N-Acetylcysteine Attenuates the Increasing Severity of Distant Organ Liver Dysfunction after Acute Kidney Injury in Rats Exposed to Bisphenol A


Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Preparation
2.2. Induction of Acute Kidney Injury
2.3. Experimental Designs
2.4. Assessments of Renal and Liver Functions
2.5. Assessments of Systemic as well as Liver Oxidative Stress and Inflammation
2.6. Light Microscopic Studies
2.7. Electron Microscopic Studies
2.8. Preparations of Liver Mitochondrial Fractions and Proteins
2.9. Determination of Liver Mitochondrial Reactive Oxygen Species (ROS)
2.10. Determination of Liver Mitochondrial Membrane Potential Change (ΔΨm)
2.11. Determination of Liver Mitochondrial Swelling
2.12. Western Blot Analysis
2.13. Statistical Analysis
3. Results
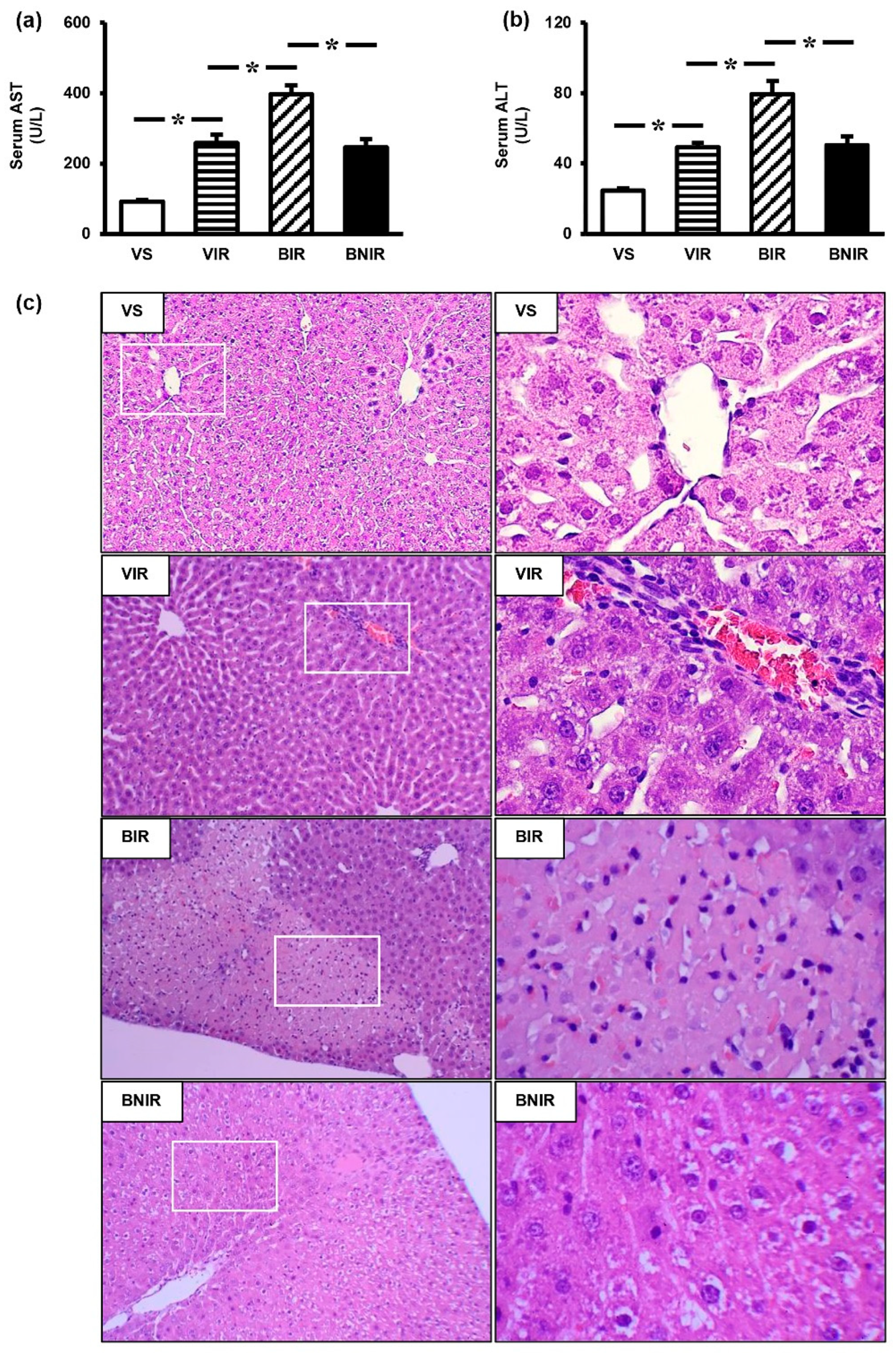
3.1. RIR Induces Remote Organ Liver Injury and the Severity is Increasing upon BPA Exposure
3.2. NAC Therapy Attenuates the Increased Severity of Remote Liver Injury after RIR under BPA-Exposed Condition
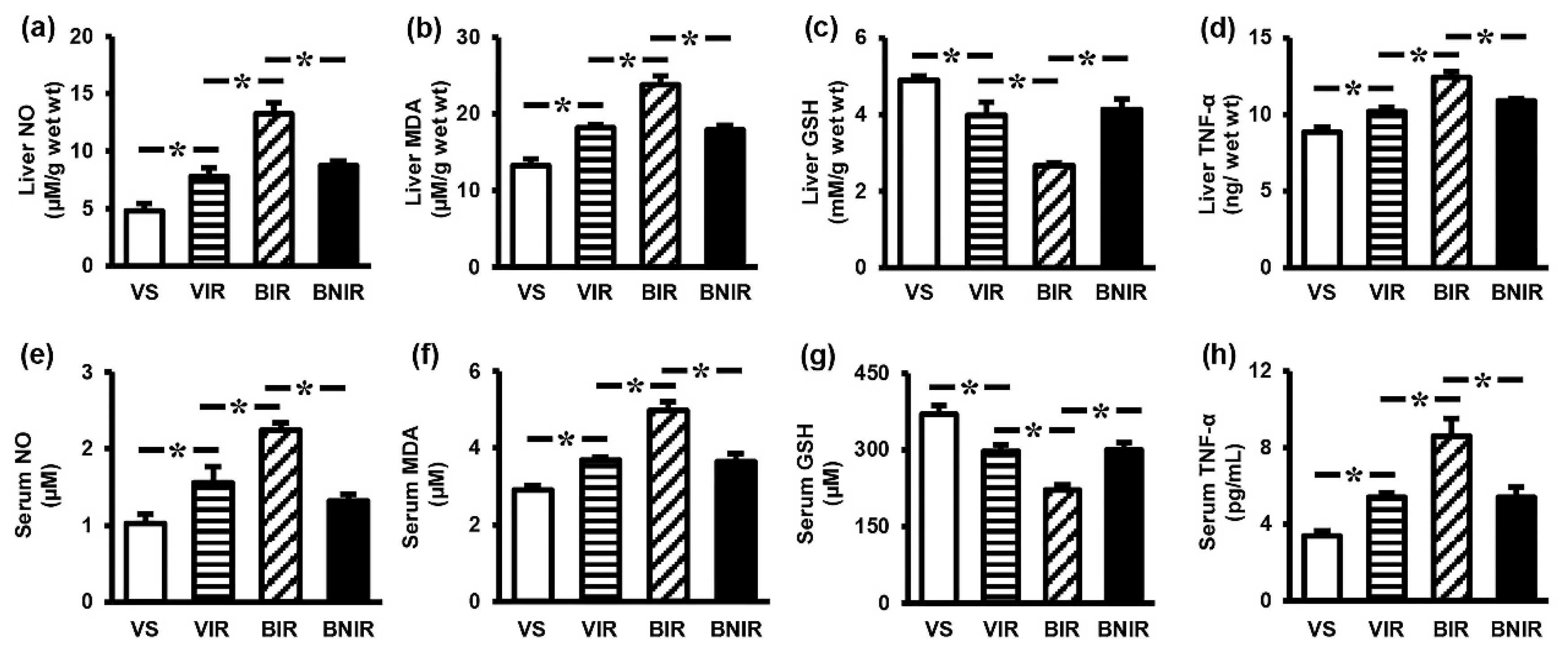
3.3. NAC Reduces Oxidative Stress and Inflammation in the Systemic Circulation as well as Remote Liver Organ after RIR under BPA-Exposed Condition
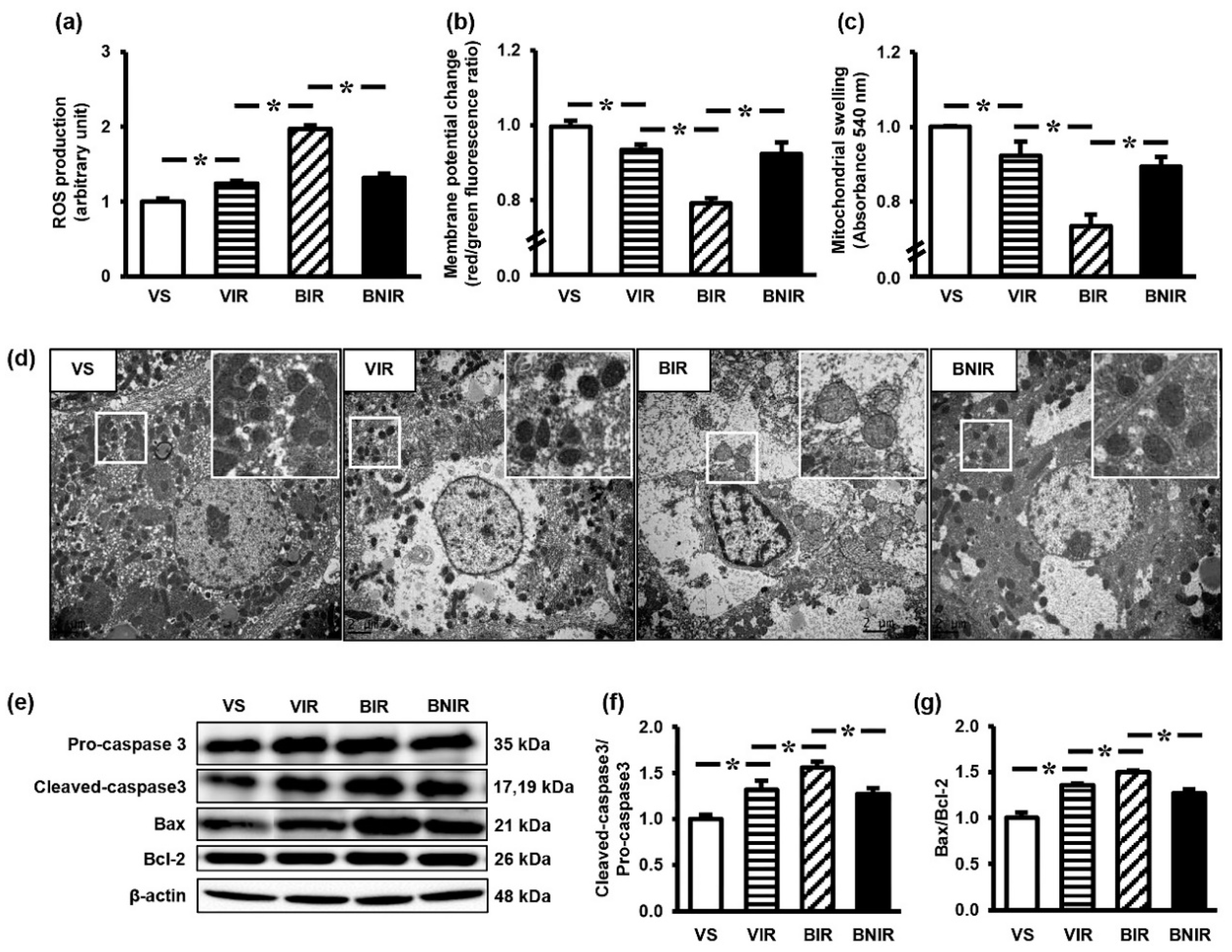
3.4. Maintenance of Remote Liver Mitochondrial Function Contributes to the Protection by NAC after RIR under BPA-Exposed Condition
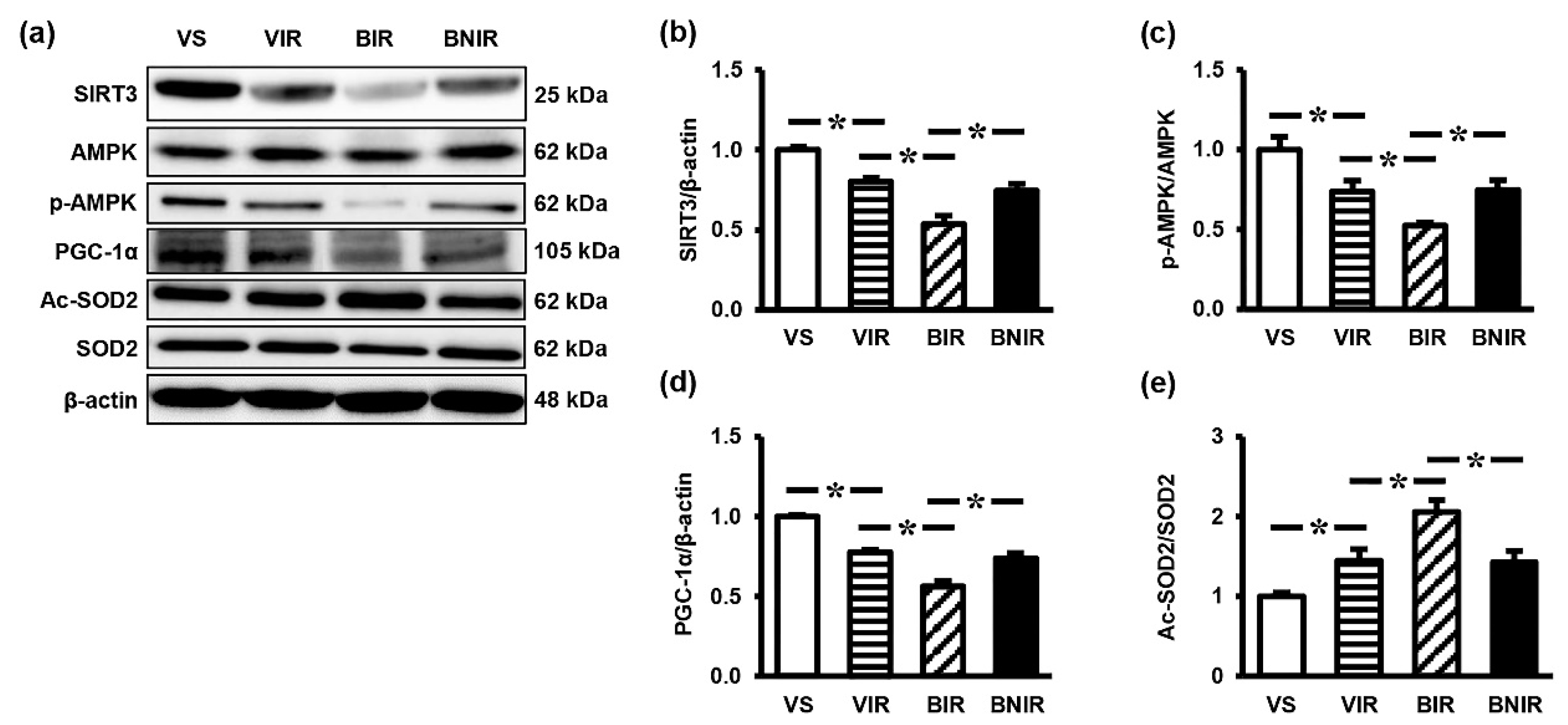
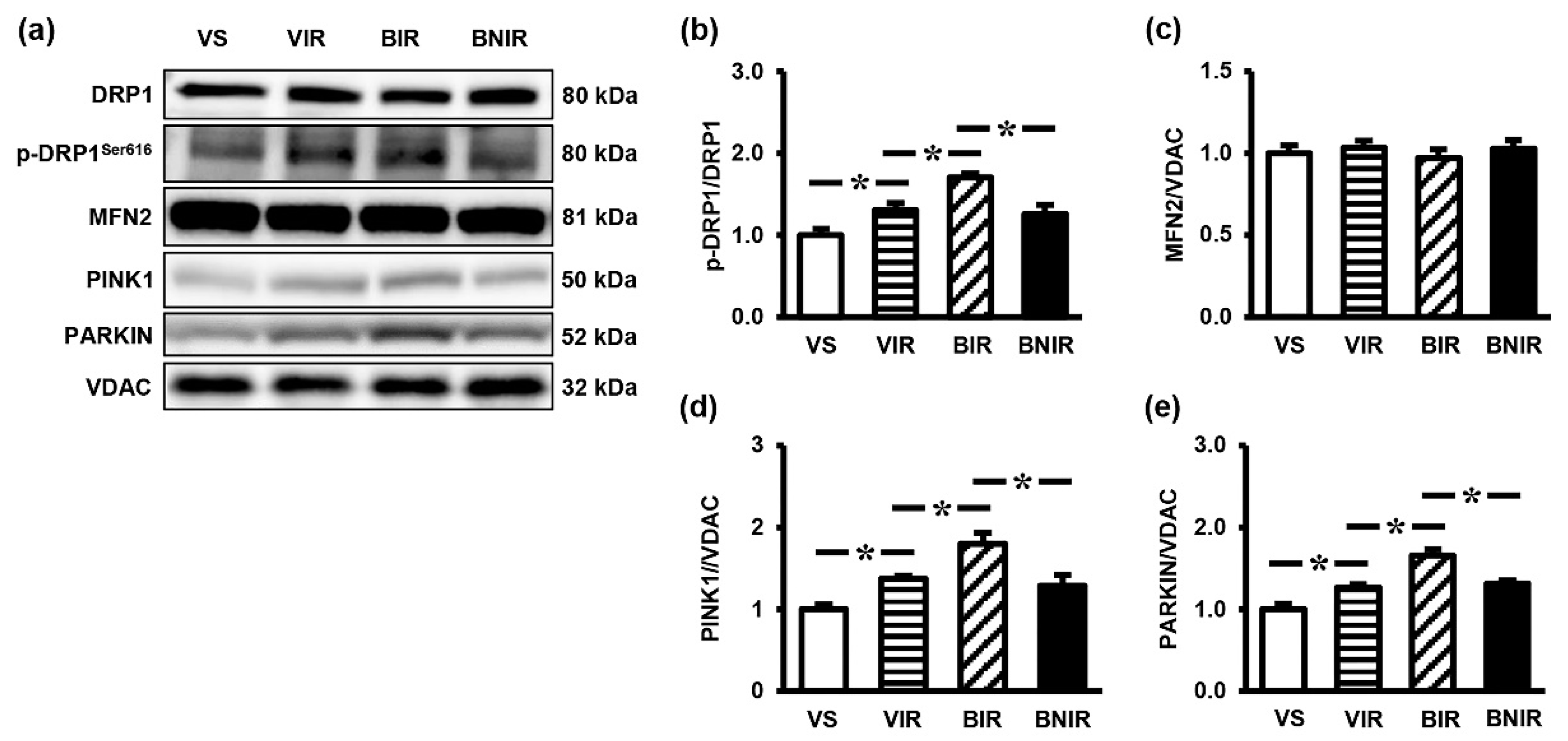
3.5. Signaling Transmission Through AMPK-PGC-1α-SIRT3 is Involved in the Remote Liver Mitochondrial Protection by NAC after RIR under BPA-Exposed Condition
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Doi, K.; Rabb, H. Impact of acute kidney injury on distant organ function: Recent findings and potential therapeutic targets. Kidney Int. 2016, 89, 555–564. [Google Scholar] [CrossRef]
- Sumida, M.; Doi, K.; Ogasawara, E.; Yamashita, T.; Hamasaki, Y.; Kariya, T.; Takimoto, E.; Yahagi, N.; Nangaku, M.; Noiri, E. Regulation of Mitochondrial Dynamics by Dynamin-Related Protein-1 in Acute Cardiorenal Syndrome. J. Am. Soc. Nephrol. 2015, 26, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Gardner, D.S.; De Brot, S.; Dunford, L.J.; Grau-Roma, L.; Welham, S.J.; Fallman, R.; O’Sullivan, S.E.; Oh, W.; Devonald, M.A. Remote effects of acute kidney injury in a porcine model. Am. J. Physiol. Renal Physiol. 2016, 310, F259–F271. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Siow, Y.L.; Isaak, C.K. Downregulation of Glutathione Biosynthesis Contributes to Oxidative Stress and Liver Dysfunction in Acute Kidney Injury. Oxidative Med. Cell. Longev. 2016, 2016, 9707292. [Google Scholar] [CrossRef] [PubMed]
- Lane, K.; Dixon, J.J.; MacPhee, I.A.; Philips, B.J. Renohepatic crosstalk: Does acute kidney injury cause liver dysfunction? Nephrol. Dial. Transplant. 2013, 28, 1634–1647. [Google Scholar] [CrossRef]
- Jalal, N.; Surendranath, A.R.; Pathak, J.L.; Yu, S.; Chung, C.Y. Bisphenol A (BPA) the mighty and the mutagenic. Toxicol. Rep. 2018, 5, 76–84. [Google Scholar] [CrossRef]
- Warner, G.R.; Flaws, J.A. Bisphenol A and Phthalates: How Environmental Chemicals Are Reshaping Toxicology. Toxicol. Sci. 2018, 166, 246–249. [Google Scholar] [CrossRef]
- Trasande, L.; Attina, T.M.; Trachtman, H. Bisphenol A exposure is associated with low-grade urinary albumin excretion in children of the United States. Kidney Int. 2013, 83, 741–748. [Google Scholar] [CrossRef]
- Hines, C.J.; Jackson, M.V.; Deddens, J.A.; Clark, J.C.; Ye, X.; Christianson, A.L.; Meadows, J.W.; Calafat, A.M. Urinary Bisphenol A (BPA) Concentrations among Workers in Industries that Manufacture and Use BPA in the USA. Ann. Work Expo. Health 2017, 61, 164–182. [Google Scholar] [CrossRef]
- Peerapanyasut, W.; Kobroob, A.; Palee, S.; Chattipakorn, N.; Wongmekiat, O. Activation of Sirtuin 3 and Maintenance of Mitochondrial Integrity by N-Acetylcysteine Protects Against Bisphenol A-Induced Kidney and Liver Toxicity in Rats. Int. J. Mol. Sci. 2019, 20, 267. [Google Scholar] [CrossRef]
- Cusumano, G.; Romagnoli, J.; Liuzzo, G.; Ciavarella, L.P.; Severino, A.; Copponi, G.; Manchi, M.; Giubilato, S.; Zannoni, G.F.; Stigliano, E.; et al. N-Acetylcysteine and High-Dose Atorvastatin Reduce Oxidative Stress in an Ischemia-Reperfusion Model in the Rat Kidney. Transplant. Proc. 2015, 47, 2757–2762. [Google Scholar] [CrossRef] [PubMed]
- Sen, H.; Deniz, S.; Yedekci, A.E.; Inangil, G.; Muftuoglu, T.; Haholu, A.; Ozkan, S. Effects of dexpanthenol and N-acetylcysteine pretreatment in rats before renal ischemia/reperfusion injury. Ren. Fail. 2014, 36, 1570–1574. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Trejo, O.E.; Reyes-Fermin, L.M.; Briones-Herrera, A.; Tapia, E.; Leon-Contreras, J.C.; Hernandez-Pando, R.; Sanchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective effects of N-acetyl-cysteine in mitochondria bioenergetics, oxidative stress, dynamics and S-glutathionylation alterations in acute kidney damage induced by folic acid. Free Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Tsuji, T.; Yasuda, H.; Sun, Y.; Fujigaki, Y.; Hishida, A. The molecular mechanisms of the attenuation of cisplatin-induced acute renal failure by N-acetylcysteine in rats. Nephrol. Dial. Transplant. 2008, 23, 2198–2205. [Google Scholar] [CrossRef] [PubMed]
- Wongjaikam, S.; Kumfu, S.; Khamseekaew, J.; Sripetchwandee, J.; Srichairatanakool, S.; Fucharoen, S.; Chattipakorn, S.C.; Chattipakorn, N. Combined Iron Chelator and Antioxidant Exerted Greater Efficacy on Cardioprotection Than Monotherapy in Iron-Overloaded Rats. PLoS ONE 2016, 11, e0159414. [Google Scholar] [CrossRef] [PubMed]
- Kobroob, A.; Peerapanyasut, W.; Chattipakorn, N.; Wongmekiat, O. Damaging Effects of Bisphenol A on the Kidney and the Protection by Melatonin: Emerging Evidences from In Vivo and In Vitro Studies. Oxidative Med. Cell. Longev. 2018, 2018, 3082438. [Google Scholar] [CrossRef] [PubMed]
- Peerapanyasut, W.; Thamprasert, K.; Wongmekiat, O. Ubiquinol supplementation protects against renal ischemia and reperfusion injury in rats. Free Radic. Res. 2014, 48, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Tyl, R.W.; Myers, C.B.; Marr, M.C.; Thomas, B.F.; Keimowitz, A.R.; Brine, D.R.; Veselica, M.M.; Fail, P.A.; Chang, T.Y.; Seely, J.C.; et al. Three-generation reproductive toxicity study of dietary bisphenol A in CD Sprague-Dawley rats. Toxicol. Sci. 2002, 68, 121–146. [Google Scholar] [CrossRef]
- Khodayar, M.J.; Kalantari, H.; Mahdavinia, M.; Khorsandi, L.; Alboghobeish, S.; Samimi, A.; Alizadeh, S.; Zeidooni, L. Protective effect of naringin against BPA-induced cardiotoxicity through prevention of oxidative stress in male Wistar rats. Drug Chem. Toxicol. 2018, 1–11. [Google Scholar] [CrossRef]
- Othman, A.I.; Edrees, G.M.; El-Missiry, M.A.; Ali, D.A.; Aboel-Nour, M.; Dabdoub, B.R. Melatonin controlled apoptosis and protected the testes and sperm quality against bisphenol A-induced oxidative toxicity. Toxicol. Ind. Health 2016, 32, 1537–1549. [Google Scholar] [CrossRef]
- Jain, S.; Kumar, C.H.; Suranagi, U.D.; Mediratta, P.K. Protective effect of N-acetylcysteine on bisphenol A-induced cognitive dysfunction and oxidative stress in rats. Food Chem. Toxicol. 2011, 49, 1404–1409. [Google Scholar] [CrossRef] [PubMed]
- Taslipinar, M.Y.; Aydin, I.; Kaldirim, U.; Aydin, F.N.; Agilli, M.; Eyi, Y.E.; Tuncer, S.K.; Altayli, E.; Ucar, F.; Macit, E.; et al. Hyperbaric oxygen treatment and N-acetylcysteine ameliorate acetaminophen-induced liver injury in a rat model. Hum. Exp. Toxicol. 2013, 32, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, I.; Parvez, S.; Winkler-Stuck, K.; Seitz, G.; Trieu, I.; Wallesch, C.W.; Schonfeld, P.; Siemen, D. Patch clamp reveals powerful blockade of the mitochondrial permeability transition pore by the D2-receptor agonist pramipexole. FASEB J. 2006, 20, 556–558. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.E. The regulation of catalysis in ATP synthase. Curr. Opin. Struct. Biol. 1994, 4, 912–918. [Google Scholar] [CrossRef]
- Kobroob, A.; Chattipakorn, N.; Wongmekiat, O. Caffeic acid phenethyl ester ameliorates cadmium-induced kidney mitochondrial injury. Chem. Biol. Interact. 2012, 200, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Beavis, A.D.; Brannan, R.D.; Garlid, K.D. Swelling and contraction of the mitochondrial matrix. I. A structural interpretation of the relationship between light scattering and matrix volume. J. Biol. Chem. 1985, 260, 13424–13433. [Google Scholar]
- Farag, M.M.; Khalifa, A.A.; Elhadidy, W.F.; Rashad, R.M. Hepatorenal protection in renal ischemia/reperfusion by celecoxib and pentoxifylline. J. Surg. Res. 2016, 204, 183–191. [Google Scholar] [CrossRef]
- Golab, F.; Kadkhodaee, M.; Zahmatkesh, M.; Hedayati, M.; Arab, H.; Schuster, R.; Zahedi, K.; Lentsch, A.B.; Soleimani, M. Ischemic and non-ischemic acute kidney injury cause hepatic damage. Kidney Int. 2009, 75, 783–792. [Google Scholar] [CrossRef]
- Sharma, M.; Kaur, T.; Singla, S.K. Protective effects of N-acetylcysteine against hyperoxaluria induced mitochondrial dysfunction in male wistar rats. Mol. Cell. Biochem. 2015, 405, 105–114. [Google Scholar] [CrossRef]
- Wright, D.J.; Renoir, T.; Smith, Z.M.; Frazier, A.E.; Francis, P.S.; Thorburn, D.R.; McGee, S.L.; Hannan, A.J.; Gray, L.J. N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl. Psychiatry 2015, 5, e492. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, Y.; Inagi, R. Mitochondria: A therapeutic target in acute kidney injury. Nephrol. Dial. Transplant. 2016, 31, 1062–1069. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, X.; Li, N.; Zhang, J.; Yang, J.; Bu, P. Sirtuin 3 deficiency aggravates contrast-induced acute kidney injury. J. Transl. Med. 2018, 16, 313. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Perry, H.M.; Huang, L.; Wilson, R.J.; Bajwa, A.; Sesaki, H.; Yan, Z.; Rosin, D.L.; Kashatus, D.F.; Okusa, M.D. Dynamin-Related Protein 1 Deficiency Promotes Recovery from AKI. J. Am. Soc. Nephrol. 2018, 29, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Yadav, A.; Tiwari, S.K.; Seth, B.; Chauhan, L.K.; Khare, P.; Ray, R.S.; Chaturvedi, R.K. Dynamin-related Protein 1 Inhibition Mitigates Bisphenol A-mediated Alterations in Mitochondrial Dynamics and Neural Stem Cell Proliferation and Differentiation. J. Biol. Chem. 2016, 291, 15923–15939. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, H.; Jiang, C.; Zhang, M. Renal ischemia/reperfusion-induced mitophagy protects against renal dysfunction via Drp1-dependent-pathway. Exp. Cell Res. 2018, 369, 27–33. [Google Scholar] [CrossRef]
- Agarwal, S.; Tiwari, S.K.; Seth, B.; Yadav, A.; Singh, A.; Mudawal, A.; Chauhan, L.K.; Gupta, S.K.; Choubey, V.; Tripathi, A.; et al. Activation of Autophagic Flux against Xenoestrogen Bisphenol-A-induced Hippocampal Neurodegeneration via AMP kinase (AMPK)/Mammalian Target of Rapamycin (mTOR) Pathways. J. Biol. Chem. 2015, 290, 21163–21184. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Sanchez-Ramos, C.; Prieto, I.; Tierrez, A.; Laso, J.; Valdecantos, M.P.; Bartrons, R.; Rosello-Catafau, J.; Monsalve, M. PGC-1alpha Downregulation in Steatotic Liver Enhances Ischemia-Reperfusion Injury and Impairs Ischemic Preconditioning. Antioxid. Redox Signal. 2017, 27, 1332–1346. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Diaz, F.; Iommarini, L.; Aure, K.; Lombes, A.; Moraes, C.T. PGC-1alpha/beta induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Hum. Mol. Genet. 2009, 18, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Gong, B.; Duan, W.; Fan, C.; Zhang, J.; Li, Z.; Xue, X.; Xu, Y.; Meng, D.; Li, B.; et al. Melatonin ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by preserving mitochondrial function: Role of AMPK-PGC-1alpha-SIRT3 signaling. Sci. Rep. 2017, 7, 41337. [Google Scholar] [CrossRef]
- Guan, Y.; Cui, Z.J.; Sun, B.; Han, L.P.; Li, C.J.; Chen, L.M. Celastrol attenuates oxidative stress in the skeletal muscle of diabetic rats by regulating the AMPK-PGC1alpha-SIRT3 signaling pathway. Int. J. Mol. Med. 2016, 37, 1229–1238. [Google Scholar] [CrossRef]
- Song, C.; Zhao, J.; Fu, B.; Li, D.; Mao, T.; Peng, W.; Wu, H.; Zhang, Y. Melatonin-mediated upregulation of Sirt3 attenuates sodium fluoride-induced hepatotoxicity by activating the MT1-PI3K/AKT-PGC-1alpha signaling pathway. Free Radic. Biol. Med. 2017, 112, 616–630. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef]
- Li, X.; Song, S.; Xu, M.; Hua, Y.; Ding, Y.; Shan, X.; Meng, G.; Wang, Y. Sirtuin3 deficiency exacerbates carbon tetrachloride-induced hepatic injury in mice. J. Biochem. Mol. Toxicol. 2019, 33, e22249. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Sham | RIR | |||||
|---|---|---|---|---|---|---|---|
| Veh | BPA5 | BPA50 | Veh | BPA5 | BPA50 | ||
| BUN (mg/dL) | 21.95 ± 0.81 a | 22.53 ± 0.92 a | 22.88 ± 0.83 a | 67.33 ± 4.38 b | 90.11 ± 0.92 c | 105.52 ± 5.65 d | |
| SCr (mg/dL) | 0.29 ± 0.00 a | 0.30 ± 0.01 a | 0.29 ± 0.01 a | 1.36 ± 0.14 b | 2.15 ± 0.11 c | 3.06 ± 0.20 d | |
| AST (U/L) | 80.25 ± 2.75 a | 78.50 ± 1.94 a | 115.00 ± 3.49 b | 233.50 ± 26.93 c | 311.50 ± 15.19 d | 384.25 ± 19.75 e | |
| ALT (U/L) | 23.75 ± 1.31 a | 24.25 ± 1.89 a | 31.75 ± 1.31 b | 45.50 ± 1.55 c | 56.50 ± 3.59 d | 70.75 ± 3.61 e | |
| NO (µM) | 0.93 ± 0.13 a | 0.94 ± 0.11 a | 0.97 ± 0.10 a | 1.32 ± 0.11 b | 1.87 ± 0.08 c | 2.35 ± 0.10 d | |
| MDA (µM) | 2.95 ± 0.12 a | 3.06 ± 0.12 a | 3.01 ± 0.21 a | 3.63 ± 0.04 b | 4.40 ± 0.35 c | 5.50 ± 0.17 d | |
| GSH (µM) | 363.50 ± 11.09 a | 365.00 ± 12.15 a | 356.33 ± 14.70 a | 294.50 ± 10.22 b | 250.00 ± 3.74 c | 210.33 ± 11.26 d | |
| TNF-α (pg/mL) | 3.24 ± 0.34 a | 3.09 ± 0.40 a | 3.31 ± 0.47 a | 5.52 ± 0.34 b | 7.51 ± 0.27 c | 9.98 ± 0.42 d |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peerapanyasut, W.; Kobroob, A.; Palee, S.; Chattipakorn, N.; Wongmekiat, O. N-Acetylcysteine Attenuates the Increasing Severity of Distant Organ Liver Dysfunction after Acute Kidney Injury in Rats Exposed to Bisphenol A. Antioxidants 2019, 8, 497. https://doi.org/10.3390/antiox8100497
Peerapanyasut W, Kobroob A, Palee S, Chattipakorn N, Wongmekiat O. N-Acetylcysteine Attenuates the Increasing Severity of Distant Organ Liver Dysfunction after Acute Kidney Injury in Rats Exposed to Bisphenol A. Antioxidants. 2019; 8(10):497. https://doi.org/10.3390/antiox8100497
Chicago/Turabian StylePeerapanyasut, Wachirasek, Anongporn Kobroob, Siripong Palee, Nipon Chattipakorn, and Orawan Wongmekiat. 2019. "N-Acetylcysteine Attenuates the Increasing Severity of Distant Organ Liver Dysfunction after Acute Kidney Injury in Rats Exposed to Bisphenol A" Antioxidants 8, no. 10: 497. https://doi.org/10.3390/antiox8100497
APA StylePeerapanyasut, W., Kobroob, A., Palee, S., Chattipakorn, N., & Wongmekiat, O. (2019). N-Acetylcysteine Attenuates the Increasing Severity of Distant Organ Liver Dysfunction after Acute Kidney Injury in Rats Exposed to Bisphenol A. Antioxidants, 8(10), 497. https://doi.org/10.3390/antiox8100497