Protein Carbonylation in Patients with Myelodysplastic Syndrome: An Opportunity for Deferasirox Therapy

 , , , , ,
, , , , ,  and
and
Abstract
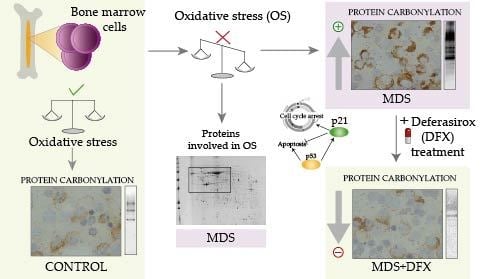
1. Introduction
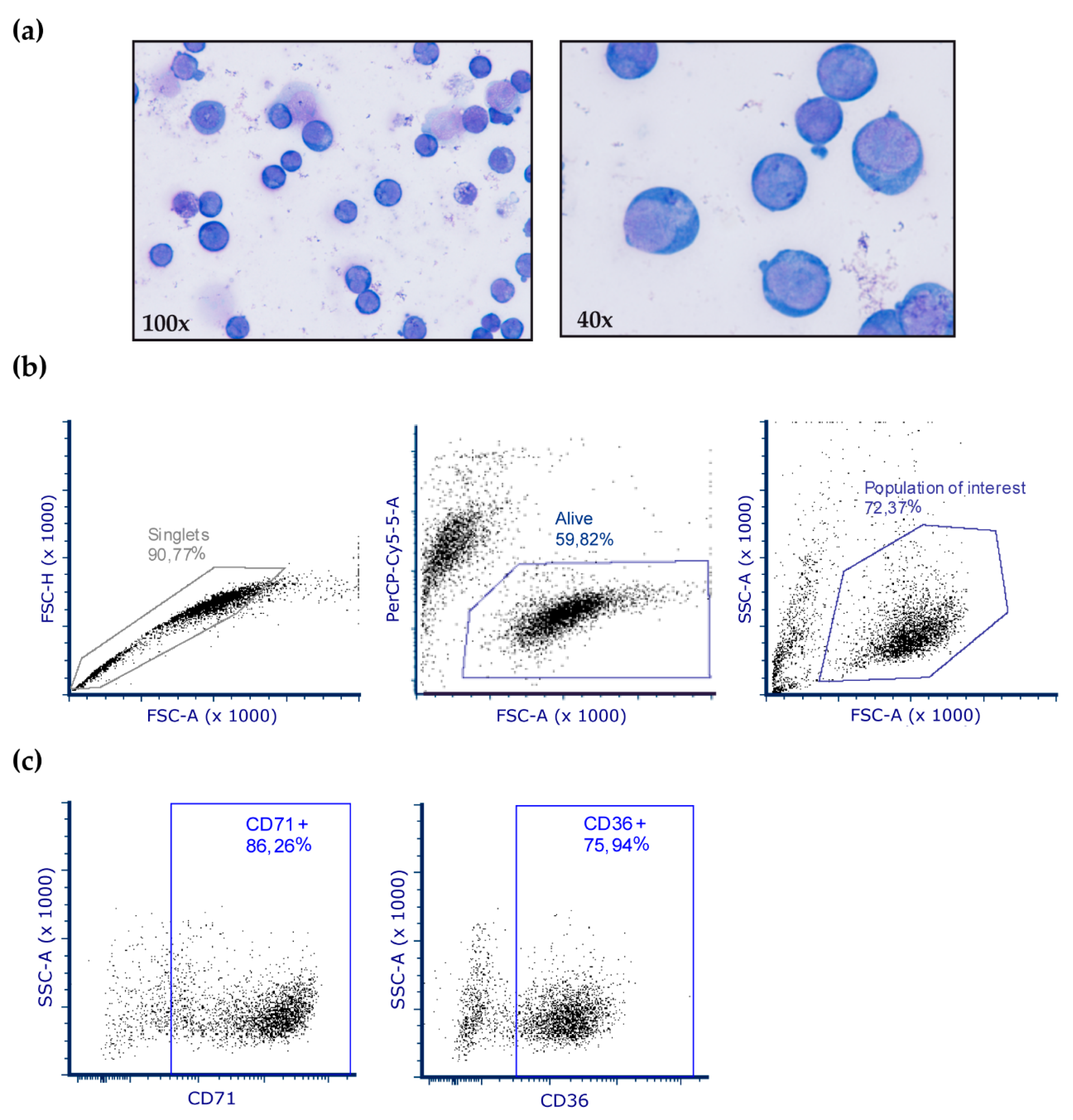
2. Materials and Methods
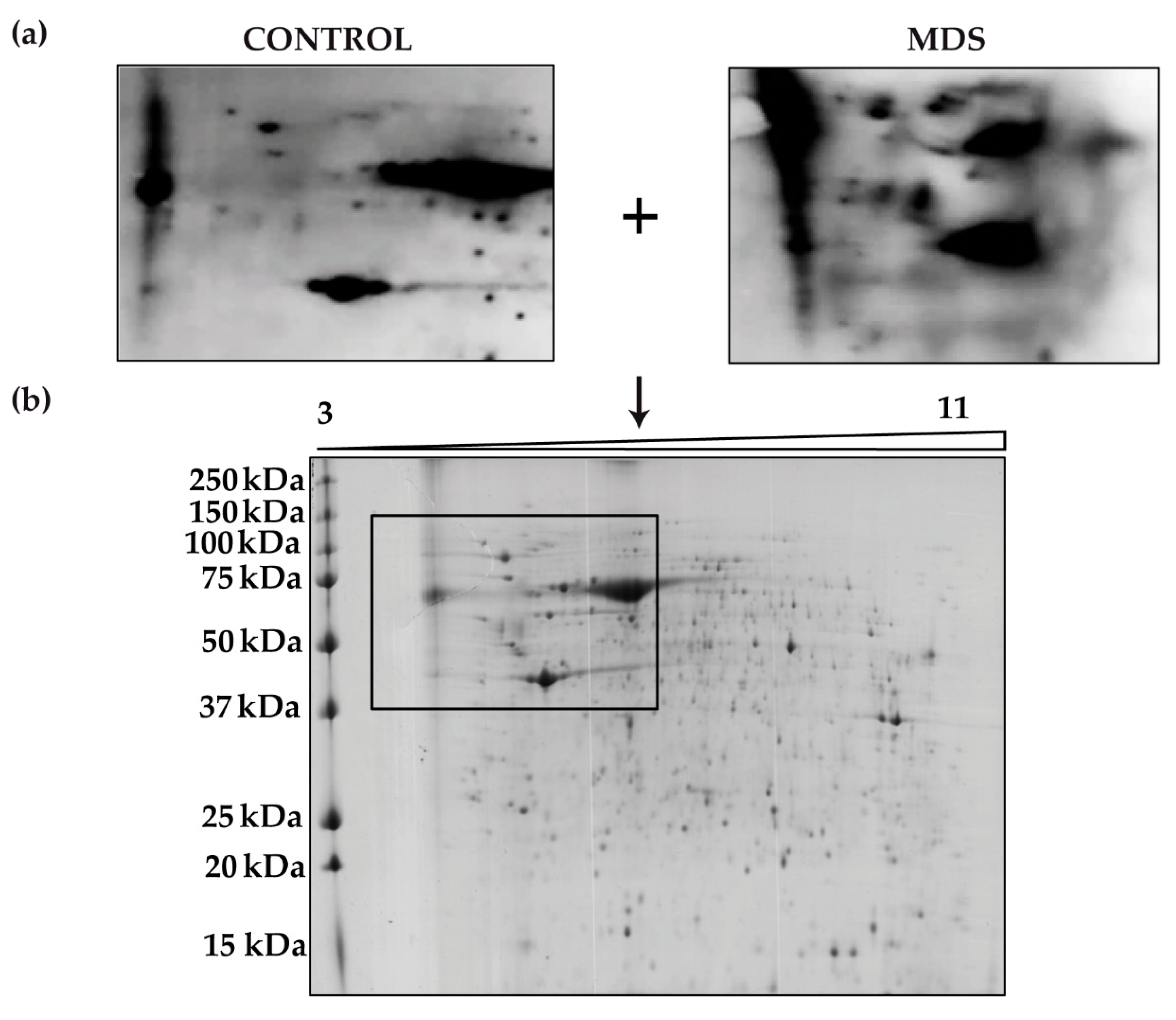
3. Results
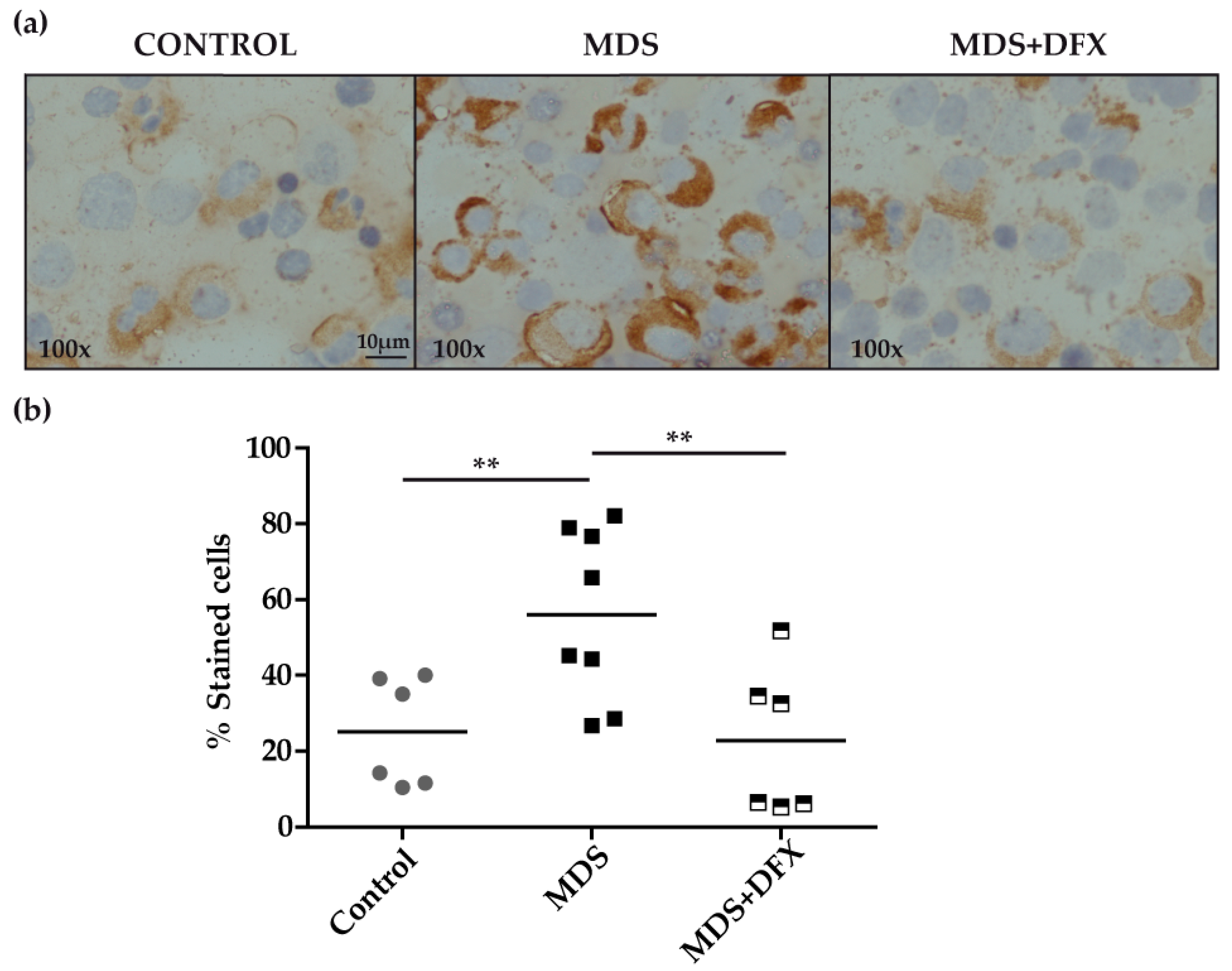
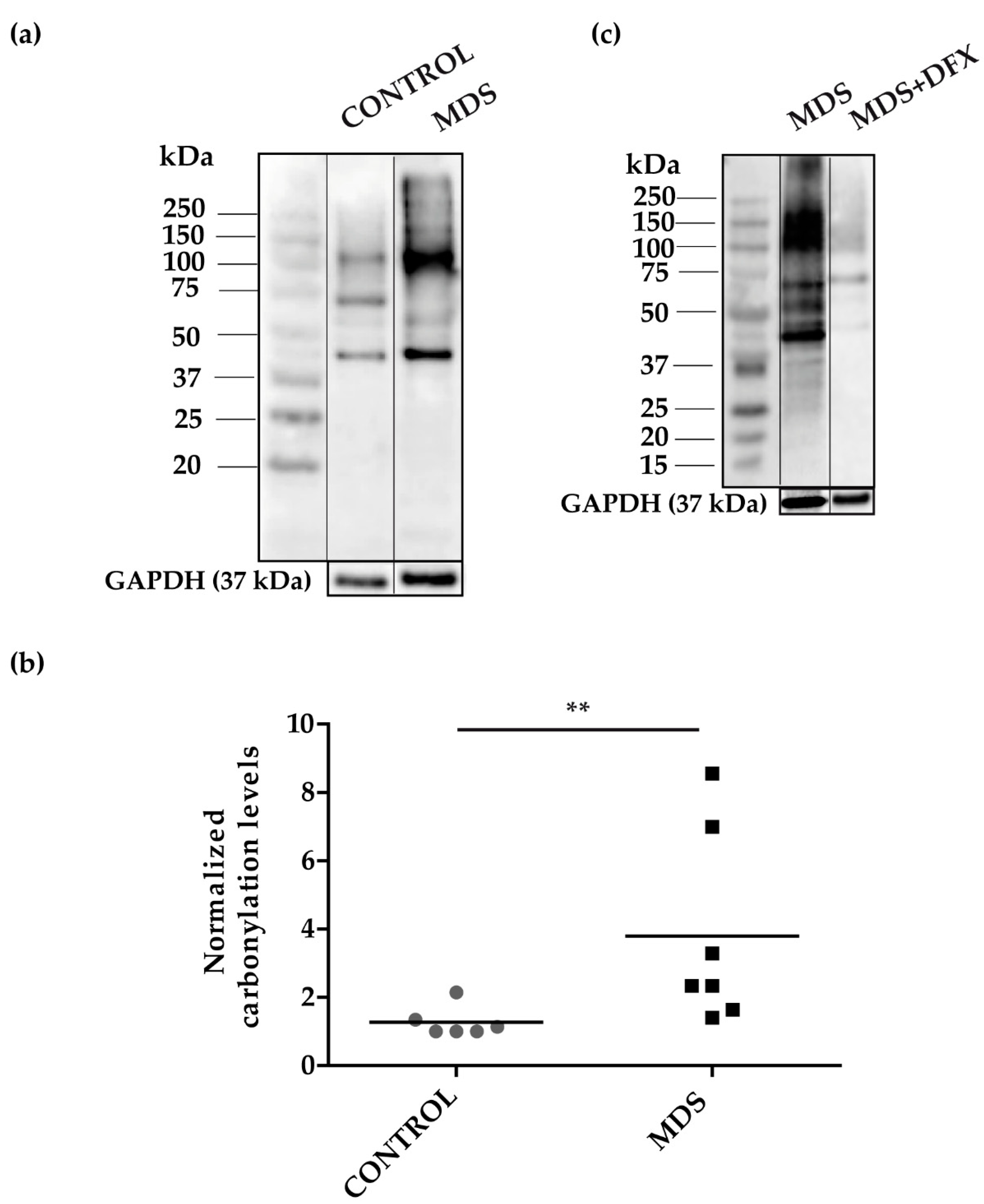
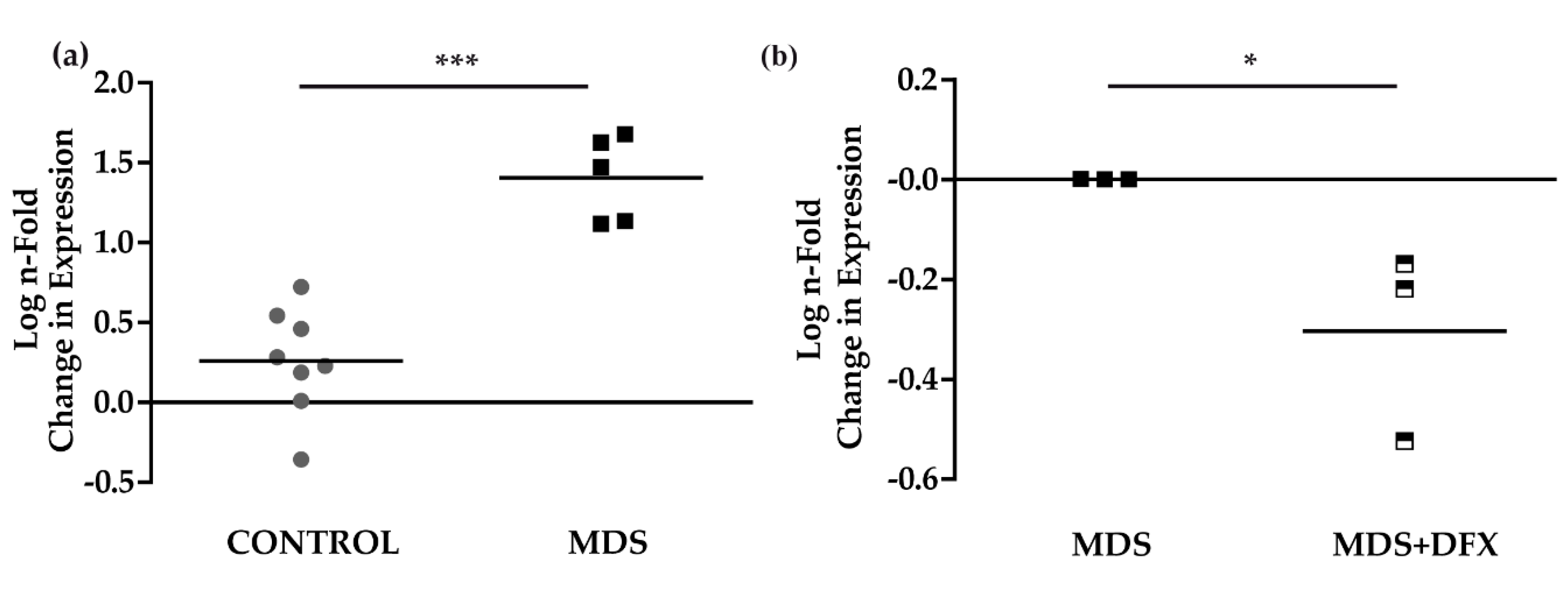
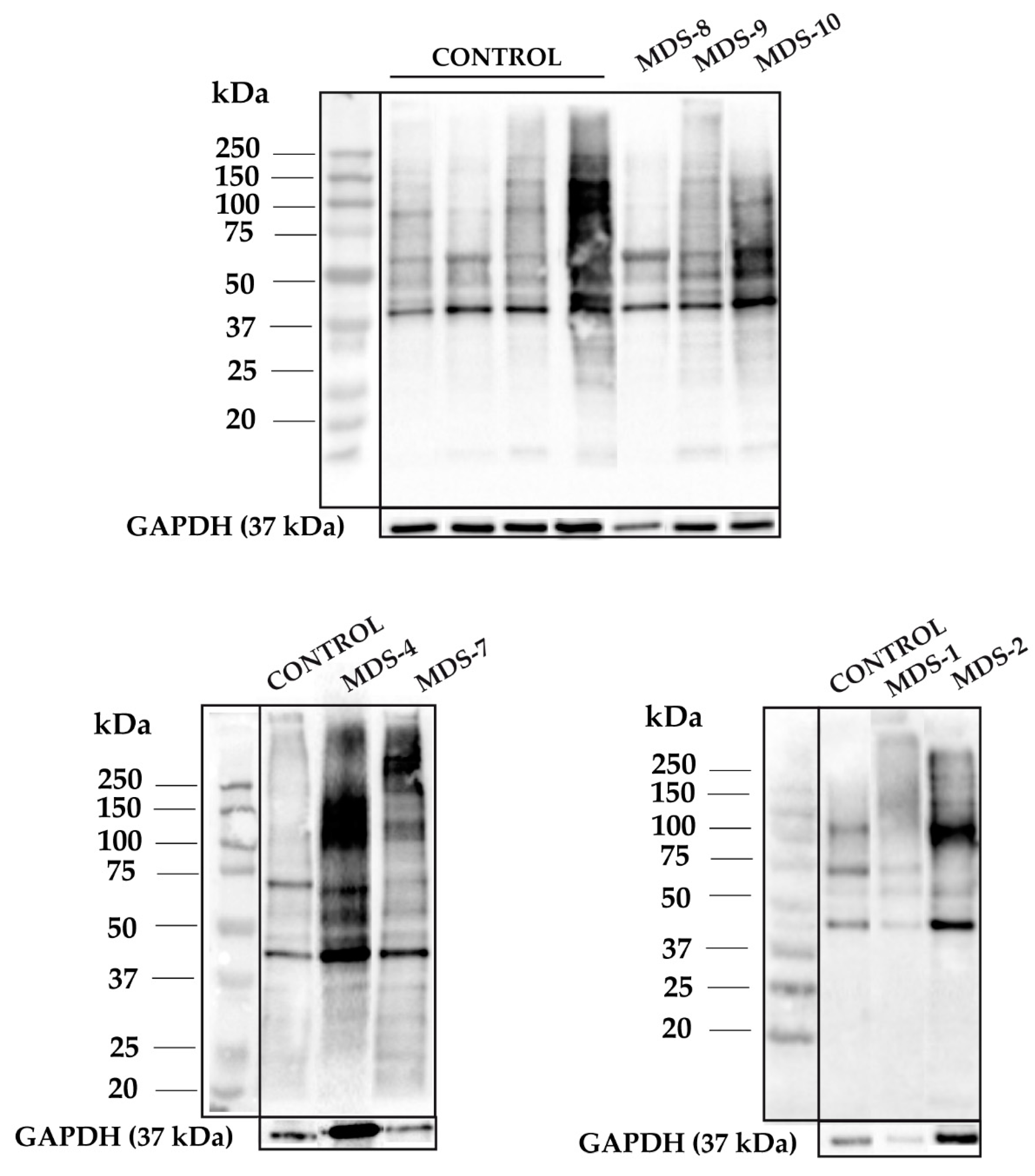
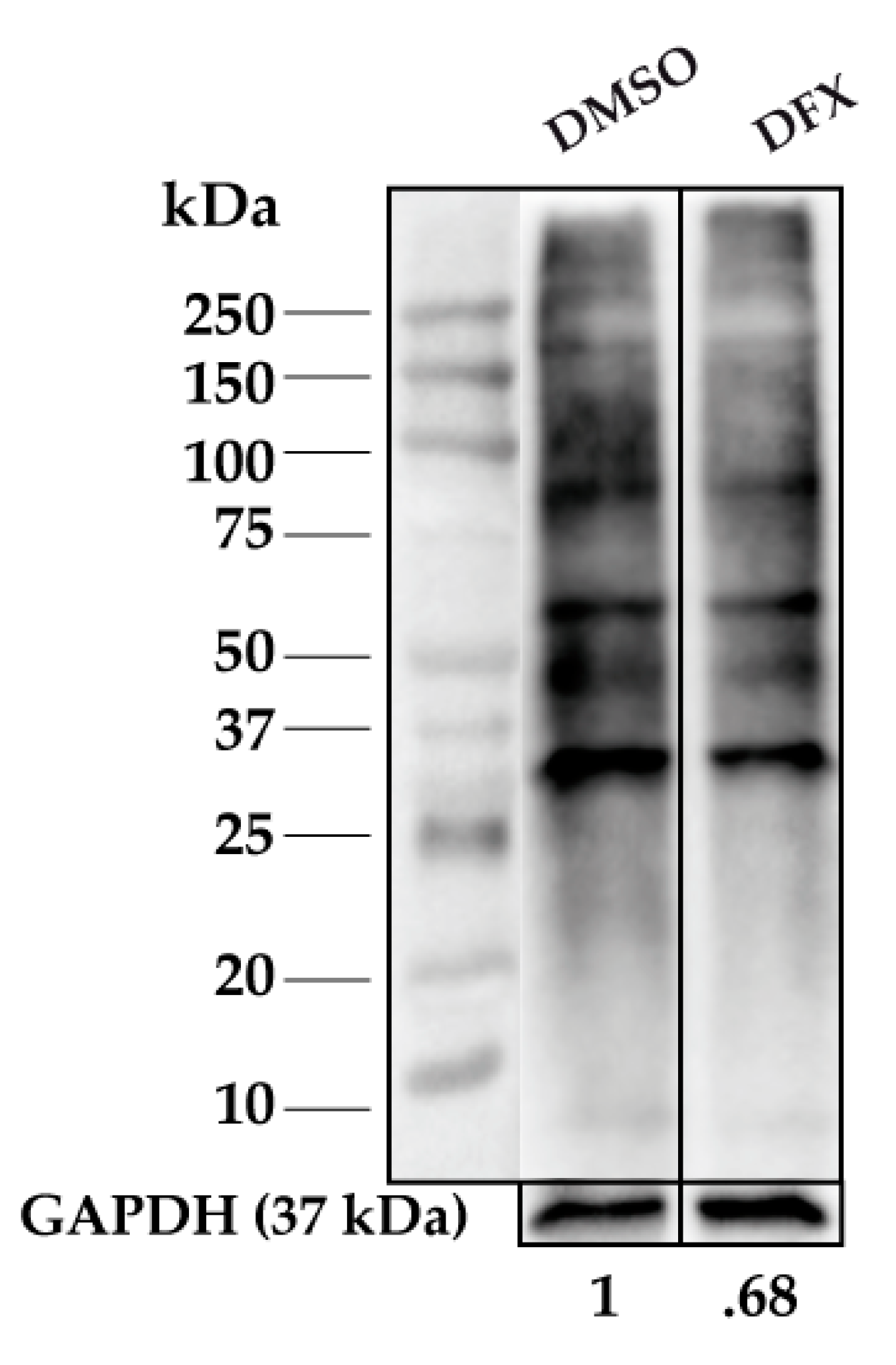
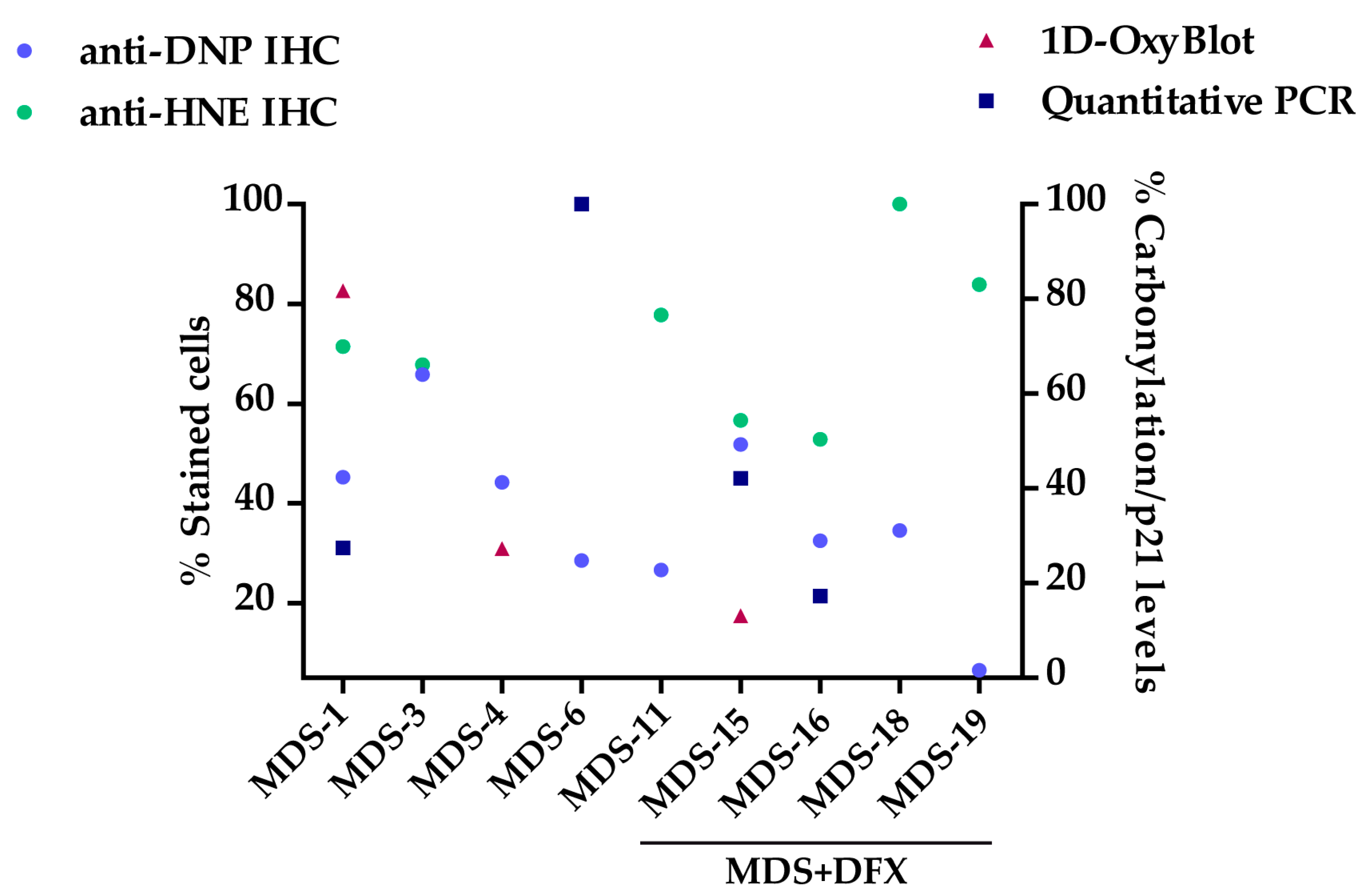
3.1. Protein Carbonylation is Increased in Myeloid Series from MDS Patients and is Decreased in Patients Treated with DFX
3.2. Protein Carbonylation is Increased in MDS Erythroid Precursors and is Decreased by DFX Treatment
3.3. Upregulation of p21 in MDS is Controlled by DFX
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A




References
- Richardson, C.; Yan, S.; Vestal, C. Oxidative Stress, Bone Marrow Failure, and Genome Instability in Hematopoietic Stem Cells. Int. J. Mol. Sci. 2015, 16, 2366–2385. [Google Scholar] [CrossRef] [PubMed]
- Gangat, N.; Patnaik, M.M.; Tefferi, A. Myelodysplastic syndromes: Contemporary review and how we treat: MDS treatment. Am. J. Hematol. 2016, 91, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.J.; Robert, C.; Gough, S.M.; Rassool, F.V.; Aplan, P.D. Oxidative stress leads to increased mutation frequency in a murine model of myelodysplastic syndrome. Leuk. Res. 2014, 38, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Peddie, C.M.; Wolf, C.R.; Mclellan, L.I.; Collins, A.R.; Bowen, D.T. Oxidative DNA damage in CD34+ myelodysplastic cells is associated with intracellular redox changes and elevated plasma tumour necrosis factor-α concentration. Br. J. Haematol. 1997, 99, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Ivars, D.; Orero, M.T.; Javier, K.; Díaz-Vico, L.; García-Giménez, J.L.; Mena, S.; Tormos, C.; Egea, M.; Pérez, P.L.; Arrizabalaga, B.; et al. Oxidative imbalance in low/intermediate-1-risk myelodysplastic syndrome patients: The influence of iron overload. Clin. Biochem. 2017, 50, 911–917. [Google Scholar] [CrossRef]
- Jiménez-Solas, T.; López-Cadenas, F.; Aires-Mejía, I.; Caballero-Berrocal, J.C.; Ortega, R.; Redondo, A.M.; Sánchez-Guijo, F.; Muntión, S.; García-Martín, L.; Albarrán, B.; et al. Deferasirox reduces oxidative DNA damage in bone marrow cells from myelodysplastic patients and improves their differentiation capacity. Br. J Haematol. 2019, 187, 93–104. [Google Scholar] [CrossRef]
- Dalle-Donne, I. Biomarkers of Oxidative Damage in Human Disease. Clinical Chemistry 2006, 52, 601–623. [Google Scholar] [CrossRef]
- Evans, J.L.; Maddux, B.A.; Goldfine, I.D. The Molecular Basis for Oxidative Stress-Induced Insulin Resistance. Antioxid. Redox Signal. 2005, 7, 1040–1052. [Google Scholar] [CrossRef]
- De Souza, G.F.; Ribeiro, H.L.; De Sousa, J.C.; Heredia, F.F.; De Freitas, R.M.; Martins, M.R.A.; Gonçalves, R.P.; Pinheiro, R.F.; Magalhaes, S.M.M. HFE gene mutation and oxidative damage biomarkers in patients with myelodysplastic syndromes and its relation to transfusional iron overload: An observational cross-sectional study. BMJ Open 2015, 5, e006048. [Google Scholar] [CrossRef]
- Gonçalves, A.C.; Cortesão, E.; Oliveiros, B.; Alves, V.; Espadana, A.I.; Rito, L.; Magalhães, E.; Lobão, M.J.; Pereira, A.; Nascimento Costa, J.M.; et al. Oxidative stress and mitochondrial dysfunction play a role in myelodysplastic syndrome development, diagnosis, and prognosis: A pilot study. Free Radic. Res. 2015, 49, 1081–1094. [Google Scholar] [CrossRef]
- Gonçalves, A.C.; Cortesão, E.; Oliveiros, B.; Alves, V.; Espadana, A.I.; Rito, L.; Magalhães, E.; Pereira, S.; Pereira, A.; Costa, J.M.N.; et al. Oxidative stress levels are correlated with P15 and P16 gene promoter methylation in myelodysplastic syndrome patients. Clin. Exp. Med. 2016, 16, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Ghoti, H.; Amer, J.; Winder, A.; Rachmilewitz, E.; Fibach, E. Oxidative stress in red blood cells, platelets and polymorphonuclear leukocytes from patients with myelodysplastic syndrome. Eur. J. Haematol. 2007, 79, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Cassarino, D.S.; Fall, C.P.; Swerdlow, R.H.; Smith, T.S.; Halvorsen, E.M.; Miller, S.W.; Parks, J.P.; Parker, W.D.; Bennett, J.P. Elevated reactive oxygen species and antioxidant enzyme activities in animal and cellular models of Parkinson’s disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1997, 1362, 77–86. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.-J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; DesMarais, T.L.; Tong, Z.; Yao, Y.; Costa, M. Oxidative stress alters global histone modification and DNA methylation. Free Radic. Biol. Med. 2015, 82, 22–28. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders — A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Afiuni-Zadeh, S.; Rogers, J.C.; Snovida, S.I.; Bomgarden, R.D.; Griffin, T.J. AminoxyTMT: A novel multi-functional reagent for characterization of protein carbonylation. BioTechniques 2016, 60, 186–196. [Google Scholar] [CrossRef]
- Linares, M.; Marín-García, P.; Méndez, D.; Puyet, A.; Diez, A.; Bautista, J.M. Proteomic Approaches to Identifying Carbonylated Proteins in Brain Tissue. J. Proteome Res. 2011, 10, 1719–1727. [Google Scholar] [CrossRef]
- Frank, J.; Pompella, A.; Biesalski, H.K. Immunohistochemical Detection of Protein Oxidation. In Oxidants and Antioxidants. Methods in Molecular BiologyTM.; Armstrong, D., Ed.; Humana Press: Totowa, NJ, USA, 2002; Volume 196, pp. 35–40. ISBN 978-0-89603-851-6. [Google Scholar]
- Barrera, G.; Pizzimenti, S.; Daga, M.; Dianzani, C.; Arcaro, A.; Cetrangolo, G.P.; Giordano, G.; Cucci, M.A.; Graf, M.; Gentile, F. Lipid Peroxidation-Derived Aldehydes, 4-Hydroxynonenal and Malondialdehyde in Aging-Related Disorders. Antioxidants 2018, 7, 102. [Google Scholar] [CrossRef]
- Schneider, R.K.; Schenone, M.; Ferreira, M.V.; Kramann, R.; Joyce, C.E.; Hartigan, C.; Beier, F.; Brümmendorf, T.H.; Germing, U.; Platzbecker, U.; et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat. Med. 2016, 22, 288–297. [Google Scholar] [CrossRef]
- Liggins, J.; Furth, A.J. Role of protein-bound carbonyl groups in the formation of advanced glycation endproducts. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1997, 1361, 8. [Google Scholar] [CrossRef]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do reactive oxygen species play a role in myeloid leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef] [PubMed]
- Fei, C.; Zhao, Y.; Guo, J.; Gu, S.; Li, X.; Chang, C. Senescence of bone marrow mesenchymal stromal cells is accompanied by activation of p53/p21 pathway in myelodysplastic syndromes. Eur. J. Haematol. 2014, 93, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Deville, L.; Hillion, J.; Ségal-Bendirdjian, E. Telomerase regulation in hematological cancers: A matter of stemness? Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2009, 1792, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Pilo, F.; Angelucci, E. A storm in the niche: Iron, oxidative stress and haemopoiesis. Blood Rev. 2018, 32, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Guo, J.; Zhang, X.; Zhang, Z.; Gu, S.; Fei, C.; Li, X.; Chang, C. Downregulation of p21 in Myelodysplastic Syndrome Is Associated With p73 Promoter Hypermethylation and Indicates Poor Prognosis. Am. J. Clin. Pathol. 2013, 140, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Li, D.; Cao, X.; Zhang, Y.; Mu, J.; Lu, W.; Xiao, X.; Li, C.; Meng, J.; Chen, J.; et al. ROS-mediated iron overload injures the hematopoiesis of bone marrow by damaging hematopoietic stem/progenitor cells in mice. Sci. Rep. 2015, 5, 10181. [Google Scholar] [CrossRef]
- Silva, B.; Faustino, P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 1347–1359. [Google Scholar] [CrossRef]
- Leitch, H.A. Improving clinical outcome in patients with myelodysplastic syndrome and iron overload using iron chelation therapy. Leuk. Res. 2007, 31, S7–S9. [Google Scholar] [CrossRef]
- Rose, C.; Brechignac, S.; Vassilief, D.; Pascal, L.; Stamatoullas, A.; Guerci, A.; Larbaa, D.; Dreyfus, F.; Beyne-Rauzy, O.; Chaury, M.P.; et al. Does iron chelation therapy improve survival in regularly transfused lower risk MDS patients? A multicenter study by the GFM. Leuk. Res. 2010, 34, 864–870. [Google Scholar] [CrossRef]
- Neukirchen, J.; Fox, F.; Kündgen, A.; Nachtkamp, K.; Strupp, C.; Haas, R.; Germing, U.; Gattermann, N. Improved survival in MDS patients receiving iron chelation therapy—A matched pair analysis of 188 patients from the Düsseldorf MDS registry. Leuk. Res. 2012, 36, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Pullarkat, V.; Sehgal, A.; Li, L.; Meng, Z.; Lin, A.; Forman, S.; Bhatia, R. Deferasirox exposure induces reactive oxygen species and reduces growth and viability of myelodysplastic hematopoietic progenitors. Leuk. Res. 2012, 36, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Tataranni, T.; Agriesti, F.; Mazzoccoli, C.; Ruggieri, V.; Scrima, R.; Laurenzana, I.; D’Auria, F.; Falzetti, F.; Di Ianni, M.; Musto, P.; et al. The iron chelator deferasirox affects redox signalling in haematopoietic stem/progenitor cells. Br. J. Haematol. 2015, 170, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Gattermann, N.; Finelli, C.; Porta, M.D.; Fenaux, P.; Ganser, A.; Guerci-Bresler, A.; Schmid, M.; Taylor, K.; Vassilieff, D.; Habr, D.; et al. Deferasirox in iron-overloaded patients with transfusion-dependent myelodysplastic syndromes: Results from the large 1-year EPIC study. Leuk. Res. 2010, 34, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, H.C.; Singh, R.N.; Torti, F.M.; Torti, S.V. Synthetic and natural iron chelators: Therapeutic potential and clinical use. Futur. Med. Chem. 2009, 1, 1643–1670. [Google Scholar] [CrossRef] [PubMed]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellstrom-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef]
- Caceres, G.; McGraw, K.; Yip, B.H.; Pellagatti, A.; Johnson, J.; Zhang, L.; Liu, K.; Zhang, L.M.; Fulp, W.J.; Lee, J.-H.; et al. TP53 suppression promotes erythropoiesis in del(5q) MDS, suggesting a targeted therapeutic strategy in lenalidomide-resistant patients. Proc. Natl. Acad. Sci. USA 2013, 110, 16127–16132. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucl. Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Hlaváčková, A.; Štikarová, J.; Pimková, K.; Chrastinová, L.; Májek, P.; Kotlín, R.; Čermák, J.; Suttnar, J.; Dyr, J.E. Enhanced plasma protein carbonylation in patients with myelodysplastic syndromes. Free Radic. Biol. Med. 2017, 108, 1–7. [Google Scholar] [CrossRef]
- Meunier, M.; Ancelet, S.; Lefebvre, C.; Arnaud, J.; Garrel, C.; Pezet, M.; Wang, Y.; Faure, P.; Szymanski, G.; Duployez, N.; et al. Reactive oxygen species levels control NF-κB activation by low dose deferasirox in erythroid progenitors of low risk myelodysplastic syndromes. Oncotarget 2017, 8, 105510–105524. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.P.; Jung, T.; Grune, T.; Almeida, H. Actin carbonylation: From cell dysfunction to organism disorder. J. Proteom. 2013, 92, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Gagliano, N.; Lusini, L.; Milzani, A.; Di Simplicio, P.; Colombo, R. Actin carbonylation: From a simple marker of protein oxidation to relevant signs of severe functional impairment. Free Radic. Biol. Med. 2001, 31, 1075–1083. [Google Scholar] [CrossRef]
- Shonnard, G.C.; Hud, N.V.; Mohrenweiser, H.W. Arginine to tryptophan substitution in the active site of a human lactate dehydrogenase variant-LDHB GUA1: Postulated effects on subunit structure and catalysis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1996, 1315, 6. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Boike, L. An Analysis of Oxidative Damage to Lactate Dehydrogenase in Context of Neurodegeneration and Catechol-Based Phenolic Antioxidant Chemistry. Bachelor’s Thesis, College of William & Mary, Williamsburg, VA, USA, December 2017. [Google Scholar]
- Di Stefano, G.; Manerba, M.; Di Ianni, L.; Fiume, L. Lactate dehydrogenase inhibition: Exploring possible applications beyond cancer treatment. Futur. Med. Chem. 2016, 8, 713–725. [Google Scholar] [CrossRef]
- Fiume, L.; Manerba, M.; Vettraino, M.; Di Stefano, G. Inhibition of lactate dehydrogenase activity as an approach to cancer therapy. Futur. Med. Chem. 2014, 6, 429–445. [Google Scholar] [CrossRef]
- Ros, J. Protein carbonylation: Principles, analysis, and biological implications. In Wiley Series on Mass Spectrometry; Wiley: Hoboken, NJ, USA, 2017; pp. 110–123. ISBN 978-1-119-07491-5. [Google Scholar]
- Gardino, A.K.; Yaffe, M.B. 14-3-3 proteins as signaling integration points for cell cycle control and apoptosis. Semin. Cell Dev. Boil. 2011, 22, 688–695. [Google Scholar] [CrossRef]
- Yang, X.; Lee, W.H.; Sobott, F.; Papagrigoriou, E.; Robinson, C.V.; Grossmann, J.G.; Sundstrom, M.; Doyle, D.A.; Elkins, J.M. Structural basis for protein-protein interactions in the 14-3-3 protein family. Proc. Natl. Acad. Sci. USA 2006, 103, 17237–17242. [Google Scholar] [CrossRef]
- Jayaraman, T.; Tejero, J.; Chen, B.B.; Blood, A.B.; Frizzell, S.; Shapiro, C.; Tiso, M.; Hood, B.L.; Wang, X.; Zhao, X.; et al. 14-3-3 Binding and Phosphorylation of Neuroglobin during Hypoxia Modulate Six-to-Five Heme Pocket Coordination and Rate of Nitrite Reduction to Nitric Oxide. J. Biol. Chem. 2011, 286, 42679–42689. [Google Scholar] [CrossRef]
- Watanabe, K.; Thandavarayan, R.; Gurusamy, N.; Zhang, S.; Muslin, A.; Suzuki, K.; Tachikawa, H.; Kodama, M.; Aizawa, Y. Role of 14-3-3 protein and oxidative stress in diabetic cardiomyopathy. Acta Physiol. Hung. 2009, 96, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Zoila, I. The Role of Nox1 and 14-3-3 in the Regulation of Slingshot Phosphatase in Vascular Smooth Muscle Cells. Bachelor’s Thesis, Faculty of Emory Collegue of Arts and Sciences, Atlanta, GA, USA, April 2012. [Google Scholar]
- Kim, H.S.; Ullevig, S.L.; Nguyen, H.N.; Vanegas, D.; Asmis, R. Redox Regulation of 14-3-3 Controls Monocyte Migration. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Ravasi, T. Systematic Characterization of the Zinc-Finger-Containing Proteins in the Mouse Transcriptome. Genome Res. 2003, 13, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Kröncke, K.-D.; Klotz, L.-O. Zinc Fingers as Biologic Redox Switches? Antioxid. Redox Signal. 2009, 11, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Oteiza, P.I. Zinc and the modulation of redox homeostasis. Free Radic. Biol. Med. 2012, 53, 1748–1759. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographic Data | Clinical Features | Performed Analysis | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient (No) | Sex (M/F) | Age (years) | MDS Subtypes(WHO) | Hb (g/dL) | ANC (109 /L) | PTLs (109 /L) | Blasts (%) | Cytogenetics (FISH) | Risk Groups (IPSS) | Iron Chelation Therapy | Ferritin (ng/m L) | A | B | C | D |
| 1 | M | 83 | RCMD-RS | 10.5 | 3 | 250 | 1 | N | Low | No | 779 | ✔ | ✔ | ✔ | |
| 2 | F | 75 | CMML | 12.3 | 4.6 | 163 | 2 | N | Low | No | 45 | ✔ | |||
| 3 | M | 84 | RCMD | 12.8 | 1.1 | 99 | 2 | N | Low | No | 88 | ✔ | |||
| 4 | M | 59 | RCMD-RS | 9.4 | 1.5 | 177 | 1 | N | Low | No | 582 | ✔ | ✔ | ✔ | |
| 5 | M | 75 | RAEB-1 | 14 | 1.7 | 164 | 6 | N | Int-1 | No | 168 | ✔ | |||
| 6 | M | 83 | RAEB-1 | 11.4 | 1.2 | 80 | 9 | Mon (7, 20). Del (5q, 12p. 17q) | Low | No | 180 | ✔ | ✔ | ||
| 7 | F | 84 | RCMD | 11.4 | 2.9 | 144 | 4 | N | Low | No | 136 | ✔ | ✔ | ||
| 8 | M | 81 | RCMD-RS | 9.9 | 3.2 | 363 | 1 | N | Low | No | 437 | ✔ | |||
| 9 | F | 73 | RAEB-2 | 11.5 | 3.4 | 440 | 15 | N | Int-1 | No | 1123 | ✔ | |||
| 10 | F | 90 | RCMD | 9.1 | 4.4 | 166 | 4 | N | Int-2 | No | ✔ | ||||
| 11 | F | 87 | RARS | 8.8 | 4.2 | 306 | 1 | N | Low | No | 1547 | ✔ | |||
| 12 | F | 70 | RAEB-1 | 10.6 | 1 | 133 | 5 | N | Low | No | 24 | ✔ | |||
| 13 | M | 82 | RCMD-RS | 9.9 | 3.2 | 363 | 1 | N | Low | No | 319 | ✔ | |||
| 14 | F | 81 | RARS | 10.9 | 4.3 | 165 | 2 | N | Low | No | 157 | ✔ | |||
| 15 | M | 66 | RARS | 10.3 | 4.4 | 414 | 2 | N | Low | Yes | 1137.6 | ✔ | ✔ | ✔ | |
| 16 | M | 70 | RARS | 6.7 | 6 | 313 | 2 | Del (20q) | Low | Yes | 884 | ✔ | ✔ | ||
| 17 | F | 66 | RAEB-2 | 10.2 | 9 | 161 | 16 | Del (5q) | Int-1 | Yes | 163 | ✔ | |||
| 18 | M | 71 | RCMD-RS | 7.8 | 7.8 | 185 | 4 | N | Int-1 | Yes | 643 | ✔ | |||
| 19 | M | 71 | RCMD | 8.2 | 1.8 | 31 | 1 | N | Low | Yes | 206.2 | ✔ | |||
| 20 | M | 57 | RCMD | 7.5 | 8 | 22 | <1 | N | Int-1 | Yes | 767 | ✔ | |||
| 21 | F | 82 | RAEB-1 | 11.3 | 8.6 | 26 | 7 | Del (5q) | Int-1 | Yes | 1179 | ✔ | |||
| Protein ID | Accesion No. | No. of Peptides Matched/Searched | %Coverage | %Score | Nominal Mass (Mr)/Pi |
|---|---|---|---|---|---|
| Actin, cytoplasmic 1 | sp|P60709|ACTB_HUMAN | 21/65 | 49 | 147 | 42,052/5.29 |
| Zinc finger protein 846 | sp|Q147U1|ZN846_HUMAN | 12/65 | 26 | 68 | 62,109/9.21 |
| 14-3-3 protein zeta/delta | sp|P63104|1433Z_HUMAN | 12/65 | 47 | 90 | 27,899/4.73 |
| L-lactate dehydrogenase A chain | sp|P00338|LDHA_HUMAN | 14/65 | 45 | 104 | 36,950/8.44 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-García, A.; Morales, M.L.; Garrido-García, V.; García-Baquero, I.; Leivas, A.; Carreño-Tarragona, G.; Sánchez, R.; Arenas, A.; Cedena, T.; Ayala, R.M.; et al. Protein Carbonylation in Patients with Myelodysplastic Syndrome: An Opportunity for Deferasirox Therapy. Antioxidants 2019, 8, 508. https://doi.org/10.3390/antiox8110508
Rodríguez-García A, Morales ML, Garrido-García V, García-Baquero I, Leivas A, Carreño-Tarragona G, Sánchez R, Arenas A, Cedena T, Ayala RM, et al. Protein Carbonylation in Patients with Myelodysplastic Syndrome: An Opportunity for Deferasirox Therapy. Antioxidants. 2019; 8(11):508. https://doi.org/10.3390/antiox8110508
Chicago/Turabian StyleRodríguez-García, Alba, María Luz Morales, Vanesa Garrido-García, Irene García-Baquero, Alejandra Leivas, Gonzalo Carreño-Tarragona, Ricardo Sánchez, Alicia Arenas, Teresa Cedena, Rosa María Ayala, and et al. 2019. "Protein Carbonylation in Patients with Myelodysplastic Syndrome: An Opportunity for Deferasirox Therapy" Antioxidants 8, no. 11: 508. https://doi.org/10.3390/antiox8110508
APA StyleRodríguez-García, A., Morales, M. L., Garrido-García, V., García-Baquero, I., Leivas, A., Carreño-Tarragona, G., Sánchez, R., Arenas, A., Cedena, T., Ayala, R. M., Bautista, J. M., Martínez-López, J., & Linares, M. (2019). Protein Carbonylation in Patients with Myelodysplastic Syndrome: An Opportunity for Deferasirox Therapy. Antioxidants, 8(11), 508. https://doi.org/10.3390/antiox8110508