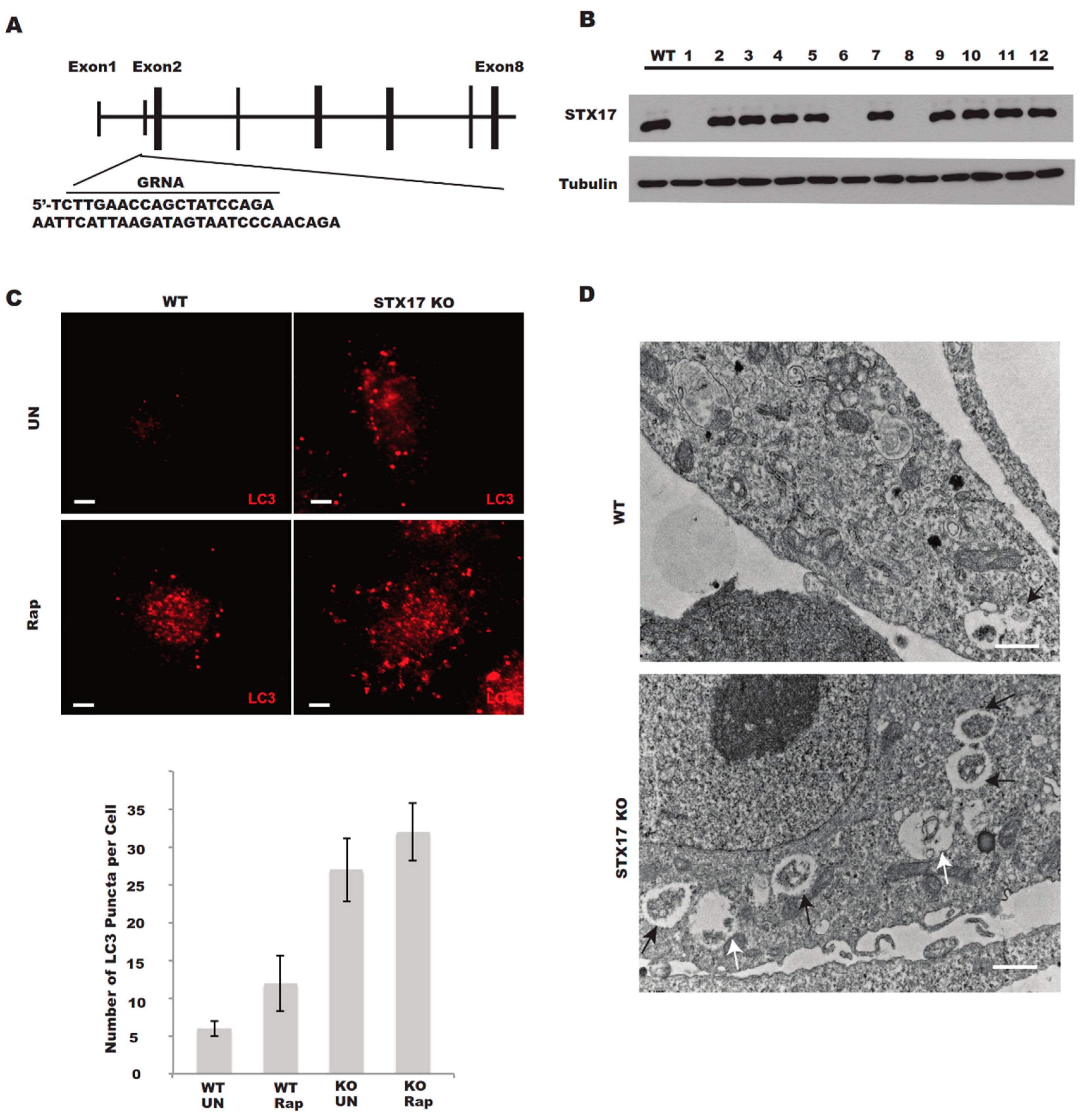
2.4. Construction of Stable Knockout Cell Lines Using the CRISPR/Cas9 Technique
2.5. Statistical Analysis
3. Results
3.1. Generation of STX17 Knockout U2OS Cells
3.2. Autophagosome–Lysosome Fusion is Partially Defective in STX17 KO Cells
3.3. The Role of STX17 in Organelle Dynamics
3.4. STX17 is Crucial in Cellular Responses to Divergent Stresses
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Gomes, L.C.; Benedetto, G.D.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; de Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Arasaki, K.; Shimizu, H.; Mogari, H.; Nishida, N.; Hirota, N.; Furuno, A.; Kudo, Y.; Baba, M.; Baba, N.; Cheng, J.; et al. A role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev. Cell 2015, 32, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Takáts, S.; Nagy, P.; Varga, A.; Pircs, K.; Kárpáti, M.; Varga, K.; Kovács, A.L.; Hegedűs, K.; Juhász, C. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J. Cell Biol. 2013, 201, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Vivona, S. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Nishimura, T.; Sakamaki, Y.; Itakura, E.; Hatta, T.; Natsume, T.; Mizushima, N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell 2014, 25, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Li, W. Impairment of autophagosome-lysosome fusion in the buff mutant mice with the VPS33A(D251E) mutation. Autophagy 2015, 11, 1608–1622. [Google Scholar] [CrossRef] [PubMed]
- Muppirala, M.; Gupta, V.; Swarup, G. Syntaxin 17 cycles between the ER and ERGIC and is required to maintain the architecture of ERGIC and Golgi. Biol. Cell 2011, 103, 333–350. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Nassiri, A.; Zhong, Q. Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L). Proc. Natl. Acad. Sci. USA 2011, 108, 7769–7774. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Fan, W.; Lu, Y.; Ding, X.; Chen, S.; Zhong, Q. A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12-Atg5 conjugate. Mol. Cell 2012, 45, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Jiang, P.; Nakano, S.; Sakamaki, Y.; Yamamoto, H.; Mizushima, N. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J. Cell Biol. 2018, 217, 2633–2645. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, M.; Nishimura, T.; Sakamaki, Y.; Yamamoto, H.; Mizushima, N. Accumulation of undegraded autophagosomes by expression of dominant-negative STX17 (syntaxin 17) mutants. Autophagy 2017, 13, 1452–1464. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Xiao, X.; Huang, F.; Liu, R. Syntaxin-17-Dependent Mitochondrial Dynamics Is Essential for Protection against Oxidative-Stress-Induced Apoptosis. Antioxidants 2019, 8, 522. https://doi.org/10.3390/antiox8110522
Wang B, Xiao X, Huang F, Liu R. Syntaxin-17-Dependent Mitochondrial Dynamics Is Essential for Protection against Oxidative-Stress-Induced Apoptosis. Antioxidants. 2019; 8(11):522. https://doi.org/10.3390/antiox8110522
Chicago/Turabian StyleWang, Binran, Xiaoyue Xiao, Fanwei Huang, and Rong Liu. 2019. "Syntaxin-17-Dependent Mitochondrial Dynamics Is Essential for Protection against Oxidative-Stress-Induced Apoptosis" Antioxidants 8, no. 11: 522. https://doi.org/10.3390/antiox8110522
APA StyleWang, B., Xiao, X., Huang, F., & Liu, R. (2019). Syntaxin-17-Dependent Mitochondrial Dynamics Is Essential for Protection against Oxidative-Stress-Induced Apoptosis. Antioxidants, 8(11), 522. https://doi.org/10.3390/antiox8110522