
Treating Cancer by Targeting Telomeres and Telomerase

{kind=link}
{kind=link}
{kind=link}
Abstract
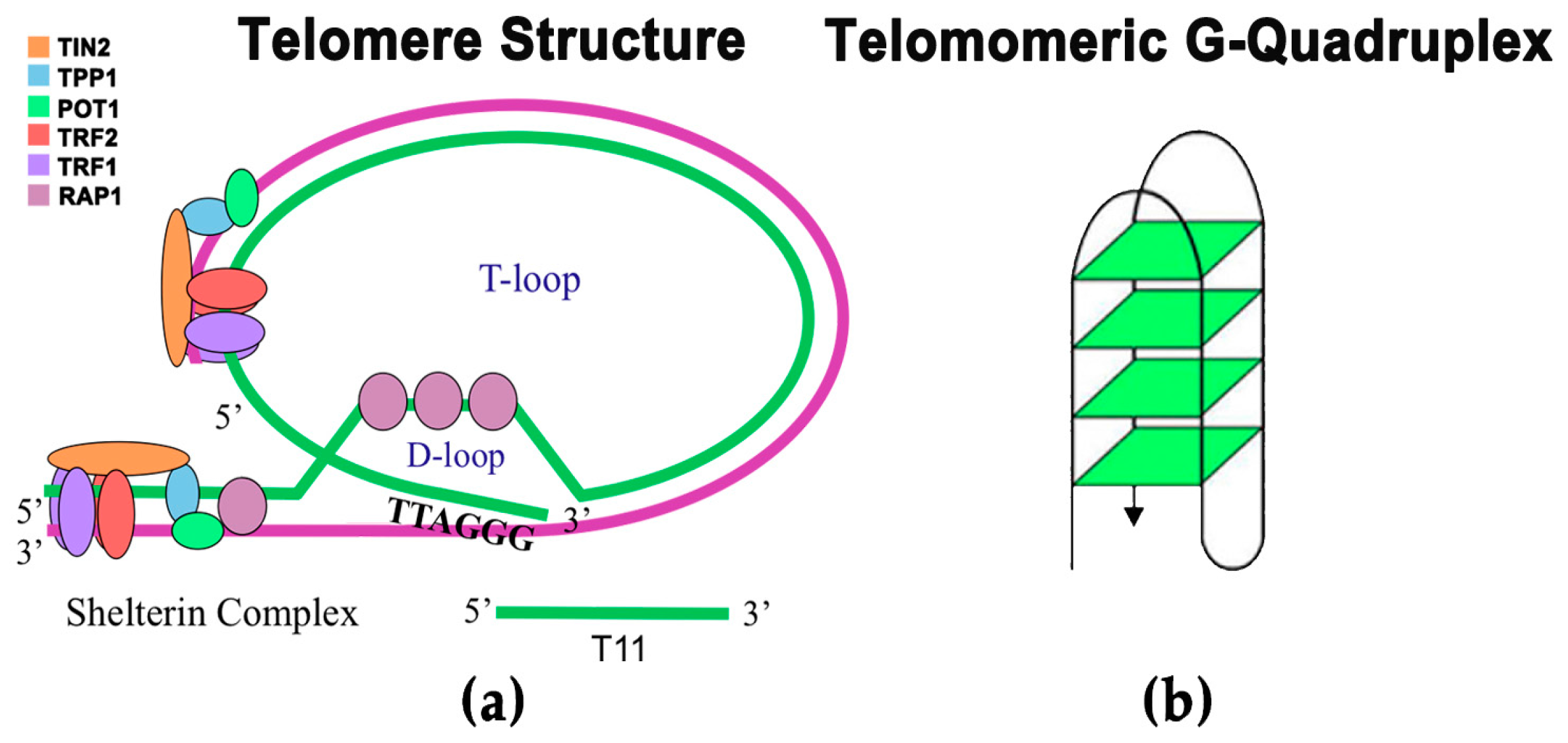
:1. Introduction
2. Current Therapies Related to Telomeres and Telomerase
3. Telomere Homolog Oligonucleotides
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Crees, Z.; Girard, J.; Rios, Z.; Botting, G.M.; Harrington, K.; Shearrow, C.; Wojdyla, L.; Stone, A.L.; Uppada, S.B.; Devito, J.T.; et al. Oligonucleotides and g-quadruplex stabilizers: Targeting telomeres and telomerase in cancer therapy. Curr. Pharm. Des. 2014, 20, 6422–6437. [Google Scholar] [CrossRef] [PubMed]
- Ruden, M.; Puri, N. Novel anticancer therapeutics targeting telomerase. Cancer Treat. Rev. 2013, 39, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Thilagavathi, J.; Venkatesh, S.; Dada, R. Telomere length in reproduction. Andrologia 2013, 45, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Reig-Viader, R.; Garcia-Caldes, M.; Ruiz-Herrera, A. Telomere homeostasis in mammalian germ cells: A review. Chromosoma 2016, 125, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.M.; Kalmbach, K.H.; Wang, F.; Dracxler, R.C.; Seth-Smith, M.L.; Kramer, Y.; Buldo-Licciardi, J.; Kohlrausch, F.B.; Keefe, D.L. A single-cell assay for telomere DNA content shows increasing telomere length heterogeneity, as well as increasing mean telomere length in human spermatozoa with advancing age. J. Assist. Reprod. Genet. 2015, 32, 1685–1690. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Protection of mammalian telomeres. Oncogene 2002, 21, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, N.; de Lange, T. Cell cycle-dependent role of MRN at dysfunctional telomeres: ATM signaling-dependent induction of nonhomologous end joining (NHEJ) in G1 and resection-mediated inhibition of NHEJ in G2. Mol. Cell. Biol. 2009, 29, 5552–5563. [Google Scholar] [CrossRef] [PubMed]
- Ribes-Zamora, A.; Indiviglio, S.M.; Mihalek, I.; Williams, C.L.; Bertuch, A.A. TRF2 interaction with Ku heterotetramerization interface gives insight into c-NHEJ prevention at human telomeres. Cell Rep. 2013, 5, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere maintenance by recombination in human cells. Nat. Genet. 2000, 26, 447–450. [Google Scholar] [PubMed]
- Luke-Glaser, S.; Poschke, H.; Luke, B. Getting in (and out of) the loop: Regulating higher order telomere structures. Front. Oncol. 2012, 2, 180. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Goldkorn, A. Telomere and telomerase therapeutics in cancer. Genes 2016, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Zurek, M.; Altschmied, J.; Kohlgruber, S.; Ale-Agha, N.; Haendeler, J. Role of telomerase in the cardiovascular system. Genes 2016, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Pech, M.F.; Garbuzov, A.; Hasegawa, K.; Sukhwani, M.; Zhang, R.J.; Benayoun, B.A.; Brockman, S.A.; Lin, S.; Brunet, A.; Orwig, K.E.; et al. High telomerase is a hallmark of undifferentiated spermatogonia and is required for maintenance of male germline stem cells. Genes Dev. 2015, 29, 2420–2434. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.; Mitchell, J.R. Telomerase in the human organism. Oncogene 2002, 21, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase in normal and cancer stem cells. FEBS Lett. 2010, 584, 3819–3825. [Google Scholar] [CrossRef] [PubMed]
- Shigeishi, H.; Sugiyama, M.; Tahara, H.; Ono, S.; Kumar Bhawal, U.; Okura, M.; Kogo, M.; Shinohara, M.; Shindoh, M.; Shintani, S.; et al. Increased telomerase activity and htert expression in human salivary gland carcinomas. Oncol. Lett. 2011, 2, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Poncet, D.; Belleville, A.; t’kint de Roodenbeke, C.; Roborel de Climens, A.; Ben Simon, E.; Merle-Beral, H.; Callet-Bauchu, E.; Salles, G.; Sabatier, L.; Delic, J.; et al. Changes in the expression of telomere maintenance genes suggest global telomere dysfunction in B-chronic lymphocytic leukemia. Blood 2008, 111, 2388–2391. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Chiappori, A.A.; Kolevska, T.; Spigel, D.R.; Hager, S.; Rarick, M.; Gadgeel, S.; Blais, N.; Von Pawel, J.; Hart, L.; Reck, M.; et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Asai, A.; Oshima, Y.; Yamamoto, Y.; Uochi, T.A.; Kusaka, H.; Akinaga, S.; Yamashita, Y.; Pongracz, K.; Pruzan, R.; Wunder, E.; et al. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res. 2003, 63, 3931–3939. [Google Scholar] [PubMed]
- Gellert, G.C.; Dikmen, Z.G.; Wright, W.E.; Gryaznov, S.; Shay, J.W. Effects of a novel telomerase inhibitor, GRN163L, in human breast cancer. Breast Cancer Res. Treat. 2006, 96, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter, A.E.; Xiao, H.; Goldblatt, E.M.; Gryaznov, S.M.; Miller, K.D.; Badve, S.; Sledge, G.W.; Herbert, B.S. Telomerase template antagonist GRN163L disrupts telomere maintenance, tumor growth, and metastasis of breast cancer. Clin. Cancer Res. 2006, 12, 3184–3192. [Google Scholar] [CrossRef]
- Herbert, B.; Pitts, A.E.; Baker, S.I.; Hamilton, S.E.; Wright, W.E.; Shay, J.W.; Corey, D.R. Inhibition of human telomerase in immortal human cells leads to progressive telomere shortening and cell death. Proc. Natl. Acad. Sci. USA 1999, 96, 14276–14281. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Wei, J.; Wunderlich, M.; Chou, F.S.; Mulloy, J.C. Immortalization of human ae pre-leukemia cells by htert allows leukemic transformation. Oncotarget 2016, 7, 55939–55950. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cook, B.D.; Dynek, J.N.; Chang, W.; Shostak, G.; Smith, S. Role for the related poly(ADP-Ribose) polymerases tankyrase 1 and 2 at human telomeres. Mol. Cell. Biol. 2002, 22, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Riffell, J.L.; Lord, C.J.; Ashworth, A. Tankyrase-targeted therapeutics: Expanding opportunities in the parp family. Nat. Rev. Drug Discov. 2012, 11, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Waaler, J.; Machon, O.; Tumova, L.; Dinh, H.; Korinek, V.; Wilson, S.R.; Paulsen, J.E.; Pedersen, N.M.; Eide, T.J.; Machonova, O.; et al. A novel tankyrase inhibitor decreases canonical wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012, 72, 2822–2832. [Google Scholar] [CrossRef] [PubMed]
- Dregalla, R.C.; Zhou, J.; Idate, R.R.; Battaglia, C.L.; Liber, H.L.; Bailey, S.M. Regulatory roles of tankyrase 1 at telomeres and in DNA repair: Suppression of T-SCE and stabilization of DNA-PKcs. Aging 2010, 2, 691–708. [Google Scholar] [CrossRef] [PubMed]
- Seimiya, H.; Muramatsu, Y.; Ohishi, T.; Tsuruo, T. Tankyrase 1 as a target for telomere-directed molecular cancer therapeutics. Cancer Cell 2005, 7, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, A.; Safa, M.; Kazemi, A. MST-312 induces G2/M cell cycle arrest and apoptosis in APL cells through inhibition of telomerase activity and suppression of NF-κB pathway. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 8425–8437. [Google Scholar] [CrossRef] [PubMed]
- Tauchi, T.; Shin-ya, K.; Sashida, G.; Sumi, M.; Okabe, S.; Ohyashiki, J.H.; Ohyashiki, K. Telomerase inhibition with a novel G-quadruplex-interactive agent, telomestatin: In vitro and in vivo studies in acute leukemia. Oncogene 2006, 25, 5719–5725. [Google Scholar] [CrossRef] [PubMed]
- Maestroni, L.; Matmati, S.; Coulon, S. Solving the telomere replication problem. Genes 2017, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Burger, A.M.; Dai, F.; Schultes, C.M.; Reszka, A.P.; Moore, M.J.; Double, J.A.; Neidle, S. The G-quadruplex-interactive molecule BRACO-19 inhibits tumor growth, consistent with telomere targeting and interference with telomerase function. Cancer Res. 2005, 65, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Gunaratnam, M.; Greciano, O.; Martins, C.; Reszka, A.P.; Schultes, C.M.; Morjani, H.; Riou, J.F.; Neidle, S. Mechanism of acridine-based telomerase inhibition and telomere shortening. Biochem. Pharmacol. 2007, 74, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Cookson, J.C.; Dai, F.; Smith, V.; Heald, R.A.; Laughton, C.A.; Stevens, M.F.; Burger, A.M. Pharmacodynamics of the G-quadruplex-stabilizing telomerase inhibitor 3,11-difluoro-6,8,13-trimethyl-8H-quino[4,3,2-kl]acridinium methosulfate (RHPS4) in vitro: Activity in human tumor cells correlates with telomere length and can be enhanced, or antagonized, with cytotoxic agents. Mol. Pharmacol. 2005, 68, 1551–1558. [Google Scholar] [PubMed]
- Salvati, E.; Leonetti, C.; Rizzo, A.; Scarsella, M.; Mottolese, M.; Galati, R.; Sperduti, I.; Stevens, M.F.; D’Incalci, M.; Blasco, M.; et al. Telomere damage induced by the G-quadruplex ligand RHPS4 has an antitumor effect. J. Clin. Investig. 2007, 117, 3236–3247. [Google Scholar] [CrossRef] [PubMed]
- Tahara, H.; Shin-Ya, K.; Seimiya, H.; Yamada, H.; Tsuruo, T.; Ide, T. G-Quadruplex stabilization by telomestatin induces TRF2 protein dissociation from telomeres and anaphase bridge formation accompanied by loss of the 3’ telomeric overhang in cancer cells. Oncogene 2006, 25, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tergaonkar, V. Noncanonical functions of telomerase: Implications in telomerase-targeted cancer therapies. Cancer Res. 2014, 74, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Low, K.C.; Tergaonkar, V. Telomerase: Central regulator of all of the hallmarks of cancer. Trends Biochem. Sci. 2013, 38, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Indran, I.R.; Hande, M.P.; Pervaiz, S. hTERT overexpression alleviates intracellular ROS production, improves mitochondrial function, and inhibits ROS-mediated apoptosis in cancer cells. Cancer Res. 2011, 71, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Masutomi, K.; Possemato, R.; Wong, J.M.; Currier, J.L.; Tothova, Z.; Manola, J.B.; Ganesan, S.; Lansdorp, P.M.; Collins, K.; Hahn, W.C. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc. Natl. Acad. Sci. USA 2005, 102, 8222–8227. [Google Scholar] [CrossRef] [PubMed]
- Park, J.I.; Venteicher, A.S.; Hong, J.Y.; Choi, J.; Jun, S.; Shkreli, M.; Chang, W.; Meng, Z.; Cheung, P.; Ji, H.; et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009, 460, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Khattar, E.; Leow, S.C.; Liu, C.Y.; Muller, J.; Ang, W.X.; Li, Y.; Franzoso, G.; Li, S.; Guccione, E.; et al. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J. Clin. Investig. 2015, 125, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Khattar, E.; Kumar, P.; Liu, C.Y.; Akincilar, S.C.; Raju, A.; Lakshmanan, M.; Maury, J.J.; Qiang, Y.; Li, S.; Tan, E.Y.; et al. Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting tRNA expression. J. Clin. Investig. 2016, 126, 4045–4060. [Google Scholar] [CrossRef] [PubMed]
- Hahn, W.C.; Stewart, S.A.; Brooks, M.W.; York, S.G.; Eaton, E.; Kurachi, A.; Beijersbergen, R.L.; Knoll, J.H.; Meyerson, M.; Weinberg, R.A. Inhibition of telomerase limits the growth of human cancer cells. Nat. Med. 1999, 5, 1164–1170. [Google Scholar] [PubMed]
- Li, Y.; Zhou, Q.L.; Sun, W.; Chandrasekharan, P.; Cheng, H.S.; Ying, Z.; Lakshmanan, M.; Raju, A.; Tenen, D.G.; Cheng, S.Y.; et al. Non-canonical NF-kappaB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat. Cell biol. 2015, 17, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Akincilar, S.C.; Khattar, E.; Boon, P.L.; Unal, B.; Fullwood, M.J.; Tergaonkar, V. Long-range chromatin interactions drive mutant tert promoter activation. Cancer Discov. 2016, 6, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Telomeres and telomerase: Their mechanisms of action and the effects of altering their functions. FEBS Lett. 2005, 579, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Vallarelli, A.F.; Rachakonda, P.S.; Andre, J.; Heidenreich, B.; Riffaud, L.; Bensussan, A.; Kumar, R.; Dumaz, N. TERT promoter mutations in melanoma render TERT expression dependent on MAPK pathway activation. Oncotarget 2016, 7, 53127–53136. [Google Scholar] [CrossRef] [PubMed]
- Queisser, A.; Heeg, S.; Thaler, M.; von Werder, A.; Opitz, O.G. Inhibition of telomerase induces alternative lengthening of telomeres during human esophageal carcinogenesis. Cancer Genet. 2013, 206, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Kyo, S.; Takakura, M.; Fujiwara, T.; Inoue, M. Understanding and exploiting htert promoter regulation for diagnosis and treatment of human cancers. Cancer Sci. 2008, 99, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
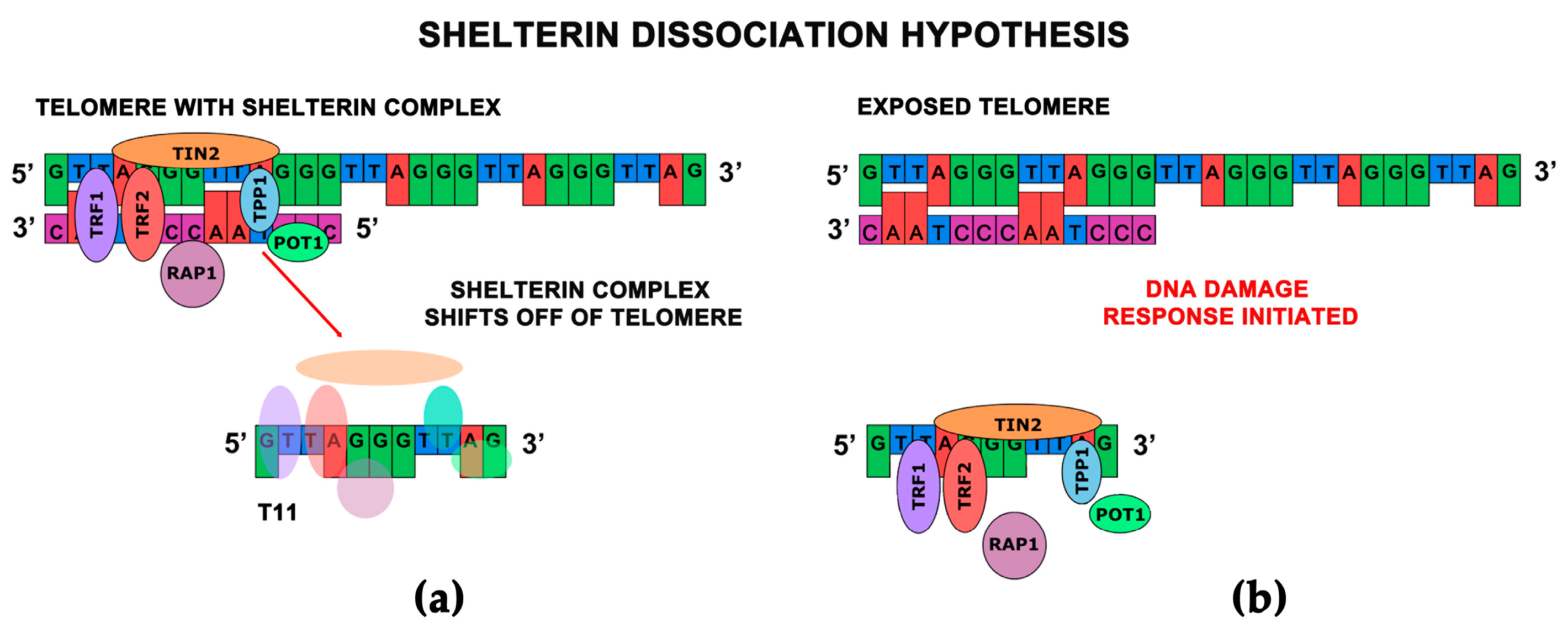
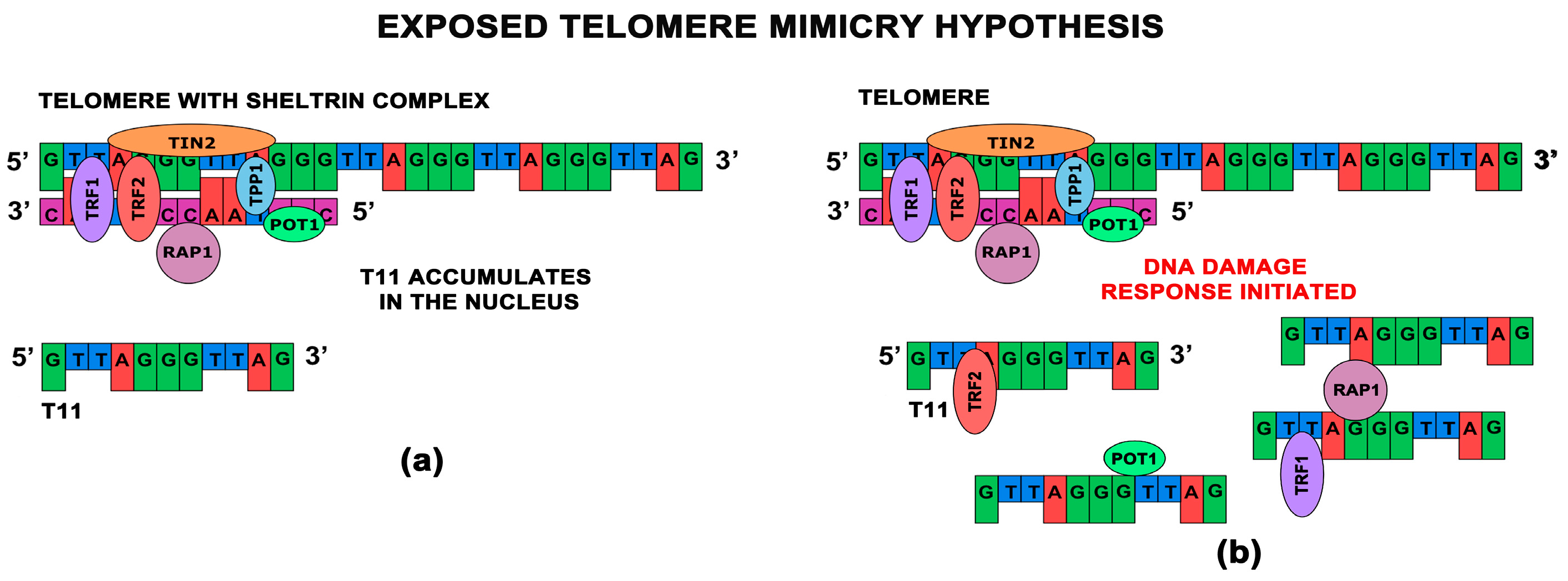
- Li, G.Z.; Eller, M.S.; Firoozabadi, R.; Gilchrest, B.A. Evidence that exposure of the telomere 3’ overhang sequence induces senescence. Proc. Natl. Acad. Sci. USA 2003, 100, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Saretzki, G.; Sitte, N.; Merkel, U.; Wurm, R.E.; von Zglinicki, T. Telomere shortening triggers a p53-dependent cell cycle arrest via accumulation of G-rich single stranded DNA fragments. Oncogene 1999, 18, 5148–5158. [Google Scholar] [CrossRef] [PubMed]
- Gnanasekar, M.; Thirugnanam, S.; Zheng, G.; Chen, A.; Ramaswamy, K. T-oligo induces apoptosis in advanced prostate cancer cells. Oligonucleotides 2009, 19, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Rankin, A.M.; Forman, L.; Sarkar, S.; Faller, D.V. Enhanced cytotoxicity from deoxyguanosine-enriched T-oligo in prostate cancer cells. Nucleic Acid Ther. 2013, 23, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.J.; Laber, D.A.; Miller, D.M.; Thomas, S.D.; Trent, J.O. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp. Mol. Pathol. 2009, 86, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Weng, D.; Cunin, M.C.; Song, B.; Price, B.D.; Eller, M.S.; Gilchrest, B.A.; Calderwood, S.K.; Gong, J. Radiosensitization of mammary carcinoma cells by telomere homolog oligonucleotide pretreatment. Breast Cancer Res. 2010, 12, R71. [Google Scholar] [CrossRef] [PubMed]
- Longe, H.O.; Romesser, P.B.; Rankin, A.M.; Faller, D.V.; Eller, M.S.; Gilchrest, B.A.; Denis, G.V. Telomere homolog oligonucleotides induce apoptosis in malignant but not in normal lymphoid cells: Mechanism and therapeutic potential. Int. J. Cancer 2009, 124, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Wojdyla, L.; Stone, A.L.; Sethakorn, N.; Uppada, S.B.; Devito, J.T.; Bissonnette, M.; Puri, N. T-oligo as an anticancer agent in colorectal cancer. Biochem. Biophys. Res. Commun. 2014, 446, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Faller, D.V. Telomere-homologous g-rich oligonucleotides sensitize human ovarian cancer cells to trail-induced growth inhibition and apoptosis. Nucleic Acid Ther. 2013, 23, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Puri, N.; Eller, M.S.; Byers, H.R.; Dykstra, S.; Kubera, J.; Gilchrest, B.A. Telomere-based DNA damage responses: A new approach to melanoma. FASEB J. 2004, 18, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Puri, N.; Pitman, R.T.; Mulnix, R.E.; Erickson, T.; Iness, A.N.; Vitali, C.; Zhao, Y.; Salgia, R. Non-small cell lung cancer is susceptible to induction of DNA damage responses and inhibition of angiogenesis by telomere overhang oligonucleotides. Cancer Lett. 2014, 343, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.; Levine, D.; Kishore, R.; Qin, G.; Thorne, T.; Lambers, E.; Sasi, S.P.; Yaar, M.; Gilchrest, B.A.; Goukassian, D.A. Inhibition of melanoma angiogenesis by telomere homolog oligonucleotides. J. Oncol. 2010, 2010, 928628. [Google Scholar] [CrossRef] [PubMed]
- Uppada, S.B.; Erickson, T.; Wojdyla, L.; Moravec, D.N.; Song, Z.; Cheng, J.; Puri, N. Novel delivery system for T-oligo using a nanocomplex formed with an alpha helical peptide for melanoma therapy. Int. J. Nanomed. 2014, 9, 43–53. [Google Scholar]
- Yaar, M.; Eller, M.S.; Panova, I.; Kubera, J.; Wee, L.H.; Cowan, K.H.; Gilchrest, B.A. Telomeric DNA induces apoptosis and senescence of human breast carcinoma cells. Breast Cancer Res. 2007, 9, R13. [Google Scholar] [CrossRef] [PubMed]
- Rankin, A.M.; Faller, D.V.; Spanjaard, R.A. Telomerase inhibitors and ‘T-oligo’ as cancer therapeutics: Contrasting molecular mechanisms of cytotoxicity. Anti-Cancer Drugs 2008, 19, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Karlseder, J.; Smogorzewska, A.; de Lange, T. Senescence induced by altered telomere state, not telomere loss. Science 2002, 295, 2446–2449. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, B.; Smogorzewska, A.; de Lange, T. TRF2 protects human telomeres from end-to-end fusions. Cell 1998, 92, 401–413. [Google Scholar] [CrossRef]
- Pitman, R.T.; Wojdyla, L.; Puri, N. Mechanism of DNA damage responses induced by exposure to an oligonucleotide homologous to the telomere overhang in melanoma. Oncotarget 2013, 4, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Eller, M.S.; Li, G.Z.; Firoozabadi, R.; Puri, N.; Gilchrest, B.A. Induction of a p95/Nbs1-mediated s phase checkpoint by telomere 3’ overhang specific DNA. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Page, T.J.; Mata, J.E.; Bridge, J.A.; Siebler, J.C.; Neff, J.R.; Iversen, P.L. The cytotoxic effects of single-stranded telomere mimics on OMA-BL1 cells. Exp. Cell Res. 1999, 252, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Calado, R.T.; Dumitriu, B. Telomere dynamics in mice and humans. Semin. Hematol. 2013, 50, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.M.; Ryder, O.A.; Houck, M.L.; Charter, S.J.; Walker, W.; Forsyth, N.R.; Austad, S.N.; Venditti, C.; Pagel, M.; Shay, J.W.; et al. Comparative biology of mammalian telomeres: Hypotheses on ancestral states and the roles of telomeres in longevity determination. Aging Cell 2011, 10, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Parrinello, S.; Kim, J.; Campisi, J. Mus musculus and mus spretus homologues of the human telomere-associated protein TIN2. Genomics 2003, 81, 422–432. [Google Scholar] [CrossRef]
- Broccoli, D.; Chong, L.; Oelmann, S.; Fernald, A.A.; Marziliano, N.; van Steensel, B.; Kipling, D.; Le Beau, M.M.; de Lange, T. Comparison of the human and mouse genes encoding the telomeric protein, TRF1: Chromosomal localization, expression and conserved protein domains. Hum. Mol. Genet. 1997, 6, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; Hockemeyer, D.; Kibe, T.; de Lange, T. Functional dissection of human and mouse POT1 proteins. Mol. Cell. Biol. 2009, 29, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Dai, S.; Sun, J.; Jiang, S.; Sui, C.; Meng, F.; Li, Y.; Fu, L.; Jiang, T.; Wang, Y.; et al. Bufalin induces mitochondria-dependent apoptosis in pancreatic and oral cancer cells by downregulating hTERT expression via activation of the JNK/p38 pathway. Evide-Based Complement. Altern. Med. 2015, 2015, 546210. [Google Scholar] [CrossRef] [PubMed]
- Eller, M.S.; Liao, X.; Liu, S.; Hanna, K.; Backvall, H.; Opresko, P.L.; Bohr, V.A.; Gilchrest, B.A. A role for WRN in telomere-based DNA damage responses. Proc. Natl. Acad. Sci. USA 2006, 103, 15073–15078. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Wang, J.; Bai, Y.; Lang, J.W.; Liu, S.; Lin, Y.; Cheng, J. Ionic polypeptides with unusual helical stability. Nat. Commun. 2011, 2, 206. [Google Scholar] [CrossRef] [PubMed]
- Yen, J.; Zhang, Y.; Gabrielson, N.P.; Yin, L.; Guan, L.; Chaudhury, I.; Lu, H.; Wang, F.; Cheng, J. Cationic, helical polypeptide-based gene delivery for IMR-90 fibroblasts and human embryonic stem cells. Biomater. Sci. 2013, 1, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Allen, T.M.; Cullis, P.R. Lipid nanoparticle delivery systems for siRNA-based therapeutics. Drug Deliv. Transl. Res. 2014, 4, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Wissing, S.A.; Kayser, O.; Muller, R.H. Solid lipid nanoparticles for parenteral drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 1257–1272. [Google Scholar] [CrossRef] [PubMed]
- Rajanna, S.; Rastogi, I.; Wojdyla, L.; Furo, H.; Kulesza, A.; Lin, L.; Sheu, B.; Frakes, M.; Ivanovich, M.; Puri, N. Current molecularly targeting therapies in NSCLC and melanoma. Anti-Cancer Agents Med. Chem. 2015, 15, 856–868. [Google Scholar] [CrossRef]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivancich, M.; Schrank, Z.; Wojdyla, L.; Leviskas, B.; Kuckovic, A.; Sanjali, A.; Puri, N. Treating Cancer by Targeting Telomeres and Telomerase. Antioxidants 2017, 6, 15. https://doi.org/10.3390/antiox6010015
Ivancich M, Schrank Z, Wojdyla L, Leviskas B, Kuckovic A, Sanjali A, Puri N. Treating Cancer by Targeting Telomeres and Telomerase. Antioxidants. 2017; 6(1):15. https://doi.org/10.3390/antiox6010015
Chicago/Turabian StyleIvancich, Marko, Zachary Schrank, Luke Wojdyla, Brandon Leviskas, Adijan Kuckovic, Ankita Sanjali, and Neelu Puri. 2017. "Treating Cancer by Targeting Telomeres and Telomerase" Antioxidants 6, no. 1: 15. https://doi.org/10.3390/antiox6010015
APA StyleIvancich, M., Schrank, Z., Wojdyla, L., Leviskas, B., Kuckovic, A., Sanjali, A., & Puri, N. (2017). Treating Cancer by Targeting Telomeres and Telomerase. Antioxidants, 6(1), 15. https://doi.org/10.3390/antiox6010015