
Mycosporine-Like Amino Acids and Their Derivatives as Natural Antioxidants

Abstract
:1. Introduction
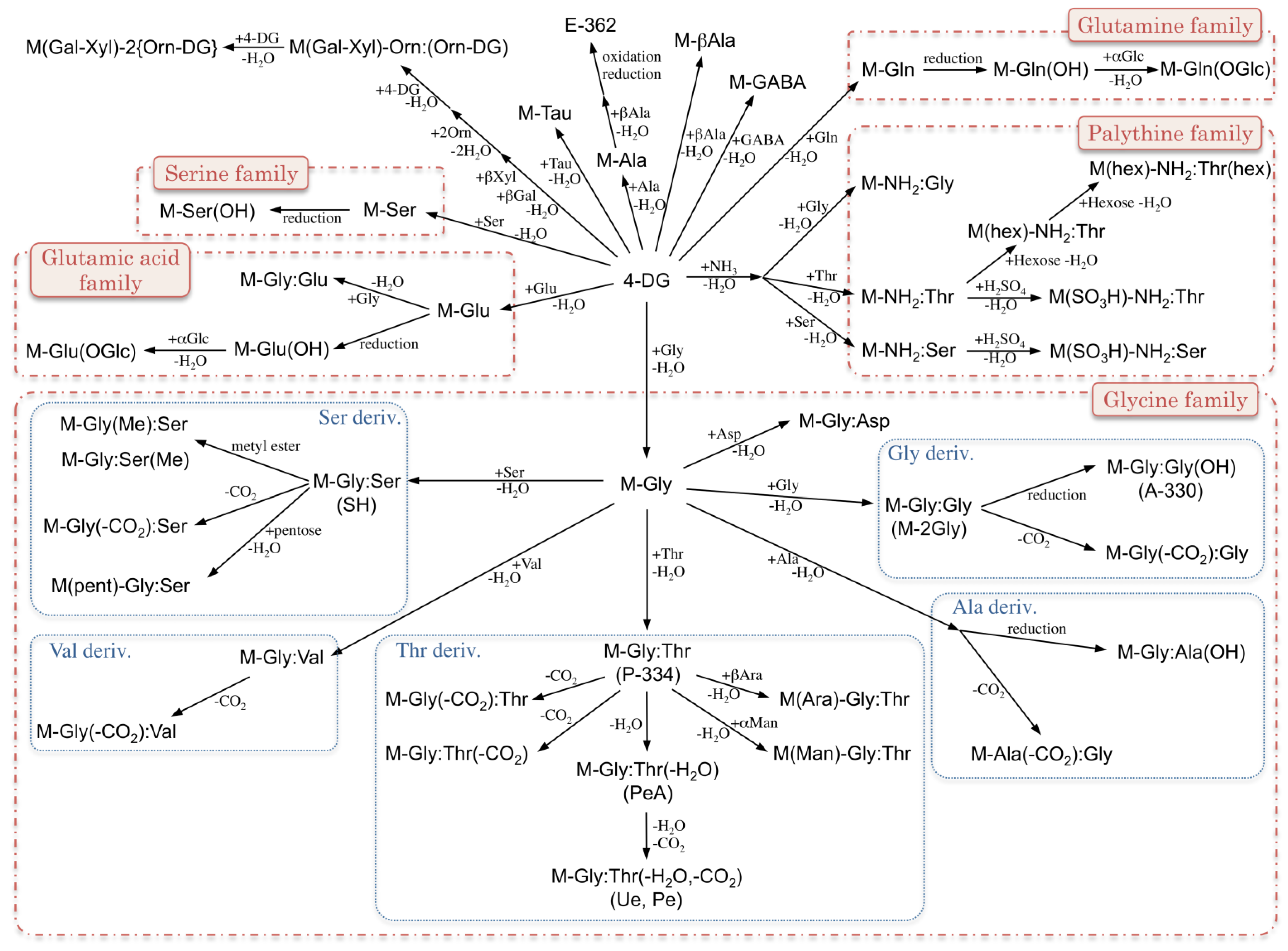
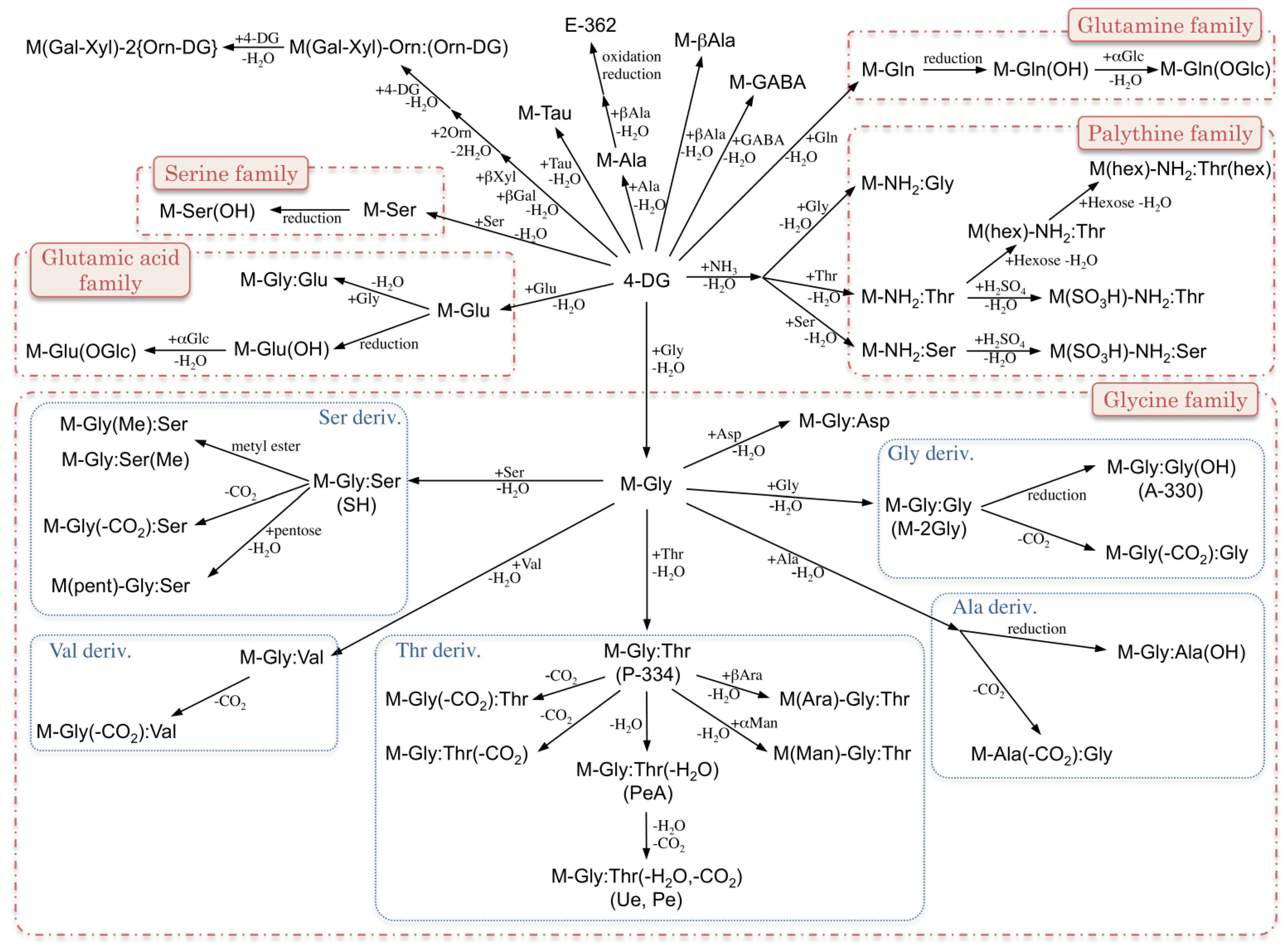
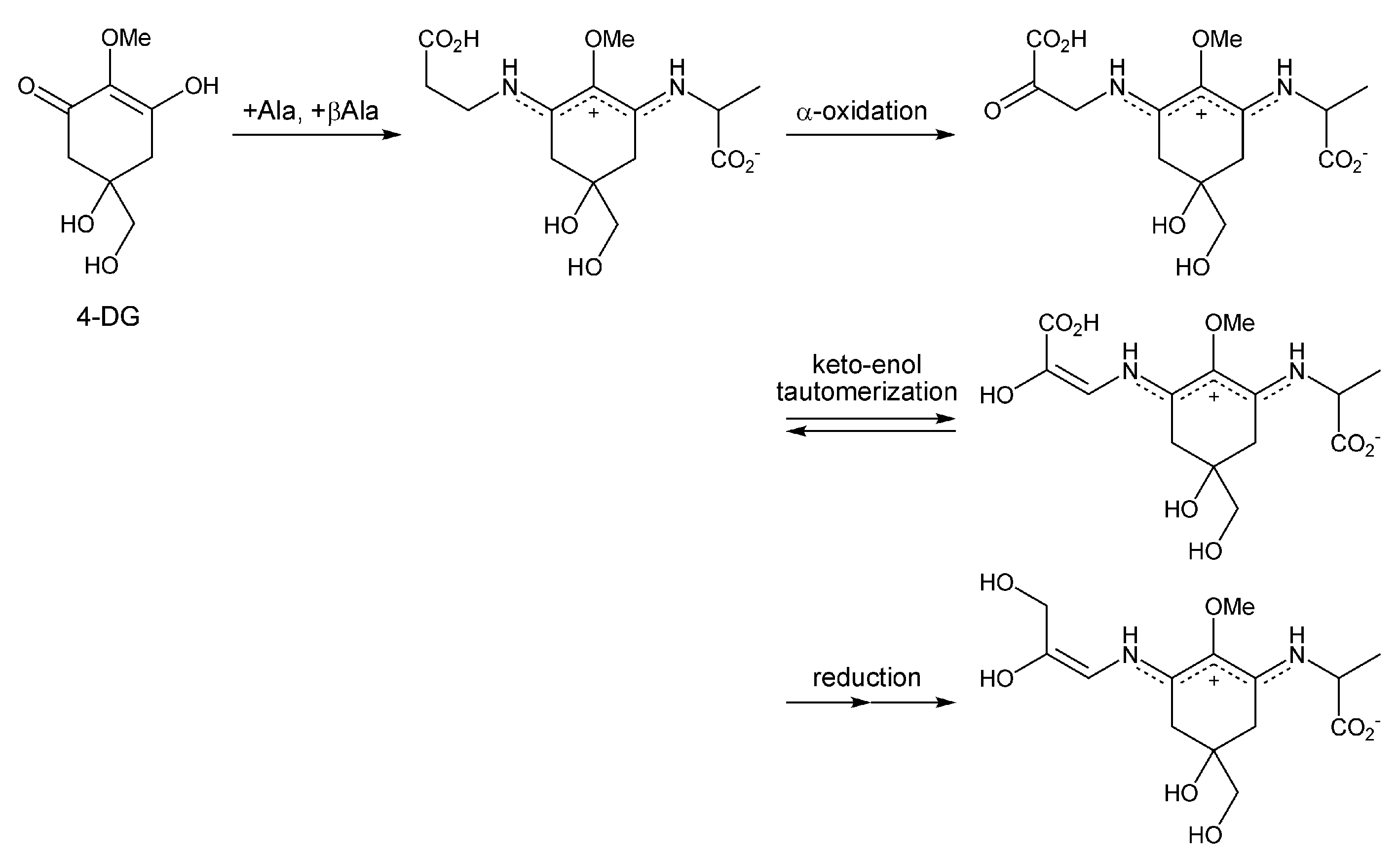
2. MAA Precursors (Figure 2)
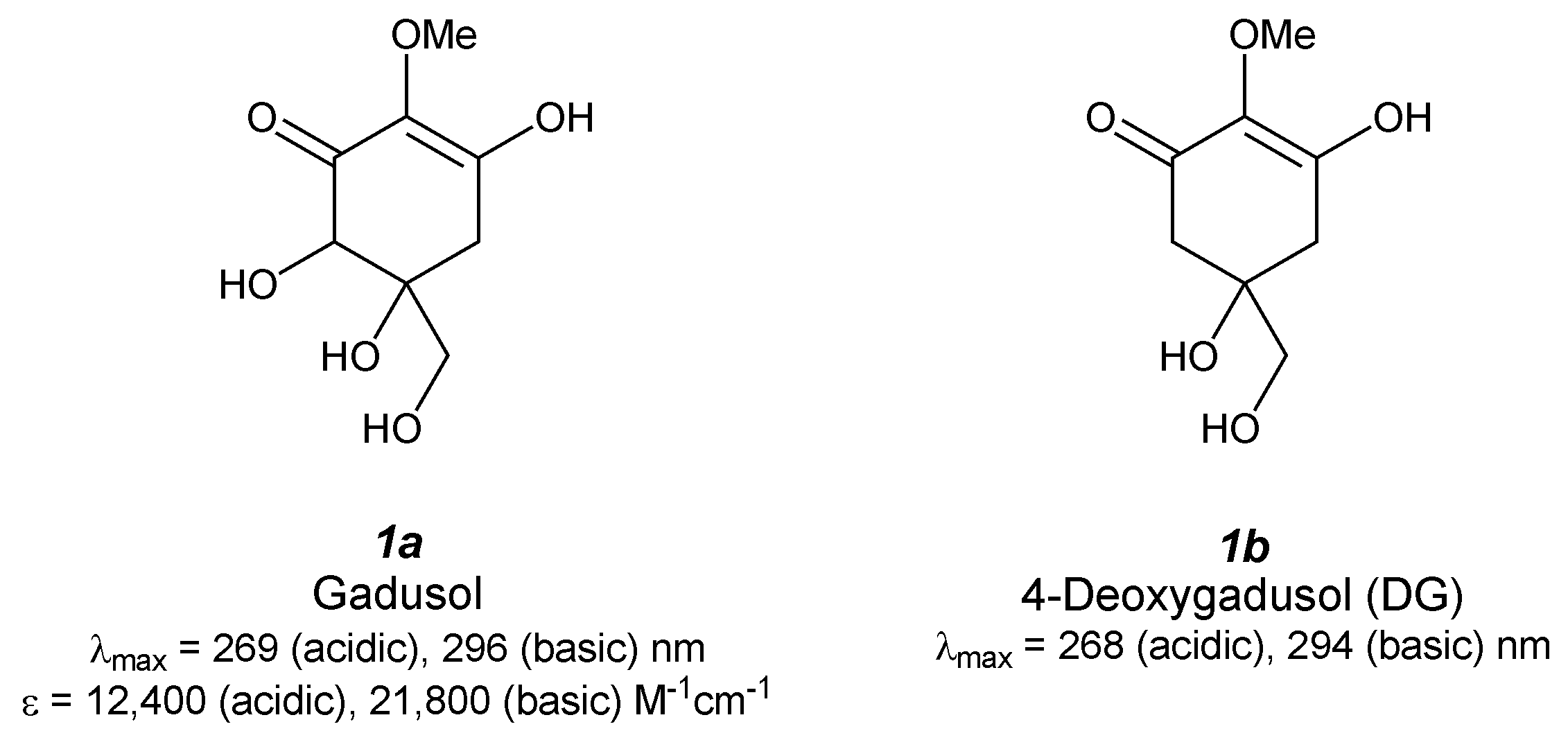
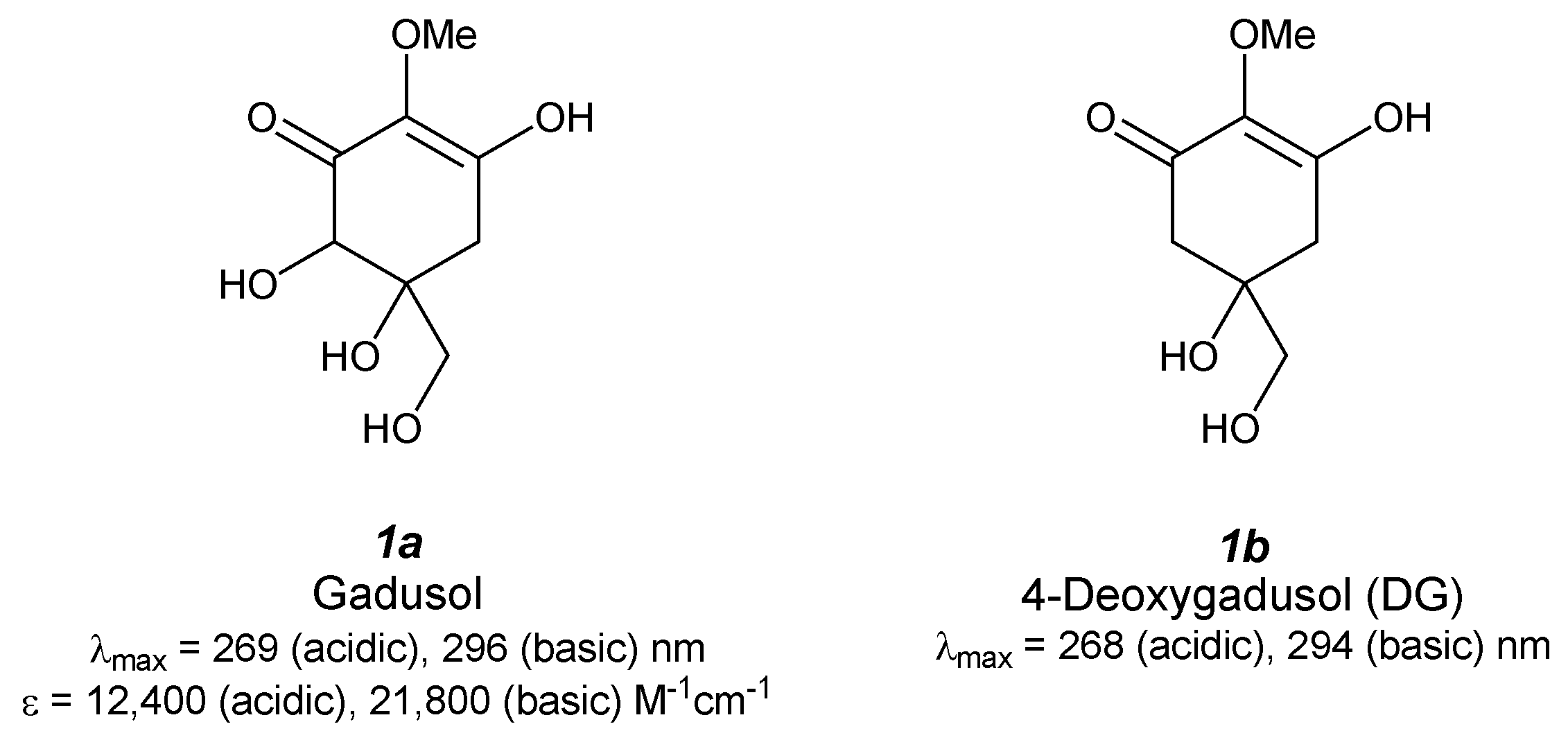
2.1. Gadusol (Figure 2(1a))
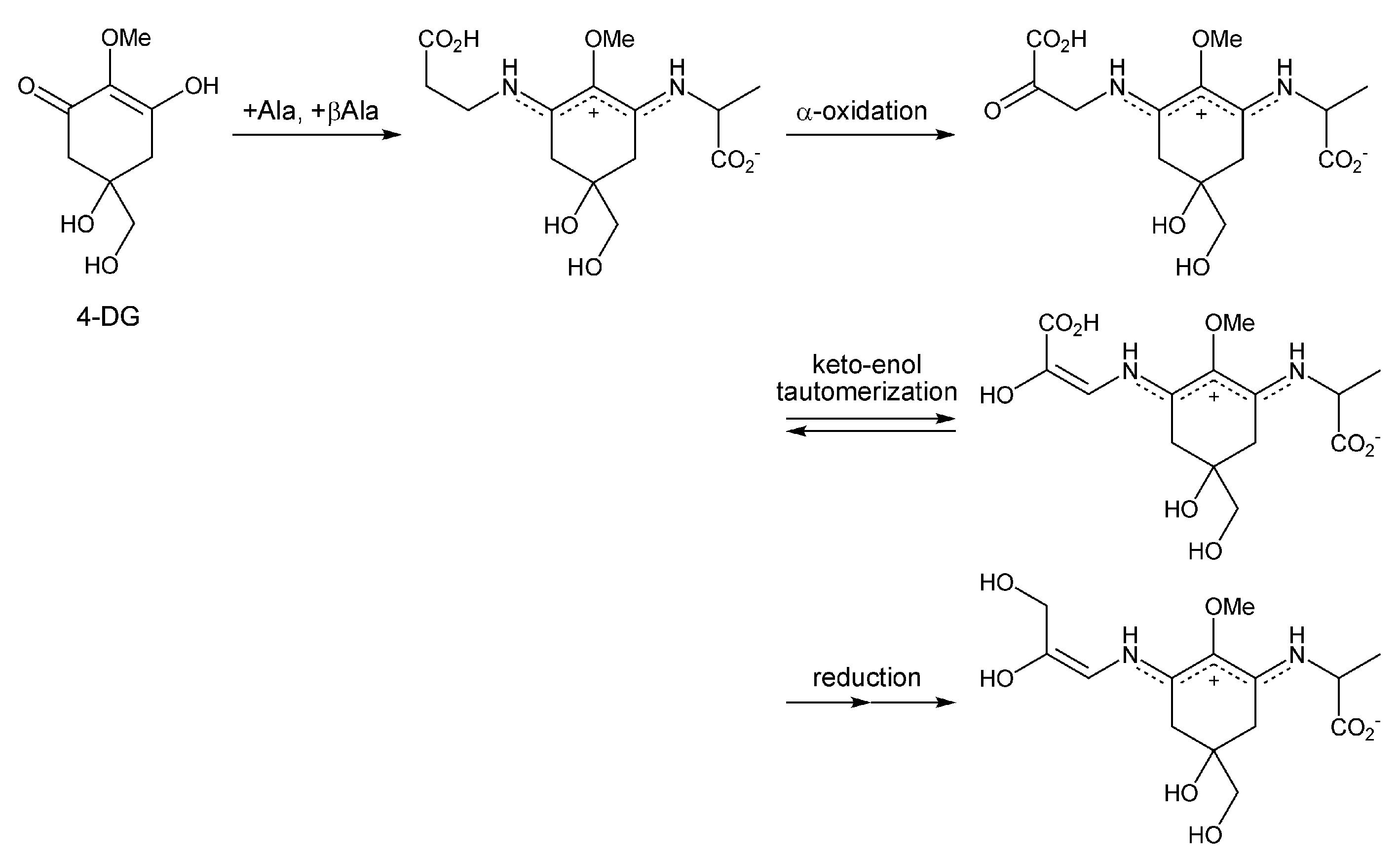
2.2. 4-Deoxygadusol (Figure 2(1b))
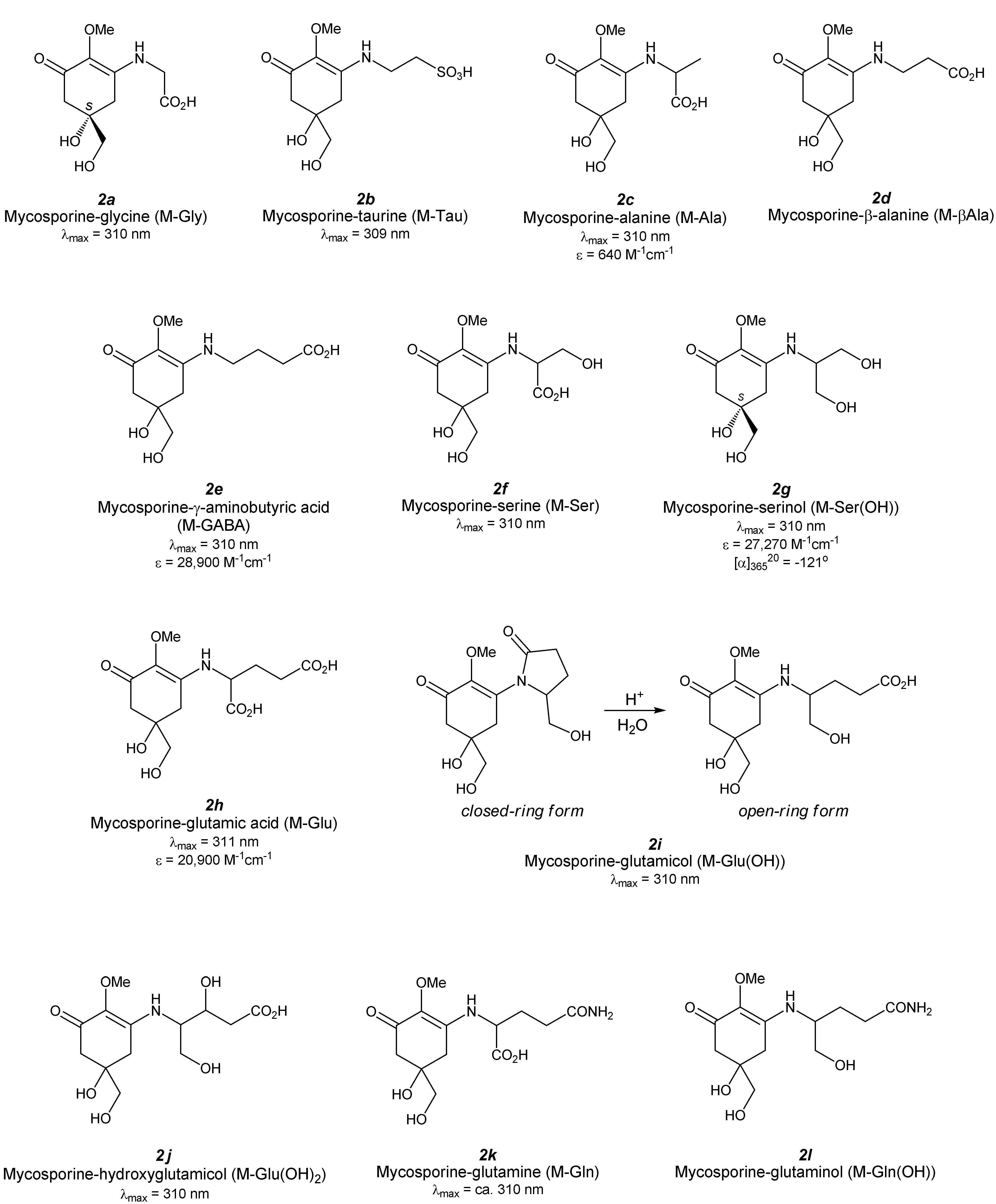
3. Mono-Substituted MAAs (Aminocyclohexenone-Type MAAs, Figure 3)

3.1. Mycosporine-Glycine (Figure 3(2a))

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH Value | M-Gly | A-330 (+Pi 14%) | P-334 (+SH) | SH | Asc |
|---|---|---|---|---|---|
| 6.0 | 20 | 1000 | 1000 | – | 11 |
| 7.5 | 4 | 60 | 400 | – | 4 |
| 8.5 | 3 | 10 | 80 | 100 | 26 |
3.2. Mycosporine-Taurine (Figure 3(2b))
3.3. Mycosporine-Alanine (Figure 3(2c))
3.4. Mycosporine-β-Alanine (Figure 3(2d))
3.5. Mycosporine-γ-Aminobutyric Acid (Figure 3(2e))
3.6. Mycosporine-Serine (Figure 3(2f))
3.7. Mycosporine-Serinol (Figure 3(2g))
3.8. Mycosporine-Glutamic Acid (Figure 3(2h))
3.9. Mycosporine-Glutamicol (Figure 3(2i))
3.10. Mycosporine-Hydroxyglutamicol (Figure 3(2j))
3.11. Mycosporine-Glutamine (Figure 3(2k))
3.12. Mycosporine-Glutaminol (Figure 3(2l))
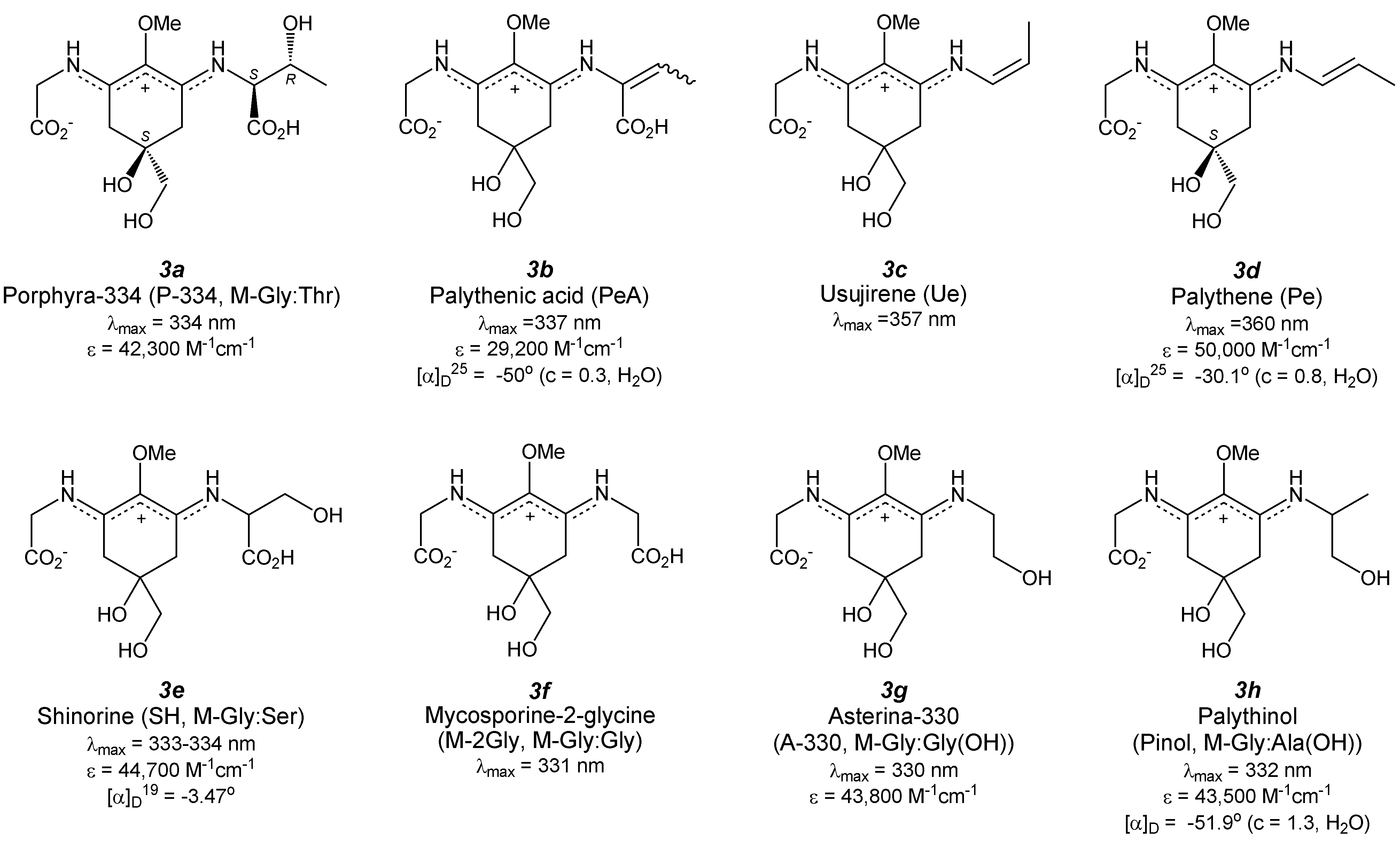
4. Di-Substituted MAAs (Aminocyclohexene Imine-Type MAAs, Figure 4)
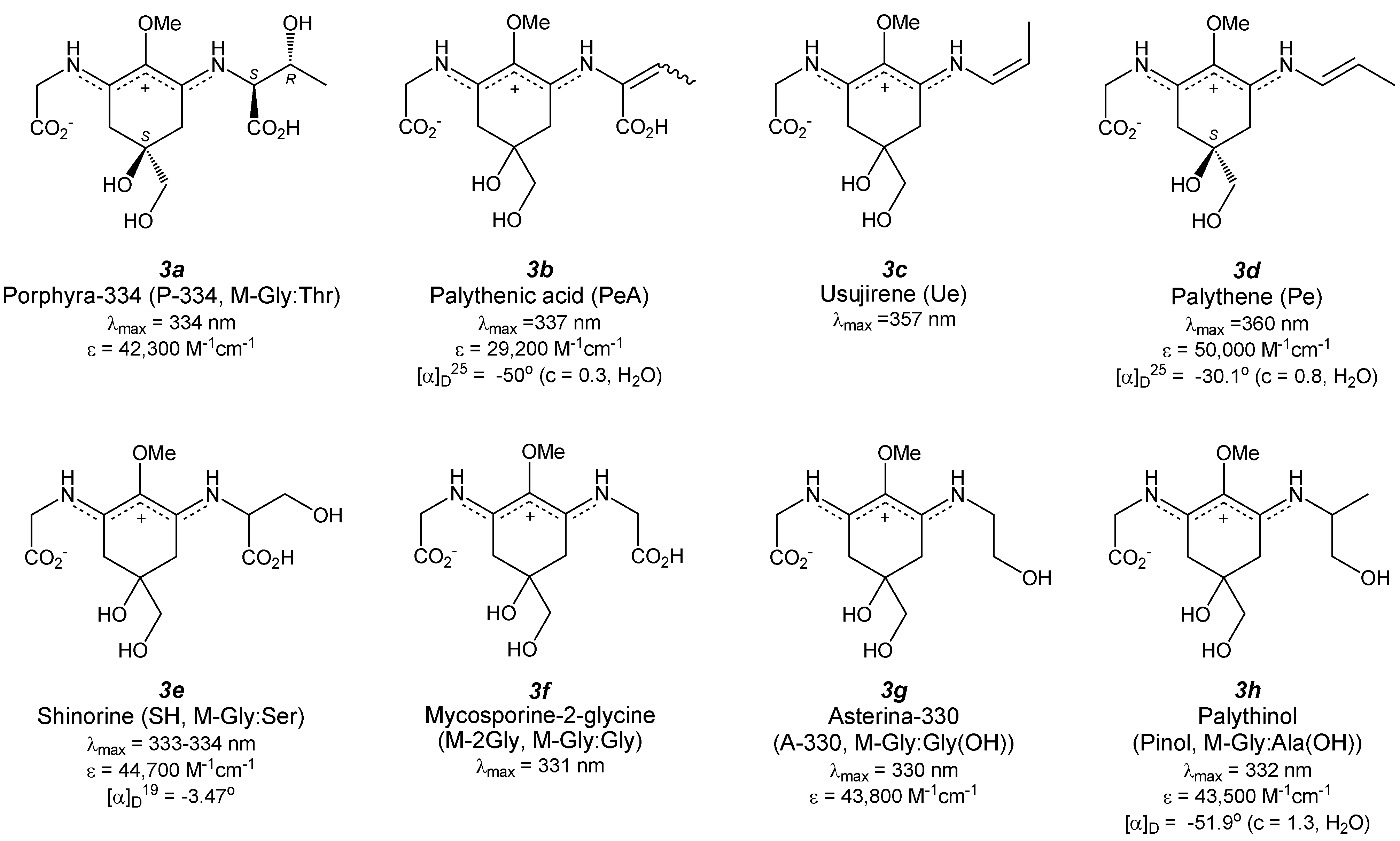
4.1. Porphyra-334 (Figure 4(3a))
4.2. Palythenic Acid (Figure 4(3b))
4.3. Usujirene (Figure 4(3c)) and Palythene (Figure 4(3d))
4.4. Shinorine (Figure 4(3e)) and Its Methyl Ester
4.5. Mycosporine-2-Glycine (Figure 4(3f))
4.6. Asterina-330 (Figure 4(3g))
4.7. Palythinol (Figure 4(3h))
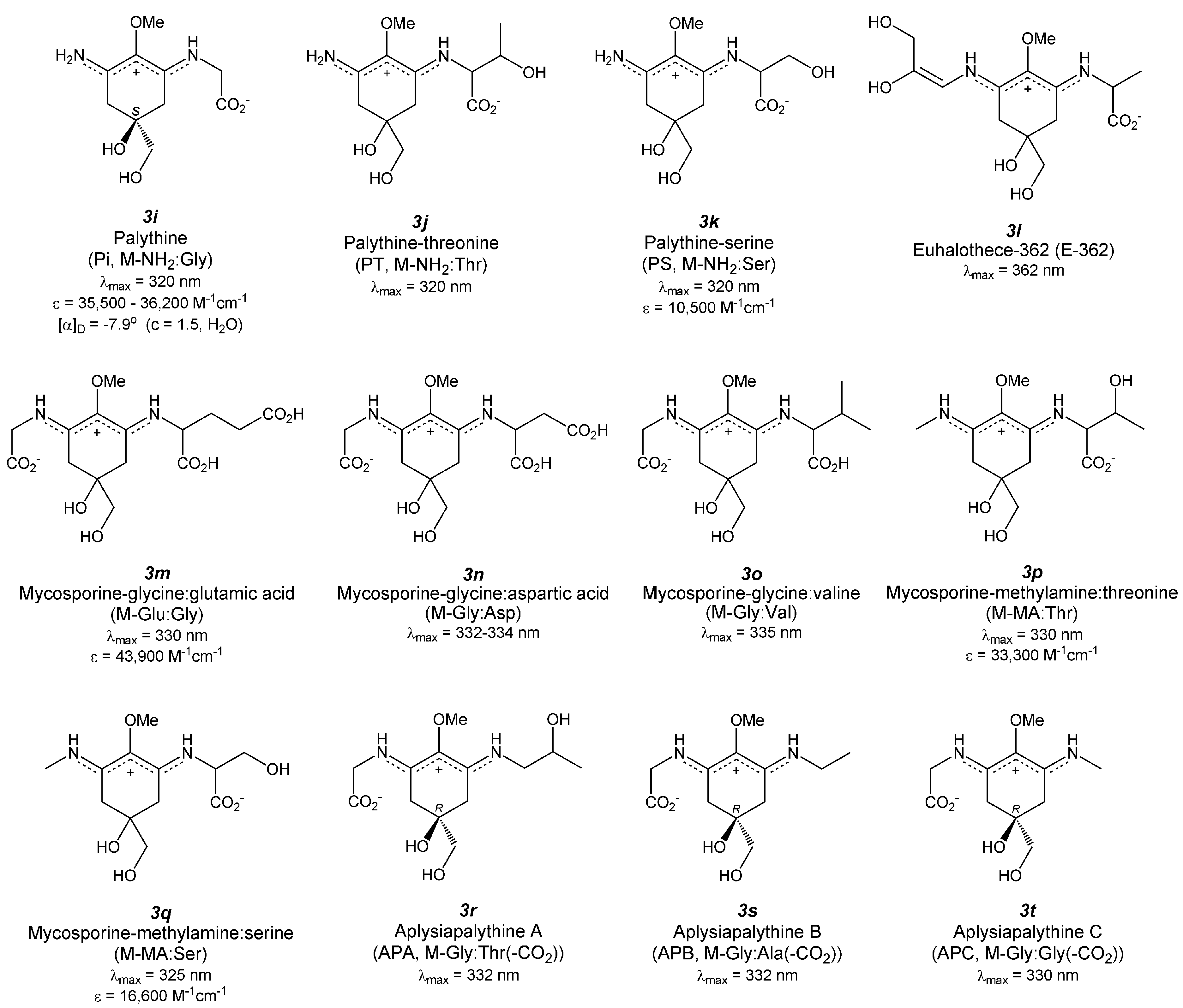
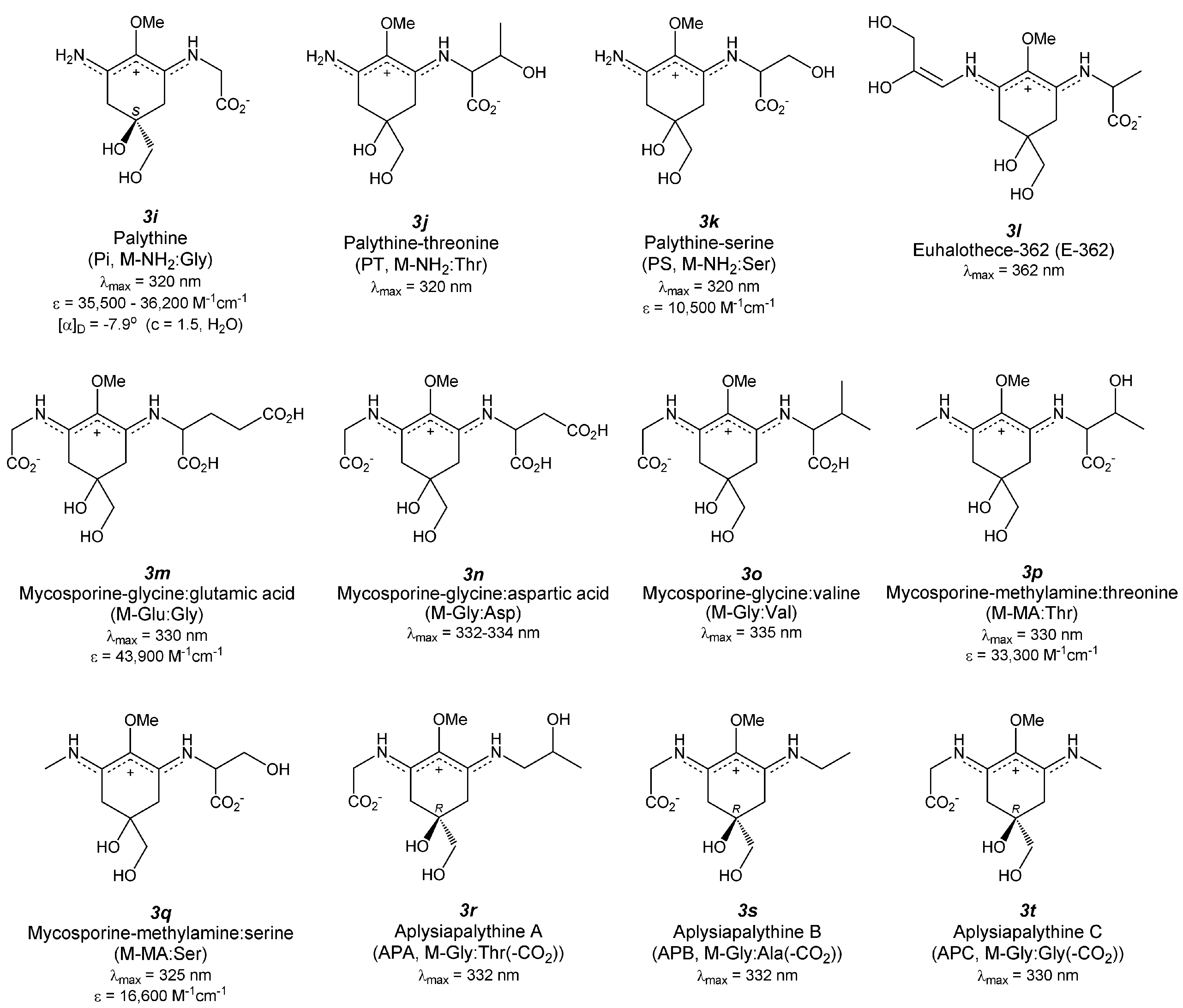
4.8. Palythine (Figure 4(3i))
4.9. Palythine-Threonine (Figure 4(3j))
4.10. Palythine-Serine (Figure 4(3k))
4.11. Euhalothece-362 (Figure 4(3l))
4.12. Mycosporine-Glycine: Glutamic Acid (Figure 4(3m))
4.13. Mycosporine-Glycine: Aspartic Acid (Figure 4(3n))
4.14. Mycosporine-Glycine: Valine (Figure 4(3o))
4.15. Decarboxylated MAA Derivatives
4.16. Hybrid MAAs (MAAs Consisting of Multiple Chromophores)
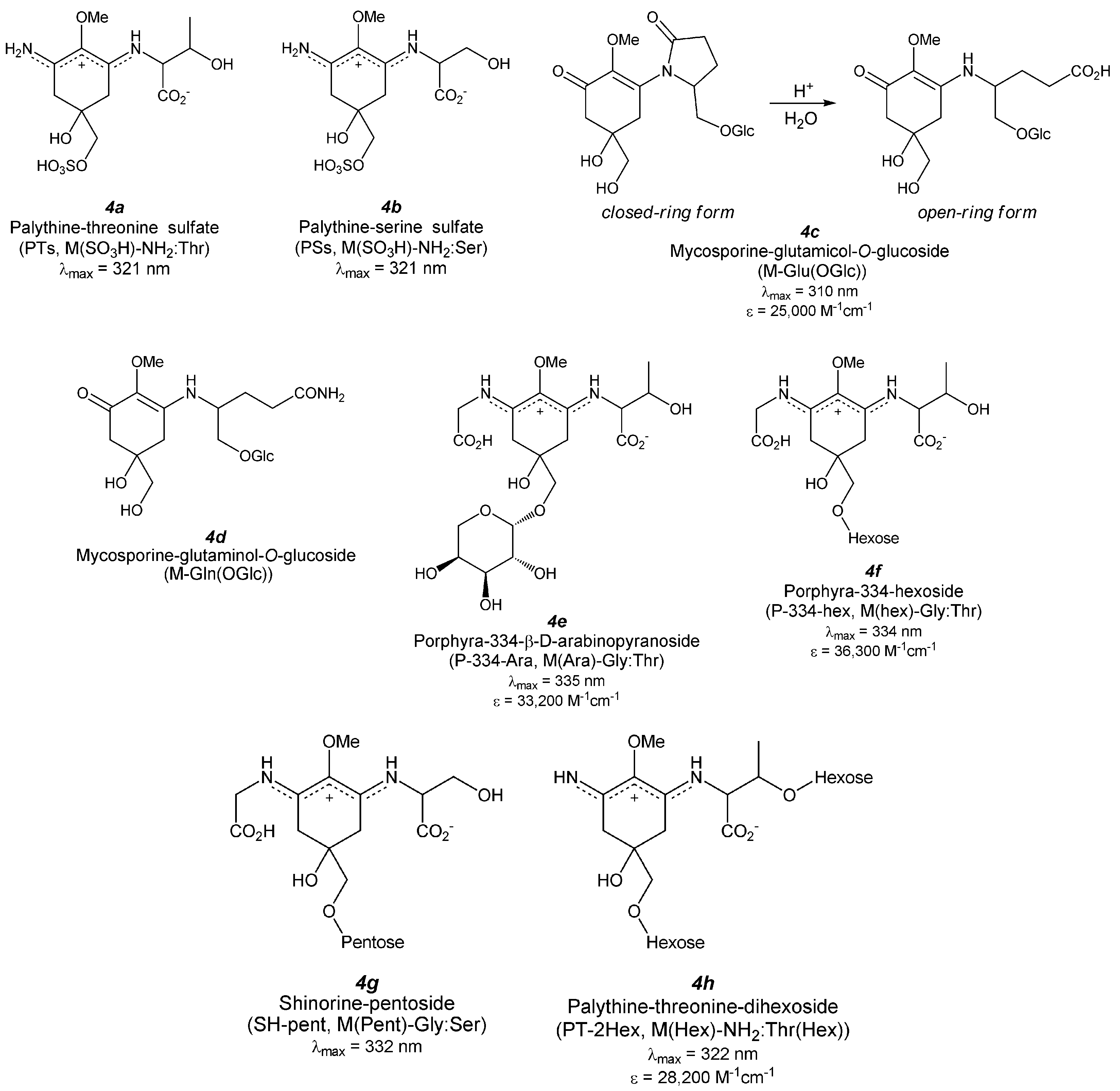
5. Derivatized MAAs (Figure 5)
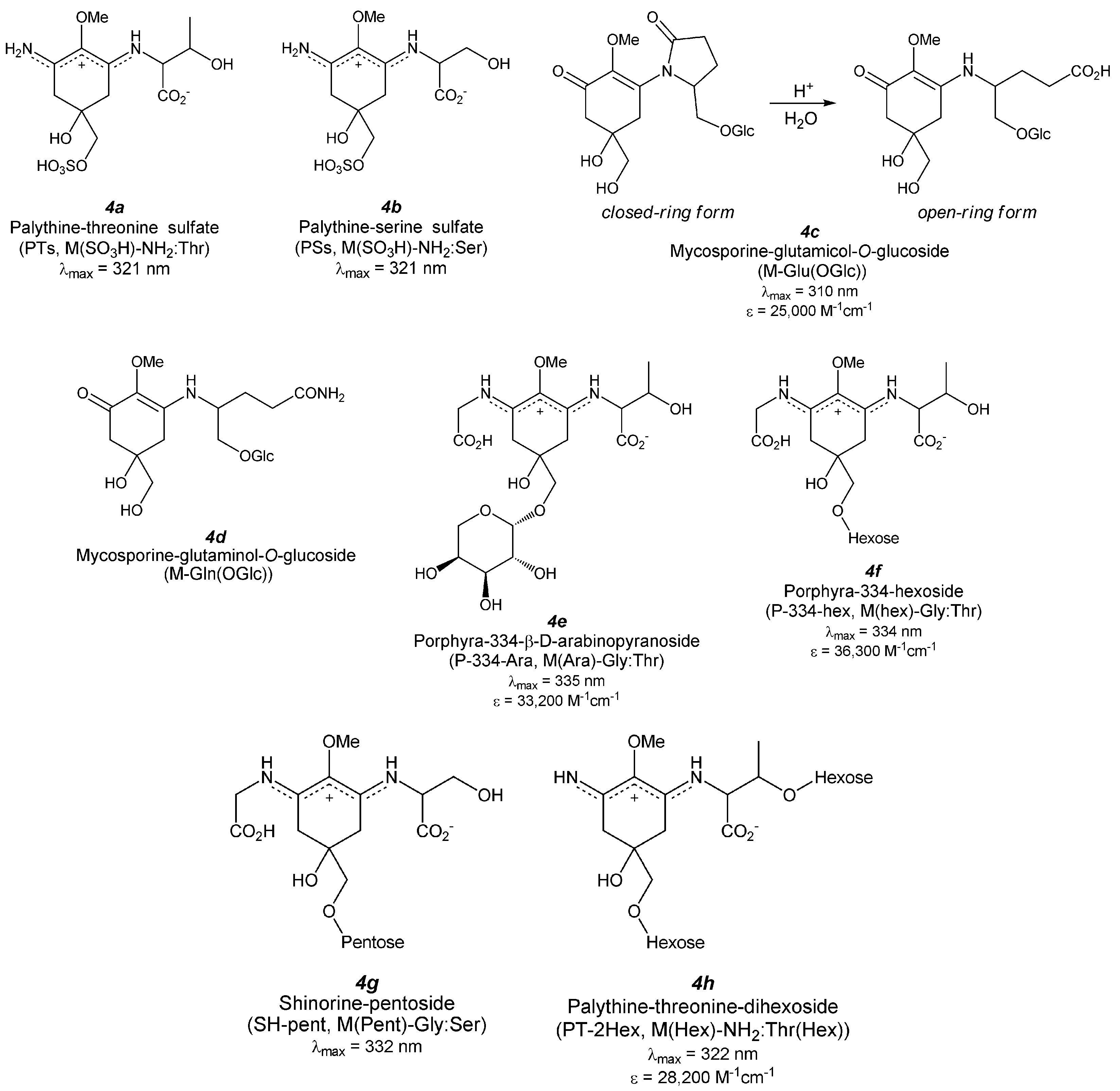
5.1. MAA Sulfates
5.1.1. Palythine-Threonine Sulfate (Figure 5(4a))
5.1.2. Palythine-Serine Sulfate (Figure 5(4b))
5.2. MAA Glycosides
5.2.1. Mono-Substituted MAA Glycosides (Aminocyclohexenone-Type MAA Glycosides)
5.2.1.1. Mycosporine-Glutamicol-O-Glucoside (Figure 5(4c))
5.2.1.2. Mycosporine-Glutaminol-O-Glucoside (Figure 5(4d))
5.2.2. Di-Substituted MAA Glycosides (Aminocyclohexene Imine-Type MAA Glycosides)
5.2.2.1. Porphyra-334 Glycosides (Figure 5(4e,f))
5.2.2.2. Shinorine Glycoside (Figure 5(4g))
5.2.2.3. Palythine-Threonine Glycoside (Figure 5(4h))
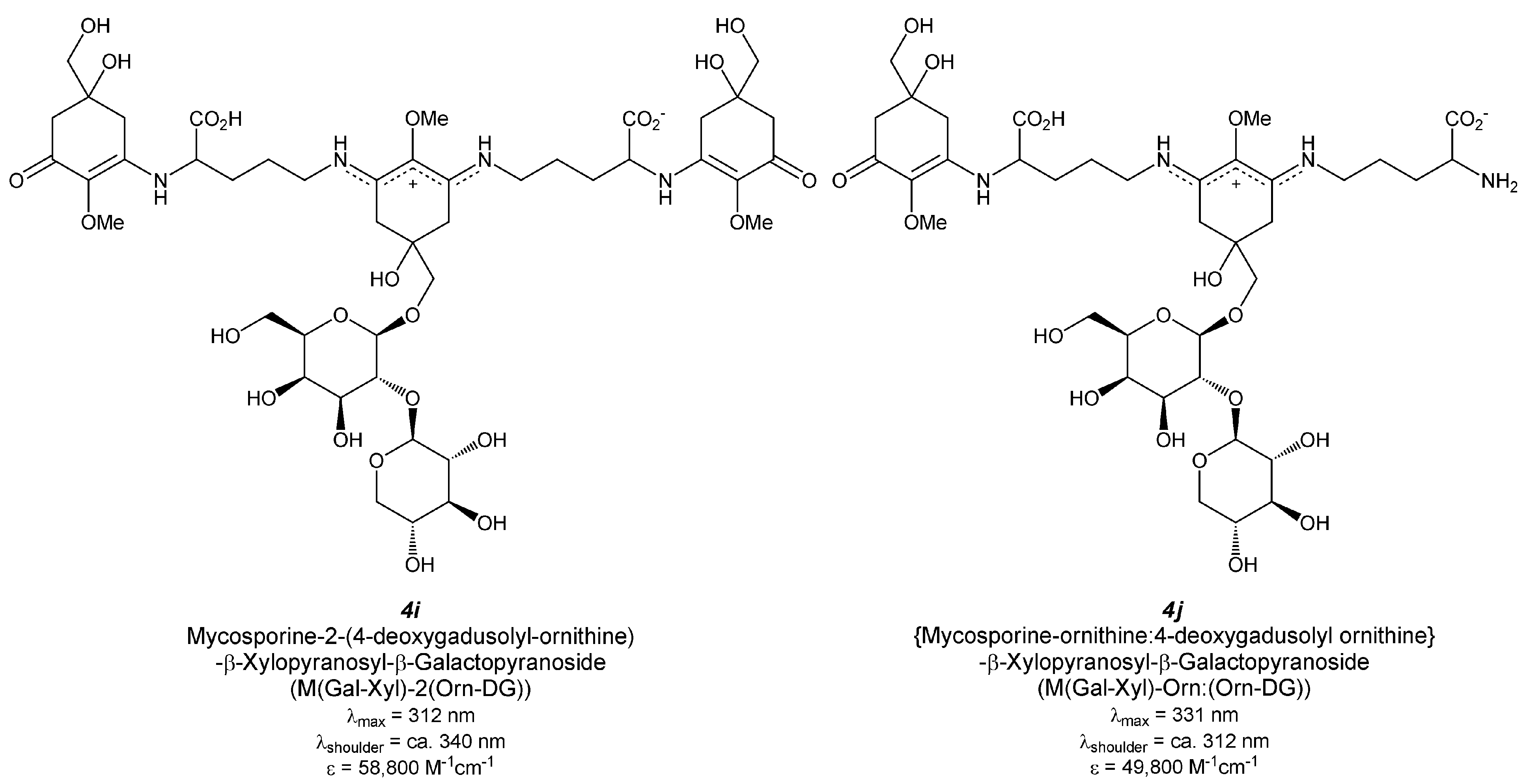
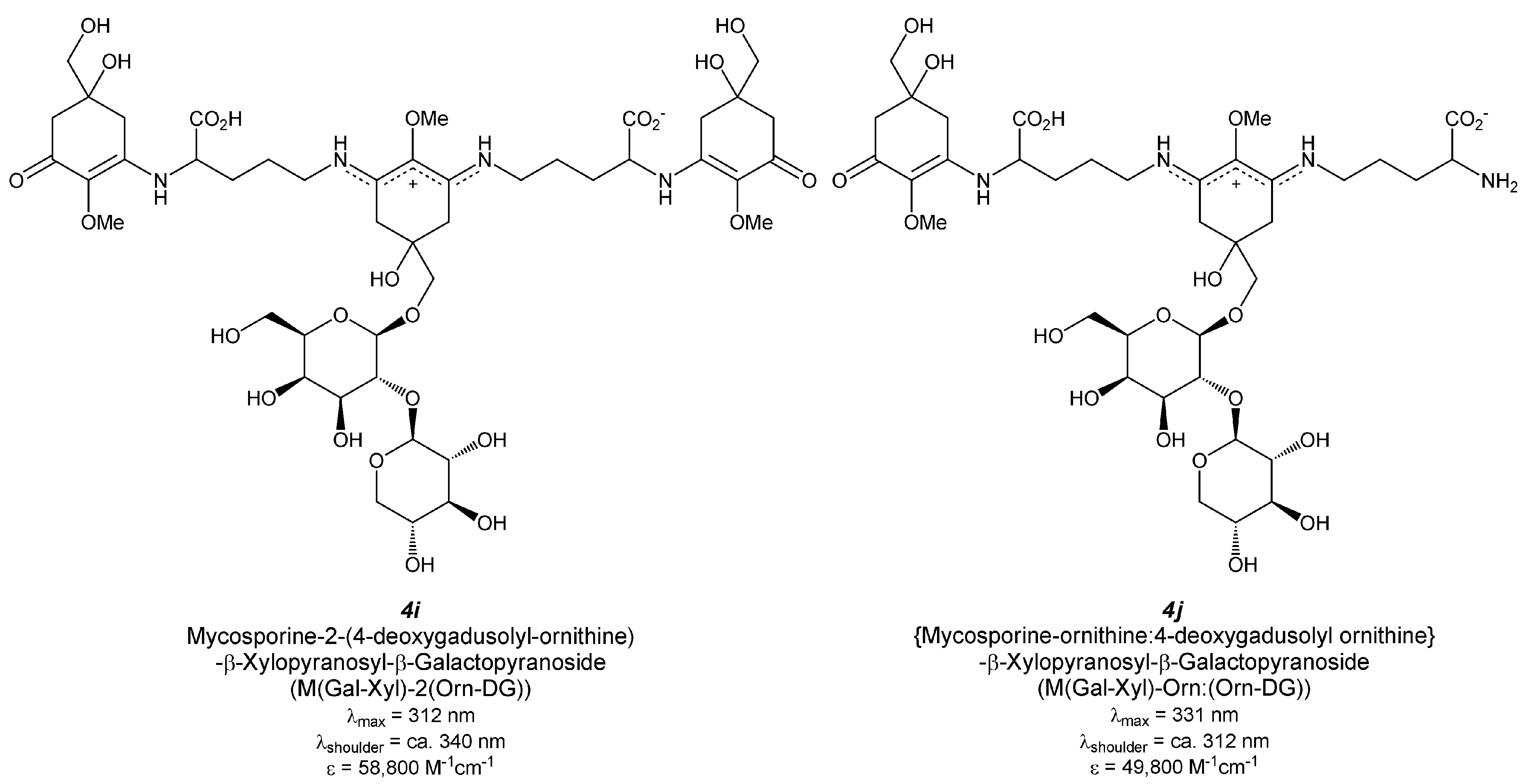
5.2.2.4. Hybrid MAA Glycoside (Figure 5(4i,j))
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix
| Compound Name | Antioxidant Activity | Physiological Property | ||||||
|---|---|---|---|---|---|---|---|---|
| ORAC | Lipid Peroxidation Inhibition | Radical Quenching Capacity | Singlet Oxygen Quenching | Hydrogen Peroxide Quenching | Protecting Effect on Living Cell | |||
| DPPH | ABTS | Superoxide | ||||||
| MAA precursors | ||||||||
| Gadusol Figure 2(1a) | yes [21] a | – | – | yes [21] a | – | – | – | – |
| 4-Deoxygadusol Figure 2(1b) | – | yes [26] | – | – | – | yes [27] b | – | – |
| Aminocyclohexenone-type MAAs | ||||||||
| Mycosporine-glycine Figure 3(2a) | – | yes [30] c yes [31] d | yes [32] e | yes [31] d | no [31] d | yes [30] c | – | yes [34] f |
| Mycosporine-GABA Figure 3(2e) | – | – | – | yes [39] g | – | – | – | – |
| Aminocyclohexene imine-type MAAs | ||||||||
| Porphyra-334 Figure 4(3a) | – | no [30] h yes [31] i yes [64] j | weak [32] e weak [65] j | weak [31] i | moderate [31] i | no [61] yes [64] j | – | yes [31] i yes [32] e yes [72] yes [74] j yes [75] j |
| dehydrated porphyra-334 | – | – | yes [65] j | – | – | – | – | – |
| Palythenic acid Figure 4(3b) | – | – | – | – | – | no [61] | – | – |
| Palythene, Usujirene Figure 4(3c, 3d) | – | yes [82] j | – | – | – | no [61] | – | – |
| Shinorine Figure 4(3e) | – | no [30] k yes [31] l | no [32] e | weak [31] l | moderate [31] l | no [61] | – | – |
| Asterina-330 Figure 4(3g) | – | no [30] m yes [31] n | – | yes [31] n | yes [31] n | no [61] | yes [97] o | – |
| Palythinol Figure 4(3h) | – | no [30] c | – | – | – | – | – | – |
| Palythine Figure 4(3i) | – | no [30] c | yes [97] ° | – | – | very weak [61] | weak [97] o | – |
| Glycosylated MAAs | ||||||||
| P-334 arabinoside Figure 5(4e) | – | – | no [67] g | yes [67] g | – | – | – | – |
| P-334 hexoside Figure 5(4f) | – | – | – | yes [69] g | – | – | – | – |
| Palythine-threonine-dihexoside Figure 5(4h) | – | – | – | yes [69] g | – | – | – | – |
| Hybrid MAA-1 (1050Da) Figure 5(4i) | – | – | yes [67] g | yes [67] g | – | – | – | – |
| Hybrid MAA-2 (880Da) Figure 5(4j) | – | – | – | yes [39] g | – | – | – | – |
| Compound Name | References |
|---|---|
| MAA precursors | |
| Gadusol Figure 2(1a) | [17,18] |
| 4-Deoxygadusol Figure 2(1b) | [24] |
| Aminocyclohexenone-type MAAs | |
| Mycosporine-glycine Figure 3(2a) | [24,29] |
| Mycosporine-taurine Figure 3(2b) | [35] |
| Mycosporine-alanine Figure 3(2c) | [37] |
| Mycosporine-β-alanine Figure 3(2d) | [38] |
| Mycosporine-GABA Figure 3(2e) | [39] |
| Mycosporine-serine Figure 3(2f) | [40] |
| Mycosporine-serinol Figure 3(2g) | [43,44] |
| Mycosporine-glutamic acid Figure 3(2h) | [46] |
| Mycosporine-glutamicol Figure 3(2i) | [41,47] |
| Mycosporine-hydroxyglutamicol Figure 3(2j) | [49] |
| Mycosporine-glutamine Figure 3(2k) | [50] |
| Mycosporine-glutaminol Figure 3(2l) | [51,52] |
| Aminocyclohexene imine-type MAAs | |
| Porphyra-334 Figure 4(3a) | [53] |
| Palythenic acid Figure 4(3b) | [76] |
| Usujirene Figure 4(3c) | [80] |
| Palythene Figure 4(3d) | [78,79] |
| Shinorine Figure 4(3e) | [83,84] |
| Mycosporine-2-glycine Figure 4(3f) | [91] |
| Asterina-330 Figure 4(3g) | [93] |
| Palythinol Figure 4(3h) | [78] |
| Palythine Figure 4(3i) | [78,95] |
| Palythine-threonine Figure 4(3j) | [77,99] |
| Palythine-serine Figure 4(3k) | [100] |
| Euhalothece-362 Figure 4(3l) | [101] |
| Mycosporine-glycine:glutamic acid Figure 4(3m) | [102] |
| Mycosporine-glycine:aspartic acid Figure 4(3n) | [103] |
| Mycosporine-glycine:valine Figure 4(3o) | [104] |
| Mycosporine-methylamine:threonine Figure 4(3p) | [105] |
| Mycosporine-methylamine:serine Figure 4(3q) | [100] |
| Aplysiapalythine A Figure 4(3r), B Figure 4(3s), and C Figure 4(3t) | [94] |
| Derivatized MAAs | |
| Palythine-threonine sulfate Figure 5(4a) | [106] |
| Palythine-serine sulfate Figure 5(4b) | [106] |
| Mycosporine-glutamicol-O-glucoside Figure 5(4c) | [48] |
| Mycosporine-glutaminol-O-glucoside Figure 5(4d) | [50,51] |
| Porphyra-334-β-d-arabinopyranoside Figure 5(4e) | [39,67] |
| Porphyra-334-hexoside Figure 5(Figure 5) | [69] |
| Shinorine-pentoside Figure 5(4g) | [39] |
| Palythine-threonine-dihexoside Figure 5(4h) | [69] |
| Mycosporine-2-(4-deoxygadusolyl-ornithine)-β-xylopyranosyl-β-galactopyranoside Figure 5(4i) | [39,67] |
| (Mycosporine-ornithine:4-deoxygadusolyl ornithine)-β-xylopyranosyl-β-galactopyranoside Figure 5(4j) | [39] |
References
- Sinha, R.P.; Singh, S.P.; Häder, D.-P. Database on mycosporines and mycosporine-like amino acids (MAAs) in fungi, cyanobacteria, macroalgae, phytoplankton and animals. J. Photochem. Photobiol. B 2007, 89, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Favre-Bonvin, J.; Bernillon, J.; Salin, N.; Arpin, N. Biosynthesis of mycosporines: Mycosporine glutaminol in Trichothecium roseum. Phytochemistry 1987, 26, 2509–2514. [Google Scholar] [CrossRef]
- Balskus, E.P.; Walsh, C.T. The genetic and molecular basis for sunscreen biosynthesis in cyanobacteria. Science 2010, 329, 1653–1656. [Google Scholar] [CrossRef] [PubMed]
- Portwich, A.; Garcia-Pichel, F. Biosynthetic pathway of mycosporines (mycosporine-like amino acids) in the cyanobacterium Chlorogloeopsis sp. strain PCC 6912. Phycologia 2003, 42, 384–392. [Google Scholar] [CrossRef]
- Sinha, R.P.; Klisch, M.; Gröniger, A.; Häder, D.-P. Ultraviolet-absorbing/screening substances in cyanobacteria, phytoplankton and macroalgae. J. Photochem. Photobiol. B 1998, 47, 83–94. [Google Scholar] [CrossRef]
- Rozema, J.; Björn, L.O.; Bornman, J.F.; Bornman, J.F.; Gaberščik, A.; Häder, D.-P.; Trošt, T.; Germ, M.; Klisch, M.; Gröniger, A.; et al. The role of UV-B radiation in aquatic and terrestrial ecosystems—An experimental and functional analysis of the evolution of UV-absorbing compounds. J. Photochem. Photobiol. B 2002, 66, 2–12. [Google Scholar] [CrossRef]
- Shick, J.M.; Dunlap, W.C. Mycosporine-like amino acids and related gadusols: Biosynthesis, accumulation, and UV-protective functions in aquatic organisms. Annu. Rev. Plant Physiol. 2002, 64, 223–262. [Google Scholar] [CrossRef] [PubMed]
- Oren, A. Mycosporine-like amino acids as osmotic solutes in a community of halophilic cyanobacteria. Geomicrobiol. J. 1997, 14, 231–240. [Google Scholar] [CrossRef]
- Portwich, A.; Garcia-Pichel, F. Ultraviolet and osmotic stresses induce and regulate the synthesis of mycosporines in the cyanobacterium Chlorogloeopsis PCC 6912. Arch. Microbiol. 1999, 172, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.P.; Ambasht, N.K.; Sinha, J.P.; Häder, D.-P. Wavelength-dependent induction of a mycosporine-like amino acid in a rice-field cyanobacterium, Nostoc commune: Role of inhibitors and salt stress. Photochem. Photobiol. Sci. 2003, 2, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Kogej, T.; Gostinčar, C.; Volkmann, M.; Gorbushina, A.A.; Gunde-Cimerman, N. Mycosporines in extremophilic fungi—Novel complementary osmolytes? Environ. Chem. 2006, 3, 105–110. [Google Scholar] [CrossRef]
- Singh, S.P.; Klisch, M.; Sinha, R.P.; Häder, D.-P. Effects of abiotic stressors on synthesis of the mycosporine-like amino acid shinorine in the cyanobacterium Anabaena variabilis PCC 7937. Photochem. Photobiol. 2008, 84, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Waditee-Sirisattha, R.; Kageyama, H.; Sopun, W.; Tanaka, Y.; Takabe, T. Identification and upregulation of biosynthetic genes required for accumulation of mycosporine-2-glycine under salt stress conditions in the halotolerant cyanobacterium Aphanothece halophytica. Appl. Environ. Microbiol. 2014, 80, 1763–1769. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.N.; Zhang, Z.C.; Feng, J.L.; Qiu, B.S. Effects of UV-B radiation and periodic desiccation on the morphogenesis of the edible terrestrial cyanobacterium Nostoc flagelliforme. Appl. Environ. Microbiol. 2012, 78, 7075–7081. [Google Scholar] [CrossRef] [PubMed]
- Olsson-Francis, K.; Watson, J.S.; Cockell, C.S. Cyanobacteria isolated from the high-intertidal zone: A model for studying the physiological prerequisites for survival in low Earth orbit. Int. J. Astrobiol. 2013, 12, 292–303. [Google Scholar] [CrossRef]
- Michalek-Wagner, K. Seasonal and sex-specific variations in levels of photo-protecting mycosporine-like amino acids. Mar. Biol. 2001, 139, 651–660. [Google Scholar]
- Chioccara, F.; Gala, A.D.; de Rosa, M.; Novellino, E.; Prota, G. Mycosporine amino acids and related compounds from the eggs of fishes. Bull. Soc. Chimi. Belg. 1980, 89, 1101–1106. [Google Scholar] [CrossRef]
- Grant, P.T.; Plack, P.A.; Thomson, R.H. Gadusol, a metabolite from fish eggs. Tetrahedron Lett. 1980, 21, 4043–4044. [Google Scholar] [CrossRef]
- Plack, P.A.; Fraser, N.W.; Grant, P.T.; Middleton, C.; Mitchell, A.I.; Thomson, R.H. Gadusol, an enolic derivative of cyclohexane-1,3-dione present in the roes cod and other marine fish. Biochem. J. 1981, 199, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Arbeloa, E.M.; Ramirez, C.L.; Procaccini, R.A.; Churio, M.S. Electrochemical characterization of the marine antioxidant gadusol. Nat. Prod. Commun. 2012, 7, 1211–1214. [Google Scholar] [PubMed]
- Arbeloa, E.M.; Uez, M.J.; Bertolotti, S.G.; Churio, M.S. Antioxidant activity of gadusol and occurrence in fish roes from Argentine sea. Food Chem. 2010, 119, 586–591. [Google Scholar] [CrossRef]
- Naguib, Y.M.A. A Fluorometric method for measurement of oxygen radical-scavenging activity of water-soluble antioxidants. Anal. Biochem. 2000, 284, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Arbeloa, E.M.; Bertolotti, S.G.; Churio, M.S. Photophysics and reductive quenching reactivity of gadusol in solution. Photochem. Photobiol. Sci. 2011, 10, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Hirata, Y. Isolation and structure of a mycosporine from the zoanthid Palythoa tuberculosa. Tetrahedron Lett. 1977, 28, 2429–2430. [Google Scholar] [CrossRef]
- Chioccara, F.; Zeuli, L.; Novellino, E. Occurrence of mycosporine related compounds in sea urchin eggs. Comp. Biochem. Phys. B. 1986, 85B, 459–461. [Google Scholar] [CrossRef]
- Dunlap, W.C.; Masaki, K.; Yamamoto, Y.; Larsen, R.M.; Karube, I. New Developments in Marine Biotechnology; Le Gal, Y., Halvorson, H.O., Eds.; Springer: Berlin, Germany, 1998; pp. 33–35. [Google Scholar]
- Suh, H.J.; Lee, H.W.; Jung, J. Singlet oxygen quenching by deoxygadusol and related mycosporine-like amino acids from phytoplankton Prorocentrum micans. J. Photosci. 2004, 11, 77–81. [Google Scholar]
- Řezanka, T.; Temina, M.; Tolstikov, A.G.; Dembitsky, V.M. Natural microbial UV radiation filters—Mycosporine-like amino acids. Folia Microbiol. 2004, 49, 339–352. [Google Scholar] [CrossRef]
- White, J.D.; Cammack, J.H.; Sakuma, K. The synthesis and absolute configuration of mycosporins. A novel application of the Staudinger reaction. J. Am. Chem. Soc. 1989, 111, 8970–8972. [Google Scholar] [CrossRef]
- Dunlap, W.C.; Yamamoto, Y. Small-molecule antioxidants in marine organisms: Antioxidant activity of mycosporine-glycine. Comp. Biochem. Physiol. 1995, 112B, 105–114. [Google Scholar] [CrossRef]
- De La Coba, F.; Aguilera, J.; Figueroa, F.L.; de Gálvez, M.V.; Herrera, E. Antioxidant activity of mycosporine-like amino acids isolated from three red macroalgae and one marine lichen. J. Appl. Phycol. 2009, 21, 161–169. [Google Scholar] [CrossRef]
- Suh, S.S.; Hwang, J.; Park, M.; Seo, H.H.; Kim, H.S.; Lee, J.H.; Moh, S.H.; Lee, T.K. Anti-inflammation activities of mycosporine-like amino acids (MAAs) in response to UV radiation suggest potential anti-skin aging activity. Mar. Drugs 2014, 12, 5174–5187. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.J.; Lee, H.W.; Jung, J. Mycosporine glycine protects biological systems against photodynamic damage by quenching singlet oxygen with a high efficiency. Photochem. Photobiol. 2003, 78, 109–113. [Google Scholar] [CrossRef]
- Oyamada, C.; Kaneniwa, M.; Ebitani, K.; Murata, M.; Ishihara, K. Mycosporine-like amino acids extracted from scallop (Patinopecten yessoensis) ovaries: UV protection and growth stimulation activities on human cells. Mar. Biotechnol. 2008, 10, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Stochaj, W.R.; Dunlap, W.C.; Shick, J.M. Two new UV-absorbing mycosporine-like amino acids from the sea anemone Anthopleura elegantissima and the effects of zooxanthellae and spectral irradiance on chemical composition and content. Mar. Biol. 1994, 118, 149–156. [Google Scholar] [CrossRef]
- Zhang, L.; Li, L.; Wu, Q. Protective effects of mycosporine-like amino acids of Synechocystis sp. PCC 6803 and their partial characterization. J. Photochem. Photobiol. B 2007, 86, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Leite, B.; Nicholson, R.L. Mycosporine-alanine: A self-inhibitor of germination from the conidial mucilage of Colletotrichum graminicola. Exp. Mycol. 1992, 16, 76–86. [Google Scholar] [CrossRef]
- Nakamura, E.; Kobayashi, J.; Abe, R. Mycosporin-Like Amino Acid. JP Patent 1984137450 A, 7 August 1984. [Google Scholar]
- Nazifi, E.; Wada, N.; Asano, T.; Nishiuchi, T.; Iwamuro, Y.; Chinaka, S.; Matsugo, S.; Sakamoto, T. Characterization of the chemical diversity of glycosylated mycosporine-like amino acids in the terrestrial cyanobacterium Nostoc commune. J. Photochem. Photobiol. B 2015, 142, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Arpin, N.; Curt, R.; Favre-Bonvin, J. Mycosporines: Review and new data concerning their structure. Revue de Mycologie 1979, 43, 247–257. [Google Scholar]
- Lunel, M.-C.; Arpin, N.; Favre-Bonvin, J. Structure of Normycosporin Glutamine, a new compound isolated from Pyronema omphalodes (Bull ex Fr.) fuckel. Tetrahedron Lett. 1980, 21, 4715–4716. [Google Scholar] [CrossRef]
- Leach, C.M. Ultraviolet-absorbing substances associated with light-induced sporulation in fungi. Can. J. Botany 1965, 43, 185–200. [Google Scholar] [CrossRef]
- Favre-Bonvin, J.; Arpin, N.; Brevard, C. Chemotaxonomic research on mushrooms. XXXI. Structure of mycosporine (P 310). Can. J. Chem. 1976, 54, 1105–1113. [Google Scholar] [CrossRef]
- White, J.D.; Cammack, J.H.; Sakuma, K.; Rewcastle, G.W.; Widener, R.K. Transformation of quinic acid. Asymmetric synthesis and absolute configulation of mycosporin I and mycosporin-gly. J. Org. Chem. 1995, 60, 3600–3611. [Google Scholar] [CrossRef]
- Bernillon, J.; Parussini, E.; Letoublon, R.; Favre-Bovin, J.; Arpin, N. Flavin-mediated photolysis of mycosporines. Phytochemistry 1990, 29, 81–84. [Google Scholar] [CrossRef]
- Young, H.; Patterson, V.J. A UV-protective compound from Glomerella cingulata. A mycosporine. Phytochem. 1982, 21, 1075–1077. [Google Scholar] [CrossRef]
- Fayret, J.; Bernillon, J.; Bouillant, M.-L.; Favre-Bonvin, J.; Arpin, N. Open and ring forms of mycosporin-2 from the ascomycete Gnomonia leptostyla. Phytochemistry 1981, 20, 2709–2710. [Google Scholar] [CrossRef]
- Bouillant, M.L.; Pittet, J.L.; Bernillon, J.; Favre-Bonvin, J.; Arpin, N. Mycosporins from Ascochyta pisi, Cladosporium herbarum, and Septoria nodorum. Phytochemistry 1981, 20, 2705–2707. [Google Scholar] [CrossRef]
- Roullier, C.; Chollet-Krugler, M.; Pferschy-Wenzig, E.M.; Maillard, A.; Rechberger, G.N.; Legouin-Gargadennec, B.; Bauer, R.; Boustie, J. Characterization and identification of mycosporines-like compounds in cyanolichens. Isolation of mycosporine hydroxyglutamicol from Nephroma laevigatum Ach. Phytochemistry 2011, 72, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Bernillon, J.; Bouillant, M.L.; Pittet, J.L.; Favre-Bonvin, J.; Arpin, N. Mycosporine glutamine and related mycosporines in the fungus Pyronema omphalodes. Phytochemistry 1984, 23, 1083–1087. [Google Scholar] [CrossRef]
- Pittet, J.L.; Bouillant, M.L.; Bernillon, J.; Arpin, N.; Favre-Bonvin, J. The presence of reduced-glutamine mycosporines, new molecules, in several Deuteromycetes. Tetrahedron Lett. 1983, 24, 65–68. [Google Scholar] [CrossRef]
- Lemoyne, F.; Bernillon, J.; Favre-Bonvin, J.; Bouillant, M.L.; Arpin, N. Occurrence and characteristics of amino alcohols and cyclohexenone. Components of fungal mycosporines. Z. Naturforsch C 1985, 40C, 612–616. [Google Scholar]
- Takano, S.; Nakanishi, A.; Uemura, D.; Hirata, Y. Isolation and structure of a 334 nm UV-absorbing substance, porphyra-334 from the red alga Porphyra tenera Kjellman. Chem. Lett. 1979, 4, 419–420. [Google Scholar] [CrossRef]
- Hirata, Y.; Uemura, D.; Ueda, K.; Takano, S. Several compounds from Palythoa tuberculosa (Coelenterata). Pure Appl. Chem. 1979, 51, 1875–1883. [Google Scholar] [CrossRef]
- Chioccara, F.; Misuraca, G.; Novellino, E.; Prota, G. Occurrence of two new mycosporine-like amino acids, mytilins A and B in the edible mussel, Mytilus galloprovincialis. Tetrahedron Lett. 1979, 34, 3181–3182. [Google Scholar] [CrossRef]
- Klisch, M.; Richter, P.; Puchta, R.; Häder, D.-P.; Bauer, W. The stereostructure of porphyra-334: An experimental and calculational NMR investigation. Evidence for an efficient “proton sponge”. Helv. Chim. Acta 2007, 90, 488–511. [Google Scholar] [CrossRef]
- Zhang, Z.; Gao, X.; Tashiro, Y.; Matsukawa, S.; Ogawa, H. Researches on the stability of porphyra-334 solution and its influence factors. J. Ocean U. China 2004, 3, 166–170. [Google Scholar] [CrossRef]
- Zhang, Z.; Tashiro, Y.; Matsukawa, S.; Ogawa, H. Influence of pH and temperature on the ultraviolet-absorbing properties of porphyra-334. Fisheries Sci. 2005, 71, 1382–1384. [Google Scholar] [CrossRef]
- Sinha, R.P.; Klisch, M.; Gröniger, A.; Häder, D.-P. Mycosporine-like amino acids in the marine red alga Gracilaria cornea—Effects of UV and heat. Environ. Exp. Bot. 2000, 43, 33–43. [Google Scholar] [CrossRef]
- Conde, F.R.; Churio, M.S.; Previtali, C.M. The photoprotector mechanism of mycosporine-like amino acids. Excited-state properties and photostability of porphyra-334 in aqueous solution. J. Photochem. Photobiol. B 2000, 56, 139–144. [Google Scholar] [CrossRef]
- Whitehead, K.; Hedges, J.I. Photodegradation and photosensitization of mycosporine-like amino acids. J. Photochem. Photobiol. B 2005, 80, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Conde, F.R.; Churio, M.S.; Previtali, C.M. The deactivation pathways of the excited-states of the mycosporine-like amino acids shinorine and porphyra-334 in aqueous solution. Photochem. Photobiol. Sci. 2004, 3, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Lee, J.H.; Seo, H.H.; Kim, H.S.; Cho, M.J.; Shin, D.S.; Kim, T.; Moh, S.H. Photoinduced conductivity in mycosporine-like amino acids. Mater. Chem. Phys. 2015, 151, 1–4. [Google Scholar] [CrossRef]
- Tao, C.; Sugawara, T.; Maeda, S.; Wang, X.; Hirata, T. Antioxidative activities of a mycosporine-like amino acid, porphyra-334. Fisheries Sci. 2008, 74, 1166–1172. [Google Scholar] [CrossRef]
- Yoshiki, M.; Tsuge, K.; Tsuruta, Y.; Yoshimura, T.; Koganemaru, K.; Sumi, T.; Matsui, T.; Matsumoto, K. Production of new antioxidant compound from mycosporine-like amino acid, porphyra-334 by heat treatment. Food Chem. 2009, 113, 1127–1132. [Google Scholar] [CrossRef]
- Tsuge, K.; Yoshimura, T.; Tsuruta, H.; Koganemaru, K.; Yoshiki, M.; Abe, S.; Tsuruhashi, T. Antioxidant Compound, Antioxidant Algae Extract and Method for Producing the Same. JP Patent 2008247901 A, 16 October 2008. [Google Scholar]
- Matsui, K.; Nazifi, E.; Kunita, S.; Wada, N.; Matsugo, S.; Sakamoto, T. Novel glycocylated mycosporine-like amino acids with radical scavenging activity from the cyanobacterium Nostoc commune. J. Photochem. Photobiol. B 2011, 105, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Sivalingam, P.M.; Ikawa, T.; Nisizawa, K. Isolation and physico-chemical properties of a substance 334 from the red alga, Porphyra yezoensis Ueda. Bot. Mar. 1976, 19, 1–7. [Google Scholar] [CrossRef]
- Nazifi, E.; Wada, N.; Yamaba, M.; Asano, T.; Nishiuchi, T.; Matsugo, S.; Sakamoto, T. Glycosylated porphyra-334 and palythine-threnonine from the terrestrial cyanobacterium Nostoc commune. Mar. Drugs 2013, 11, 3124–3154. [Google Scholar] [CrossRef] [PubMed]
- De La Coba, F.; Aguilera, J.; Figueroa, F.L. Use of a Mycosporin-Type Amino Acid (Porphyra 334) As an Antioxidant. PCT International Application Patent WO2007026035, 10 May 2007. [Google Scholar]
- Misonou, T.; Saitoh, J.; Oshiba, S.; Tokitomo, Y.; Maegawa, M.; Inoue, Y.; Hori, H.; Sakurai, T. UV-absorbing substance in the red alga Porphyra yezoensis (Bangiales, Rhodophyta) block thymine photodimer production. Mar. Biotechnol. 2003, 5, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Schürch, C.; Zülli, F.; Nissen, H.-P.; Prieur, H. Mycosporine-like amino acids: Natural UV-screening compounds from red algae to protect the skin against photoaging. SÖFW J. 2003, 129, 38–41. [Google Scholar]
- De La Coba, F.; Aguilera, J.; de Gálves, M.V.; Álvarez, M.; Gallego, E.; Figueroa, F.L.; Herrera, E. Prevention of the ultraviolet effects on clinical and histopathological changes, as well as the heat shock protein-70 expression in mouse skin by topical application of algal UV-absorbing compounds. J. Dermatol. Sci. 2009, 55, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; You, D.H.; Han, T.; Choi, E.-M. Modulation of viability and apoptosis of UVB-exposed human keratinocyte HaCaT cells by aqueous methanol extract of laver (Porphyra yezoensis). J. Photochem. Photobiol. B 2014, 141, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.; Park, S.J.; Kim, I.H.; Choi, Y.H.; Nam, T.J. Protective effect of porphyra-334 on UVA-induced photoaging in human skin fibroblasts. Int. J. Mol. Med. 2014, 34, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Nakamura, H.; Hirata, Y. Isolation and structure of a UV-absorbing substance 337 from the ascidian Halocynthia roretzi. Tetrahedron Lett. 1981, 22, 3001–3002. [Google Scholar] [CrossRef]
- Carreto, J.I.; Carignan, M.O.; Montoya, N.G. A high-resolution reverse-phase liquid chromatography method for the analysis of mycosporine-like amino acids (MAAs) in marine organisms. Marine Biology 2005, 146, 236–252. [Google Scholar] [CrossRef]
- Takano, S.; Uemura, D.; Hirata, Y. Isolation and structure of two new amino acids, palythinol and palythene, from the Zoanthid Palythoa tuberculosa. Tetrahedron Lett. 1978, 49, 4909–4912. [Google Scholar] [CrossRef]
- Uemura, D.; Katayama, C.; Wada, A.; Hirata, Y. Crystal and molecular structure of palythene possessing a novel 360 nm chromophore. Chem. Lett. 1980, 755–756. [Google Scholar] [CrossRef]
- Sekikawa, I.; Kubota, C.; Hiraoki, T.; Tsujino, I. Isolation and structure of a 357 nm UV-absorbing substance, usujirene, from the red alga Palmaria palmata (L.) O. Kuntze. Jpn. J. Phycol. 1986, 34, 185–188. [Google Scholar]
- Conde, F.R.; Carignan, M.O.; Churio, M.S.; Carreto, J.I. In vitro cis-trans photoisomerization of palythene and usujirene. Implications on the in vivo transformation of mycosporine-like amino acids. Photochem. Photobiol. 2003, 77, 146–150. [Google Scholar] [CrossRef]
- Nakayama, R.; Tamura, Y.; Kikuzaki, H.; Nakatani, N. Antioxidant effect of the constituents of Susabinori (Porphyra yezoensis). J. Am. Oil Chem. Soc. 1999, 76, 649–653. [Google Scholar] [CrossRef]
- Tsujino, I.; Yabe, K.; Sekikawa, I. Isolation and structure of a new amino acid, shinorine, from the red alga Chondrus yendoi. Bot. Mar. 1980, 23, 65–67. [Google Scholar]
- Yabe, K.; Sekikawa, I.; Tsujino, I. Isolation and Structure of a 333–334 nm Absorbing Substance(Y) from A Red Alga, Trichocarpus crinitus. Avaibable online: http://ci.nii.ac.jp/naid/110004520699 (accessed on 21 August 2015).
- Yabe, K. Purification and crystallization of UV-absorbing compounds: Mycosporine-like amino acids—Palythine, shinorine and porphyra-334 from marine red algae. Photomed. Photobiol. 2002, 24, 39–42. [Google Scholar]
- Carreto, J.I.; Carignan, M.O.; Montoya, N.G. Comparative studies on mycosporine-like amino acids, paralytic shellfish toxins and pigment profiles of the toxic dinoflagellates Alexandrium tamarense, A. catenella and A. minutum. Mar. Ecol. Prog. Ser. 2001, 223, 49–60. [Google Scholar] [CrossRef]
- Callone, A.I.; Carignan, M.; Montoya, N.G.; Carreto, J.I. Biotransformation of mycosporine-like amino acids (MAAs) in the toxic dinoflagellate Alexandrium tamarense. J. Photochem. Photobiol. B 2006, 84, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Laurion, I.; Roy, S. Growth and photoprotection in three dinoflagellates (including two strains of Alexandrium tamarense) and one diatom exposed to four weeks of natural and enhanced ultraviolet-B radiation. J. Phycol. 2009, 45, 16–33. [Google Scholar] [CrossRef]
- Carignan, M.O.; Carreto, J.I. Characterization of mycosporine-serine-glycine methyl ester, a major mycosporine-like amino acid from dinoflagellates: A mass spectrometry study. J. Phycol. 2013, 49, 680–688. [Google Scholar] [CrossRef]
- Dunlap, W.C.; Shick, J.M. Ultraviolet radiation-absorbing mycosporine-like amino acids in coral reef organisms: A biochemical and environmental perspective. J. Phycol. 1998, 34, 418–430. [Google Scholar] [CrossRef]
- Shick, J.M.; Dunlap, W.C.; Chalker, B.E.; Banaszak, A.T.; Rosenzweig, T.K. Survey of ultraviolet radiation-absorbing mycosporine-like amino acids in organs of coral reef holothuroids. Mar. Ecol. Prog. Ser. 1992, 90, 139–148. [Google Scholar] [CrossRef]
- Kedar, L.; Kashman, Y.; Oren, A. Mycosporine-2-glycine is the major mycosporine-like amino acid in a unicellular cyanobacterium (Euhalothece sp.) isolated from a gypsum crust in a hypersaline saltern pond. FEMS Microbiol. Lett. 2002, 208, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Kobayashi, J.; Hirata, Y. Isolation and structure of a 330 nm UV-absorbing substance, Asterina-330 from the starfish Asterina pectinifera. Chem. Lett. 1981, 1413–1414. [Google Scholar] [CrossRef]
- Kamio, M.; Kicklighter, C.E.; Nguyen, L.; Germann, M.W.; Derby, C.D. Isolation and structural elucidation of novel mycosporine-like amino acids as alarm cues in the defensive ink secretion of the sea hare Aplysia colifornica. Helv. Chim. Acta 2011, 94, 1012–1018. [Google Scholar] [CrossRef]
- Tsujino, I.; Yabe, K.; Sekikawa, I.; Hamanaka, N. Isolation and structure of a mycosporine from the red alga Chondrus yendoi. Tetrahedron Lett. 1978, 19, 1401–1402. [Google Scholar] [CrossRef]
- Furusaki, A.; Matsumoto, T.; Tsujino, I.; Sekikawa, I. The crystal and molecular structure of palythine trihydrate. Bull. Chem. Soc. Jap. 1980, 53, 319–323. [Google Scholar] [CrossRef]
- Rastogi, R.P.; Incharoensakdi, A. Characterization of UV-screening compounds, mycosporine-like amino acids, and scytonemin in the cyanobacterium Lyngbya sp. CU2555. FEMS Microbiol. Ecol. 2014, 87, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Conde, F.R.; Churio, M.S.; Previtali, C.M. Experimental study of the excited-state properties and photostability of the mycosporine-like amino acid palythine in aqueous solution. Photochem Photobiol. Sci. 2007, 6, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Carignan, M.O.; Cardozo, K.H.M.; Oliveira-Silva, D.; Colepicolo, P.; Carreto, J.I. Palythine-threonine, a major novel mycosporine-like amino acid (MAA) isolated from the hermatypic coral Pocillopora capitata. J. Photochem. Photobiol. B 2009, 94, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Teai, T.T.; Raharivelomanana, P.; Bianchini, J.P.; Faure, R.; Martin, P.M.V.; Cambon, A. Structure of two new iminomycosporines isolated from Pocillopora eydouxi. Tetrahedron Lett. 1997, 38, 5799–5800. [Google Scholar] [CrossRef]
- Volkmann, M.; Gorbushina, A.A.; Kedar, L.; Oren, A. Structure of euhalothece-362, a novel red-shifted mycosporine-like amino acid, from a halophilic cyanobacterium (Euhalothece sp.). FEMS Microbiol. Lett. 2006, 258, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Bandaranayake, W.M.; Bemis, J.E.; Bourne, D.J. Ultraviolet absorbing pigments from the marine sponge Dysidea herbacea: Isolation and structure of a new mycosporine. Comp. Biochem. Phys. C 1996, 115C, 281–286. [Google Scholar] [CrossRef]
- Grant, P.T.; Middleton, C.; Plack, P.A.; Thomson, R.H. The isolation of four aminocyclohexenimines (mycosporines) and a structurally related derivative of cyclohexane-1: 3-dione (gadusol) from the brine shrimp, Artemia. Comp. Biochem. Physiol. B 1985, 80B, 755–759. [Google Scholar] [CrossRef]
- Karentz, D.; Mc Euen, F.S.; Land, M.C.; Dunlap, W.C. Survey of mycosporin-like amino acid compounds in Antarctic marine organisms: potential protection from ultraviolet exposure. Mar. Biol. 1991, 108, 157–166. [Google Scholar] [CrossRef]
- Won, J.J.W.; Rideout, J.A.; Chalker, B.E. Isolation and structure of a novel mycosporine-like amino acid from the reef-building corals Pocillopora damicornis and Stylophora pistillata. Tetrahedron Lett. 1995, 36, 5255–5256. [Google Scholar] [CrossRef]
- Won, J.J.W.; Chalker, B.E.; Rideout, J.A. Two new UV-absorbing compounds from Stylophora pistillata: Sulfate esters of mycosporine-like amino acids. Tetrahedron Lett. 1997, 38, 2525–2526. [Google Scholar] [CrossRef]
- Volkmann, M.; Whitehead, K.; Rütters, H.; Rullkötter, J.; Gorbushina, A.A. Mycosporine-glutamicol-glucoside: A natural UV-absorbing secondary metabolite of rock-inhabiting microcolonial fungi. Rapid Commun. Mass Spectrom. 2003, 17, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, M.; Whitehead, K.; Rütters, H.; Rullkötter, J.; Gorbushina, A.A. Erratum: Mycosporine-glutamicol-glucoside: A natural UV-absorbing secondary metabolite of rock-inhabiting microcolonial fungi. Rapid Commun. Mass Spectrom. 2006, 20, 2520. [Google Scholar] [CrossRef]
- Garcia-Pichel, F.; Castenholz, R.W. Occurance of UV-absorbing, mycosporine-like compounds among cyanobacterial isolates and an estimate of their screening capacity. Appl. Environ. Microbiol. 1993, 59, 163–169. [Google Scholar] [PubMed]
- Scherer, S.; Chen, T.W.; Böger, P. A new UV-A/B protecting pigment in the terrestrial cyanobacterium Nostoc commune. Plant Physiol. 1988, 88, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Böhm, G.A.; Pfleiderer, W.; Böger, P.; Scherer, S. Structure of novel oligosaccharide-mycosporine-amino acid ultraviolet A/B sunscreen pigment from the terrestrial cyanobacterium Nostoc commune. J. Biol. Chem. 1995, 270, 8536–8539. [Google Scholar] [CrossRef] [PubMed]
- Multifunctional Sunscreen Pigments from the Terrestrial Cyanobacterium Nostoc commune. Available online: http://dspace.lib.kanazawa-u.ac.jp/dspace/handle/2297/37263 (accessed on 21 August 2015).
- Arima, H.; Horiguchi, N.; Takaichi, S.; Kofuji, R.; Ishida, K.; Wada, K.; Sakamoto, T. Molecular genetic and chemotaxonomic characterization of the terrestrial cyanobacterium Nostoc commune and its neighboring species. FEMS Microbiol. Ecol. 2012, 79, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Potts, M. Nostoc. In The Ecology of Cyanobacteria; Whitton, B.A., Potts, M., Eds.; Kluwer Academic Publishers: Dordrecht, the Netherlands, 2000; pp. 465–504. [Google Scholar]
- Scherer, S.; Ernst, A.; Chen, T.W.; Böger, P. Rewetting of drought-resistant blue-green algae: Time course of water uptake and reappearance of respiration, photosynthesis, and nitrogen fixation. Oecologia 1984, 62, 418–423. [Google Scholar] [CrossRef]
- Tamaru, Y.; Takani, Y.; Yoshida, T.; Sakamoto, T. Crucial role of extracellular polysaccharides in desiccation and freezing tolerance in the terrestrial cyanobacterium Nostoc commune. Appl. Environ. Microbiol. 2005, 71, 7327–7333. [Google Scholar] [CrossRef] [PubMed]
- Clegg, J.S.; van Hoa, N.; Sorgeloos, P. Thermal tolerance and heat shock proteins in encysted embryos of Artemia from widely different thermal habitats. Hydrobiologia 2001, 466, 221–229. [Google Scholar] [CrossRef]
- Billi, D.; Potts, M. Life and death of dried prokaryotes. Res. Microbiol. 2002, 153, 7–12. [Google Scholar] [CrossRef]
- Lipman, C.B. The successful revival of Nostoc commune from a herbarium specimen eighty-seven years old. Bull. Torr. Bot. Club 1941, 68, 664–666. [Google Scholar] [CrossRef]
- Cameron, R.E. Species of Nostoc vaucher occurring in the Sonoran Desert in Arizona. Trans. Am. Microsc. Soc. 1962, 81, 379–384. [Google Scholar] [CrossRef]
- Gao, Q.; Garcia-Pichel, F. Microbial ultraviolet sunscreens. Nat. Rev. 2011, 9, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Wada, N.; Sakamoto, T.; Matsugo, S. Multiple roles of photosynthetic and sunscreen pigments in cyanobacteria focusing on the oxidative stress. Metabolites 2013, 3, 463–483. [Google Scholar] [CrossRef] [PubMed]
- Oren, A.; Gunde-Cimerman, N. Mycosporines and mycosporine-like amino acids: UV protectants or multipurpose secondary metabolites? FEMS Microbiol. Lett. 2007, 269, 1–10. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wada, N.; Sakamoto, T.; Matsugo, S. Mycosporine-Like Amino Acids and Their Derivatives as Natural Antioxidants. Antioxidants 2015, 4, 603-646. https://doi.org/10.3390/antiox4030603
Wada N, Sakamoto T, Matsugo S. Mycosporine-Like Amino Acids and Their Derivatives as Natural Antioxidants. Antioxidants. 2015; 4(3):603-646. https://doi.org/10.3390/antiox4030603
Chicago/Turabian StyleWada, Naoki, Toshio Sakamoto, and Seiichi Matsugo. 2015. "Mycosporine-Like Amino Acids and Their Derivatives as Natural Antioxidants" Antioxidants 4, no. 3: 603-646. https://doi.org/10.3390/antiox4030603
APA StyleWada, N., Sakamoto, T., & Matsugo, S. (2015). Mycosporine-Like Amino Acids and Their Derivatives as Natural Antioxidants. Antioxidants, 4(3), 603-646. https://doi.org/10.3390/antiox4030603