
Na/K-ATPase Signaling and Salt Sensitivity: The Role of Oxidative Stress



{kind=link}
Abstract
:1. Introduction
2. The Concept of “Natriuretic Hormone” and Renal Sodium Handling
3. The Role of CTS in Na/K-ATPase Signaling Mediated RPT Sodium Handling
4. The Role of ROS in Na/K-ATPase Signaling Mediated RPT Sodium Handling
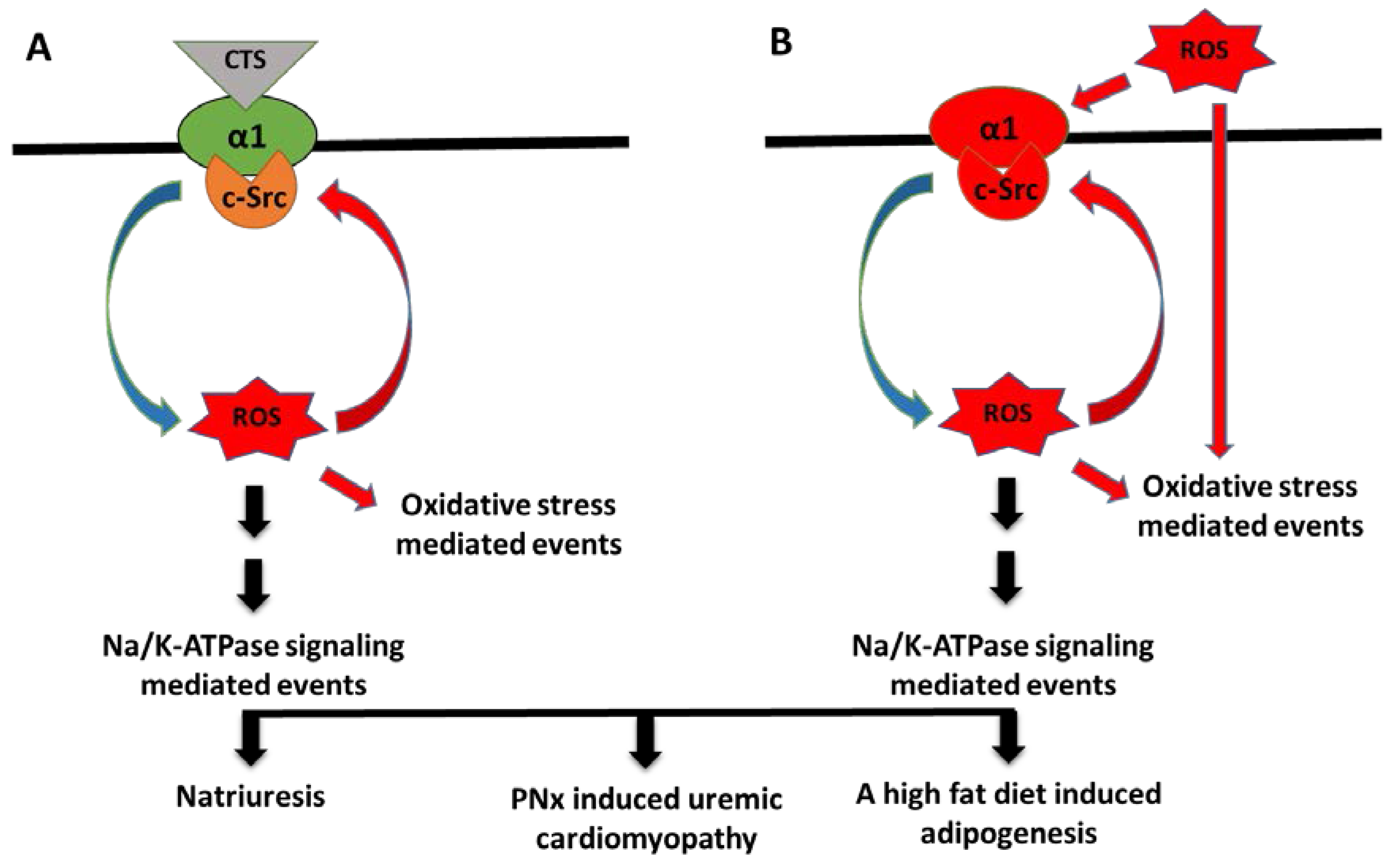
5. The ROS Paradox
6. Conclusions and Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Després, J.-P.; Fullerton, H.J.; et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016, 133, e38–e360. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, D.A.; Jones, D.; Textor, S.; Goff, D.C.; Murphy, T.P.; Toto, R.D.; White, A.; Cushman, W.C.; White, W.; Sica, D.; et al. Resistant Hypertension: Diagnosis, Evaluation, and Treatment: A Scientific Statement From the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2008, 51, 1403–1419. [Google Scholar] [CrossRef] [PubMed]
- Overlack, A.; Ruppert, M.; Kolloch, R.; Kraft, K.; Stumpe, K.O. Age is a major determinant of the divergent blood pressure responses to varying salt intake in essential hypertension. Am. J. Hypertens. 1995, 8, 829–836. [Google Scholar] [CrossRef]
- Weinberger, M.H.; Miller, J.Z.; Luft, F.C. Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension 1986, 8, II127–II134. [Google Scholar] [CrossRef] [PubMed]
- Boudville, N.; Ward, S.; Benaroia, M.; House, A.A. Increased sodium intake correlates with greater use of antihypertensive agents by subjects with chronic kidney disease. Am. J. Hypertens. 2005, 18, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Elijovich, F.; Weinberger, M.H.; Anderson, C.A.M.; Appel, L.A.; Bursztyn, M.; Cook, N.R.; Dart, R.A.; Newton-Cheh, C.H.; Sacks, F.M.; Laffer, C.L.; et al. Salt Sensitivity of Blood Pressure: A Scientific Statement From the American Heart Association. Hypertension 2016, 68, e7–e46. [Google Scholar] [CrossRef] [PubMed]
- Dahl, L.K.; Heine, M.; Thompson, K. Genetic influence of the kidneys on blood pressure. Evidence from chronic renal homografts in rats with opposite predispositions to hypertension. Circ. Res. 1974, 40, 94–101. [Google Scholar] [CrossRef]
- Bianchi, G.; Fox, U.; di Francesco, G.F.; Bardi, U. The hypertensive role of the kidney in spontaneously hypertensive rats. Clin. Sci. Mol. Med. Suppl. 1973, 45, 135s–139s. [Google Scholar] [CrossRef] [PubMed]
- Heller, J.; Schubert, G.; Havlíčkova, J.; Thurau, K. The role of the kidney in the development of hypertension: A transplantation study in the Prague hypertensive rat. Pflugers Arch. 1993, 425, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.A.; DiBona, G.F.; Mark, A.L. Effects of interstrain renal transplantation on NaCl-induced hypertension in Dahl rats. Hypertension 1990, 15, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.J.; Luke, R.G.; Dustan, H.P.; Kashgarian, M.; Whelchel, J.D.; Jones, P.; Diethelm, A.G. Remission of essential hypertension after renal transplantation. N. Engl. J. Med. 1983, 309, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- He, F.J.; MacGregor, G.A. Effect of longer-term modest salt reduction on blood pressure. Cochrane Database Syst. Rev. 2004. [Google Scholar] [CrossRef]
- Guyton, A.C. Blood pressure control—Special role of the kidneys and body fluids. Science 1991, 252, 1813–1816. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; Granger, J.P.; do Carmo, J.M.; da Silva, A.A.; Dubinion, J.; George, E.; Hamza, S.; Speed, J.; Hall, M.E. Hypertension: Physiology and pathophysiology. Compr. Physiol. 2012, 2, 2393–2442. [Google Scholar] [PubMed]
- Doris, P.A. Renal proximal tubule sodium transport and genetic mechanisms of essential hypertension. J. Hypertens. 2000, 18, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Burnier, M.; Bochud, M.; Maillard, M. Proximal tubular function and salt sensitivity. Curr. Hypertens. Rep. 2006, 8, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Skrabal, F.; Herholz, H.; Neumayr, M.; Hamberger, L.; Ledochowski, M.; Sporer, H.; Hörtnagl, H.; Schwarz, S.; Schönitzer, D. Salt sensitivity in humans is linked to enhanced sympathetic responsiveness and to enhanced proximal tubular reabsorption. Hypertension 1984, 6, 152–158. [Google Scholar] [PubMed]
- Chiolero, A.; Wurzner, G.; Burnier, M. Renal determinants of the salt sensitivity of blood pressure. Nephrol. Dial. Transplant. 2001, 16, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Chiolero, A.; Maillard, M.; Nussberger, J.; Brunner, H.R.; Burnier, M. Proximal sodium reabsorption: An independent determinant of blood pressure response to salt. Hypertension 2000, 36, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Barba, G.; Cappuccio, F.P.; Russo, L.; Stinga, F.; Iacone, R.; Strazzullo, P. Renal function and blood pressure response to dietary salt restriction in normotensive men. Hypertension 1996, 27, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Strazzullo, P.; Barba, G.; Cappuccio, F.P.; Siani, A.; Trevisan, M.; Farinaro, E.; Pagano, E.; Barbato, A.; Iacone, R.; Galletti, F. Altered renal sodium handling in men with abdominal adiposity: A link to hypertension. J. Hypertens. 2001, 19, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Harris, P.J.; Morgan, T.O. Age-related changes in angiotensin II-stimulated proximal tubule fluid reabsorption in the spontaneously hypertensive rat. J. Hypertens. Suppl. 1988, 6, S449–S451. [Google Scholar] [CrossRef] [PubMed]
- Biollaz, J.; Waeber, B.; Diezi, J.; Burnier, M.; Brunner, H.R. Lithium infusion to study sodium handling in unanesthetized hypertensive rats. Hypertension 1986, 8, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Boer, P.A.; Morelli, J.M.; Figueiredo, J.F.; Gontijo, J.A.R. Early altered renal sodium handling determined by lithium clearance in spontaneously hypertensive rats (SHR): Role of renal nerves. Life Sci. 2005, 76, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.C.; Kirchner, K.A.; Abernethy, J.D.; Langford, H.G. Differential effect of salt loading on sodium and lithium excretion in Dahl salt-resistant and -sensitive rats. Hypertension 1984, 6, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yan, Y.; Liu, L.; Xie, Z.; Malhotra, D.; Joe, B.; Shapiro, J.I. Impairment of Na/K-ATPase signaling in renal proximal tubule contributes to Dahl salt-sensitive hypertension. J. Biol. Chem. 2011, 286, 22806–22813. [Google Scholar] [CrossRef] [PubMed]
- Ferrandi, M.; Salardi, S.; Tripodi, G.; Barassi, P.; Rivera, R.; Manunta, P.; Goldshleger, R.; Ferrari, P.; Bianchi, P.; Karlish, S.J.D. Evidence for an interaction between adducin and Na+-K+-ATPase: Relation to genetic hypertension. Am. J. Physiol. 1999, 277, H1338–H1349. [Google Scholar] [PubMed]
- Blaustein, M.P. Sodium ions, calcium ions, blood pressure regulation, and hypertension: A reassessment and a hypothesis. Am. J. Physiol. Cell Physiol. 1977, 232, C165–C173. [Google Scholar]
- Haddy, F.J.; Pamnani, M.B.; Clough, D.L. Humoral factors and the sodium-potassium pump in volume expanded hypertension. Life Sci. 1979, 24, 2105–2117. [Google Scholar] [CrossRef]
- De Wardener, H.E.; Clarkson, E.M. Concept of natriuretic hormone. Physiol. Rev. 1985, 65, 658–759. [Google Scholar] [PubMed]
- Kelly, R.A.; Smith, T.W. The search for the endogenous digitalis: An alternative hypothesis. Am. J. Physiol. 1989, 256, C937–C950. [Google Scholar] [PubMed]
- Bagrov, A.Y.; Shapiro, J.I. Endogenous digitalis: Pathophysiologic roles and therapeutic applications. Nat. Clin. Pract. Nephrol. 2008, 4, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Buckalew, V.M. Endogenous digitalis-like factors. An historical overview. Front. Biosci. 2005, 10, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Shapiro, J.I.; Bagrov, A.Y. Endogenous cardiotonic steroids and salt-sensitive hypertension. Biochim. Biophys. Acta 2010, 1802, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xie, Z.J. The sodium pump and cardiotonic steroids-induced signal transduction protein kinases and calcium-signaling microdomain in regulation of transporter trafficking. Biochim. Biophys. Acta 2010, 1802, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Schoner, W.; Scheiner-Bobis, G. Role of endogenous cardiotonic steroids in sodium homeostasis. Nephrol. Dial. Transplant. 2008, 23, 2723–2729. [Google Scholar] [CrossRef] [PubMed]
- McDougall, J.G.; Yates, N.A. Natriuresis and inhibition of Na+/K+-ATPase: Modulation of response by physiological manipulation. Clin. Exp. Pharmacol. Physiol. Suppl. 1998, 25, S57–S60. [Google Scholar] [CrossRef] [PubMed]
- Yates, N.A.; McDougall, J.G. Effects of direct renal arterial infusion of bufalin and ouabain in conscious sheep. Br. J. Pharmacol. 1993, 108, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Yates, N.A.; McDougall, J.G. Interaction of exogenous ouabain and chronic mineralocorticoid treatment in the kidney of the conscious sheep. Clin. Exp. Pharmacol. Physiol. 1997, 24, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, M.A.; Sandberg, S.M.; Edwards, B.S. Role of renal Na+,K(+)-ATPase in the regulation of sodium excretion under normal conditions and in acute congestive heart failure. Circulation 1992, 85, 1912–1917. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Agalakova, N.I.; Morrell, C.H.; Lakatta, E.G.; Bagrov, A.Y. ANP differentially modulates marinobufagenin-induced sodium pump inhibition in kidney and aorta. Hypertension 2006, 48, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shapiro, J.I. Regulation of sodium pump endocytosis by cardiotonic steroids: Molecular mechanisms and physiological implications. Pathophysiology 2007, 14, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Lakatta, E.G.; Bagrov, A.Y. Endogenous Na,K Pump Ligands Are Differentially Regulated During Acute NaCl Loading of Dahl Rats. Circulation 2000, 102, 3009–3014. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Talan, M.I.; Agalakova, N.I.; Lakatta, E.G.; Bagrov, A.Y. Endogenous Ligand of {alpha}1 Sodium Pump, Marinobufagenin, Is a Novel Mediator of Sodium Chloride-Dependent Hypertension. Circulation 2002, 105, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Haddy, F.J.; Pamnani, M.B. Role of ouabain-like factors and Na-K-ATPase inhibitors in hypertension—Some old and recent findings. Clin. Exp. Hypertens. 1998, 20, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Manunta, P.; Hamilton, B.P.; Hamlyn, J.M. Salt intake and depletion increase circulating levels of endogenous ouabain in normal men. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R553–R559. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Haller, S.; Periyasamy, S.; Brewster, P.; Zhang, H.; Adlakha, S.; Fedorova, O.V.; Xie, Z.; Bagrov, A.Y.; Shapiro, J.I. Renal Ischemia Regulates Marinobufagenin Release in Humans. Hypertension 2010, 56, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Schoner, W.; Scheiner-Bobis, G. Endogenous and exogenous cardiac glycosides: Their roles in hypertension, salt metabolism, and cell growth. Am. J. Physiol. Cell Physiol. 2007, 293, C509–C536. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, J.M.; Blaustein, M.P.; Bova, S.; DuCharme, D.W.; Harris, D.W.; Mandel, F.; Mathews, W.R.; Ludens, J.H. Identification and characterization of a ouabain-like compound from human plasma. Proc. Natl. Acad. Sci. USA 1991, 88, 6259–6263. [Google Scholar] [CrossRef] [PubMed]
- Laredo, J.; Shah, J.R.; Lu, Z.; Hamilton, B.P.; Hamlyn, J.M. Angiotensin II stimulates secretion of endogenous ouabain from bovine adrenocortical cells via angiotensin type 2 receptors. Hypertension 1997, 29, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Fedorova, O.V.; Dmitriev, R.I.; French, A.W.; Anderson, D.E. Plasma marinobufagenin-like and ouabain-like immunoreactivity during saline volume expansion in anesthetized dogs. Cardiovasc. Res. 1996, 31, 296–305. [Google Scholar] [CrossRef]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Kolodkin, N.I.; Agalakova, N.I.; Lakatta, E.G.; Bagrov, A.Y. Marinobufagenin, an endogenous alpha-1 sodium pump ligand, in hypertensive Dahl salt-sensitive rats. Hypertension 2001, 37, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Dostanic-Larson, I.; van Huysse, J.W.; Lorenz, J.N.; Lingrel, J.B. The highly conserved cardiac glycoside binding site of Na,K-ATPase plays a role in blood pressure regulation. Proc. Natl. Acad. Sci. USA 2005, 102, 15845–15850. [Google Scholar] [CrossRef] [PubMed]
- Loreaux, E.L.; Kaul, B.; Lorenz, J.N.; Lingrel, J.B. Ouabain-Sensitive alpha1 Na,K-ATPase enhances natriuretic response to saline load. J. Am. Soc. Nephrol. 2008, 19, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ren, Y.P.; Lv, Z.R. Ouabain induces cardiac remodeling in rats independent of blood pressure. Acta Pharmacol. Sin. 2007, 28, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Skoumal, R.; Szokodi, I.; Aro, J.; Földes, G.; Göőz, M.; Seres, L.; Sármán, B.; Lakó-Futó, Z.; Papp, L.; Vuolteenaho, O.; et al. Involvement of endogenous ouabain-like compound in the cardiac hypertrophic process in vivo. Life Sci. 2007, 80, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Vetteth, S.; Periyasamy, S.M.; Kanj, M.; Fedorova, L.; Khouri, S.; Kahaleh, M.B.; Xie, Z.; Malhotra, D.; Kolodkin, N.I. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 2006, 47, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Ferrandi, M.; Molinari, I.; Barassi, P.; Minotti, E.; Bianchi, G.; Ferrari, P. Organ hypertrophic signaling within caveolae membrane subdomains triggered by ouabain and antagonized by PST 2238. J. Biol. Chem. 2004, 279, 33306–33314. [Google Scholar] [CrossRef] [PubMed]
- Feraille, E.; Doucet, A. Sodium-potassium-adenosinetriphosphatase-dependent sodium transport in the kidney: Hormonal control. Physiol. Rev. 2001, 81, 345–418. [Google Scholar] [PubMed]
- Chibalin, A.V.; Ogimoto, G.; Pedemonte, C.H.; Pressley, T.A.; Katz, A.I.; Féraille, E.; Berggren, P.-O.; Bertorello, A.M. Dopamine-induced endocytosis of Na+,K+-ATPase is initiated by phosphorylation of Ser-18 in the rat alpha subunit and Is responsible for the decreased activity in epithelial cells. J. Biol. Chem. 1999, 274, 1920–1927. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Fan, L.; Crowder, L.A.; Karim-Jimenez, Z.; Murer, H.; Moe, O.W. Dopamine Acutely Stimulates Na+/H+Exchanger (NHE3) Endocytosis via Clathrin-coated Vesicles. J. Biol. Chem. 2001, 276, 26906–26915. [Google Scholar] [CrossRef] [PubMed]
- Bacic, D.; Kaissling, B.; McLeroy, P.; Zou, L.; Baum, M.; Moe, O.W. Dopamine acutely decreases apical membrane Na//H exchanger NHE3 protein in mouse renal proximal tubule. Kidney Int. 2003, 64, 2133–2141. [Google Scholar] [CrossRef] [PubMed]
- Dada, L.A.; Chandel, N.S.; Ridge, K.M.; Pedemonte, C.; Bertorello, A.M.; Sznajder, J.I. Hypoxia-induced endocytosis of Na,K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-zeta. J. Clin. Investig. 2003, 111, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- McDonough, A.A. Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R851–R861. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.P. Why isn’t endogenous ouabain more widely accepted? Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H635–H639. [Google Scholar] [CrossRef] [PubMed]
- Lingrel, J.B. The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Annu. Rev. Physiol. 2010, 72, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xie, Z. The Na/K-ATPase/Src complex and cardiotonic steroid-activated protein kinase cascades. Pflugers Arch. 2009, 457, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Xie, Z.J. The Na-K-ATPase and calcium-signaling microdomains. Physiology (Bethesda) 2008, 23, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Cai, T. Na+-K+-ATPase-mediated signal transduction: From protein interaction to cellular function. Mol. Interv. 2003, 3, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Askari, A. Na(+)/K(+)-ATPase as a signal transducer. Eur. J. Biochem. 2002, 269, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Xie, J. The Na/K-ATPase-mediated signal transduction as a target for new drug development. Front. Biosci. 2005, 10, 3100–3109. [Google Scholar] [CrossRef] [PubMed]
- Aperia, A. New roles for an old enzyme: Na,K-ATPase emerges as an interesting drug target. J. Intern. Med. 2007, 261, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.V.; Xie, Z. The Na,K-ATPase receptor complex: Its organization and membership. Cell Biochem. Biophys. 2006, 46, 303–316. [Google Scholar] [CrossRef]
- Schoner, W. Endogenous cardiac glycosides, a new class of steroid hormones. Eur. J. Biochem. 2002, 269, 2440–2448. [Google Scholar] [CrossRef] [PubMed]
- Schoner, W.; Scheiner-Bobis, G. Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am. J. Cardiovasc. Drugs 2007, 7, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Ferrandi, M.; Molinari, I.; Bianchi, G.; Ferrari, P. Ouabain-dependent signaling in caveolae as a novel therapeutic target for hypertension. Cell. Mol. Biol. (Noisy-le-Grand) 2006, 52, 15–18. [Google Scholar]
- Liu, J.; Tian, J.; Haas, M.; Shapiro, J.I.; Askari, A.; Xie, Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J. Biol. Chem. 2000, 275, 27838–27844. [Google Scholar] [PubMed]
- Tian, J.; Liu, J.; Garlid, K.D.; Shapiro, J.I.; Xie, Z. Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial K(ATP) channels. Mol. Cell. Biochem. 2003, 242, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Shapiro, A.P.; Haller, S.; Katragadda, V.; Liu, L.; Tian, J.; Basrur, V.; Malhotra, B.; Xie, Z.; Abraham, N.G. Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J. Biol. Chem. 2013, 288, 34249–34258. [Google Scholar] [CrossRef] [PubMed]
- Petrushanko, I.Y.; Yakushev, S.; Mitkevich, V.A.; Kamanina, Y.V.; Ziganshin, R.H.; Meng, X.; Anashkina, A.A.; Makhro, A.; Lopina, O.D.; Gassmann, M. S-glutathionylation of the Na,K-ATPase catalytic alpha subunit is a determinant of the enzyme redox sensitivity. J. Biol. Chem. 2012, 287, 32195–32205. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Garcia, A.; Mahmmoud, Y.A.; Hamilton, E.J.; Galougahi, K.K.; Fry, N.A.S.; Figtree, G.A.; Cornelius, F.; Clarke, R.J.; Rasmussen, H.H. Susceptibility of β1 Na+-K+ Pump Subunit to Glutathionylation and Oxidative Inhibition Depends on Conformational State of Pump. J. Biol. Chem. 2012, 287, 12353–12364. [Google Scholar] [CrossRef] [PubMed]
- Kajikawa, M.; Fujimo, S.; Tsuura, Y.; Mukai, E.; Takeda, T.; Hamamoto, Y.; Takehiro, M.; Fujita, J.; Yamada, Y.; Seino, Y. Ouabain suppresses glucose-induced mitochondrial ATP production and insulin release by generating reactive oxygen species in pancreatic islets. Diabetes 2002, 51, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Kometiani, P.; Liu, J.; Li, J.; Shapiro, J.I.; Askari, A. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J. Biol. Chem. 1999, 274, 19323–19328. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Wu, L.; Qu, W.; Malhotra, D.; Xie, Z.; Shapiro, J.I.; Liu, J. Regulation of apical NHE3 trafficking by ouabain-induced activation of the basolateral Na+-K+-ATPase receptor complex. Am. J. Physiol. Cell Physiol. 2008, 294, C555–C563. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Periyasamy, S.M.; Gunning, W.; Fedorova, O.V.; Bagrov, A.Y.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Effects of cardiac glycosides on sodium pump expression and function in LLC-PK1 and MDCK cells. Kidney Int. 2002, 62, 2118–2125. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kesiry, R.; Periyasamy, S.M.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Ouabain induces endocytosis of plasmalemmal Na/K-ATPase in LLC-PK1 cells by a clathrin-dependent mechanism. Kidney Int. 2004, 66, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liang, M.; Liu, L.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Ouabain-induced endocytosis of the plasmalemmal Na/K-ATPase in LLC-PK1 cells requires caveolin-1. Kidney Int. 2005, 67, 1844–1854. [Google Scholar] [CrossRef]
- Periyasamy, S.M.; Liu, J.; Tanta, F.; Kabak, B.; Wakefield, B.; Malhotra, D.; Kennedy, D.J.; Nadoor, A.; Fedorova, O.V.; Gunning, W.; Xie, Z.; et al. Salt loading induces redistribution of the plasmalemmal Na/K-ATPase in proximal tubule cells. Kidney Int. 2005, 67, 1868–1877. [Google Scholar] [CrossRef]
- Biemesderfer, D.; Pizzonia, J.; Abu-Alfa, A.; Exner, M.; Reilly, R.; Igarashi, P.; Aronson, P.S. NHE3: A Na+/H+ exchanger isoform of renal brush border. Am. J. Physiol. 1993, 265, F736–F742. [Google Scholar] [PubMed]
- Schultheis, P.J.; Clarke, L.L.; Meneton, P.; Miller, M.L.; Soleimani, M.; Gawenis, L.R.; Riddle, T.M.; Duffy, J.J.; Doetschman, T.; Wang, T.; et al. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat. Genet. 1998, 19, 282–285. [Google Scholar] [PubMed]
- Preisig, P.A.; Rector, F.C., Jr. Role of Na+-H+ antiport in rat proximal tubule NaCl absorption. Am. J. Physiol. 1988, 255, F461–F465. [Google Scholar] [PubMed]
- Yang, L.; Leong, P.K.K.; Chen, J.O.; Patel, N.; Hamm-Alvarez, S.F.; McDonough, A.A. Acute hypertension provokes internalization of proximal tubule NHE3 without inhibition of transport activity. Am. J. Physiol. Renal Physiol. 2002, 282, F730–F740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mircheff, A.K.; Hensley, C.B.; Magyar, C.E.; Warnock, D.G.; Chambrey, R.; Yip, K.P.; Marsh, D.J.; Holstein-Rathlou, N.H.; McDonough, A.A. Rapid redistribution and inhibition of renal sodium transporters during acute pressure natriuresis. Am. J. Physiol. 1996, 270, F1004–F1014. [Google Scholar] [PubMed]
- Amlal, H.; Chen, Q.; Greeley, T.; Pavelic, L.; Soleimani, M. Coordinated down-regulation of NBC-1 and NHE-3 in sodium and bicarbonate loading. Kidney Int. 2001, 60, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.; Garcia-Cabado, A.; Yu, F.; Teter, K.; Lukacs, G.; Skorecki, K.; Moore, H.P.; Orlowski, J.; Grinstein, S. The Epithelial Sodium-Hydrogen Antiporter Na+/H+ Exchanger 3 Accumulates and Is Functional in Recycling Endosomes. J. Biol. Chem. 1998, 273, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Janecki, A.J.; Montrose, M.H.; Zimniak, P.; Zweibaum, A.; Tse, C.M.; Khurana, S.; Donowitz, M. Subcellular Redistribution Is Involved in Acute Regulation of the Brush Border Na+/H+ Exchanger Isoform 3 in Human Colon Adenocarcinoma Cell Line Caco-2. Protein Kinase C-Mediated Inhibition of the exchanger. J. Biol. Chem. 1998, 273, 8790–8798. [Google Scholar] [CrossRef] [PubMed]
- Akhter, S.; Cavet, M.E.; Tse, C.M.; Donowitz, M. C-terminal domains of Na(+)/H(+) exchanger isoform 3 are involved in the basal and serum-stimulated membrane trafficking of the exchanger. Biochemistry 2000, 39, 1990–2000. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Amemiya, M.; Peng, Y.; Moe, O.W.; Preisig, P.A.; Alpern, R.J. Acid incubation causes exocytic insertion of NHE3 in OKP cells. Am. J. Physiol. Cell Physiol. 2000, 279, C410–C419. [Google Scholar] [PubMed]
- Li, X.; Galli, T.; Leu, S.; Wade, J.B.; Weinman, E.J.; Leung, G.; Cheong, A.; Louvard, D.; Donowitz, M. Na+-H+ exchanger 3 (NHE3) is present in lipid rafts in the rabbit ileal brush border: A role for rafts in trafficking and rapid stimulation of NHE3. J. Physiol. 2001, 537, 537–552. [Google Scholar] [CrossRef] [PubMed]
- Khundmiri, S.J.; Lederer, E. PTH and DA regulate Na-K ATPase through divergent pathways. Am. J. Physiol. Renal Physiol. 2002, 282, F512–F522. [Google Scholar] [CrossRef] [PubMed]
- Chibalin, A.V.; Zierath, J.R.; Katz, A.I.; Berggren, P.O.; Bertorello, A.M. Phosphatidylinositol 3-kinase-mediated endocytosis of renal Na+, K+-ATPase alpha subunit in response to dopamine. Mol. Biol. Cell 1998, 9, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Norian, J.M.; Magyar, C.E.; Holstein-Rathlou, N.H.; Mircheff, A.K.; McDonough, A.A. In vivo PTH provokes apical NHE3 and NaPi2 redistribution and Na-K-ATPase inhibition. Am. J. Physiol. 1999, 276, F711–F719. [Google Scholar] [PubMed]
- Vaziri, N.D.; Rodriguez-Iturbe, B. Mechanisms of disease: Oxidative stress and inflammation in the pathogenesis of hypertension. Nat. Clin. Pract. Nephrol. 2006, 2, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.S. Oxidative stress and nitric oxide deficiency in the kidney: A critical link to hypertension? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R913–R935. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: What is the clinical significance? Hypertension 2004, 44, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Welch, W.J. Intrarenal oxygen and hypertension. Clin. Exp. Pharmacol. Physiol. 2006, 33, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Kitiyakara, C.; Chabrashvili, T.; Chen, Y.; Blau, J.; Karber, A.; Aslam, S.; Welch, W.J.; Wilcox, C.S. Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J. Am. Soc. Nephrol. 2003, 14, 2775–2782. [Google Scholar] [CrossRef] [PubMed]
- Schreck, C.; O’Connor, P.M. NAD(P)H oxidase and renal epithelial ion transport. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1023–R1029. [Google Scholar] [CrossRef] [PubMed]
- Panico, C.; Luo, Z.; Damiano, S.; Artigiano, F.; Gill, P.; Welch, W.J. Renal Proximal Tubular Reabsorption Is Reduced In Adult Spontaneously Hypertensive Rats: Roles of Superoxide and Na+/H+ Exchanger 3. Hypertension 2009, 54, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Han, H.J.; Lee, Y.J.; Park, S.H.; Lee, J.H.; Taub, M. High glucose-induced oxidative stress inhibits Na+/glucose cotransporter activity in renal proximal tubule cells. Am. J. Physiol. Renal Physiol. 2005, 288, F988–F996. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Imam, S.Z.; Ali, S.F.; Mayeux, P.R. Peroxynitrite and the regulation of Na(+),K(+)-ATPase activity by angiotensin II in the rat proximal tubule. Nitric Oxide 2002, 7, 30–35. [Google Scholar] [CrossRef]
- Paravicini, T.M.; Touyz, R.M. Redox signaling in hypertension. Cardiovasc. Res. 2006, 71, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.S.; Gutterman, D. Focus on oxidative stress in the cardiovascular and renal systems. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H3–H6. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.E.; Leong, P.K.K.; McDonough, A.A. Reducing blood pressure in SHR with enalapril provokes redistribution of NHE3, NaPi2, and NCC and decreases NaPi2 and ACE abundance. Am. J. Physiol. Renal Physiol. 2007, 293, F1197–F1208. [Google Scholar] [CrossRef] [PubMed]
- Crajoinas, R.O.; Lessa, L.M.A.; Carraro-Lacroix, L.R.; Davel, A.P.C.; Pacheco, B.P.M.; Rossoni, L.V.; Malnic, G.; Girardi, A.C.C. Posttranslational mechanisms associated with reduced NHE3 activity in adult vs. young prehypertensive SHR. Am. J. Physiol. Renal Physiol. 2010, 299, F872–F881. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.A.; Lee, S.H.; Walker, J.; Dileto-Fang, C.; Ginsberg, L.; Stapleton, S.R. Regulation of proximal tubule sodium/hydrogen antiporter with chronic volume contraction. Am. J. Physiol. Renal Physiol. 2001, 280, F922–F926. [Google Scholar] [PubMed]
- Banday, A.A.; Lokhandwala, M.F. Loss of Biphasic Effect on Na/K-ATPase Activity by Angiotensin II Involves Defective Angiotensin Type 1 Receptor-Nitric Oxide Signaling. Hypertension 2008, 52, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Knox, F.G. Nitric oxide activates PKCalpha and inhibits Na+-K+-ATPase in opossum kidney cells. Am. J. Physiol. 1999, 277, F859–F865. [Google Scholar] [PubMed]
- Sabuhi, R.; Asghar, M.; Hussain, T. Inhibition of NAD(P)H oxidase potentiates AT2 receptor agonist-induced natriuresis in Sprague-Dawley rats. Am. J. Physiol. Renal Physiol. 2010, 299, F815–F820. [Google Scholar] [CrossRef]
- Silva, E.; Soares-da-Silva, P. Reactive oxygen species and the regulation of renal Na+-K+-ATPase in opossum kidney cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1764–R1770. [Google Scholar] [CrossRef] [PubMed]
- Petrushanko, I.; Bogdanov, N.; Bulygina, E.; Grenacher, B.; Leinsoo, T.; Boldyrev, A.; Gassmann, M.; Bogdanova, A. Na-K-ATPase in rat cerebellar granule cells is redox sensitive. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R916–R925. [Google Scholar] [CrossRef] [PubMed]
- Petrushanko, I.Y.; Bogdanov, N.B.; Lapina, N.; Boldyrev, A.A.; Gassmann, M.; Bogdanova, A.Y. Oxygen-induced Regulation of Na/K ATPase in cerebellar granule cells. J. Gen. Physiol. 2007, 130, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P. Src redox regulation: There is more than meets the eye. Mol. Cells 2008, 26, 329–337. [Google Scholar] [PubMed]
- Giannoni, E.; Buricchi1, F.; Raugei1, G.; Ramponi1, G.; Chiarugi, P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell. Biol. 2005, 25, 6391–6403. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Kemble, D.J. To C or not to C: Direct and indirect redox regulation of Src protein tyrosine kinase. Cell Cycle 2009, 8, 2353–2355. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, J.; Liu, J.; Yuan, Z.; Pierre, S.V.; Qu, W. Involvement of Na+/K+-ATPase in hydrogen peroxide-induced hypertrophy in cardiac myocytes. Free Radic. Biol. Med. 2006, 41, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.T.; Martin, R.; Yan, Y.; Shapiro, J.I.; Liu, J. Carbonylation Modification Regulates Na/K-ATPase Signaling and Salt Sensitivity: A Review and a Hypothesis. Front. Physiol. 2016, 7, 256. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Shapiro, A.P.; Mopidevi, B.R.; Chaudhry, M.A.; Maxwell, K.; Haller, S.T.; Drummond, C.A.; Kennedy, D.J.; Tian, J.; Malhotra, D.; et al. Protein Carbonylation of an Amino Acid Residue of the Na/K-ATPase alpha1 Subunit Determines Na/K-ATPase Signaling and Sodium Transport in Renal Proximal Tubular Cells. J. Am. Heart Assoc. 2016, 5, e003675. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Cheema, A.K.; Zhang, L.; Suzuki, Y.J. Protein Carbonylation as a Novel Mechanism in Redox Signaling. Circ. Res. 2008, 102, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Cattaruzza, M.; Hecker, M. Protein Carbonylation and Decarboylation. Circ. Res. 2008, 102, 273–274. [Google Scholar] [CrossRef] [PubMed]
- Elkareh, J.; Kennedy, D.J.; Yashaswi, B.; Vetteth, S.; Shidyak, A.; Kim, E.G.; Smaili, S.; Periyasamy, S.M.; Hariri, I.M.; Fedorova, L.; et al. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension 2007, 49, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Chaudhry, M.; Maxwell, K.; Yan, Y.; Wang, X.; Shah, P.T.; Khawaja, A.A.; Martin, R.; Robinette, T.J.; et al. Attenuation of Na/K-ATPase Mediated Oxidant Amplification with pNaKtide Ameliorates Experimental Uremic Cardiomyopathy. Sci. Rep. 2016, 6, 34592. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Maxwell, K.; Yan, Y.; Liu, J.; Chaudhry, M.A.; Getty, M.; Xie, Z.; Abraham, N.G.; Shapiro, J.I. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci. Adv. 2015, 1, e1500781. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.-S.; Adam, A.G.; Jaimes, E.A.; Raij, L. In Salt-Sensitive Hypertension, Increased Superoxide Production Is Linked to Functional Upregulation of Angiotensin II. Hypertension 2003, 42, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.-S.; Schulman, I.H.; Pagano, P.J.; Jaimes, E.A.; Raij, L. Reduced NAD(P)H Oxidase in Low Renin Hypertension. Hypertension 2006, 47, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.E.; Fedorova, O.V.; Morrell, C.H.; Longo, D.L.; Kashkin, V.A.; Metzler, J.D.; Bagrov, A.Y.; Lakatta, E.G. Endogenous sodium pump inhibitors and age-associated increases in salt sensitivity of blood pressure in normotensives. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1248–R1254. [Google Scholar] [CrossRef] [PubMed]
- Manunta, P.; Stella, P.; Rivera, R.; Ciurlino, D.; Cusi, D.; Ferrandi, M.; Hamlyn, J.M.; Bianchi, G. Left Ventricular Mass, Stroke Volume, and Ouabain-Like Factor in Essential Hypertension. Hypertension 1999, 34, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Redon, J.; Oliva, M.R.; Tormos, C.; Giner, V.; Chaves, J.; Iradi, A.; Sáez, G.T. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension 2003, 41, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, E.A.; Zhou, M.-Z.; Pearse, D.D.; Puzis, L.; Raij, L. Upregulation of cortical COX-2 in salt-sensitive hypertension: Role of angiotensin II and reactive oxygen species. Am. J. Physiol. Renal Physiol. 2008, 294, F385–F392. [Google Scholar] [CrossRef] [PubMed]
- Geiszt, M.; Kopp, J.B.; Várnai, P.; Leto, T.L. Identification of renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Touyz, R.M. Oxidative stress, Noxs, and hypertension: Experimental evidence and clinical controversies. Ann. Med. 2012, 44, S2–S16. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, W.; Chen, W.; Jiang, X.; Zhang, Y.; Wang, Z.; Yang, J.; Jones, J.E.; Jose, P.A.; Yang, Z. Regulation of blood pressure, oxidative stress and AT1R by high salt diet in mutant human dopamine D5 receptor transgenic mice. Hypertens. Res. 2015, 38, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Appel, L.J.; Moore, T.J.; Obarzanek, E.; Vollmer, W.M.; Svetkey, L.P.; Sacks, F.M.; Bray, G.A.; Vogt, T.M.; Cutler, J.A.; Windhauser, M.M.; et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N. Engl. J. Med. 1997, 336, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R., 3rd; Appel, L.J.; Risby, T.H. Effect of dietary patterns on measures of lipid peroxidation: Results from a randomized clinical trial. Circulation 1998, 98, 2390–2395. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Svetkey, L.P.; Vollmer, W.M.; Appel, L.J.; Bray, G.A.; Harsha, D.; Obarzanek, E.; Conlin, P.R.; Miller, E.R., 3rd; Simons-Morton, D.G.; et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N. Engl. J. Med. 2001, 344, 3–10. [Google Scholar] [CrossRef] [PubMed]
- John, J.H.; Ziebland, S.; Yudkin, P.; Roe, L.S.; Neil, H.A.W. Effects of fruit and vegetable consumption on plasma antioxidant concentrations and blood pressure: A randomised controlled trial. Lancet 2002, 359, 1969–1974. [Google Scholar] [CrossRef]
- Galley, H.F.; Thornton, J.; Howdle, P.D.; Walker, B.E.; Webster, N.R. Combination oral antioxidant supplementation reduces blood pressure. Clin. Sci. (Lond.) 1997, 92, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Giugliano, D.; Quatraro, A.; Lefebvre, P.J. Anti-oxidants show an anti-hypertensive effect in diabetic and hypertensive subjects. Clin. Sci. (Lond.) 1991, 81, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Motz, E.; Giugliano, D. Hypertension and ascorbic acid. Lancet 2000, 355, 1272–1273. [Google Scholar] [CrossRef]
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet 2002, 360, 23–33. [Google Scholar]
- Huang, H.-Y.; Caballero, B.; Chang, S.; Alberg, A.J.; Semba, R.D.; Schneyer, C.R.; Wilson, R.F.; Cheng, T.-Y.; Vassy, J.; Prokopowicz, G.; et al. The Efficacy and Safety of Multivitamin and Mineral Supplement Use To Prevent Cancer and Chronic Disease in Adults: A Systematic Review for a National Institutes of Health State-of-the-Science Conference. Ann. Intern. Med. 2006, 145, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 2010, 31, 2741–2748. [Google Scholar] [CrossRef] [PubMed]
- Biesalski, H.K.; Tinz, J. Multivitamin/mineral supplements: Rationale and safety—A systematic review. Nutrition 2017, 33, 76–82. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Yan, Y.; Nie, Y.; Shapiro, J.I. Na/K-ATPase Signaling and Salt Sensitivity: The Role of Oxidative Stress. Antioxidants 2017, 6, 18. https://doi.org/10.3390/antiox6010018
Liu J, Yan Y, Nie Y, Shapiro JI. Na/K-ATPase Signaling and Salt Sensitivity: The Role of Oxidative Stress. Antioxidants. 2017; 6(1):18. https://doi.org/10.3390/antiox6010018
Chicago/Turabian StyleLiu, Jiang, Yanling Yan, Ying Nie, and Joseph I. Shapiro. 2017. "Na/K-ATPase Signaling and Salt Sensitivity: The Role of Oxidative Stress" Antioxidants 6, no. 1: 18. https://doi.org/10.3390/antiox6010018
APA StyleLiu, J., Yan, Y., Nie, Y., & Shapiro, J. I. (2017). Na/K-ATPase Signaling and Salt Sensitivity: The Role of Oxidative Stress. Antioxidants, 6(1), 18. https://doi.org/10.3390/antiox6010018