Structural and Biochemical Insights into the Reactivity of Thioredoxin h1 from Chlamydomonas reinhardtii

 ,
,  , , , , ,
, , , , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Material and Enzymes
2.2. Enzymatic Assay
2.3. Inactivation of CrTRXh1 by IAM-Dependent Alkylation Treatments
2.4. pKa Determination by Matrix-Assisted Laser Desorption/Ionization-Time of Flight (MALDI-TOF) MS
2.5. Identification of the Reactive Cysteine within the Monoalkylated Form of CrTRXh1
2.6. Circular Dichroism Analysis
2.7. Crystallization and Data Collection
2.8. Structure Solution and Refinement
2.9. Data Availability
3. Results
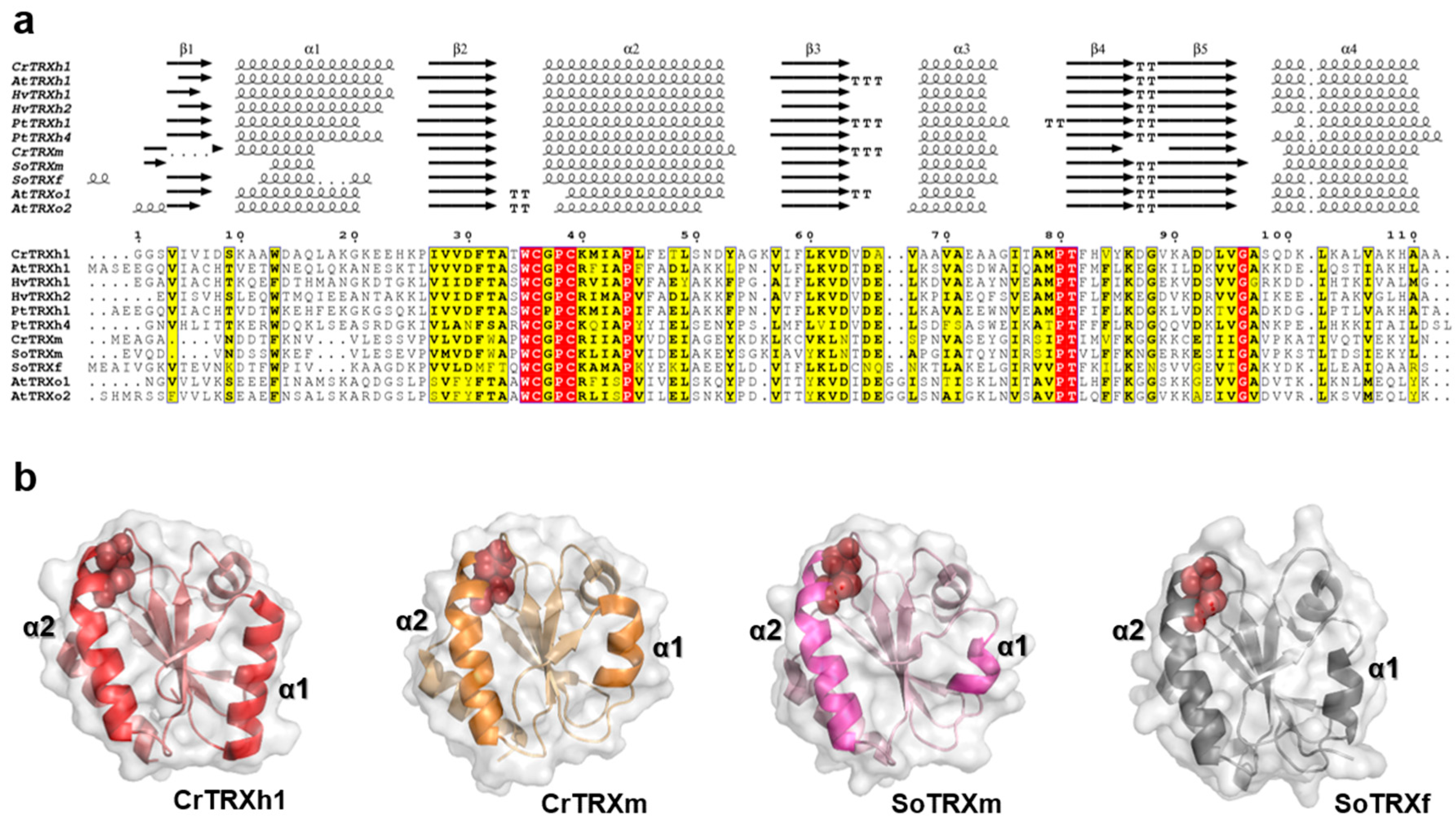
3.1. Sequence and Structural Comparison of Plant TRXs
3.2. CrTRXh1 Shares the Substrate Recognition Loop with Other h-Class TRXs
3.3. Structure Analysis of Reduced and Oxidized CrTRXh1
3.4. Active Site of Reduced and Oxidized CrTRXh1
3.5. C36S and C39S Mutants vs Wild-Type Reduced CrTRXh1
3.6. CrTRXh1 Alkylation by Iodoacetamide is pH-Sensitive
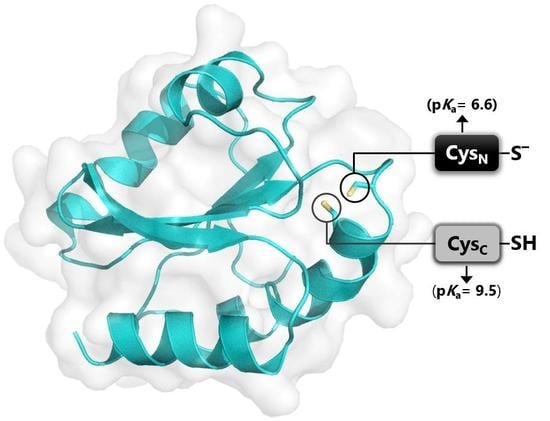
3.7. pKa Determination of CysN and CysC by Quantitative MALDI-TOF Mass Spectrometry
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
Abbreviations
| ASA | accessible surface area |
| BSA | buried surface area |
| CD | circular dichroism |
| Cys | cysteine(s) |
| DTNB | 5,5’-dithiobis-2-nitrobenzoic acid |
| IAM | iodoacetamide |
| MALDI-TOF | matrix-assisted laser desorption/ionization-time of flight |
| MS | mass spectrometry |
| MS/MS | tandem mass spectrometry |
| SRL | substrate recognition loop |
| TCEP | Tris(2-carboxyethyl)phosphine |
| TRX | thioredoxin |
References
- Lemaire, S.D.; Michelet, L.; Zaffagnini, M.; Massot, V.; Issakidis-Bourguet, E. Thioredoxins in chloroplasts. Curr. Genet. 2007, 51, 343–365. [Google Scholar] [CrossRef] [PubMed]
- Michelet, L.; Zaffagnini, M.; Morisse, S.; Sparla, F.; Pérez-Pérez, M.E.; Francia, F.; Danon, A.; Marchand, C.H.; Fermani, S.; Trost, P.; et al. Redox regulation of the Calvin-Benson cycle: Something old, something new. Front. Plant. Sci. 2013, 4, 470. [Google Scholar] [CrossRef] [PubMed]
- Geigenberger, P.; Thormählen, I.; Daloso, D.M.; Fernie, A.R. The Unprecedented Versatility of the Plant Thioredoxin System. Trends Plant. Sci. 2017, 22, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Zaffagnini, M.; Fermani, S.; Marchand, C.H.; Costa, A.; Sparla, F.; Rouhier, N.; Geigenberger, P.; Lemaire, S.D.; Trost, P. Redox Homeostasis in Photosynthetic Organisms: Novel and Established Thiol-Based Molecular Mechanisms. Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Thomas, J.A. S-glutathiolated hepatocyte proteins and insulin disulfides as substrates for reduction by glutaredoxin, thioredoxin, protein disulfide isomerase, and glutathione. Arch. Biochem. Biophys. 1996, 335, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M.; Forrester, M.T.; Hess, D.T.; Stamler, J.S. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science 2008, 320, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Greetham, D.; Vickerstaff, J.; Shenton, D.; Perrone, G.G.; Dawes, I.W.; Grant, C.M. Thioredoxins function as deglutathionylase enzymes in the yeast Saccharomyces cerevisiae. BMC Biochem. 2010, 11, 3. [Google Scholar] [CrossRef]
- Bedhomme, M.; Adamo, M.; Marchand, C.H.; Couturier, J.; Rouhier, N.; Lemaire, S.D.; Zaffagnini, M.; Trost, P. Glutathionylation of cytosolic glyceraldehyde-3-phosphate dehydrogenase from the model plant Arabidopsis thaliana is reversed by both glutaredoxins and thioredoxins in vitro. Biochem. J. 2012, 445, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Kneeshaw, S.; Gelineau, S.; Tada, Y.; Loake, G.J.; Spoel, S.H. Selective protein denitrosylation activity of Thioredoxin-h5 modulates plant Immunity. Mol. Cell 2014, 56, 153–162. [Google Scholar] [CrossRef]
- Berger, H.; De Mia, M.; Morisse, S.; Marchand, C.H.; Lemaire, S.D.; Wobbe, L.; Kruse, O. A Light Switch Based on Protein S-Nitrosylation Fine-Tunes Photosynthetic Light Harvesting in Chlamydomonas. Plant Physiol. 2016, 171, 821–832. [Google Scholar] [CrossRef]
- Subramani, J.; Kundumani-Sridharan, V.; Hilgers, R.H.; Owens, C.; Das, K.C. Thioredoxin Uses a GSH-independent Route to Deglutathionylate Endothelial Nitric-oxide Synthase and Protect against Myocardial Infarction. J. Biol. Chem. 2016, 291, 23374–23389. [Google Scholar] [CrossRef] [PubMed]
- Kallis, G.B.; Holmgren, A. Differential reactivity of the functional sulfhydryl groups of cysteine-32 and cysteine-35 present in the reduced form of thioredoxin from Escherichia coli. J. Biol. Chem. 1980, 255, 10261–10265. [Google Scholar] [PubMed]
- Brandes, H.K.; Larimer, F.W.; Geck, M.K.; Stringer, C.D.; Schurmann, P.; Hartman, F.C. Direct identification of the primary nucleophile of thioredoxin f. J. Biol. Chem. 1993, 268, 18411–18414. [Google Scholar] [PubMed]
- Holmgren, A. Thioredoxin structure and mechanism: Conformational changes on oxidation of the active-site sulfhydryls to a disulfide. Structure 1995, 3, 239–243. [Google Scholar] [CrossRef]
- Trost, P.; Fermani, S.; Calvaresi, M.; Zaffagnini, M. Biochemical basis of sulphenomics: How protein sulphenic acids may be stabilized by the protein microenvironment. Plant. Cell Environ. 2017, 40, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Roos, G.; Foloppe, N.; Messens, J. Understanding the pKa of redox cysteines: The key role of hydrogen bonding. Antioxid. Redox Signal. 2013, 18, 94–127. [Google Scholar] [CrossRef]
- Dyson, H.J.; Jeng, M.F.; Tennant, L.L.; Slaby, I.; Lindell, M.; Cui, D.S.; Kuprin, S.; Holmgren, A. Effects of buried charged groups on cysteine thiol ionization and reactivity in Escherichia coli thioredoxin: Structural and functional characterization of mutants of Asp 26 and Lys 57. Biochemistry 1997, 36, 2622–2636. [Google Scholar] [CrossRef]
- Chivers, P.T.; Raines, R.T. General acid/base catalysis in the active site of Escherichia coli thioredoxin. Biochemistry 1997, 36, 15810–15816. [Google Scholar] [CrossRef]
- Menchise, V.; Corbier, C.; Didierjean, C.; Saviano, M.; Benedetti, E.; Jacquot, J.P.; Aubry, A. Crystal structure of the wild-type and D30A mutant thioredoxin h of Chlamydomonas reinhardtii and implications for the catalytic mechanism. Biochem. J. 2001, 359, 65–75. [Google Scholar] [CrossRef]
- Roos, G.; Foloppe, N.; Van Laer, K.; Wyns, L.; Nilsson, L.; Geerlings, P.; Messens, J. How thioredoxin dissociates its mixed disulfide. PLoS Comput. Biol. 2009, 5, e1000461. [Google Scholar] [CrossRef]
- Meyer, Y.; Belin, C.; Delorme-Hinoux, V.; Reichheld, J.P.; Riondet, C. Thioredoxin and Glutaredoxin Systems in Plants: Molecular Mechanisms, Crosstalks, and Functional Significance. Antioxid. Redox Signal. 2012. [Google Scholar] [CrossRef]
- Lindahl, M.; Mata-Cabana, A.; Kieselbach, T. The Disulfide Proteome and Other Reactive Cysteine Proteomes: Analysis and Functional Significance. Antioxid. Redox Signal. 2011, 14, 2581–2642. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, B.B.; Holmgren, A.; Jacquot, J.P.; Scheibe, R. Fifty years in the thioredoxin field and a bountiful harvest. Biochim. Biophys. Acta 2012, 1820, 1822–1829. [Google Scholar] [CrossRef]
- Pérez-Pérez, M.E.; Mauriès, A.; Maes, A.; Tourasse, N.J.; Hamon, M.; Lemaire, S.D.; Marchand, C.H. The Deep Thioredoxome in Chlamydomonas reinhardtii: New Insights into Redox Regulation. Mol. Plant 2017, 10, 1107–1125. [Google Scholar] [CrossRef] [PubMed]
- Lancelin, J.M.; Guilhaudis, L.; Krimm, I.; Blackledge, M.J.; Marion, D.; Jacquot, J.P. NMR structures of thioredoxin m from the green alga Chlamydomonas reinhardtii. Proteins 2000, 41, 334–349. [Google Scholar] [CrossRef]
- Peterson, F.C.; Lytle, B.L.; Sampath, S.; Vinarov, D.; Tyler, E.; Shahan, M.; Markley, J.L.; Volkman, B.F. Solution structure of thioredoxin h1 from Arabidopsis thaliana. Protein Sci. 2005, 14, 2195–2200. [Google Scholar] [CrossRef]
- Zannini, F.; Roret, T.; Przybyla-Toscano, J.; Dhalleine, T.; Rouhier, N.; Couturier, J. Mitochondrial Arabidopsis thaliana TRXo Isoforms Bind an Iron(-)Sulfur Cluster and Reduce NFU Proteins In Vitro. Antioxidants 2018, 7. [Google Scholar] [CrossRef]
- Maeda, K.; Hägglund, P.; Finnie, C.; Svensson, B.; Henriksen, A. Crystal structures of barley thioredoxin h isoforms HvTrxh1 and HvTrxh2 reveal features involved in protein recognition and possibly in discriminating the isoform specificity. Protein Sci. 2008, 17, 1015–1024. [Google Scholar] [CrossRef]
- Coudevylle, N.; Thureau, A.; Hemmerlin, C.; Gelhaye, E.; Jacquot, J.P.; Cung, M.T. Solution structure of a natural CPPC active site variant, the reduced form of thioredoxin h1 from poplar. Biochemistry 2005, 44, 2001–2008. [Google Scholar] [CrossRef]
- Koh, C.S.; Navrot, N.; Didierjean, C.; Rouhier, N.; Hirasawa, M.; Knaff, D.B.; Wingsle, G.; Samian, R.; Jacquot, J.P.; Corbier, C.; et al. An atypical catalytic mechanism involving three cysteines of thioredoxin. J. Biol. Chem. 2008, 283, 23062–23072. [Google Scholar] [CrossRef]
- Capitani, G.; Markovic-Housley, Z.; DelVal, G.; Morris, M.; Jansonius, J.N.; Schürmann, P. Crystal structures of two functionally different thioredoxins in spinach chloroplasts. J. Mol. Biol. 2000, 302, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Zaffagnini, M.; Michelet, L.; Massot, V.; Trost, P.; Lemaire, S.D. Biochemical characterization of glutaredoxins from Chlamydomonas reinhardtii reveals the unique properties of a chloroplastic CGFS-type glutaredoxin. J. Biol. Chem. 2008, 283, 8868–8876. [Google Scholar] [CrossRef] [PubMed]
- Bedhomme, M.; Zaffagnini, M.; Marchand, C.H.; Gao, X.H.; Moslonka-Lefebvre, M.; Michelet, L.; Decottignies, P.; Lemaire, S.D. Regulation by glutathionylation of isocitrate lyase from Chlamydomonas reinhardtii. J. Biol. Chem. 2009, 284, 36282–36291. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Hägglund, P.; Finnie, C.; Svensson, B.; Henriksen, A. Structural basis for target protein recognition by the protein disulfide reductase thioredoxin. Structure 2006, 14, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Bjornberg, O.; Maeda, K.; Svensson, B.; Hagglund, P. Dissecting molecular interactions involved in recognition of target disulfides by the barley thioredoxin system. Biochemistry 2012, 51, 9930–9939. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, S.D.; Tedesco, D.; Crozet, P.; Michelet, L.; Fermani, S.; Zaffagnini, M.; Henri, J. Crystal Structure of Chloroplastic Thioredoxin f2 from Chlamydomonas reinhardtii Reveals Distinct Surface Properties. Antioxidants 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Katti, S.K.; LeMaster, D.M.; Eklund, H. Crystal structure of thioredoxin from Escherichia coli at 1.68 A resolution. J. Mol. Biol. 1990, 212, 167–184. [Google Scholar] [CrossRef]
- Saarinen, M.; Gleason, F.K.; Eklund, H. Crystal structure of thioredoxin-2 from Anabaena. Structure 1995, 3, 1097–1108. [Google Scholar] [CrossRef]
- Weichsel, A.; Gasdaska, J.R.; Powis, G.; Montfort, W.R. Crystal structures of reduced, oxidized, and mutated human thioredoxins: Evidence for a regulatory homodimer. Structure 1996, 4, 735–751. [Google Scholar] [CrossRef]
- Zaffagnini, M.; Bedhomme, M.; Marchand, C.H.; Couturier, J.R.; Gao, X.H.; Rouhier, N.; Trost, P.; Lemaire, S.D. Glutaredoxin S12: Unique properties for redox signaling. Antioxid. Redox Signal. 2012, 16, 17–32. [Google Scholar] [CrossRef]
- Nelson, K.J.; Day, A.E.; Zeng, B.B.; King, S.B.; Poole, L.B. Isotope-coded, iodoacetamide-based reagent to determine individual cysteine pK(a) values by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal. Biochem. 2008, 375, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Jeng, M.F.; Campbell, A.P.; Begley, T.; Holmgren, A.; Case, D.A.; Wright, P.E.; Dyson, H.J. High-resolution solution structures of oxidized and reduced Escherichia coli thioredoxin. Structure 1994, 2, 853–868. [Google Scholar] [CrossRef]
- Qin, J.; Clore, G.M.; Gronenborn, A.M. The high-resolution three-dimensional solution structures of the oxidized and reduced states of human thioredoxin. Structure 1994, 2, 503–522. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Red-CrTRXh1 | Ox-CrTRXh1 | Monomeric C36S | Dimeric C36S | C39S | |
|---|---|---|---|---|---|
| Data collection | |||||
| Unit cell (Å) | a = b = 48.77, c = 143.93 | a = b = 48.68, c = 143.70 | a = 60.78, b = 34.82, c = 48.16 | a = b = 48.74, c = 143.19 | a = b = 48.43, c = 143.66 |
| Space group | P3121 | P3121 | P21212 | P3121 | P3121 |
| N° molecules ASU | 2 | 2 | 1 | 2 | 2 |
| Resolution range * (Å) | 47.98–1.70 (1.76–1.70) | 42.16–1.57 (1.63–1.57) | 48.16–0.94 (0.96–0.94) | 42.21–1.22 (1.24–1.22) | 41.94–1.81 (1.84–1.81) |
| Unique reflections | 22,494 (2165) | 28,348 (2646) | 64,505 (1579) | 59,565 (2934) | 18,676 (1044) |
| Completeness * (%) | 99.3 (98.5) | 99.5 (96.7) | 96.5 (47.1) | 99.7 (98.7) | 99.6 (97.0) |
| Rmerge * | 0.145 (0.858) | 0.072 (0.804) | 0.043 (0.369) | 0.063 (1.108) | 0.111 (0.740) |
| Rpim * | 0.064 (0.398) | 0.034 (0.428) | 0.033 (0.367) | 0.028 (0.507) | 0.057 (0.345) |
| CC1/2 * | 0.980 (0.683) | 0.998 (0.661) | 0.997 (0.625) | 0.998 (0.635) | 0.990 (0.715) |
| I/(I) * | 7.9 (2.2) | 10.0 (1.5) | 18.9 (1.7) | 14.7 (1.7) | 9.7 (1.9) |
| Multiplicity * | 6.5 (6.6) | 5.7 (4.9) | 4.1 (1.6) | 6.8 (6.6) | 5.8 (5.7) |
| Refinement | |||||
| Resolution range * (Å) | 42.24–1.70 (1.78–1.70) | 42.16–1.57 (1.62–1.57) | 48.16–0.94 (0.95–0.94) | 42.21–1.22 (1.23–1.22) | 41.94–1.81 (1.85–1.81) |
| Reflection used * | 22,475 (2724) | 28,277 (2671) | 64,420 (1042) | 59,506 (3597) | 18,626 (2581) |
| R/Rfree | 0.189/0.224 | 0.187/0.216 | 0.157/0.166 | 0.168/0.174 | 0.219/0.266 |
| rmsd from ideality (Å, °) | 0.006, 0.743 | 0.006, 0.820 | 0.006, 1.007 | 0.007, 1.079 | 0.002, 0.438 |
| N° atoms | |||||
| Non-hydrogen atoms | 1867 | 1832 | 1063 | 2015 | 1799 |
| Protein atoms | 1675 | 1660 | 856 | 1675 | 1637 |
| Solvent molecules | 192 | 172 | 207 | 346 | 162 |
| Hydrogens | / | / | 897 | 1724 | / |
| B value (Å2) | |||||
| Mean | 26.8 | 26.1 | 12.6 | 16.2 | 24.7 |
| Wilson | 25.8 | 23.6 | 8.4 | 12.6 | 24.0 |
| Protein atoms | 26.1 | 25.5 | 9.3 | 14.6 | 24.2 |
| Solvent molecules | 32.8 | 32.6 | 20.7 | 23.5 | 30.1 |
| Heteroatoms | / | / | 33.2 | 37.8 | / |
| Ramachandran plot (%) § | |||||
| Most favored | 99.1 | 98.6 | 99.1 | 97.7 | 98.6 |
| Allowed | 0.9 | 1.4 | 0.9 | 2.3 | 1.4 |
| Disallowed | 0 | 0 | 0 | 0 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchand, C.H.; Fermani, S.; Rossi, J.; Gurrieri, L.; Tedesco, D.; Henri, J.; Sparla, F.; Trost, P.; Lemaire, S.D.; Zaffagnini, M. Structural and Biochemical Insights into the Reactivity of Thioredoxin h1 from Chlamydomonas reinhardtii. Antioxidants 2019, 8, 10. https://doi.org/10.3390/antiox8010010
Marchand CH, Fermani S, Rossi J, Gurrieri L, Tedesco D, Henri J, Sparla F, Trost P, Lemaire SD, Zaffagnini M. Structural and Biochemical Insights into the Reactivity of Thioredoxin h1 from Chlamydomonas reinhardtii. Antioxidants. 2019; 8(1):10. https://doi.org/10.3390/antiox8010010
Chicago/Turabian StyleMarchand, Christophe H., Simona Fermani, Jacopo Rossi, Libero Gurrieri, Daniele Tedesco, Julien Henri, Francesca Sparla, Paolo Trost, Stéphane D. Lemaire, and Mirko Zaffagnini. 2019. "Structural and Biochemical Insights into the Reactivity of Thioredoxin h1 from Chlamydomonas reinhardtii" Antioxidants 8, no. 1: 10. https://doi.org/10.3390/antiox8010010
APA StyleMarchand, C. H., Fermani, S., Rossi, J., Gurrieri, L., Tedesco, D., Henri, J., Sparla, F., Trost, P., Lemaire, S. D., & Zaffagnini, M. (2019). Structural and Biochemical Insights into the Reactivity of Thioredoxin h1 from Chlamydomonas reinhardtii. Antioxidants, 8(1), 10. https://doi.org/10.3390/antiox8010010