S-Adenosylmethionine and Superoxide Dismutase 1 Synergistically Counteract Alzheimer’s Disease Features Progression in TgCRND8 Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
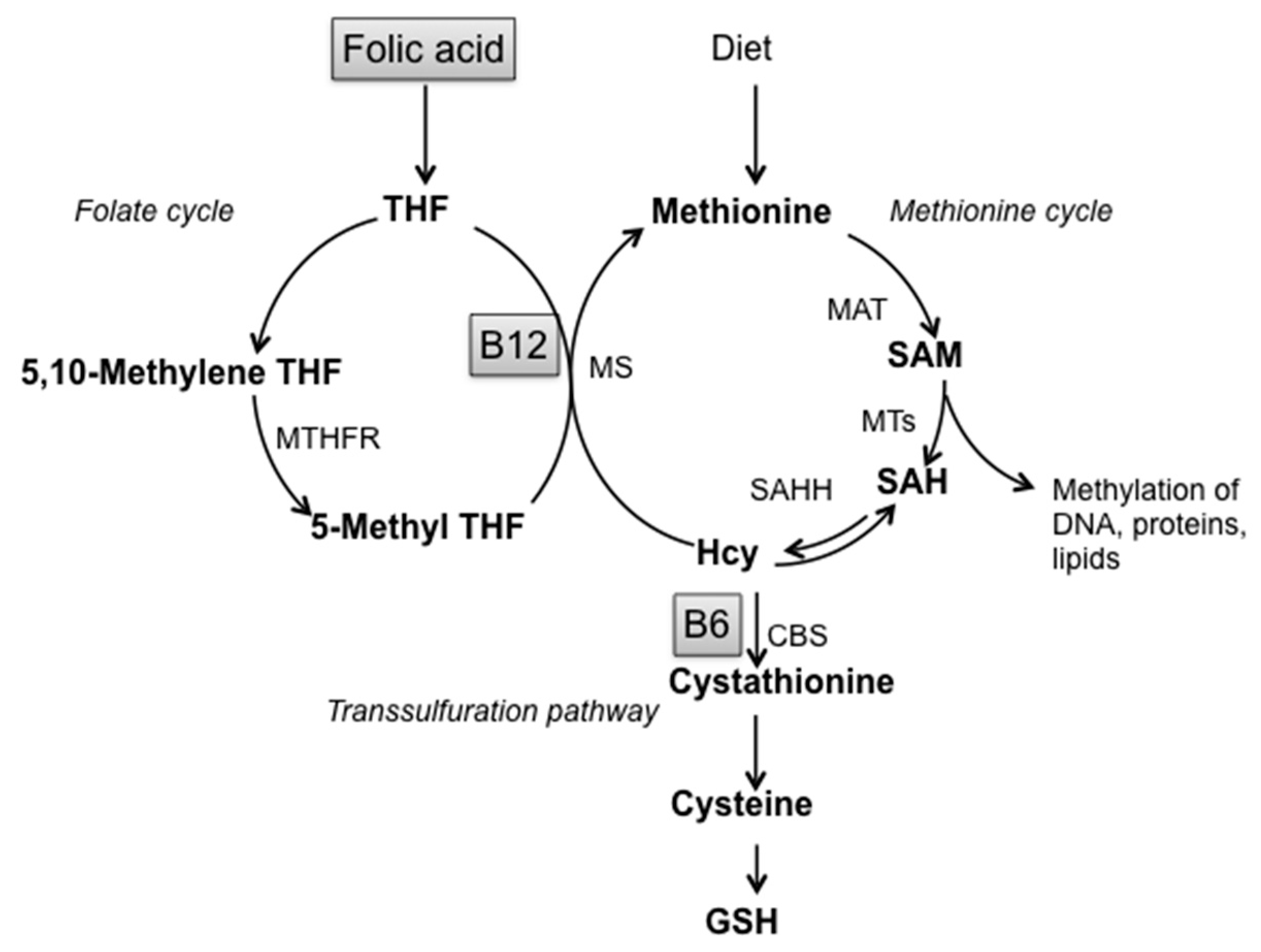
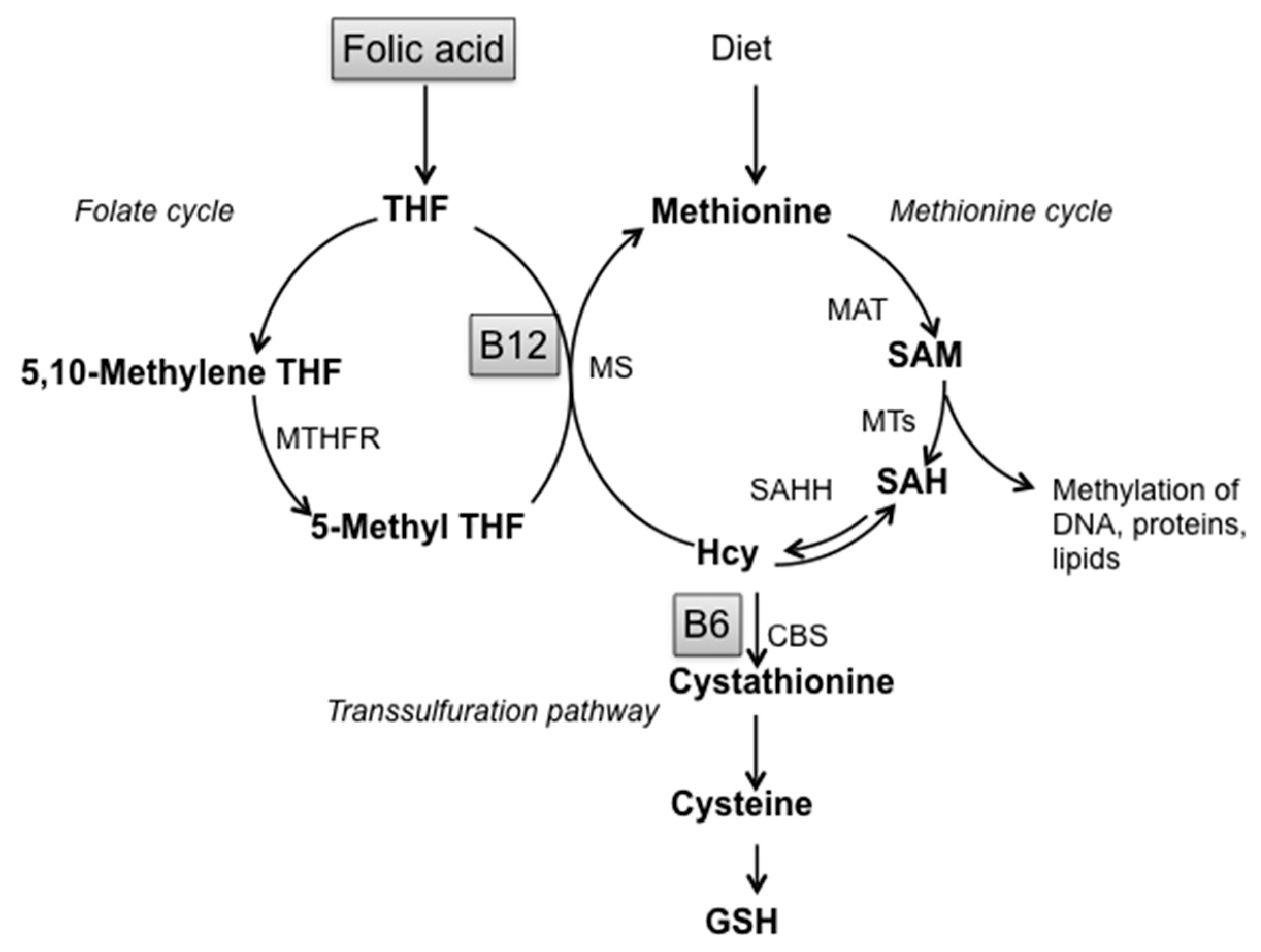
:1. Introduction
2. Materials and Methods
2.1. Mice and Diets
2.2. SOD Activity
2.3. Lipid Peroxidation (LPO) Assay
2.4. Quantitative Real-Time PCR Analysis
2.5. ELISA Protein Analysis
2.6. Immunohistochemistry
2.7. Statistical Analysis
3. Results
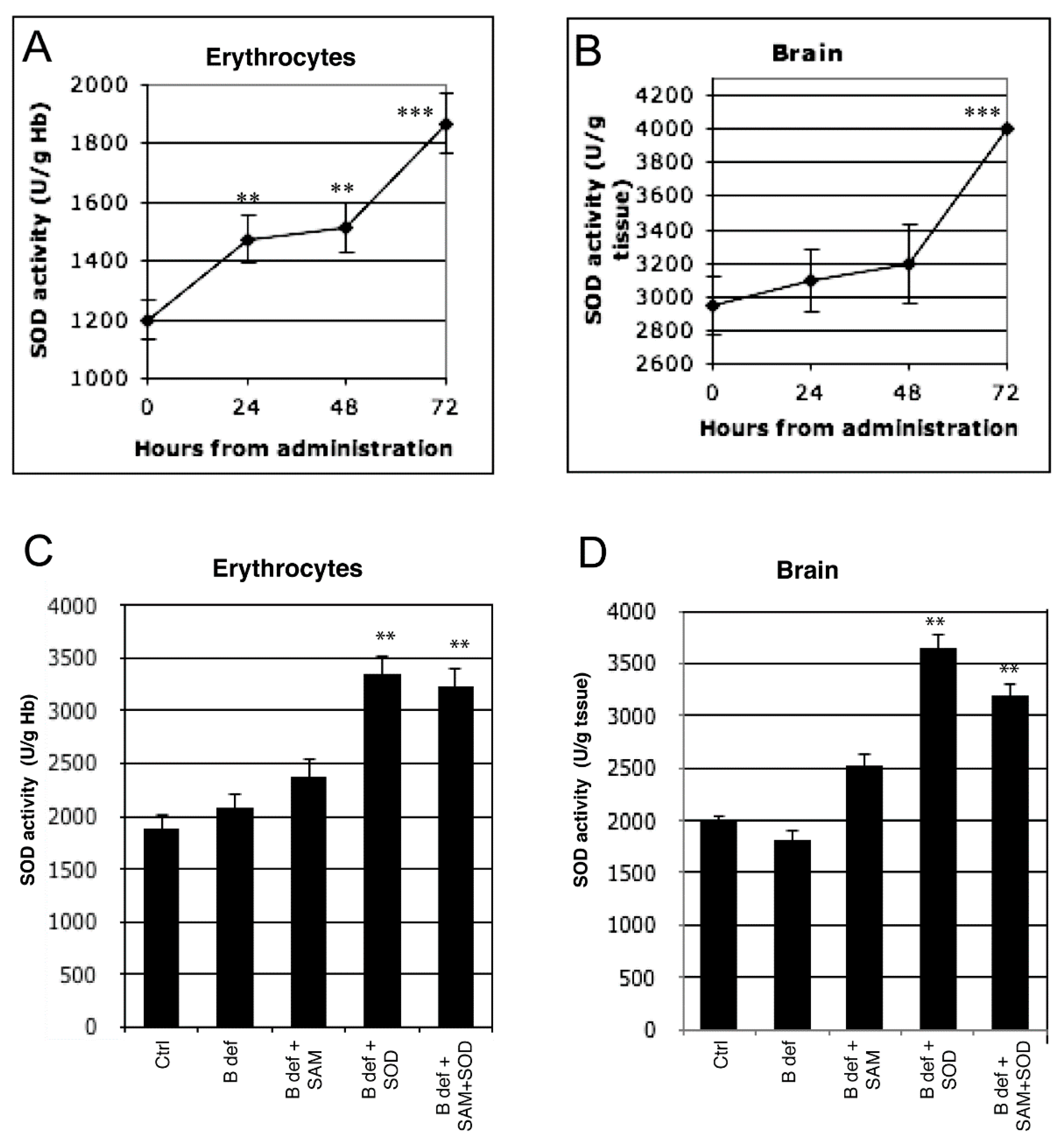
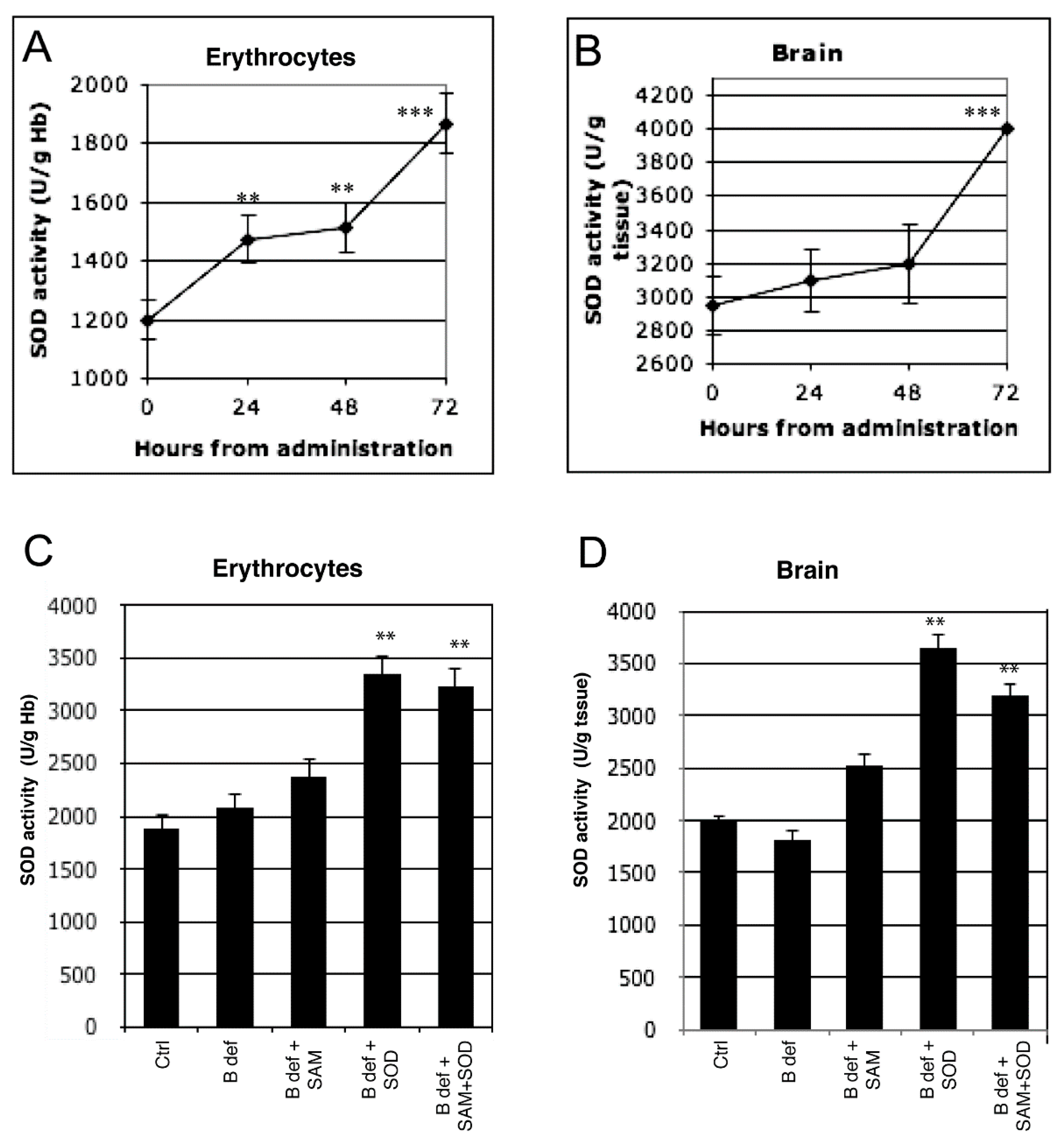
3.1. SOD Uptake
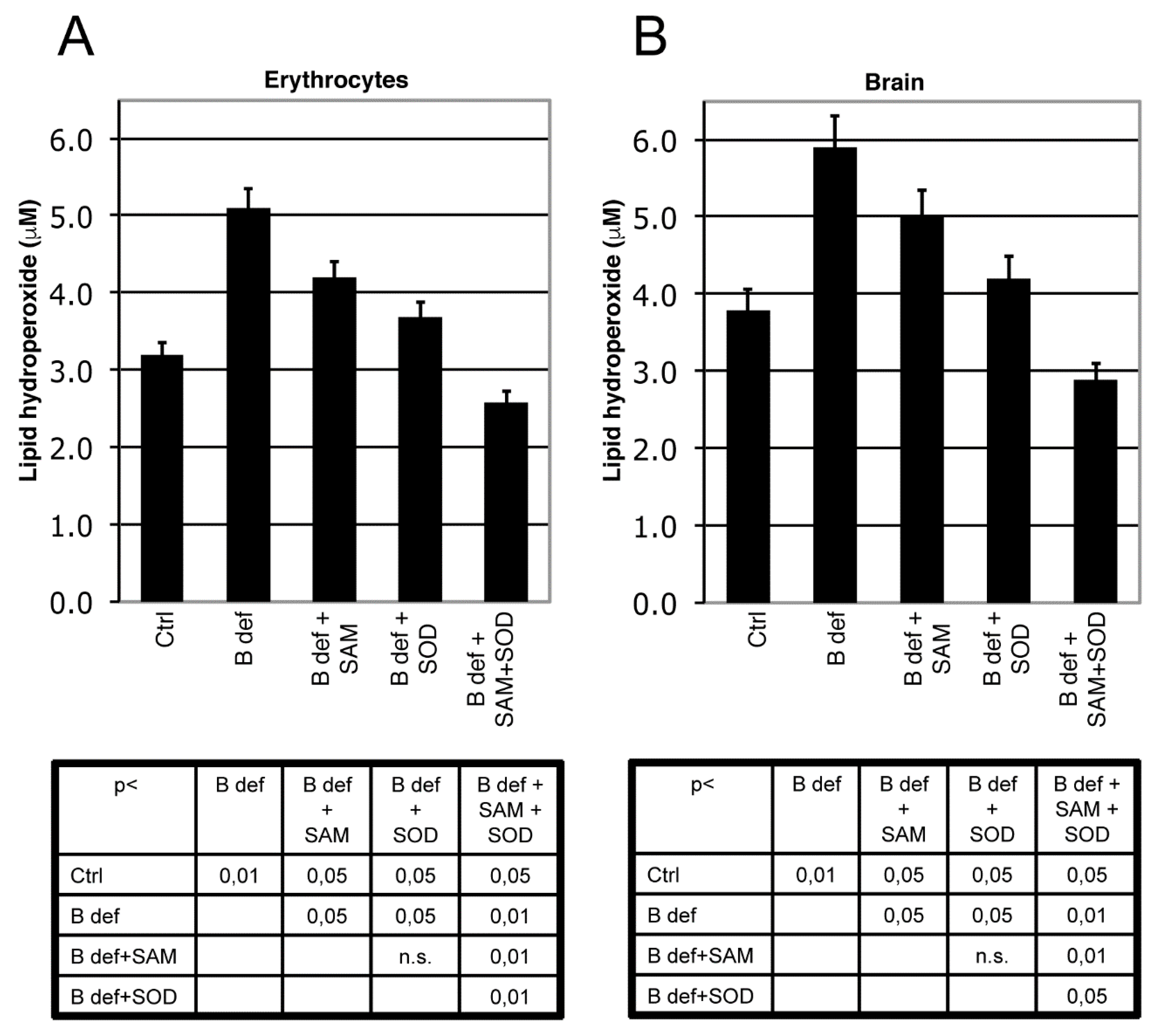
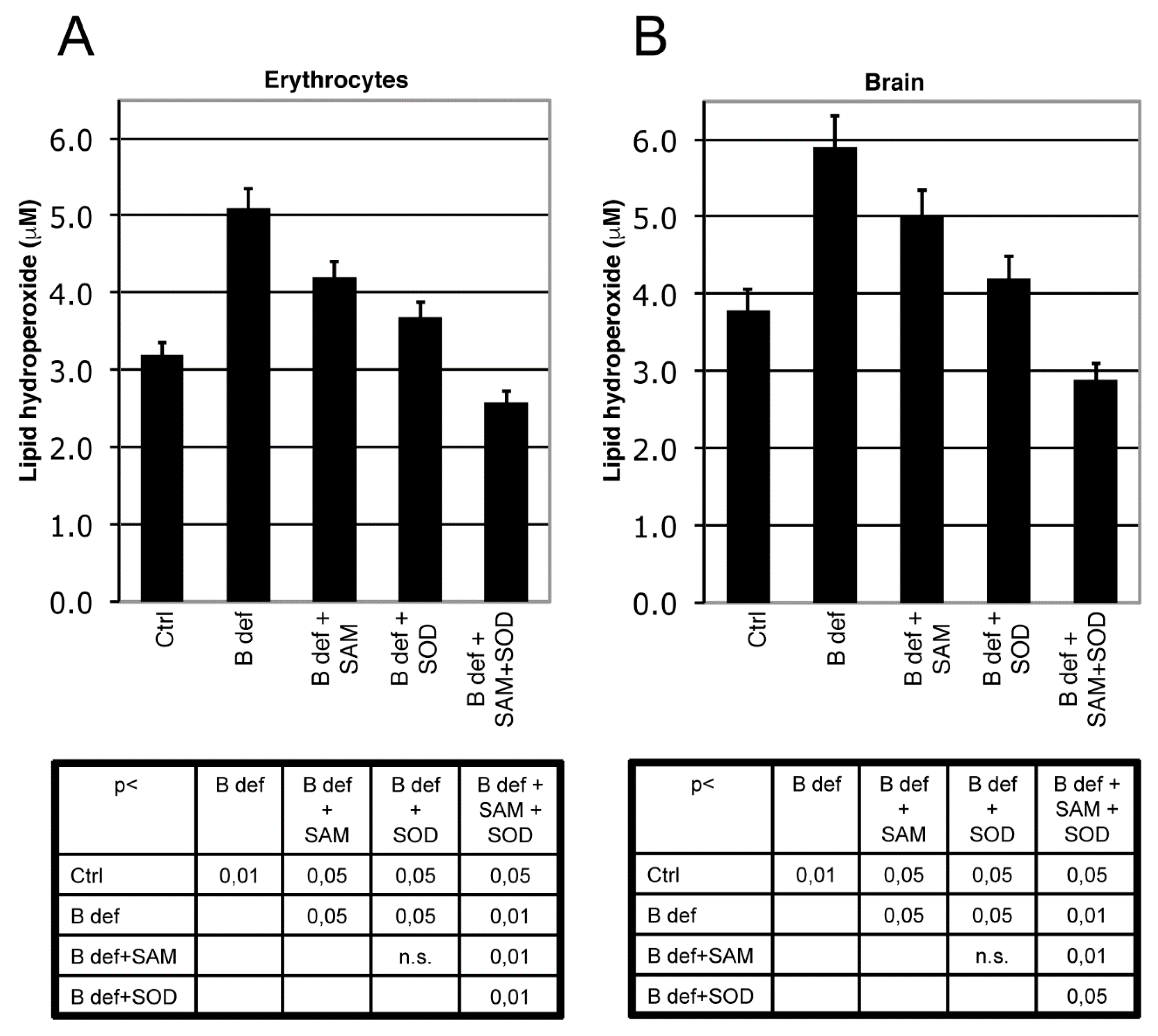
3.2. SAM and SOD Reduce Oxidative Stress
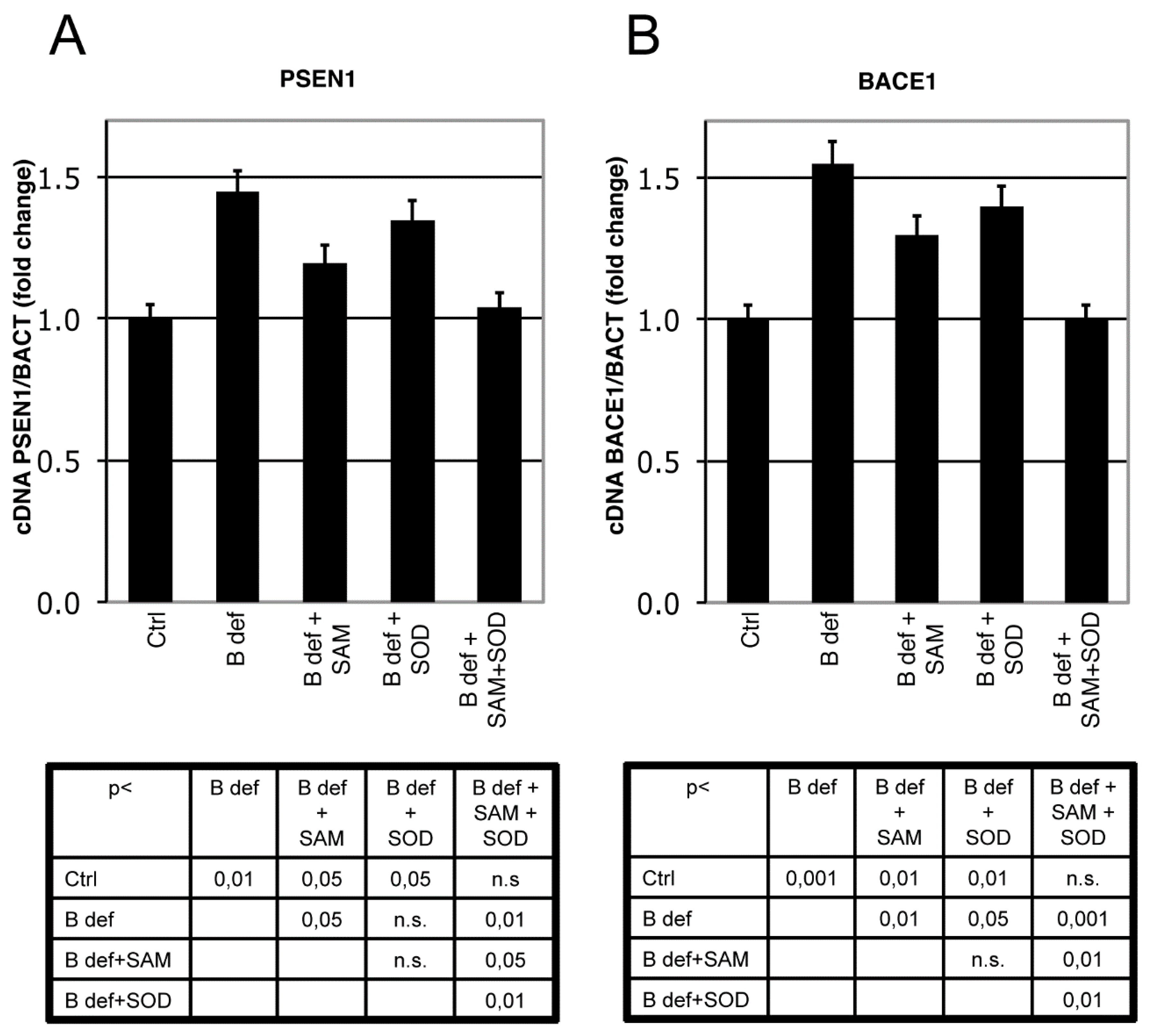
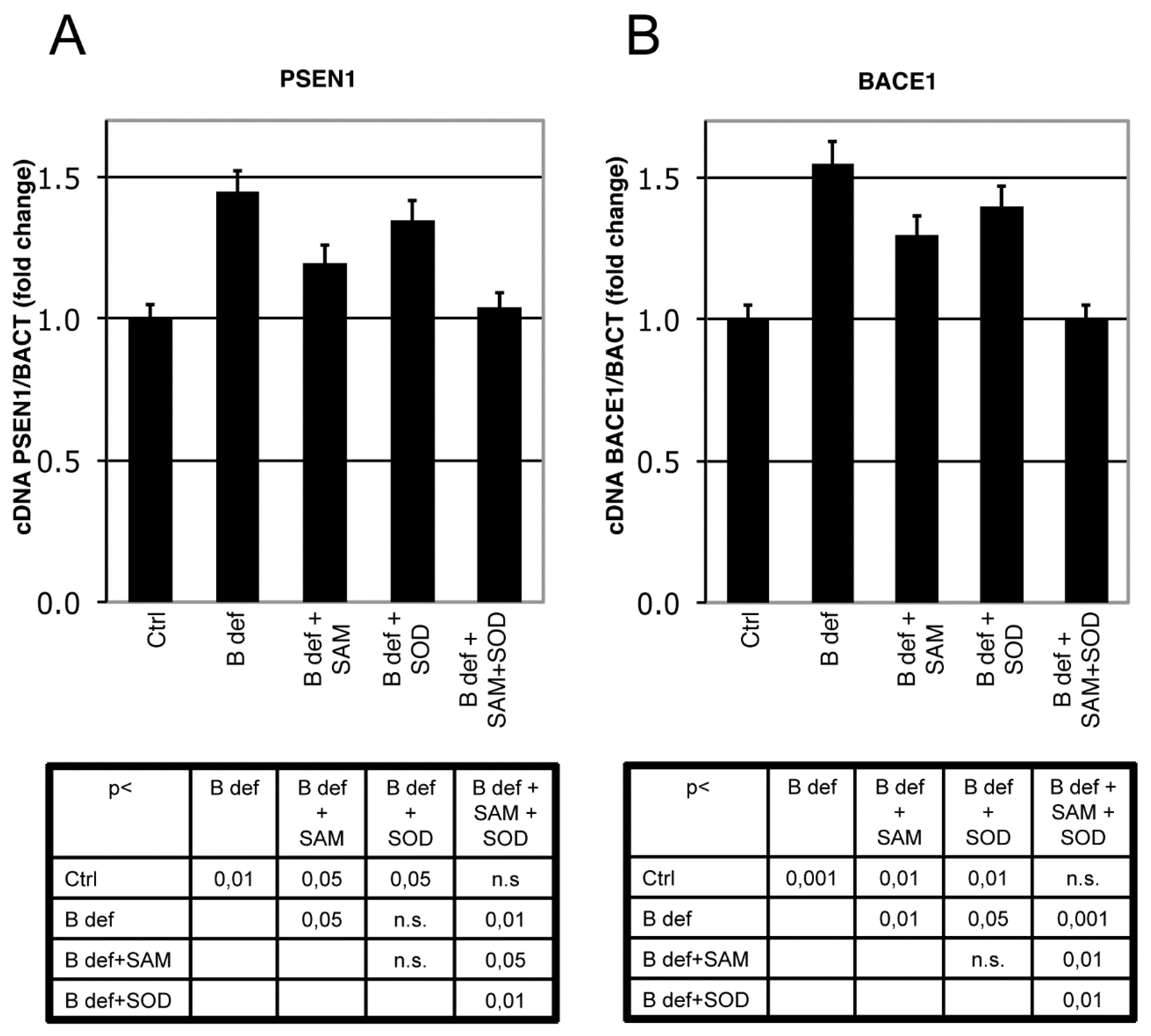
3.3. SAM and SOD Reduce PSEN1 and BACE Expression
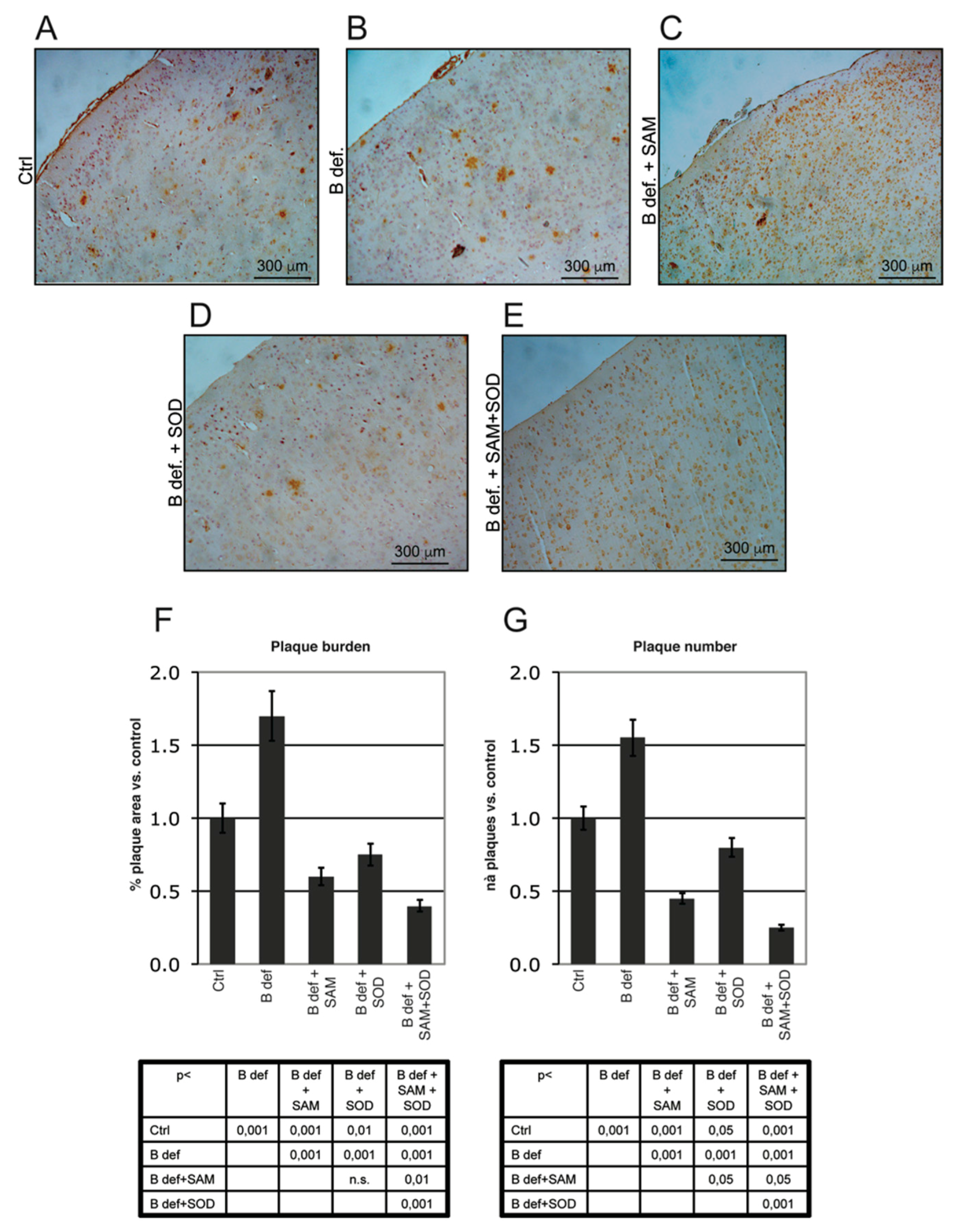
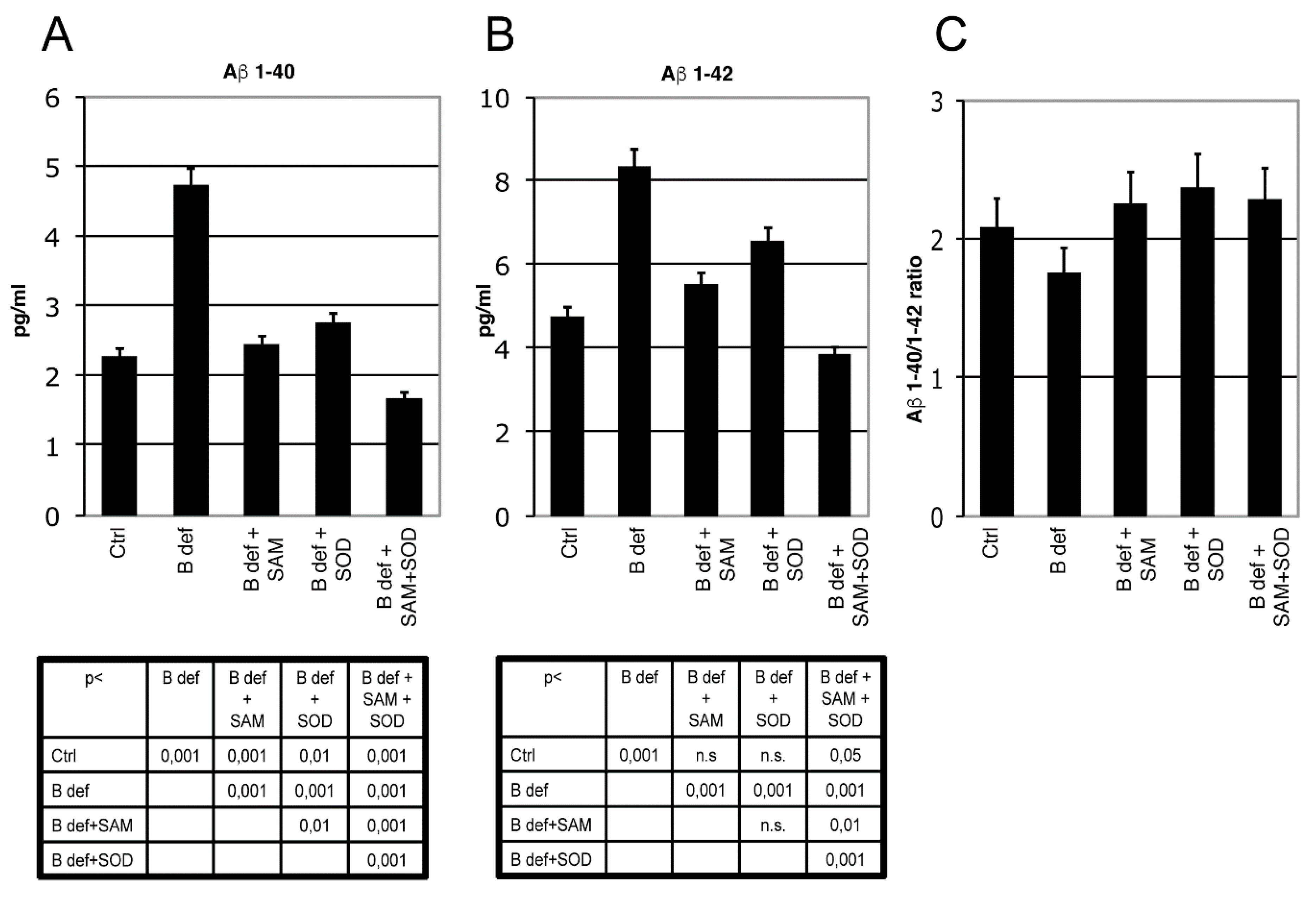
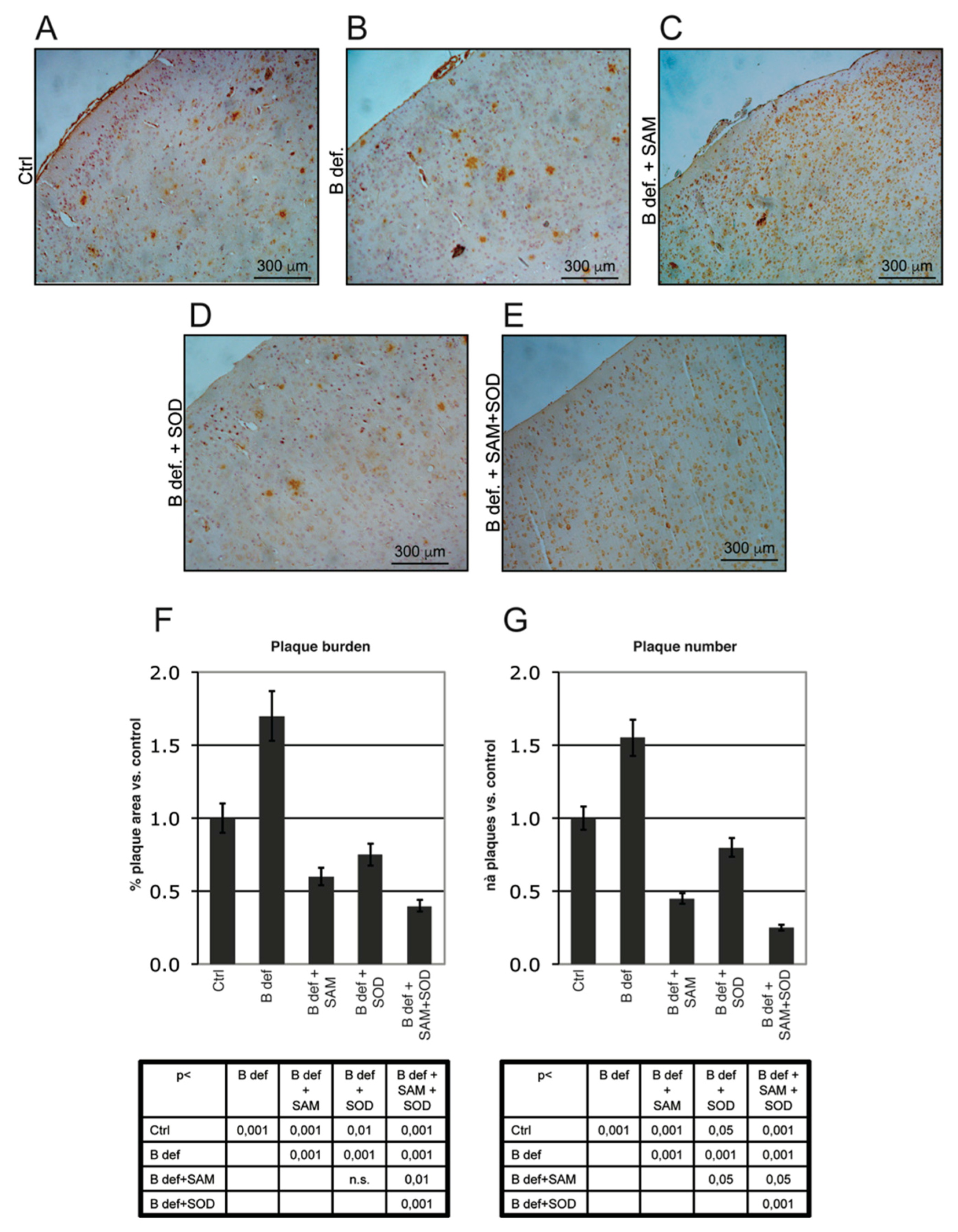
3.4. SAM and SOD Reduce Amyloid Production and Plaque Burden
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimers Dement. 2013, 9, 63–75.e2. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- García-Blanco, A.; Baquero, M.; Vento, M.; Gil, E.; Bataller, L.; Cháfer-Pericás, C. Potential oxidative stress biomarkers of mild cognitive impairment due to Alzheimer disease. J. Neurol. Sci. 2017, 373, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Giulietti, A.; Vignini, A.; Nanetti, L.; Mazzanti, L.; Di Primio, R.; Salvolini, E. Alzheimer’s Disease Risk and Progression: The Role of Nutritional Supplements and their Effect on Drug Therapy Outcome. Curr. Neuropharmacol. 2016, 14, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Grundke-Iqbal, I. Alzheimer’s disease, a multifactorial disorder seeking Multitherapies. Alzheimers Dement. 2010, 6, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Migliore, L.; Coppede, F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat. Res. 2009, 667, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.K.; Mooney, R.P. The predisposing factors, biological markers, neuroimaging techniques and medical complications associated with Alzheimer’s disease. W. Va. Med. J. 2011, 107, 26–29. [Google Scholar] [PubMed]
- Herrmann, W.; Obeid, R. Homocysteine: A biomarker in neurodegenerative diseases. Clin. Chem. Lab. Med. 2011, 49, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Castellani, R.J.; Zhu, X.; Moreira, P.I.; Perry, G.; Smith, M.A. Involvement of oxidative stress in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2006, 65, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Meraz-Ríos, M.A.; Franco-Bocanegra, D.; Toral Rios, D.; Campos-Peña, V. Early Onset Alzheimer’s Disease and Oxidative Stress. Oxid. Med. Cell. Longev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Refsum, H. Homocysteine, B Vitamins, and Cognitive Impairment. Annu. Rev. Nutr. 2016, 36, 211–239. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxid. Med. Cell. Longev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Wang, X.; Lee, H.G.; Smith, M.A.; Perry, G.; Zhu, X. Neuronal failure in Alzheimer’s disease: A view through the oxidative stress looking-glass. Neurosci. Bull. 2014, 30, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Markesbery, W.R. Oxidative damage in mild cognitive impairment and early Alzheimer’s disease. J. Neurosci. Res. 2007, 85, 3036–3040. [Google Scholar] [CrossRef] [PubMed]
- Casado, A.; Lopez-Fernandez, M.E.; Casado, M.C.; De La Torre, R. Lipid peroxidation and antioxidant enzyme activities in vascular and Alzheimer dementias. Neurochem. Res. 2008, 33, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Chang, W.N.; Tsai, N.W.; Huang, C.C.; Kung, C.T.; Su, Y.J.; Lin, W.C.; Cheng, B.C.; Su, C.M.; Chiang, Y.F.; et al. The Roles of Biomarkers of Oxidative Stress and Antioxidant in Alzheimer’s Disease: A Systematic Review. BioMed Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Bradley-Whitman, M.A.; Lovell, M.A. Biomarkers of lipid peroxidation in Alzheimer disease (AD): An update. Arch. Toxicol. 2015, 89, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F.; Migliore, L. DNA damage in neurodegenerative diseases. Mutat. Res. 2015, 776, 84–97. [Google Scholar] [CrossRef] [PubMed]
- França, M.B.; Lima, K.C.; Eleutherio, E.C. Oxidative Stress and Amyloid Toxicity: Insights from Yeast. J. Cell. Biochem. 2017, 118, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid beta-peptide (1–42)-induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Saharana, S.; Mandal, P.K. The Emerging Role of Glutathione in Alzheimer’s Disease. J. Alzheimers Dis. 2014, 40, 519–529. [Google Scholar]
- B´elanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Mariani, E.; Polidori, M.C.; Cherubini, A.; Mecocci, P. Oxidative stress in brain aging, neurodegenerative and vascular disease: An overview. J. Chromatogr. B. 2005, 827, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer’s disease. Neurology 1998, 51, 1562–1566. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of Glutathione Homeostasis in Neurodegenerative Diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K.; et al. SOD1 (Copper/Zinc Superoxide Dismutase) deficiency drives Amyloid β protein oligomerization and memory loss in mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 44557–44568. [Google Scholar] [CrossRef] [PubMed]
- Massaad, C.A.; Washington, T.M.; Pautler, R.G.; Klann, E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 13576–13581. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.D. The metabolism of homocysteine: Pathways and regulation. Eur. J. Pediatr. 1998, 157, S40–S44. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.D. Metabolic regulatory properties of S-adenosylmethionine and S-adenosylhomocysteine. Clin. Chem. Lab. Med. 2007, 45, 1694–1699. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.L.; Phiel, C.J.; Toland, A.E. The role for oxidative stress in aberrant DNA methylation in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 1077–1096. [Google Scholar] [CrossRef] [PubMed]
- Troesch, B.; Weber, P.; Mohajeri, M.H. Potential Links between Impaired One-Carbon Metabolism Due to Polymorphisms, Inadequate B-Vitamin Status, and the Development of Alzheimer’s Disease. Nutrients 2016, 8, 803. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Cavallaro, R.A.; Ricceri, L.; D’Anselmi, F.; Coluccia, P.; Calamandrei, G.; Scarpa, S. B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-beta deposition in mice. Mol. Cell. Neurosci. 2008, 37, 731–746. [Google Scholar] [CrossRef] [PubMed]
- Nicolia, V.; Fuso, A.; Cavallaro, R.A.; Di Luzio, A.; Scarpa, S. B vitamin deficiency promotes tau phosphorylation through regulation ofGSK3b and PP2A. J. Alzheimers Dis. 2010, 19, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Pasqualato, A.; Fiorenza, M.T.; Cavallaro, R.A.; Scarpa, S. Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamindeficiency. Neurobiol. Aging 2011, 32, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Cavallaro, R.A.; Scarpa, S. DNA methylase and demethylaseactivities are modulated by one-carbon metabolism in Alzheimer’s disease models. J. Nutr. Biochem. 2011, 22, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Ricceri, L.; Cavallaro, R.A.; Isopi, E.; Mangia, F.; Fiorenza, M.T.; Scarpa, S. S-adenosylmethionine reduces the progress of the Alzheimer-like features induced by B-vitamin deficiency in mice. Neurobiol. Aging 2012, 33, 1482 e1–1482.e16. [Google Scholar] [CrossRef]
- Cavallaro, R.A.; Fuso, A.; Nicolia, V.; Scarpa, S. S-adenosylmethionine prevents oxidative stress and modulates glutathione metabolism in TgCRND8 mice fed a B-vitamin deficient diet. J. Alzheimers Dis. 2010, 20, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Persichilli, S.; Gervasoni, J.; Di Napoli, A.; Fuso, A.; Nicolia, V.; Giardina, B.; Scarpa, S.; Desiderio, C.; Cavallaro, R.A. Plasma thiols levels in Alzheimer’s disease mice under diet-induced hyperhomocysteinemia: Effect of S-adenosylmethionine and superoxide-dismutase supplementation. J. Alzheimers Dis. 2015, 44, 1323–1331. [Google Scholar] [PubMed]
- Olajide, O.J.; Yawson, E.O.; Gbadamosi, I.T.; Arogundade, T.T.; Lambe, E.; Obasi, K.; Lawal, I.T.; Ibrahim, A.; Ogunrinola, K.Y. Ascorbic acid ameliorates behavioural deficits and neuropathological alterations in rat model of Alzheimer’s disease. Environ. Toxicol. Pharmacol. 2017, 50, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Persson, T.; Popescu, B.O.; Cedazo-Minguez, A. Oxidative Stress in Alzheimer’s Disease: Why Did AntioxidantTherapy Fail? Oxid. Med. Cell. Longev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Conte, V.; Uryu, K.; Fujimoto, S.; Yao, Y.; Rokach, J.; Longhi, L.; Trojanowski, J.Q.; Lee, V.M.-Y.; McIntosh, T.K.; Praticò, D. Vitamin E Reduces Amyloidosis and Improves Cognitive Function in Tg2576 Mice Following Repetitive Concussive Brain Injury. J. Neurochem. 2004, 90, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Lloret, A.; Badía, M.-C.; Mora, N.J.; Pallardó, F.V.; Alonso, M.-D.; Viña, J. Vitamin E Paradox in Alzheimer’s Disease: It Does Not Prevent Loss of Cognition and May Even Be Detrimental. J. Alzheimers Dis. 2009, 17, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Li, F.J.; Shen, L.; Ji, H.F. Dietary Intakes of Vitamin E, Vitamin C, and Β-Carotene and Risk of Alzheimer’s Disease: A Meta- Analysis. J. Alzheimers Dis. 2012, 31, 253–258. [Google Scholar] [PubMed]
- Canterini, S.; Bosco, A.; Carletti, V.; Fuso, A.; Curci, A.; Mangia, F.; Fiorenza, M.T. Subcellular TSC22D4 localization in cerebellum granule neurons of the mouse depends on development and differentiation. Cerebellum 2012, 11, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Gerbarg, P.; Bottiglieri, T.; Massoumi, L.; Carpenter, L.L.; Lavretsky, H.; Muskin, P.R.; Brown, R.P.; Mischoulon, D.; As Work Group of the American Psychiatric Association Council on Research. S-Adenosylmethionine (SAMe) for Neuropsychiatric Disorders: A Clinician-Oriented Review of Research. J. Clin. Psychiatry 2017, 78, e656–e667. [Google Scholar] [CrossRef] [PubMed]
- Romao, S. Therapeutic value of oral supplementation with melon superoxide dismutase and wheat gliadin combination. Nutrition 2015, 31, 430–436, Erratum in: Nutrition 2015, 31, 1187. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K.; Maloney, B.; Basha, M.R.; Ge, Y.W.; Zawia, N.H. How and when environmental agents and dietary factors affect the course of Alzheimer’s disease: The “LEARn” model (latent early-life associatedregulation) may explain the triggering of AD. Curr. Alzheimer Res. 2007, 4, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Basha, M.R.; Zawia, N.H. The environment, epigenetics and amyloidogenesis. J. Mol. Neurosci. 2008, 34, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F.; Tannorella, P.; Pezzini, I.; Migheli, F.; Ricci, G.; Caldarazzo lenco, E.; Piaceri, I.; Polini, A.; Nacmias, B.; Monzani, F.; Sorbi, S.; Siciliano, G.; Migliore, L. Folate, homocysteine, vitamin B12, and polymorphisms of genes participating in one-carbon metabolism in late-onset Alzheimer’s disease patients and healthy controls. Antioxid. Redox Signal. 2012, 17, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Tang, M.X.; Miller, J.; Green, R.; Mehta, P.D.; Mayeux, R. Relation of plasma homocysteine to plasma amyloid beta levels. Neurochem. Res. 2007, 32, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Haan, M.N.; Miller, J.W.; Aiello, A.E.; Whitmer, R.A.; Jagust, W.J.; Mungas, D.M.; Allen, L.H.; Green, R. Homocysteine, B vitamins, and the incidence of dementia and cognitive impairment: Results from the Sacramento Area Latino Study on Aging. Am. J. Clin. Nutr. 2007, 85, 511–517. [Google Scholar] [PubMed]
- Pocernich, C.B.; Butterfield, D.A. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim. Biophys. Acta 2011, 1822, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Rodriguez, C.; Spaulding, J.; Aw, T.Y.; Feng, J. Age-dependent and tissue-related glutathione redox status in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2012, 28, 655–666. [Google Scholar] [PubMed]
- Takumi, S.; Okamura, K.; Yanagisawa, H.; Sano, T.; Kobayashi, Y.; Nohara, K. The effect of a methyl-deficient diet on the global DNA methylation and the DNA methylation regulatory pathways. J. Appl. Toxicol. 2015, 35, 1550–1556. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.-F.S.; Hsu, Y.-C.; Lin, H.-L.; Yang, F.L. Folate depletion and elevated plasma homocysteine promo teoxidative stress in rat livers. J. Nutr. 2001, 131, 33–38. [Google Scholar] [PubMed]
- Aksenov, M.Y.; Aksenova, M.V.; Butterfield, D.A.; Geddes, J.W.; Markesbery, W.R. Protein oxidation in the brain in Alzheimer’s disease. Neuroscience 2001, 103, 373–383. [Google Scholar] [CrossRef]
- Keller, J.N.; Schmitt, F.A.; Scheff, S.W.; Ding, Q.; Chen, Q.; Butterfield, D.A.; Markesbery, W.R. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 2005, 64, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.N.; Kasahara, E.; Hiraoka, M.; Lin, L.R.; Ho, Y.S. Effects of variation in superoxide dismutases (SOD) on oxidative stress and apoptosis in lens epithelium. Exp. Eye Res. 2004, 79, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Elchuri, S.; Oberley, T.D.; Qi, W.; Eisenstein, R.S.; Jackson Roberts, L.; Van Remmen, H.; Epstein, C.J.; Huang, T.T. Cu ZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2005, 24, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.; Song, W.; Liu, Y.; Chaudhuri, A.; Pieke-Dahl, S.; Strong, R.; Huang, T.T.; Epstein, C.J.; Roberts, L.J., II; Csete, M.; et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 2006, 40, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Glass, J.D. Oxidative stress induced by loss of Cu, Zn-superoxide dismutase (SOD1) or superoxide-generating herbicides causes axonal degeneration in mouse DRG cultures. Acta Neuropath. 2010, 119, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Esposito, L.; Raber, J.; Kekonius, L.; Yan, F.; Yu, G.Q.; Bien-Ly, N.; Puoliväli, J.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J. Neurosci. 2006, 26, 5167–5179. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, X. Antioxidant therapies for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2012. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiecki, M.M.; Hall, M.N.; Liu, X.; Oka, J.; Harper, K.N.; Slavkovich, V.; Ilievski, V.; Levy, D.; van Geen, A.; Mey, J.L.; et al. Blood glutathione redox status and global methylation of peripheral blood mononuclear cell DNA in Bangladeshi adults. Epigenetics 2013, 8, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Zhou, W.; Fung, V.; Christensen, M.A.; Qing, H.; Sun, X.; Song, W. Oxidative stress potentiates BACE1 gene expression and Aβ generation. J. Neural Transm. 2005, 112, 455. [Google Scholar] [CrossRef] [PubMed]
- Tallarida, R.J. Quantitative methods for assessing drug synergism. Genes Cancer 2011, 2, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Do Carmo, S.; Hanzel, C.E.; Jacobs, M.L.; Machnes, Z.; Iulita, M.F.; Yang, J.; Yu, L.; Ducatenzeiler, A.; Danik, M.; Breuillaud, L.S.; et al. Rescue of Early bace-1 and Global DNA Demethylation by S-Adenosylmethionine Reduces Amyloid Pathology and Improves Cognition in an Alzheimer’s Model. Sci. Rep. 2016, 6, 34051. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Remington, R.; Kotyla, E.; Lepore, A.; Zemianek, J.; Shea, T.B. A vitamin/nutriceutical formulation improves memory and cognitive performance in community-dwelling adults without dementia. J. Nutr. Health Aging 2010, 14, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Paskavitz, J.; Remington, R.; Rasmussen, S.; Shea, T.B. Efficacy of a vitamin/nutriceutical formulation for early stage Alzheimer Disease: A 1 year open label pilot study with an 16 months care-giver extension. Am. J. Alzheimers Dis. Other Dement. 2008, 23, 571–585. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavallaro, R.A.; Nicolia, V.; Fiorenza, M.T.; Scarpa, S.; Fuso, A. S-Adenosylmethionine and Superoxide Dismutase 1 Synergistically Counteract Alzheimer’s Disease Features Progression in TgCRND8 Mice. Antioxidants 2017, 6, 76. https://doi.org/10.3390/antiox6040076
Cavallaro RA, Nicolia V, Fiorenza MT, Scarpa S, Fuso A. S-Adenosylmethionine and Superoxide Dismutase 1 Synergistically Counteract Alzheimer’s Disease Features Progression in TgCRND8 Mice. Antioxidants. 2017; 6(4):76. https://doi.org/10.3390/antiox6040076
Chicago/Turabian StyleCavallaro, Rosaria A., Vincenzina Nicolia, Maria Teresa Fiorenza, Sigfrido Scarpa, and Andrea Fuso. 2017. "S-Adenosylmethionine and Superoxide Dismutase 1 Synergistically Counteract Alzheimer’s Disease Features Progression in TgCRND8 Mice" Antioxidants 6, no. 4: 76. https://doi.org/10.3390/antiox6040076
APA StyleCavallaro, R. A., Nicolia, V., Fiorenza, M. T., Scarpa, S., & Fuso, A. (2017). S-Adenosylmethionine and Superoxide Dismutase 1 Synergistically Counteract Alzheimer’s Disease Features Progression in TgCRND8 Mice. Antioxidants, 6(4), 76. https://doi.org/10.3390/antiox6040076