
Restorative Effects of Synbiotics on Colonic Ultrastructure and Oxidative Stress in Dogs with Chronic Enteropathy

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Concerns
2.2. Study Design
2.3. Transmission Electron Microscopy (TEM)
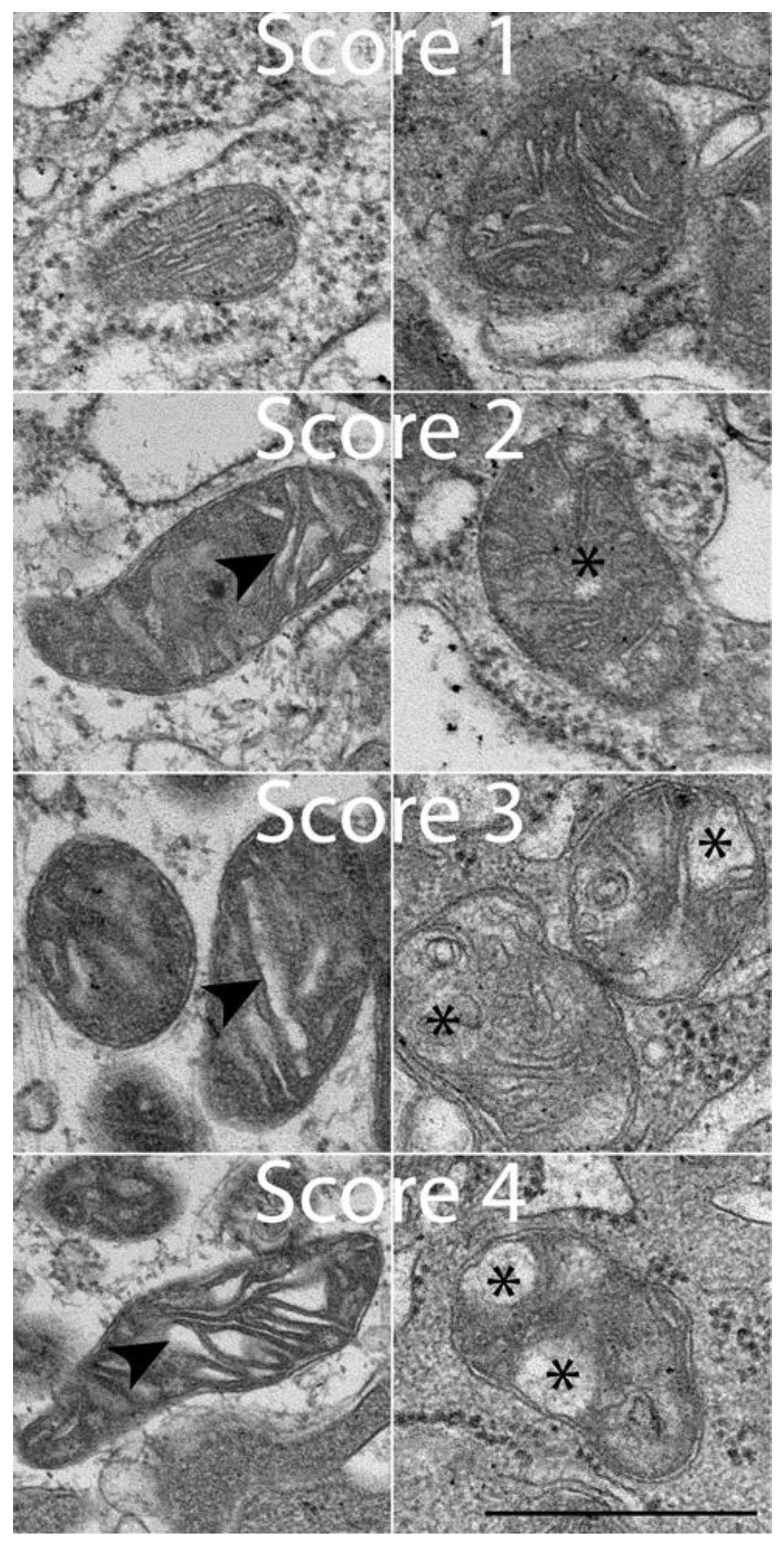
2.4. Morphologic Criteria for Assessing Ultrastructural Changes
2.5. Lipid Peroxidation Assay
2.6. Total RNA Extraction and Sequencing
2.7. Transcriptional Analysis of Genes Involved in OS
2.8. Analysis of the Enrichment of Differentially Expressed Genes (DEGs)
3. Results
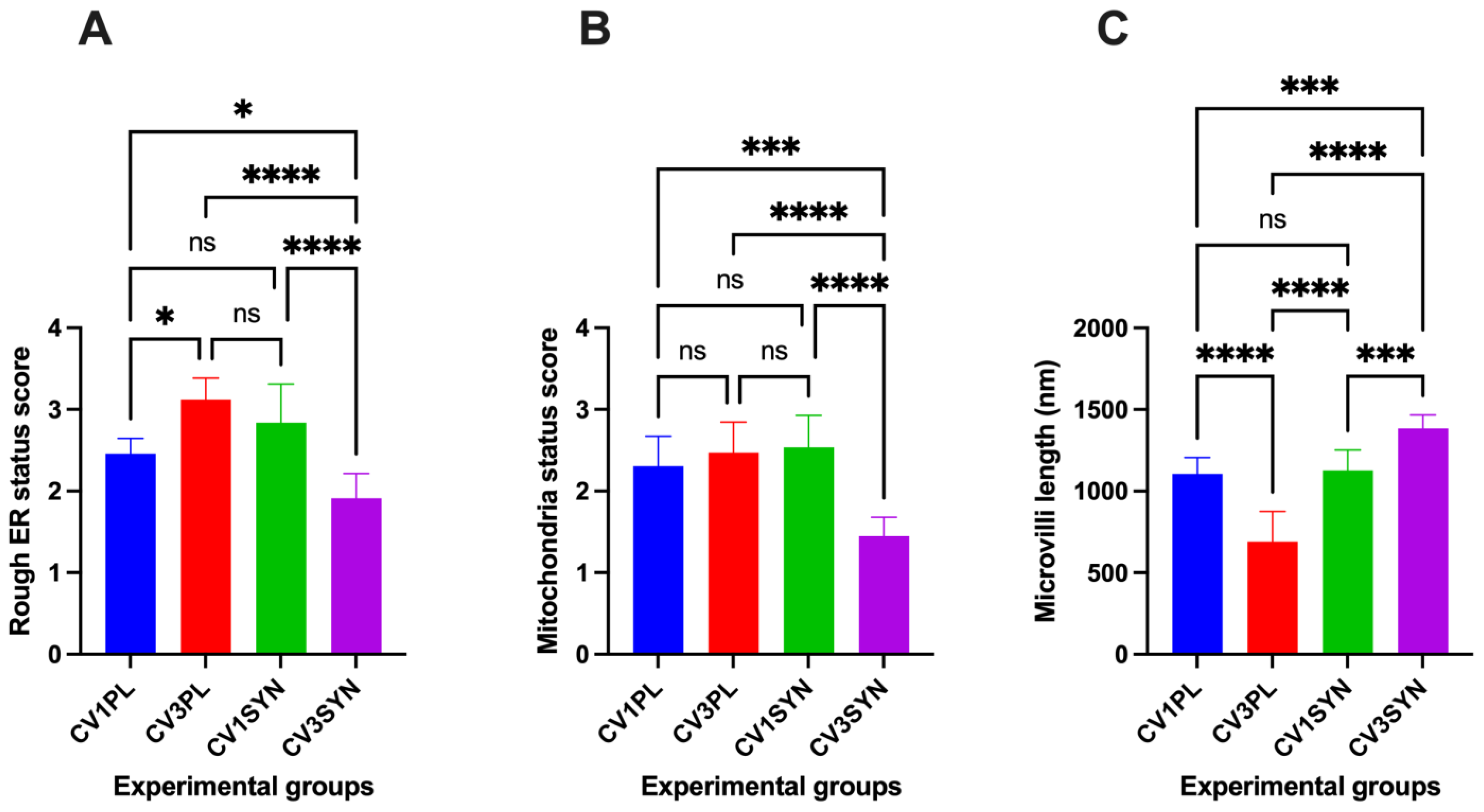
3.1. Assessment of Ultrastructural Changes
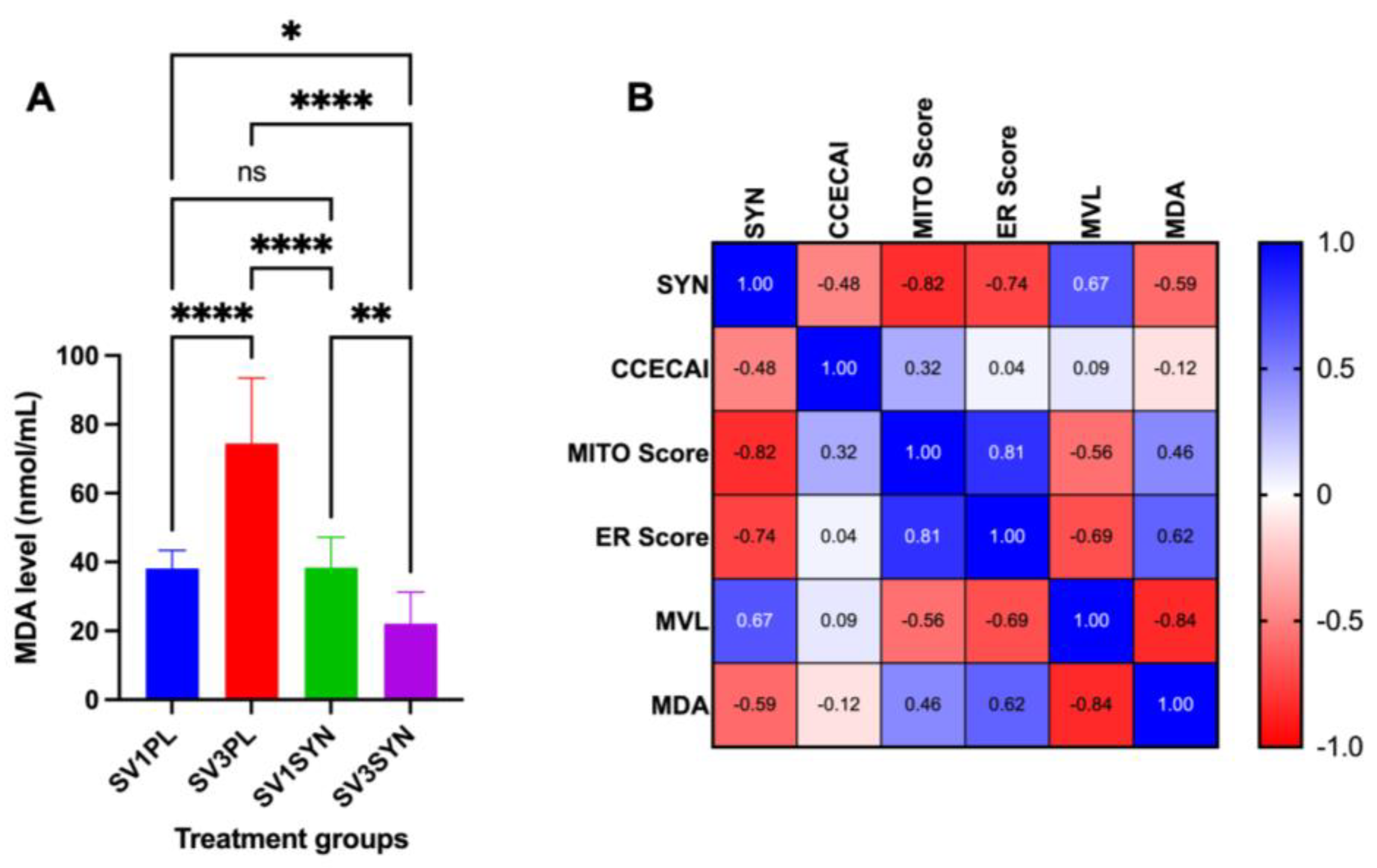
3.2. Assessment of Oxidative Stress Marker
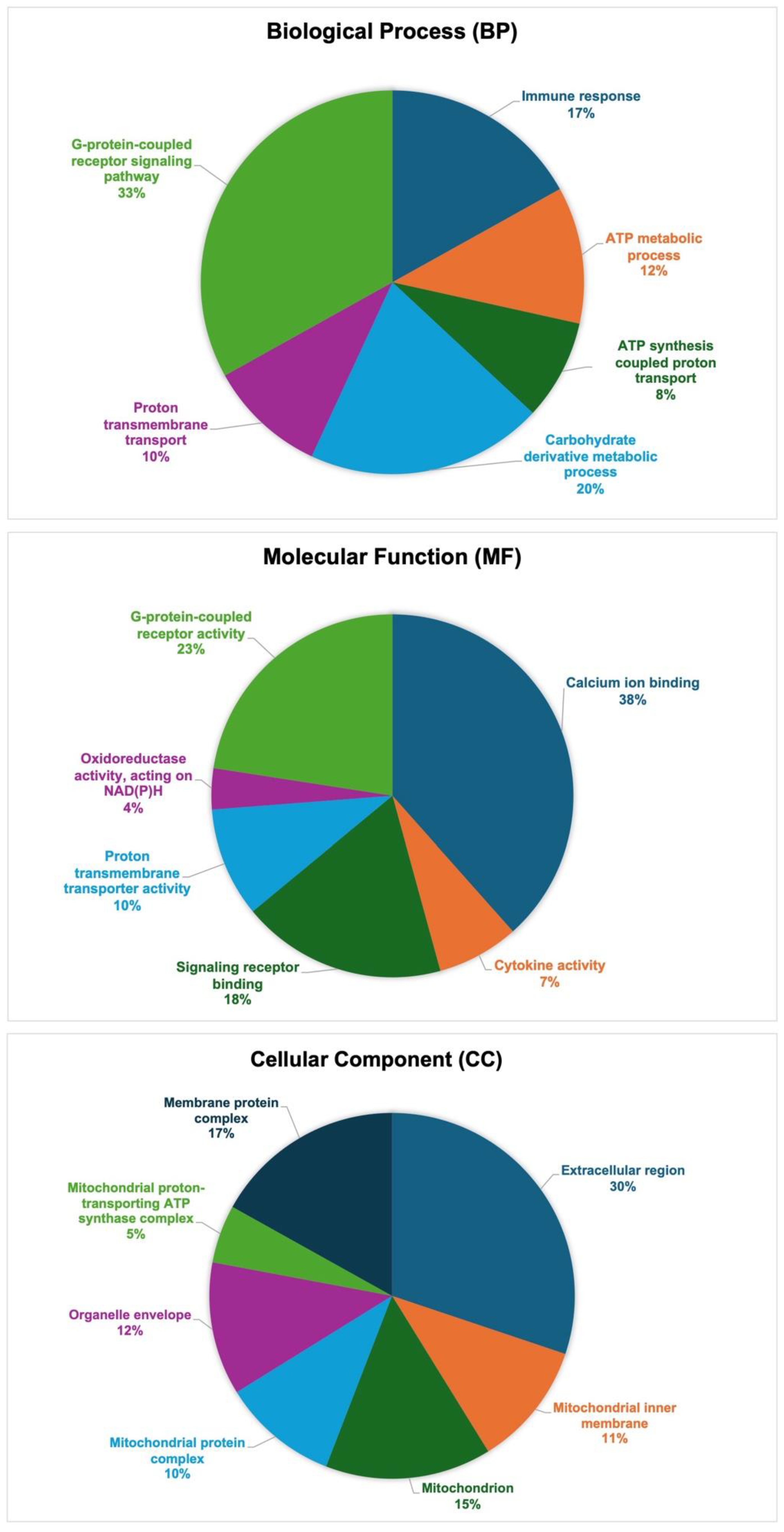
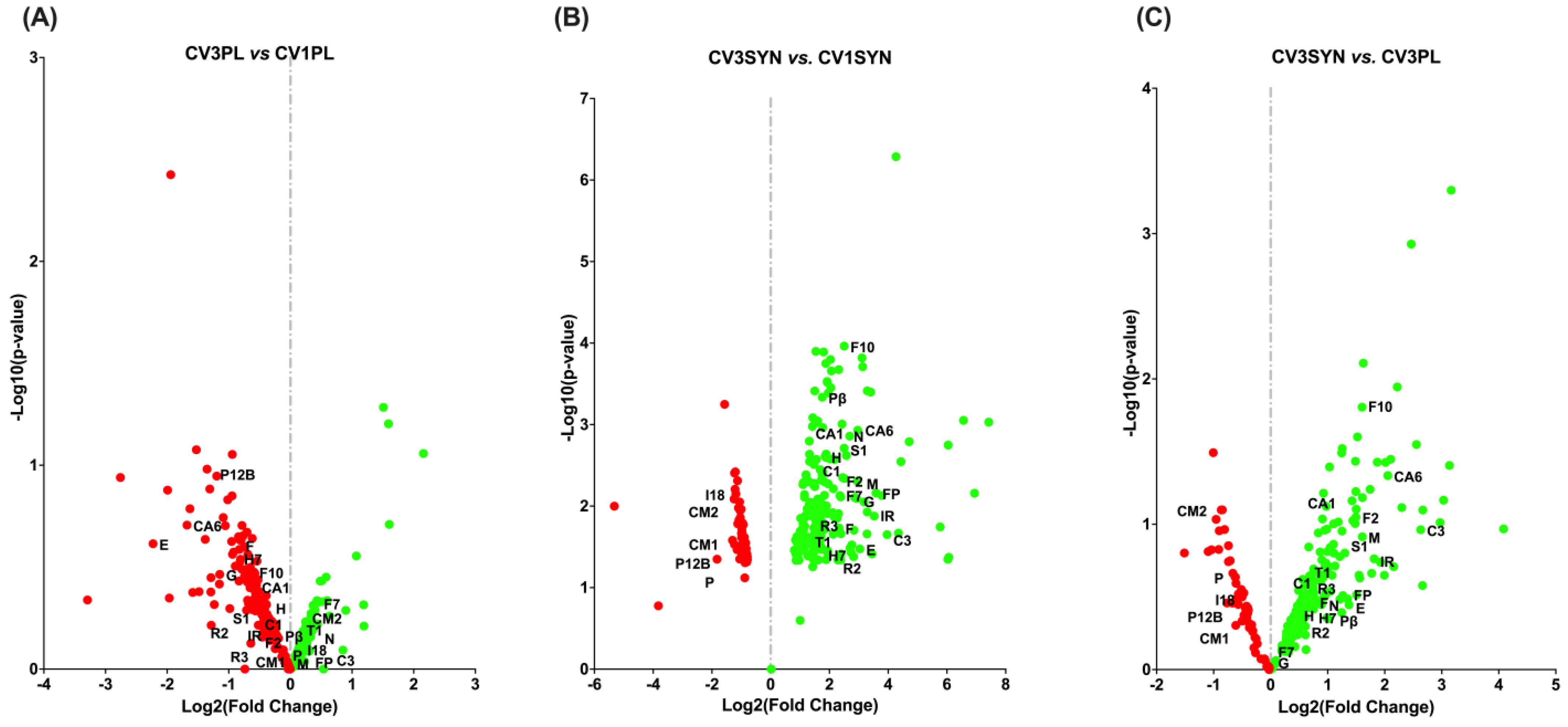
3.3. Gene Ontology (GO) Functional Annotation of DEGs
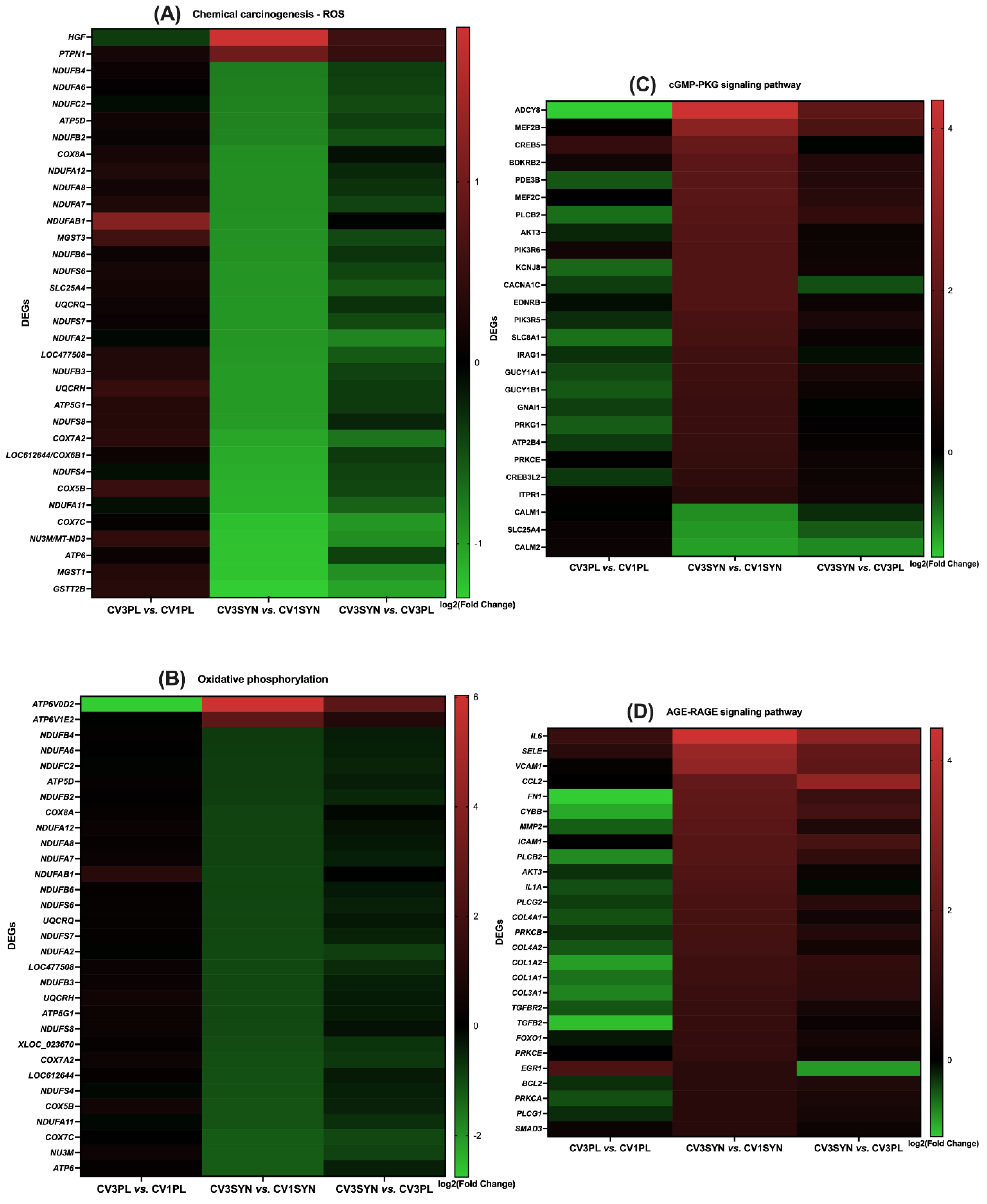
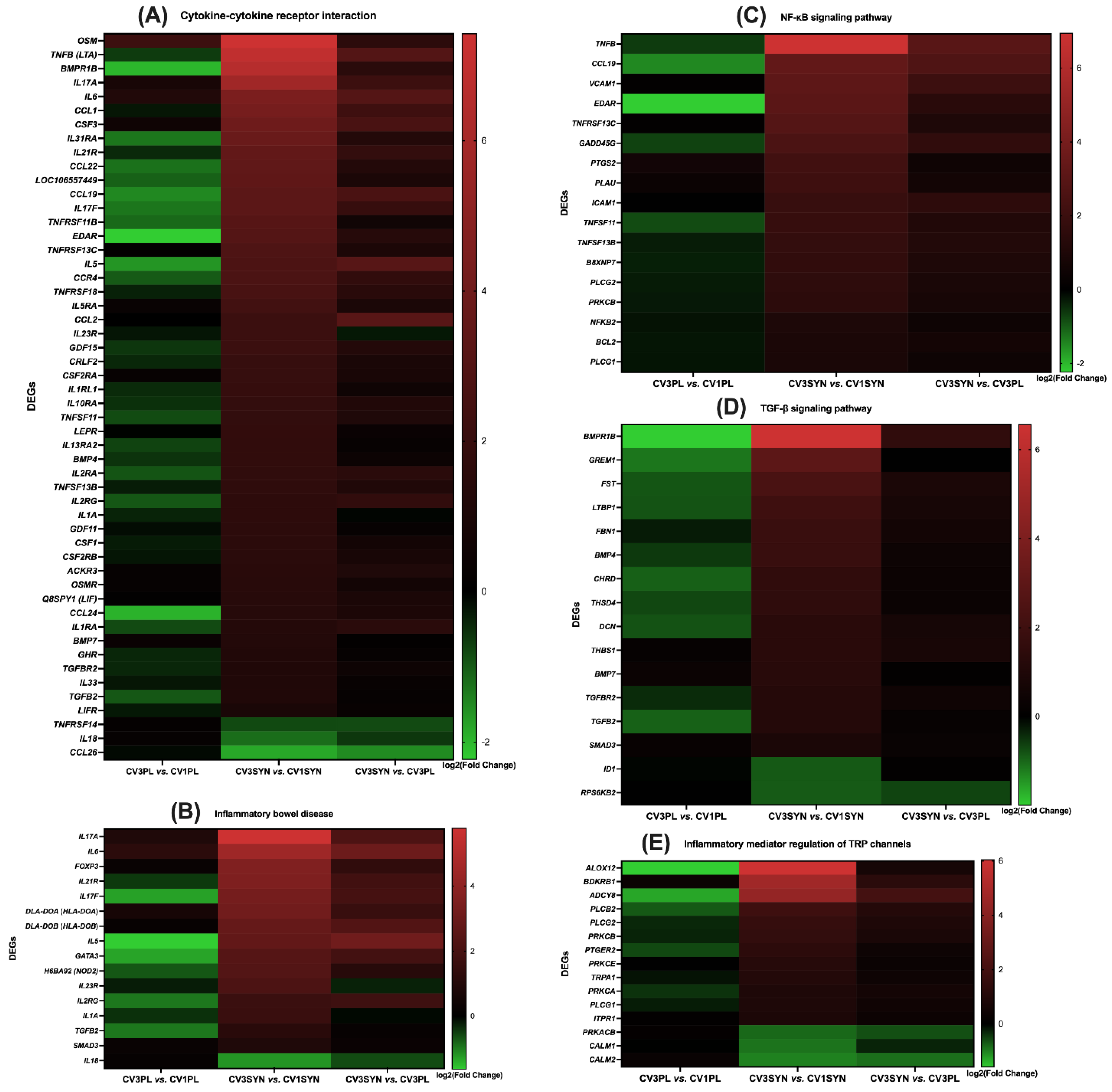
3.4. KEGG Pathway Enrichment Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jergens, A.E.; Heilmann, R.M. Canine Chronic Enteropathy—Current State-of-the-Art and Emerging Concepts. Front. Vet. Sci. 2022, 9, 923013. [Google Scholar] [CrossRef]
- Dandrieux, J.R.S.; Mansfield, C.S. Chronic Enteropathy In Canines: Prevalence, Impact And Management Strategies. Vet. Med. Res. Rep. 2019, 10, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Allenspach, K.; Wieland, B.; Gröne, A.; Gaschen, F. Chronic Enteropathies in Dogs: Evaluation of Risk Factors for Negative Outcome. J. Vet. Intern. Med. 2007, 21, 700–708. [Google Scholar] [CrossRef]
- Sahoo, D.K.; Heilmann, R.M.; Patel, A. Editorial: Understanding Molecular Mechanisms to Facilitate the Development of Biomarkers for Therapeutic Intervention in Gastrointestinal Diseases and Sepsis. Front. Genet. 2025, 16. [Google Scholar] [CrossRef]
- Xenoulis, P.G.; Palculict, B.; Allenspach, K.; Steiner, J.M.; Van House, A.M.; Suchodolski, J.S. Molecular-Phylogenetic Characterization of Microbial Communities Imbalances in the Small Intestine of Dogs with Inflammatory Bowel Disease. FEMS Microbiol. Ecol. 2008, 66, 579–589. [Google Scholar] [CrossRef]
- Honneffer, J.B.; Minamoto, Y.; Suchodolski, J.S. Microbiota Alterations in Acute and Chronic Gastrointestinal Inflammation of Cats and Dogs. World J. Gastroenterol. 2014, 20, 16489–16497. [Google Scholar] [CrossRef]
- Rossi, G.; Pengo, G.; Caldin, M.; Piccionello, A.P.; Steiner, J.M.; Cohen, N.D.; Jergens, A.E.; Suchodolski, J.S. Comparison of Microbiological, Histological, and Immunomodulatory Parameters in Response to Treatment with Either Combination Therapy with Prednisone and Metronidazole or Probiotic VSL#3 Strains in Dogs with Idiopathic Inflammatory Bowel Disease. PLoS ONE 2014, 9, e94699. [Google Scholar] [CrossRef]
- White, R.; Atherly, T.; Guard, B.; Rossi, G.; Wang, C.; Mosher, C.; Webb, C.; Hill, S.; Ackermann, M.; Sciabarra, P.; et al. Randomized, Controlled Trial Evaluating the Effect of Multi-Strain Probiotic on the Mucosal Microbiota in Canine Idiopathic Inflammatory Bowel Disease. Gut Microbes 2017, 8, 451–466. [Google Scholar] [CrossRef]
- Sahoo, D.K.; Allenspach, K.; Mochel, J.P.; Parker, V.; Rudinsky, A.J.; Winston, J.A.; Bourgois-Mochel, A.; Ackermann, M.; Heilmann, R.M.; Köller, G.; et al. Synbiotic-IgY Therapy Modulates the Mucosal Microbiome and Inflammatory Indices in Dogs with Chronic Inflammatory Enteropathy: A Randomized, Double-Blind, Placebo-Controlled Study. Vet. Sci. 2022, 10, 25. [Google Scholar] [CrossRef]
- Day, M.J.; Bilzer, T.; Mansell, J.; Wilcock, B.; Hall, E.J.; Jergens, A.; Minami, T.; Willard, M.; Washabau, R. Histopathological Standards for the Diagnosis of Gastrointestinal Inflammation in Endoscopic Biopsy Samples from the Dog and Cat: A Report from the World Small Animal Veterinary Association Gastrointestinal Standardization Group. J. Comp. Pathol. 2008, 138, S1–S43. [Google Scholar] [CrossRef]
- Willard, M.D.; Moore, G.E.; Denton, B.D.; Day, M.J.; Mansell, J.; Bilzer, T.; Wilcock, B.; Gualtieri, M.; Olivero, D.; Lecoindre, P.; et al. Effect of Tissue Processing on Assessment of Endoscopic Intestinal Biopsies in Dogs and Cats. J. Vet. Intern. Med. 2010, 24, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Allenspach, K.A.; Mochel, J.P.; Du, Y.; Priestnall, S.L.; Moore, F.; Slayter, M.; Rodrigues, A.; Ackermann, M.; Krockenberger, M.; Mansell, J.; et al. Correlating Gastrointestinal Histopathologic Changes to Clinical Disease Activity in Dogs With Idiopathic Inflammatory Bowel Disease. Vet. Pathol. 2019, 56, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Laschi, R.; Pasquinelli, G.; Versura, P. Scanning Electron Microscopy Application in Clinical Research. Scanning Microsc. 1987, 1, 1771–1795. [Google Scholar]
- Schattenfroh, S.; Bartels, M.; Nagel, E. Early Morphological Changes in Crohn’s DiseaseTransmission Electron-Microscopic Findings and Their Interpretation: An Overview. Acta Anat. 1994, 149, 237–246. [Google Scholar] [CrossRef]
- Bertini, M.; Sbarbati, A.; Canioni, D.; Schmitz, J. Scanning Electron Microscopy in Childhood Inflammatory Bowel Disease. Scan. Microsc. Int. 1998, 12, 495–502. [Google Scholar]
- Morroni, M.; Sbarbati, A.; D’Angelo, G.; Catassi, C.; Giorgi, P.; Cinti, S. Scanning Electron Microscopy of the Small Intestine Mucosa in Children with Celiac Disease After Long-Term Dietary Treatment. Scanning Microsc. 1989, 3, 1161–1166. [Google Scholar]
- Aluwihare, A.P. Electron Microscopy in Crohn’s Disease. Gut 1971, 12, 509. [Google Scholar] [CrossRef]
- Lewis, D.; Walker-Smith, J.A.; Phillips, A.D. Microvilli- and Desmosome-Associated Bodies in Crohn’s Disease and Other Disorders in Childhood: An Ultrastructural Abnormality of the Small and Large Intestine. J. Pediatr. Gastroenterol. Nutr. 1984, 3, 46–55. [Google Scholar] [CrossRef]
- Marin, M.L.; Geller, S.A.; Greenstein, A.J.; Marin, R.H.; Gordon, R.E.; Aufses, A.H. Ultrastructural Pathology of Crohn’s Disease: Correlated Transmission Electron Microscopy, Scanning Electron Microscopy, and Freeze Fracture Studies. Am. J. Gastroenterol. 1983, 78, 355–364. [Google Scholar]
- Mughal, S.; Filipe, M.I. Ultrastructural Study of Inflammatory Bowel Disease. Histol. Histopathol. 1992, 7, 599–605. [Google Scholar]
- Myllärniemi, H.; Nickels, J. Scanning Electron Microscopy of Crohn’s Disease and Ulcerative Colitis of the Colon. Virchows Arch. A Pathol. Anat. Histol. 1980, 385, 343–350. [Google Scholar] [CrossRef]
- Nagel, E.; Bartels, M.; Pichlmayr, R. Scanning Electron-Microscopic Lesions in Crohn’s Disease: Relevance for the Interpretation of Postoperative Recurrence. Gastroenterology 1995, 108, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Nyhlin, H.; Stenling, R. The Small-Intestinal Mucosa in Patients with Crohn’s Disease Assessed by Scanning Electron and Light Microscopy. Scand. J. Gastroenterol. 1984, 19, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Rickert, R.R.; Carter, H.W. The “Early” Ulcerative Lesion of Crohn’s Disease : Correlative Light and Scanning Electron-Microscopic Studies. J. Clin. Gastroenterol. 1980, 2, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Fratila, O.C.; Craciun, C. Ultrastructural Evidence of Mucosal Healing after Infliximab in Patients with Ulcerative Colitis. J. Gastrointestin. Liver Dis. 2010, 19, 147–153. [Google Scholar]
- Shields, H.M.; Bates, M.L.; Goldman, H.; Zuckerman, G.R.; Mills, B.A.; Best, C.J.; Bair, F.A.; Goran, D.A.; DeSchryver-Kecskemeti, K. Scanning Electron Microscopic Appearance of Chronic Ulcerative Colitis with and without Dysplasia. Gastroenterology 1985, 89, 62–72. [Google Scholar] [CrossRef]
- Nomura, E.; Sujino, T.; Hosoe, N.; Yoshimatsu, Y.; Tanemoto, S.; Takabayashi, K.; Mutaguchi, M.; Shimoda, M.; Naganuma, M.; Ogata, H.; et al. Characteristics of the Mucosal Surface on Scanning Electron Microscopy in Patients with Remitting Ulcerative Colitis. Dig. Dis. Sci. 2021, 66, 3141–3148. [Google Scholar] [CrossRef]
- Walker, D.; Knuchel-Takano, A.; Mccutchan, A.; Chang, Y.M.; Downes, C.; Miller, S.; Stevens, K.; Verheyen, K.; Phillips, A.D.; Miah, S.; et al. A Comprehensive Pathological Survey of Duodenal Biopsies from Dogs with Diet-Responsive Chronic Enteropathy. J. Vet. Intern. Med. 2013, 27, 862–874. [Google Scholar] [CrossRef]
- Shah, H.; Trivedi, M.; Gurjar, T.; Sahoo, D.K.; Jergens, A.E.; Yadav, V.K.; Patel, A.; Pandya, P. Decoding the Gut Microbiome in Companion Animals: Impacts and Innovations. Microorganisms 2024, 12, 1831. [Google Scholar] [CrossRef]
- Mounir, M.; Ibijbijen, A.; Farih, K.; Rabetafika, H.N.; Razafindralambo, H.L. Synbiotics and Their Antioxidant Properties, Mechanisms, and Benefits on Human and Animal Health: A Narrative Review. Biomolecules 2022, 12, 1443. [Google Scholar] [CrossRef]
- Feng, T.; Wang, J. Oxidative Stress Tolerance and Antioxidant Capacity of Lactic Acid Bacteria as Probiotic: A Systematic Review. Gut Microbes 2020, 12, 1801944. [Google Scholar] [CrossRef]
- Chaudhary, A.; Prajapati, N.; Prajapati, A.; Singh, S.; Joshi, M.; Prajapati, D.; Patani, A.; Sahoo, D.K.; Patel, A. Postbiotic Emissaries: A Comprehensive Review on the Bioprospecting and Production of Bioactive Compounds by Enterococcus Species. Int. J. Food Sci. Technol. 2024, 59, 6769–6782. [Google Scholar] [CrossRef]
- Sahoo, D.K.; Chainy, G.B.N.; Jergens, A.E. Editorial: Gastrointestinal (GI) Disorders and Antioxidant Therapeutics. Front. Endocrinol. 2025, 16. [Google Scholar] [CrossRef]
- Mohammed, A.A.; Jiang, S.; Jacobs, J.A.; Cheng, H.W. Effect of a Synbiotic Supplement on Cecal Microbial Ecology, Antioxidant Status, and Immune Response of Broiler Chickens Reared under Heat Stress. Poult. Sci. 2019, 98, 4408–4415. [Google Scholar] [CrossRef]
- Cukkemane, A.; Kumar, P.; Sathyamoorthy, B. A Metabolomics Footprint Approach to Understanding the Benefits of Synbiotics in Functional Foods and Dietary Therapeutics for Health, Communicable and Non-Communicable Diseases. Food Res. Int. 2020, 128, 108679. [Google Scholar] [CrossRef]
- Zheng, H.J.; Guo, J.; Jia, Q.; Huang, Y.S.; Huang, W.J.; Zhang, W.; Zhang, F.; Liu, W.J.; Wang, Y. The Effect of Probiotic and Synbiotic Supplementation on Biomarkers of Inflammation and Oxidative Stress in Diabetic Patients: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Pharmacol. Res. 2019, 142, 303–313. [Google Scholar] [CrossRef]
- Allenspach, K.; Culverwell, C.; Chan, D. Long-Term Outcome in Dogs with Chronic Enteropathies: 203 Cases. Vet. Rec. 2016, 178, 368. [Google Scholar] [CrossRef]
- Volkmann, M.; Steiner, J.M.; Fosgate, G.T.; Zentek, J.; Hartmann, S.; Kohn, B. Chronic Diarrhea in Dogs – Retrospective Study in 136 Cases. J. Vet. Intern. Med. 2017, 31, 1043–1055. [Google Scholar] [CrossRef]
- Ching, T.; Huang, S.; Garmire, L.X. Power Analysis and Sample Size Estimation for RNA-Seq Differential Expression. RNA 2014, 20, 1684. [Google Scholar] [CrossRef]
- Wong, D.; Sahoo, K.D.; Faivre, C.; Kopper, J.; Dersh, K.; Beachler, T.; Esser, M. Oxidative Stress in Critically Ill Neonatal Foals. J. Vet. Intern. Med. 2025, 39, e17297. [Google Scholar] [CrossRef]
- Canis Lupus Familiaris Genome Assembly Dog10K_Boxer_Tasha-NCBI-NLM. Available online: https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000002285.5/ (accessed on 5 December 2024).
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and Quantifying Mammalian Transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Krause, C.; Suwada, K.; Blomme, E.A.G.; Kowalkowski, K.; Liguori, M.J.; Mahalingaiah, P.K.; Mittelstadt, S.; Peterson, R.; Rendino, L.; Vo, A.; et al. Preclinical Species Gene Expression Database: Development and Meta-Analysis. Front. Genet. 2023, 13, 1078050. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential Expression Analysis for Sequence Count Data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene Ontology Analysis for RNA-Seq: Accounting for Selection Bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A Web Server for Annotation and Identification of Enriched Pathways and Diseases. Nucleic Acids Res. 2011, 39, W316. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated Genome Annotation and Pathway Identification Using the KEGG Orthology (KO) as a Controlled Vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- KEGG: Kyoto Encyclopedia of Genes and Genomes. Available online: https://www.genome.jp/kegg/ (accessed on 6 December 2024).
- Chhabria, S.; Mathur, S.; Vadakan, S.; Sahoo, D.K.; Mishra, P.; Paital, B. A Review on Phytochemical and Pharmacological Facets of Tropical Ethnomedicinal Plants as Reformed DPP-IV Inhibitors to Regulate Incretin Activity. Front. Endocrinol. 2022, 13, 1027237. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Sahoo, D.K.; Roy, A.; Samanta, L.; Chainy, G.B.N. Thiol Redox Status Critically Influences Mitochondrial Response to Thyroid Hormone-Induced Hepatic Oxidative Injury: A Temporal Analysis. Cell Biochem. Funct. 2010, 28, 126–134. [Google Scholar] [CrossRef]
- Sahoo, D.K.; Jena, S.; Chainy, G.B.N. Thyroid Dysfunction and Testicular Redox Status: An Intriguing Association. In Oxidants, Antioxidants, and Impact of the Oxidative Status in Male Reproduction; Academic Press: Cambridge, MA, USA, 2019; pp. 149–170. [Google Scholar] [CrossRef]
- Patani, A.; Balram, D.; Yadav, V.K.; Lian, K.Y.; Patel, A.; Sahoo, D.K. Harnessing the Power of Nutritional Antioxidants against Adrenal Hormone Imbalance-Associated Oxidative Stress. Front. Endocrinol. 2023, 14, 1271521. [Google Scholar] [CrossRef]
- Sahoo, D.K.; Samanta, L.; Kesari, K.K.; Mukherjee, S. Editorial: Hormonal Imbalance-Associated Oxidative Stress and Protective Benefits of Nutritional Antioxidants. Front. Endocrinol. 2024, 15, 1368580. [Google Scholar] [CrossRef]
- Pati, S.G.; Panda, F.; Paital, B.; Sahoo, D.K.; Jena, S. Oxidative Stress Physiology in Scylla Serrata for Environmental Health Assessment. Front. Environ. Sci. 2023, 11, 1142495. [Google Scholar] [CrossRef]
- Ladumor, R.; Pandya, H.; Thakkar, M.; Mehta, D.; Paithankar, P.; Alfarraj, S.; Ansari, M.J.; Pandya, P.; Yadav, V.K.; Sahoo, D.K.; et al. Environmentally Relevant Concentrations of Nickel and Imidacloprid Induce Reproductive Toxicity in Earthworm (Eisenia Fetida Fetida). Comp. Biochem. Physiol. Part. C Toxicol. Pharmacol. 2024, 283, 109964. [Google Scholar] [CrossRef]
- Mishra, P.; Sahoo, D.K.; Mohanty, C.; Samanta, L. Curcumin-Loaded Nanoparticles Effectively Prevent T4-Induced Oxidative Stress in Rat Heart. Cell Biochem. Funct. 2024, 42, e4070. [Google Scholar] [CrossRef]
- Sahoo, D.K.; Roy, A.; Chattopadhyay, S.; Chainy, G.B.N. Effect of T3 Treatment on Glutathione Redox Pool and Its Metabolizing Enzymes in Mitochondrial and Post-Mitochondrial Fractions of Adult Rat Testes. Indian. J. Exp. Biol. 2007, 45, 338–346. [Google Scholar]
- Sahoo, D.K.; Roy, A.; Chainy, G.B.N. Rat Testicular Mitochondrial Antioxidant Defence System and Its Modulation by Aging. Acta Biol. Hung. 2008, 59, 413–424. [Google Scholar] [CrossRef]
- Sahoo, D.K.; Chainy, G.B.N. Hormone-Linked Redox Status and Its Modulation by Antioxidants. Vitam. Horm. 2023, 121, 197–246. [Google Scholar] [CrossRef]
- Sahoo, D.K.; Wong, D.; Patani, A.; Paital, B.; Yadav, V.K.; Patel, A.; Jergens, A.E. Exploring the Role of Antioxidants in Sepsis-Associated Oxidative Stress: A Comprehensive Review. Front. Cell. Infect. Microbiol. 2024, 14, 1348713. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, D.K.; Heilmann, R.M.; Paital, B.; Patel, A.; Yadav, V.K.; Wong, D.; Jergens, A.E. Oxidative Stress, Hormones, and Effects of Natural Antioxidants on Intestinal Inflammation in Inflammatory Bowel Disease. Front. Endocrinol. 2023, 14, 1217165. [Google Scholar] [CrossRef] [PubMed]
- Muro, P.; Zhang, L.; Li, S.; Zhao, Z.; Jin, T.; Mao, F.; Mao, Z. The Emerging Role of Oxidative Stress in Inflammatory Bowel Disease. Front. Endocrinol. 2024, 15, 1390351. [Google Scholar] [CrossRef]
- Crumeyrolle-Arias, M.; Jaglin, M.; Bruneau, A.; Vancassel, S.; Cardona, A.; Daugé, V.; Naudon, L.; Rabot, S. Absence of the Gut Microbiota Enhances Anxiety-like Behavior and Neuroendocrine Response to Acute Stress in Rats. Psychoneuroendocrinology 2014, 42, 207–217. [Google Scholar] [CrossRef] [PubMed]
- So, S.Y.; Savidge, T.C. Gut Feelings: The Microbiota-Gut-Brain Axis on Steroids. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 322, G1–G20. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.N.; Kubo, C.; Koga, Y. Postnatal Microbial Colonization Programs the Hypothalamic-Pituitary-Adrenal System for Stress Response in Mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef]
- Vagnerová, K.; Vodička, M.; Hermanová, P.; Ergang, P.; Šrůtková, D.; Klusoňová, P.; Balounová, K.; Hudcovic, T.; Pácha, J. Interactions Between Gut Microbiota and Acute Restraint Stress in Peripheral Structures of the Hypothalamic–Pituitary–Adrenal Axis and the Intestine of Male Mice. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Gareau, M.G.; Jury, J.; MacQueen, G.; Sherman, P.M.; Perdue, M.H. Probiotic Treatment of Rat Pups Normalises Corticosterone Release and Ameliorates Colonic Dysfunction Induced by Maternal Separation. Gut 2007, 56, 1522–1528. [Google Scholar] [CrossRef]
- Lutgendorff, F.; Akkermans, L.; Soderholm, J. The Role of Microbiota and Probiotics in Stress-Induced Gastrointestinal Damage. Curr. Mol. Med. 2008, 8, 282–298. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Marchesi, J.R.; Scully, P.; Codling, C.; Ceolho, A.M.; Quigley, E.M.M.; Cryan, J.F.; Dinan, T.G. Early Life Stress Alters Behavior, Immunity, and Microbiota in Rats: Implications for Irritable Bowel Syndrome and Psychiatric Illnesses. Biol. Psychiatry 2009, 65, 263–267. [Google Scholar] [CrossRef]
- Yang, K.; Jian, S.; Wen, C.; Guo, D.; Liao, P.; Wen, J.; Kuang, T.; Han, S.; Liu, Q.; Deng, B. Gallnut Tannic Acid Exerts Anti-Stress Effects on Stress-Induced Inflammatory Response, Dysbiotic Gut Microbiota, and Alterations of Serum Metabolic Profile in Beagle Dogs. Front. Nutr. 2022, 9, 847966. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, B.; Sun, S.; Liu, N.; Li, Y.; Lan, M.; Wang, X.; Zhang, Y.; Wu, A.; Yang, S.; et al. Immunoprotection Effects of Chicken Egg Yolk Immunoglobulins (IgY) against Aeromonas Veronii Infection in Sinocyclocheilus Grahami. Aquaculture 2023, 563, 738935. [Google Scholar] [CrossRef]
- Zhang, L.; Bai, Y.; Tao, J.; Yang, S.; Tu, C.; Liu, L.; Huang, X.; Li, L.; Qin, Z. Effects of Feeding Chicken Egg Yolk Antibodies on Intestinal Cell Apoptosis, Oxidative Stress and Microbial Flora of Tilapia (Oreochromis Niloticus) Infected with Streptococcus Agalactiae. Fish. Shellfish. Immunol. 2024, 150, 109596. [Google Scholar] [CrossRef]
- Lu, Y.; Xiong, Y.; Zhang, S.; Wang, B.; Feng, Y.; Pu, Z.; Wei, K.; Chen, J.; Chen, D.; Zhang, P. D-Mannose Reduces Oxidative Stress, Inhibits Inflammation, and Increases Treg Cell Proportions in Mice with Ulcerative Colitis. Front. Pharmacol. 2024, 15, 1454713. [Google Scholar] [CrossRef]
- Joshi, C.S.; Salazar, A.M.; Wang, C.; Ligon, M.M.; Chappidi, R.R.; Fashemi, B.E.; Felder, P.A.; Mora, A.; Grimm, S.L.; Coarfa, C.; et al. D-Mannose Reduces Cellular Senescence and NLRP3/GasderminD/IL-1β-Driven Pyroptotic Uroepithelial Cell Shedding in the Murine Bladder. Dev. Cell 2024, 59, 33–47.e5. [Google Scholar] [CrossRef]
- Wang, J.; Motlagh, N.J.; Wang, C.; Wojtkiewicz, G.R.; Schmidt, S.; Chau, C.; Narsimhan, R.; Kullenberg, E.G.; Zhu, C.; Linnoila, J.; et al. D-Mannose Suppresses Oxidative Response and Blocks Phagocytosis in Experimental Neuroinflammation. Proc. Natl. Acad. Sci. USA 2021, 118, e2107663118. [Google Scholar] [CrossRef]
- Dong, L.; Xie, J.; Wang, Y.; Jiang, H.; Chen, K.; Li, D.; Wang, J.; Liu, Y.; He, J.; Zhou, J.; et al. Mannose Ameliorates Experimental Colitis by Protecting Intestinal Barrier Integrity. Nat. Commun. 2022, 13, 4804. [Google Scholar] [CrossRef]
- Novak, E.A.; Mollen, K.P. Mitochondrial Dysfunction in Inflammatory Bowel Disease. Front. Cell Dev. Biol. 2015, 3. [Google Scholar] [CrossRef]
- Sharma, N. Exploring the Potential of Mannan Oligosaccharides in Enhancing Animal Growth, Immunity, and Overall Health: A Review. Carbohydr. Polym. Technol. Appl. 2025, 9, 100603. [Google Scholar] [CrossRef]
- Lu, Z.; Feng, L.; Jiang, W.; Wu, P.; Liu, Y.; Jiang, J.; Kuang, S.; Tang, L.; Li, S.; Zhong, C.; et al. Mannan Oligosaccharides Alleviate Oxidative Injury in the Head Kidney and Spleen in Grass Carp (Ctenopharyngodon Idella) via the Nrf2 Signaling Pathway after Aeromonas Hydrophila Infection. J. Anim. Sci. Biotechnol. 2023, 14, 58. [Google Scholar] [CrossRef]
- Shaki, F.; Pourahmad, J. Mitochondrial Toxicity of Depleted Uranium: Protection by Beta-Glucan. Iran. J. Pharm. Res. 2013, 12, 131. [Google Scholar] [PubMed]
- Stothers, C.L.; Burelbach, K.R.; Owen, A.M.; Patil, N.K.; McBride, M.A.; Bohannon, J.K.; Luan, L.; Hernandez, A.; Patil, T.K.; Williams, D.L.; et al. Beta-Glucan Induces Distinct and Protective Innate Immune Memory in Differentiated Macrophages. J. Immunol. 2021, 207, 2785. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Olejar, K.J.; On, S.L.W.; Chelikani, V. The Potential of Lactobacillus Spp. for Modulating Oxidative Stress in the Gastrointestinal Tract. Antioxidants 2020, 9, 610. [Google Scholar] [CrossRef] [PubMed]
- Finamore, A.; Ambra, R.; Nobili, F.; Garaguso, I.; Raguzzini, A.; Serafini, M. Redox Role of Lactobacillus Casei Shirota against the Cellular Damage Induced by 2,2’-Azobis (2-Amidinopropane) Dihydrochloride-Induced Oxidative and Inflammatory Stress in Enterocytes-like Epithelial Cells. Front. Immunol. 2018, 9, 363322. [Google Scholar] [CrossRef]
- Serata, M.; Kiwaki, M.; Iino, T. Functional Analysis of a Novel Hydrogen Peroxide Resistance Gene in Lactobacillus Casei Strain Shirota. Microbiology 2016, 162, 1885–1894. [Google Scholar] [CrossRef]
- Yamazaki, T.; Yamada, S.; Ohshio, K.; Sugamata, M.; Morita, Y. Lactobacillus Paracasei KW3110 Prevents Inflammatory-Stress-Induced Mitochondrial Dysfunction in Mouse Macrophages. Int. J. Mol. Sci. 2022, 23, 1443. [Google Scholar] [CrossRef]
- Tang, C.; Meng, F.; Pang, X.; Chen, M.; Zhou, L.; Lu, Z.; Lu, Y. Protective Effects of Lactobacillus Acidophilus NX2-6 against Oleic Acid-Induced Steatosis, Mitochondrial Dysfunction, Endoplasmic Reticulum Stress and Inflammatory Responses. J. Funct. Foods 2020, 74, 104206. [Google Scholar] [CrossRef]
- Cuevas-González, P.F.; Aguilar-Toalá, J.E.; García, H.S.; González-Córdova, A.F.; Vallejo-Cordoba, B.; Hernández-Mendoza, A. Protective Effect of the Intracellular Content from Potential Probiotic Bacteria against Oxidative Damage Induced by Acrylamide in Human Erythrocytes. Probiotics Antimicrob. Proteins 2020, 12, 1459–1470. [Google Scholar] [CrossRef]
- Li, B.; Du, P.; Smith, E.E.; Wang, S.; Jiao, Y.; Guo, L.; Huo, G.; Liu, F. In Vitro and in Vivo Evaluation of an Exopolysaccharide Produced by Lactobacillus Helveticus KLDS1.8701 for the Alleviative Effect on Oxidative Stress. Food Funct. 2019, 10, 1707–1717. [Google Scholar] [CrossRef]
- Petruk, G.; Donadio, G.; Lanzilli, M.; Isticato, R.; Monti, D.M. Alternative Use of Bacillus Subtilis Spores: Protection against Environmental Oxidative Stress in Human Normal Keratinocytes. Sci. Rep. 2018, 8, 1745. [Google Scholar] [CrossRef]
- Zou, X.Y.; Zhang, M.; Tu, W.J.; Zhang, Q.; Jin, M.L.; Fang, R.D.; Jiang, S. Bacillus Subtilis Inhibits Intestinal Inflammation and Oxidative Stress by Regulating Gut Flora and Related Metabolites in Laying Hens. Animal 2022, 16, 100474. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Sun, J.; Li, S.; Wei, L.; Sun, R.; Feng, X.; Zhang, H.; Chen, T.; Xi, Q.; Zhang, Y.; et al. Protective Effect of Enterococcus Faecium against Ethanol-Induced Gastric Injury via Extracellular Vesicles. Microbiol. Spectr. 2024, 12, e0389423. [Google Scholar] [CrossRef]
- Merino de Paz, N.; Carrillo-Palau, M.; Hernández-Camba, A.; Abreu-González, P.; de Vera-González, A.; González-Delgado, A.; Martín-González, C.; González-Gay, M.; Ferraz-Amaro, I. Association of Serum Malondialdehyde Levels with Lipid Profile and Liver Function in Patients with Inflammatory Bowel Disease. Antioxidants 2024, 13. [Google Scholar] [CrossRef]
- Sahoo, D.K.; Roy, A.; Chainy, G.B.N. Protective Effects of Vitamin E and Curcumin on L-Thyroxine-Induced Rat Testicular Oxidative Stress. Chem. Biol. Interact. 2008, 176, 121–128. [Google Scholar] [CrossRef]
- Candellone, A.; Girolami, F.; Badino, P.; Jarriyawattanachaikul, W.; Odore, R. Changes in the Oxidative Stress Status of Dogs Affected by Acute Enteropathies. Vet. Sci. 2022, 9, 276. [Google Scholar] [CrossRef]
- Dvorak, A.M.; Dickersin, G.R. Crohn’s Disease: Electron Microscopic Studies. Pathol. Annu. 1979, 14 Pt 2, 259–306. [Google Scholar]
- Shiner, M.; Birbeck, M.S.C. The Microvilli of the Small Intestinal Surface Epithelium in Coeliac Disease and in Idiopathic Steatorrhoea. Gut 1961, 2, 277. [Google Scholar] [CrossRef]
- Sbarbati, A.; Valletta, E.; Bertini, M.; Cipolli, M.; Morroni, M.; Pinelli, L.; Tatò, L. Gluten Sensitivity and “normal” Histology: Is the Intestinal Mucosa Really Normal? Dig. Liver Dis. 2003, 35, 768–773. [Google Scholar] [CrossRef]
- Bochimoto, H.; Kondoh, D.; Nagata, R.; Ishihara, Y.; Tomiyasu, J.; Han, K.H.; Shimada, K.; Sasaki, M.; Kitamura, N.; Fukushima, M. Ultrastructural Changes in Colonic Epithelial Cells in a Rat Model of Inflammatory Bowel Disease. Microsc. Res. Tech. 2019, 82, 1339–1344. [Google Scholar] [CrossRef]
- Bou-Fersen, A.M.; Anim, J.T.; Khan, I. Experimental Colitis Is Associated with Ultrastructural Changes in Inflamed and Uninflamed Regions of the Gastrointestinal Tract. Med. Princ. Pract. 2008, 17, 190–196. [Google Scholar] [CrossRef]
- Manners, H.K.; Hart, C.A.; Getty, B.; Kelly, D.F.; Sørensen, S.H.; Batt, R.M. Characterization of Intestinal Morphologic, Biochemical, and Ultrastructural Features in Gluten-Sensitive Irish Setters during Controlled Oral Gluten Challenge Exposure after Weaning. Am. J. Vet. Res. 1998, 59, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Bermea, K.C.; Kumar, D.; Singh, A.; Verma, A.; Kaileh, M.; Sen, R.; Lakatta, E.G.; Adamo, L. RelA-Mediated Signaling Connects Adaptation to Chronic Cardiomyocyte Stress with Myocardial and Systemic Inflammation in the ADCY8 Model of Accelerated Aging. Geroscience 2024, 46, 4243. [Google Scholar] [CrossRef] [PubMed]
- Han, W.Q.; Xu, L.; Tang, X.F.; Chen, W.D.; Wu, Y.J.; Gao, P.J. Membrane Rafts–Redox Signalling Pathway Contributes to Renal Fibrosis via Modulation of the Renal Tubular Epithelial–Mesenchymal Transition. J. Physiol. 2018, 596, 3603. [Google Scholar] [CrossRef]
- Liu, Y.S.; Hsu, J.W.; Lin, H.Y.; Lai, S.W.; Huang, B.R.; Tsai, C.F.; Lu, D.Y. Bradykinin B1 Receptor Contributes to Interleukin-8 Production and Glioblastoma Migration through Interaction of STAT3 and SP-1. Neuropharmacology 2019, 144, 143–154. [Google Scholar] [CrossRef]
- Vilotić, A.; Nacka-Aleksić, M.; Pirković, A.; Bojić-Trbojević, Ž.; Dekanski, D.; Jovanović Krivokuća, M. IL-6 and IL-8: An Overview of Their Roles in Healthy and Pathological Pregnancies. Int. J. Mol. Sci. 2022, 23, 14574. [Google Scholar] [CrossRef]
- Chinedu, O.; Tonassé, W.V.; Albuquerque, D.M.; Domingos, I.d.F.; Araújo, A.d.S.; Bezerra, M.A.C.; Sonati, M.d.F.; dos Santos, M.N.N. Polymorphisms in the Heme Oxygenase-1 and Bone Morphogenetic Protein Receptor Type 1b Genes and Estimated Glomerular Filtration Rate in Brazilian Sickle Cell Anemia Patients. Hematol. Transfus. Cell Ther. 2020, 43, 165. [Google Scholar] [CrossRef]
- Foret, M.K.; Orciani, C.; Welikovitch, L.A.; Huang, C.; Cuello, A.C.; Do Carmo, S. Early Oxidative Stress and DNA Damage in Aβ-Burdened Hippocampal Neurons in an Alzheimer’s-like Transgenic Rat Model. Commun. Biol. 2024, 7, 861. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, S.; Zhu, Z.; Gatt, A.; Liu, J. E-Selectin in Vascular Pathophysiology. Front. Immunol. 2024, 15, 1401399. [Google Scholar] [CrossRef]
- Lu, X.; Liu, Y.; Xuan, W.; Ye, J.; Yao, H.; Huang, C.; Li, J. Circ_1639 Induces Cells Inflammation Responses by Sponging MiR-122 and Regulating TNFRSF13C Expression in Alcoholic Liver Disease. Toxicol. Lett. 2019, 314, 89–97. [Google Scholar] [CrossRef]
- Mushtaq, U.; Bashir, M.; Nabi, S.; Khanday, F.A. Epidermal Growth Factor Receptor and Integrins Meet Redox Signaling through P66shc and Rac1. Cytokine 2021, 146, 155625. [Google Scholar] [CrossRef]
- Lin, S.J.; Shyue, S.K.; Hung, Y.Y.; Chen, Y.H.; Ku, H.H.; Chen, J.W.; Tam, K.B.; Chen, Y.L. Superoxide Dismutase Inhibits the Expression of Vascular Cell Adhesion Molecule-1 and Intracellular Cell Adhesion Molecule-1 Induced by Tumor Necrosis Factor-α in Human Endothelial Cells through the JNK/P38 Pathways. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 334–340. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892. [Google Scholar] [CrossRef] [PubMed]
- Kleinschek, M.A.; Boniface, K.; Sadekova, S.; Grein, J.; Murphy, E.E.; Turner, S.P.; Raskin, L.; Desai, B.; Faubion, W.A.; De Malefyt, R.W.; et al. Circulating and Gut-Resident Human Th17 Cells Express CD161 and Promote Intestinal Inflammation. J. Exp. Med. 2009, 206, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Kumar, P.; Monin, L.; Castillo, P.; Elsegeiny, W.; Horne, W.; Eddens, T.; Vikram, A.; Good, M.; Schoenborn, A.A.; Bibby, K.; et al. Intestinal Interleukin-17 Receptor Signaling Mediates Reciprocal Control of the Gut Microbiota and Autoimmune Inflammation. Immunity 2016, 44, 659–671. [Google Scholar] [CrossRef]
- Hueber, W.; Sands, B.E.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.R.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a Human Anti-IL-17A Monoclonal Antibody, for Moderate to Severe Crohn’s Disease: Unexpected Results of a Randomised, Double-Blindplacebo- Controlled Trial. Gut 2012, 61, 1693–1700. [Google Scholar] [CrossRef]
- Targan, S.R.; Feagan, B.; Vermeire, S.; Panaccione, R.; Melmed, G.Y.; Landers, C.; Li, D.; Russell, C.; Newmark, R.; Zhang, N.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Brodalumab in Patients with Moderate-to-Severe Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 1599–1607. [Google Scholar] [CrossRef]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulan, F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. IL-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727. [Google Scholar] [CrossRef]
- Whibley, N.; Gaffen, S.L. Gut-Busters: IL-17 Ain’t Afraid of No IL-23. Immunity 2015, 43, 620–622. [Google Scholar] [CrossRef]
- Maxwell, J.R.; Zhang, Y.; Brown, W.A.; Smith, C.L.; Byrne, F.R.; Fiorino, M.; Stevens, E.; Bigler, J.; Davis, J.A.; Rottman, J.B.; et al. Differential Roles for Interleukin-23 and Interleukin-17 in Intestinal Immunoregulation. Immunity 2015, 43, 739–750. [Google Scholar] [CrossRef]
- Puel, A.; Cypowyj, S.; Bustamante, J.; Wright, J.F.; Liu, L.; Lim, H.K.; Migaud, M.; Israel, L.; Chrabieh, M.; Audry, M.; et al. Chronic Mucocutaneous Candidiasis in Humans with Inborn Errors of Interleukin-17 Immunity. Science (1979) 2011, 332, 65–68. [Google Scholar] [CrossRef]
- Hellenthal, K.E.M.; Brabenec, L.; Gross, E.R.; Wagner, N.M. TRP Channels as Sensors of Aldehyde and Oxidative Stress. Biomolecules 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Li, Y.; Jin, G.; Huang, T.; Zou, M.; Duan, S. The Biological Role of Arachidonic Acid 12-Lipoxygenase (ALOX12) in Various Human Diseases. Biomed. Pharmacother. 2020, 129, 110354. [Google Scholar] [CrossRef]
- Catanzaro, O.; Capponi, J.A.; Michieli, J.; Labal, E.; Di Martino, I.; Sirois, P. Bradykinin B1 Antagonism Inhibits Oxidative Stress and Restores Na+K+ ATPase Activity in Diabetic Rat Peripheral Nervous System. Peptides 2013, 44, 100–104. [Google Scholar] [CrossRef]
- Huang, Z.; Zhou, L.; Duan, J.; Qin, S.; Jiang, J.; Chen, H.; Wang, K.; Liu, R.; Yuan, M.; Tang, X.; et al. Oxidative Stress Promotes Liver Cancer Metastasis via RNF25-Mediated E-Cadherin Protein Degradation. Adv. Sci. 2024, 11, 2306929. [Google Scholar] [CrossRef]
- Aravind, P.; Bulbule, S.R.; Hemalatha, N.; Babu, R.L.; Devaraju, K.S. Elevation of Gene Expression of Calcineurin, Calmodulin and Calsyntenin in Oxidative Stress Induced PC12 Cells. Genes. Dis. 2021, 8, 87–93. [Google Scholar] [CrossRef]
- Biasi, F.; Leonarduzzi, G.; Oteiza, P.I.; Poli, G. Inflammatory Bowel Disease: Mechanisms, Redox Considerations, and Therapeutic Targets. Antioxid. Redox Signal 2013, 19, 1711. [Google Scholar] [CrossRef]
- Andresen, L.; Jørgensen, V.L.; Perner, A.; Hansen, A.; Eugen-Olsen, J.; Rask-Madsen, J. Activation of Nuclear Factor KappaB in Colonic Mucosa from Patients with Collagenous and Ulcerative Colitis. Gut 2005, 54, 503–509. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, N.; Xie, X.; Bi, G.; Ba, H.; Li, L.; Zhang, J.; Deng, X.; Yao, Y.; Tang, Z.; et al. The Macrophage-Specific V-ATPase Subunit ATP6V0D2 Restricts Inflammasome Activation and Bacterial Infection by Facilitating Autophagosome-Lysosome Fusion. Autophagy 2019, 15, 960. [Google Scholar] [CrossRef]
- Hossain, M.N.; Sakemura, R.; Fujii, M.; Ayusawa, D. G-Protein γ Subunit GNG11 Strongly Regulates Cellular Senescence. Biochem. Biophys. Res. Commun. 2006, 351, 645–650. [Google Scholar] [CrossRef]
- Thierer, J.H.; Foresti, O.; Yadav, P.K.; Wilson, M.H.; Moll, T.O.C.; Shen, M.C.; Busch-Nentwich, E.M.; Morash, M.; Mohlke, K.L.; Rawls, J.F.; et al. Pla2g12b Drives Expansion of Triglyceride-Rich Lipoproteins. Nat. Commun. 2024, 15, 2095. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, E.; Friess, U. Oxidative Stress, AGE, and Atherosclerosis. Kidney Int. 2007, 72, S17–S26. [Google Scholar] [CrossRef] [PubMed]
- Cannizzaro, L.; Rossoni, G.; Savi, F.; Altomare, A.; Marinello, C.; Saethang, T.; Carini, M.; Payne, D.M.; Pisitkun, T.; Aldini, G.; et al. Regulatory Landscape of AGE-RAGE-Oxidative Stress Axis and Its Modulation by PPARγ Activation in High Fructose Diet-Induced Metabolic Syndrome. Nutr. Metab. 2017, 14, 5. [Google Scholar] [CrossRef]
- Cabrera-García, A.I.; Suchodolski, J.S.; Steiner, J.M.; Heilmann, R.M. Association between Serum Soluble Receptor for Advanced Glycation End-Products (RAGE) Deficiency and Severity of Clinicopathologic Evidence of Canine Chronic Inflammatory Enteropathy. J. Vet. Diagn. Investig. 2020, 32, 664–674. [Google Scholar] [CrossRef]
- Cabrera-García, A.I.; Protschka, M.; Alber, G.; Kather, S.; Dengler, F.; Müller, U.; Steiner, J.M.; Heilmann, R.M. Dysregulation of Gastrointestinal RAGE (Receptor for Advanced Glycation End Products) Expression in Dogs with Chronic Inflammatory Enteropathy. Vet. Immunol. Immunopathol. 2021, 234, 110216. [Google Scholar] [CrossRef]
- Heilmann, R.M.; Otoni, C.C.; Jergens, A.E.; Grützner, N.; Suchodolski, J.S.; Steiner, J.M. Systemic Levels of the Anti-Inflammatory Decoy Receptor Soluble RAGE (Receptor for Advanced Glycation End Products) Are Decreased in Dogs with Inflammatory Bowel Disease. Vet. Immunol. Immunopathol. 2014, 161, 184–192. [Google Scholar] [CrossRef]
- Fu, X.H.; Chen, C.Z.; Wang, Y.; Peng, Y.X.; Wang, W.H.; Yuan, B.; Gao, Y.; Jiang, H.; Zhang, J.B. COL1A1 Affects Apoptosis by Regulating Oxidative Stress and Autophagy in Bovine Cumulus Cells. Theriogenology 2019, 139, 81–89. [Google Scholar] [CrossRef]
- Mao, M.; Labelle-Dumais, C.; Keene, D.R.; Gould, D.B. Elevated TGFβ Signaling Contributes to Ocular Anterior Segment Dysgenesis in Col4a1 Mutant Mice. Matrix Biol. 2022, 110, 151–173. [Google Scholar] [CrossRef]
- Faustman, D.; Davis, M. TNF Receptor 2 Pathway: Drug Target for Autoimmune Diseases. Nat. Rev. Drug Discov. 2010, 9, 482–493. [Google Scholar] [CrossRef]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Defer, N.; Azroyan, A.; Pecker, F.; Pavoine, C. TNFR1 and TNFR2 Signaling Interplay in Cardiac Myocytes. J. Biol. Chem. 2007, 282, 35564–35573. [Google Scholar] [CrossRef]
- Fischer, R.; Maier, O. Interrelation of Oxidative Stress and Inflammation in Neurodegenerative Disease: Role of TNF. Oxid. Med. Cell Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Maier, O.; Siegemund, M.; Wajant, H.; Scheurich, P.; Pfizenmaier, K. A TNF Receptor 2 Selective Agonist Rescues Human Neurons from Oxidative Stress-Induced Cell Death. PLoS ONE 2011, 6, e27621. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Janknecht, R.; Hunter, T. The Kit Receptor Promotes Cell Survival via Activation of PI 3-Kinase and Subsequent Akt-Mediated Phosphorylation of Bad on Ser136. Curr. Biol. 1998, 8, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of Cell Death Protease Caspase-9 by Phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef]
- Veroni, C.; Gabriele, L.; Canini, I.; Castiello, L.; Coccia, E.; Remoli, M.E.; Columba-Cabezas, S.; Aricò, E.; Aloisi, F.; Agresti, C. Activation of TNF Receptor 2 in Microglia Promotes Induction of Anti-Inflammatory Pathways. Mol. Cell. Neurosci. 2010, 45, 234–244. [Google Scholar] [CrossRef]
- Maier, O.; Fischer, R.; Agresti, C.; Pfizenmaier, K. TNF Receptor 2 Protects Oligodendrocyte Progenitor Cells against Oxidative Stress. Biochem. Biophys. Res. Commun. 2013, 440, 336–341. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 Family Reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef]
- Baud, O.; Haynes, R.F.; Wang, H.; Folkerth, R.D.; Li, J.; Volpe, J.J.; Rosenberg, P.A. Developmental Up-Regulation of MnSOD in Rat Oligodendrocytes Confers Protection against Oxidative Injury. Eur. J. Neurosci. 2004, 20, 29–40. [Google Scholar] [CrossRef]
- Wong, G.H.W.; Goeddel, D.V. Induction of Manganous Superoxide Dismutase by Tumor Necrosis Factor: Possible Protective Mechanism. Science 1988, 242, 941–944. [Google Scholar] [CrossRef]
- Moianu, A.; Andone, S.; Stoian, A.; Bălașa, R.; Huțanu, A.; Sărmășan, E. A Potential Role of Interleukin-5 in the Pathogenesis and Progression of Amyotrophic Lateral Sclerosis: A New Molecular Perspective. Int. J. Mol. Sci. 2024, 25, 3782. [Google Scholar] [CrossRef] [PubMed]
- Hoppenot, D.; Malakauskas, K.; Lavinskiene, S.; Sakalauskas, R. P-STAT6, PU.1, and NF-ΚB Are Involved in Allergen-Induced Late-Phase Airway Inflammation in Asthma Patients. BMC Pulm. Med. 2015, 15, 122. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, C.; Li, D.; Zheng, Y.; Wang, J. IL-5 Blocks Apoptosis and Tau Hyperphosphorylation Induced by Aβ25-35 Peptide in PC12 Cells. J. Physiol. Biochem. 2017, 73, 259–266. [Google Scholar] [CrossRef]
- Marasco, M.R.; Conteh, A.M.; Reissaus, C.A.; Cupit, J.E.; Appleman, E.M.; Mirmira, R.G.; Linnemann, A.K. Interleukin-6 Reduces B-Cell Oxidative Stress by Linking Autophagy with the Antioxidant Response. Diabetes 2018, 67, 1576–1588. [Google Scholar] [CrossRef]
- El-Assal, O.; Hong, F.; Kim, W.H.; Radaeva, S.; Gao, B. IL-6-Deficient Mice Are Susceptible to Ethanol-Induced Hepatic Steatosis: IL-6 Protects against Ethanol-Induced Oxidative Stress and Mitochondrial Permeability Transition in the Liver. Cell. Mol. Immunol. 2004, 1, 205–211. [Google Scholar]
- Matsuoka, Y.; Nakayama, H.; Yoshida, R.; Hirosue, A.; Nagata, M.; Tanaka, T.; Kawahara, K.; Sakata, J.; Arita, H.; Nakashima, H.; et al. IL-6 Controls Resistance to Radiation by Suppressing Oxidative Stress via the Nrf2-Antioxidant Pathway in Oral Squamous Cell Carcinoma. Br. J. Cancer 2016, 115, 1234–1244. [Google Scholar] [CrossRef]
- Wruck, C.J.; Streetz, K.; Pavic, G.; Götz, M.E.; Tohidnezhad, M.; Brandenburg, L.O.; Varoga, D.; Eickelberg, O.; Herdegen, T.; Trautwein, C.; et al. Nrf2 Induces Interleukin-6 (IL-6) Expression via an Antioxidant Response Element within the IL-6 Promoter. J. Biol. Chem. 2011, 286, 4493–4499. [Google Scholar] [CrossRef]
- Daffu, G.; del Pozo, C.H.; O’Shea, K.M.; Ananthakrishnan, R.; Ramasamy, R.; Schmidt, A.M. Radical Roles for RAGE in the Pathogenesis of Oxidative Stress in Cardiovascular Diseases and Beyond. Int. J. Mol. Sci. 2013, 14, 19891. [Google Scholar] [CrossRef]
- Pathomthongtaweechai, N.; Chutipongtanate, S. AGE/RAGE Signaling-Mediated Endoplasmic Reticulum Stress and Future Prospects in Non-Coding RNA Therapeutics for Diabetic Nephropathy. Biomed. Pharmacother. 2020, 131, 110655. [Google Scholar] [CrossRef]
- El-Deeb, M.M.K.; El-Sheredy, H.G.; Mohammed, A.F. The Possible Role of Interleukin (IL)-18 and Nitrous Oxide and Their Relation to Oxidative Stress in the Development and Progression of Breast Cancer. Asian Pac. J. Cancer Prev. 2019, 20, 2659. [Google Scholar] [CrossRef]
- Ferrer-Torres, D.; Nancarrow, D.J.; Steinberg, H.; Wang, Z.; Kuick, R.; Weh, K.M.; Mills, R.E.; Ray, D.; Ray, P.; Lin, J.; et al. Constitutively Higher Level of GSTT2 in Esophageal Tissues From African Americans Protects Cells Against DNA Damage. Gastroenterology 2018, 156, 1404. [Google Scholar] [CrossRef] [PubMed]
- Preynat-Seauve, O.; Coudurier, S.; Favier, A.; Marche, P.N.; Villiers, C. Oxidative Stress Impairs Intracellular Events Involved in Antigen Processing and Presentation to T Cells. Cell Stress. Chaperones 2003, 8, 162. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Signorile, A.; Roca, E.; Papa, S. CAMP Response Element-Binding Protein (CREB) Is Imported into Mitochondria and Promotes Protein Synthesis. FEBS J. 2009, 276, 4325–4333. [Google Scholar] [CrossRef] [PubMed]
- Luciano-Mateo, F.; Cabré, N.; Fernández-Arroyo, S.; Baiges-Gaya, G.; Hernández-Aguilera, A.; Rodríguez-Tomàs, E.; Muñoz-Pinedo, C.; Menéndez, J.A.; Camps, J.; Joven, J. Chemokine C–C Motif Ligand 2 Overexpression Drives Tissue-Specific Metabolic Responses in the Liver and Muscle of Mice. Sci. Rep. 2020, 10, 11954. [Google Scholar] [CrossRef]
- Cescon, M.; Gattazzo, F.; Chen, P.; Bonaldo, P. Collagen VI at a Glance. J. Cell Sci. 2015, 128, 3525–3531. [Google Scholar] [CrossRef]
- Guan, Z.-H.; Yang, D.; Wang, Y.; Ma, J.-B.; Wang, G.-N. Ectodysplasin-A2 Receptor (EDA2R) Knockdown Alleviates Myocardial Ischemia/Reperfusion Injury through Inhibiting the Activation of the NF-ΚB Signaling Pathway. Exp. Anim. 2024, 73, 376. [Google Scholar] [CrossRef]
- Hirooka, Y.; Nozaki, Y.; Niki, K.; Inoue, A.; Sugiyama, M.; Kinoshita, K.; Funauchi, M.; Matsumura, I. Foxp3-Positive Regulatory T Cells Contribute to Antifibrotic Effects in Renal Fibrosis via an Interleukin-18 Receptor Signaling Pathway. Front. Med. 2020, 7, 604656. [Google Scholar] [CrossRef]
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic Value of Tumor-Infiltrating FoxP3+ Regulatory T Cells in Cancers: A Systematic Review and Meta-Analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef]
- Lin, C.; Zhao, X.; Sun, D.; Zhang, L.; Fang, W.; Zhu, T.; Wang, Q.; Liu, B.; Wei, S.; Chen, G.; et al. Transcriptional Activation of Follistatin by Nrf2 Protects Pulmonary Epithelial Cells against Silica Nanoparticle-Induced Oxidative Stress. Sci. Rep. 2016, 6, 21133. [Google Scholar] [CrossRef]
- Kaur, G.; Wang, X.; Li, X.; Ong, H.; He, X.; Cai, C. Overexpression of GREM1 Improves the Survival Capacity of Aged Cardiac Mesenchymal Progenitor Cells via Upregulation of the ERK/NRF2-Associated Antioxidant Signal Pathway. Cells 2023, 12, 1203. [Google Scholar] [CrossRef]
- Hashemi, S.; Salma, J.; Wales, S.; McDermott, J.C. Pro-Survival Function of MEF2 in Cardiomyocytes Is Enhanced by β-Blockers. Cell Death Discov. 2015, 1, 15019. [Google Scholar] [CrossRef] [PubMed]
- She, H.; Mao, Z. Regulation of Myocyte Enhancer Factor-2 Transcription Factors by Neurotoxins. Neurotoxicology 2011, 32, 563. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, H.; Liu, X.; Yu, H.; Huang, X.; Huang, W.; Wang, G. Neuregulin-1 Alleviate Oxidative Stress and Mitigate Inflammation by Suppressing NOX4 and NLRP3/Caspase-1 in Myocardial Ischaemia-Reperfusion Injury. J. Cell. Mol. Med. 2021, 25, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbacher, N.; Bar-Sagi, D.; Philips, M. Ras/MAPK Signaling from Endomembranes. Mol. Oncol. 2009, 3, 297–307. [Google Scholar] [CrossRef]
- Downward, J. Ras Signalling and Apoptosis. Curr. Opin. Genet. Dev. 1998, 8, 49–54. [Google Scholar] [CrossRef]
- Hlavatá, L.; Nyström, T. Ras Proteins Control Mitochondrial Biogenesis and Function in Saccharomyces Cerevisiae. Folia Microbiol. 2003, 48, 725–730. [Google Scholar] [CrossRef]
- Kamp, D.W.; Shacter, E.; Weitzman, S.A. Chronic Inflammation and Cancer: The Role of the Mitochondria. Oncology 2011, 25, 400–410. [Google Scholar]
- Krishnappa, V.; Boregowda, S.V.; Phinney, D.G. FGF2 Protects Mouse Mesenchymal Stem Cells from Oxidative Stress by Modulating a Twist2-P53 Signaling Axis. Cytotherapy 2014, 16, S74. [Google Scholar] [CrossRef]
- Mei, L.; Chen, Y.; Chen, P.; Chen, H.; He, S.; Jin, C.; Wang, Y.; Hu, Z.; Li, W.; Jin, L.; et al. Fibroblast Growth Factor 7 Alleviates Myocardial Infarction by Improving Oxidative Stress via PI3Kα/AKT-Mediated Regulation of Nrf2 and HXK2. Redox Biol. 2022, 56, 102468. [Google Scholar] [CrossRef]
- Zhou, D.-P.; Deng, L.-C.; Feng, X.; Xu, H.-J.; Tian, Y.; Yang, W.-W.; Zeng, P.-P.; Zou, L.-H.; Yan, X.-H.; Zhu, X.-Y.; et al. FGF10 Mitigates Doxorubicin-Induced Myocardial Toxicity in Mice via Activation of FGFR2b/PHLDA1/AKT Axis. Acta Pharmacol. Sin. 2023, 44, 2004–2018. [Google Scholar] [CrossRef]
- Gao, S.; Guo, K.; Chen, Y.; Zhao, J.; Jing, R.; Wang, L.; Li, X.; Hu, Z.; Xu, N.; Li, X. Keratinocyte Growth Factor 2 Ameliorates UVB-Induced Skin Damage via Activating the AhR/Nrf2 Signaling Pathway. Front. Pharmacol. 2021, 12, 655281. [Google Scholar] [CrossRef]
- Codocedo, J.F.; Montecinos-Oliva, C.; Inestrosa, N.C. Wnt-Related SynGAP1 Is a Neuroprotective Factor of Glutamatergic Synapses against Aβ Oligomers. Front. Cell. Neurosci. 2015, 9, 227. [Google Scholar] [CrossRef]
- Sato, T.; Takino, J.I.; Nagamine, K.; Nishio, K.; Hori, T. RASGRP2 Suppresses Apoptosis via Inhibition of ROS Production in Vascular Endothelial Cells. Sci. World J. 2019, 2019, 4639165. [Google Scholar] [CrossRef]
- Tang, S.; Chen, T.; Yu, Z.; Zhu, X.; Yang, M.; Xie, B.; Li, N.; Cao, X.; Wang, J. RasGRP3 Limits Toll-like Receptor-Triggered Inflammatory Response in Macrophages by Activating Rap1 Small GTPase. Nat. Commun. 2014, 5, 4657. [Google Scholar] [CrossRef]
- Bokoch, G.M. Biology of the P21-Activated Kinases. Annu. Rev. Biochem. 2003, 72, 743–781. [Google Scholar] [CrossRef]
- Orr, A.W.; Hahn, C.; Blackman, B.R.; Schwartz, M.A. P21-Activated Kinase Signaling Regulates Oxidant-Dependent NF-ΚB Activation by Flow. Circ. Res. 2008, 103, 671–679. [Google Scholar] [CrossRef]
- Cunha, D.A.; Cito, M.; Carlsson, P.O.; Vanderwinden, J.M.; Molkentin, J.D.; Bugliani, M.; Marchetti, P.; Eizirik, D.L.; Cnop, M. Thrombospondin 1 Protects Pancreatic β-Cells from Lipotoxicity via the PERK–NRF2 Pathway. Cell Death Differ. 2016, 23, 1995. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, J.J.; Chung, J.S.; Lee, S.R.; Lee, G.Y.; Kim, H.J.; Yoo, Y. Do RASSF1A Suppresses the Activated K-Ras-Induced Oxidative DNA Damage. Biochem. Biophys. Res. Commun. 2011, 408, 149–153. [Google Scholar] [CrossRef]
- Gordon, M.; El-Kalla, M.; Zhao, Y.; Fiteih, Y.; Law, J.; Volodko, N.; Mohamed, A.; El-Kadi, A.O.S.; Liu, L.; Odenbach, J.; et al. The Tumor Suppressor Gene, RASSF1A, Is Essential for Protection against Inflammation -Induced Injury. PLoS ONE 2013, 8, e75483. [Google Scholar] [CrossRef]
- Chung, J.; Huda, M.N.; Shin, Y.; Han, S.; Akter, S.; Kang, I.; Ha, J.; Choe, W.; Choi, T.G.; Kim, S.S. Correlation between Oxidative Stress and Transforming Growth Factor-Beta in Cancers. Int. J. Mol. Sci. 2021, 22, 13181. [Google Scholar] [CrossRef]
- Son, Y.; Kim, S.; Chung, H.T.; Pae, H.O. Reactive Oxygen Species in the Activation of MAP Kinases. Methods Enzymol. 2013, 528, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Pérez, M.; Nuño-Lámbarri, N.; Clavijo-Cornejo, D.; Luna-López, A.; Souza, V.; Bucio, L.; Miranda, R.U.; Muñoz, L.; Gomez-Quiroz, L.E.; Uribe-Carvajal, S.; et al. Hepatocyte Growth Factor Reduces Free Cholesterol-Mediated Lipotoxicity in Primary Hepatocytes by Countering Oxidative Stress. Oxid. Med. Cell. Longev. 2016, 2016, 7960386. [Google Scholar] [CrossRef] [PubMed]
- Clavijo-Cornejo, D.; Enriquez-Cortina, C.; López-Reyes, A.; Domínguez-Pérez, M.; Nuño, N.; Domínguez-Meraz, M.; Bucio, L.; Souza, V.; Factor, V.M.; Thorgeirsson, S.S.; et al. Biphasic Regulation of the NADPH Oxidase by HGF/c-Met Signaling Pathway in Primary Mouse Hepatocytes. Biochimie 2013, 95, 1177–1184. [Google Scholar] [CrossRef]
- López-Ramirez, J.; Lazzarini-Lechuga, R.; Gerardo-Ramírez, M.; Escobedo-Calvario, A.; Chávez-Rodríguez, L.; Salas-Silva, S.; Nuño-Lámbarri, N.; Massó, F.; Souza-Arroyo, V.; Miranda-Labra, R.U.; et al. The Hepatocyte Growth Factor Induces an Anti-Inflammatory and Repairing Response in the Cholestasis-Induced Colon Damage. Open Explor. 2022, 1, 40–50. [Google Scholar] [CrossRef]
- Asao, H.; Okuyama, C.; Kumaki, S.; Ishii, N.; Tsuchiya, S.; Foster, D.; Sugamura, K. Cutting Edge: The Common Gamma-Chain Is an Indispensable Subunit of the IL-21 Receptor Complex. J. Immunol. 2001, 167, 1–5. [Google Scholar] [CrossRef]
- Loschinski, R.; Böttcher, M.; Stoll, A.; Bruns, H.; Mackensen, A.; Mougiakakos, D. IL-21 Modulates Memory and Exhaustion Phenotype of T-Cells in a Fatty Acid Oxidation-Dependent Manner. Oncotarget 2018, 9, 13125. [Google Scholar] [CrossRef]
- Quintero-Villegas, A.; Valdés-Ferrer, S.I. Role of 5-HT7 Receptors in the Immune System in Health and Disease. Mol. Med. 2019, 26, 2. [Google Scholar] [CrossRef]
- Yuksel, T.N.; Yayla, M.; Halici, Z.; Cadirci, E.; Polat, B.; Kose, D. Protective Effect of 5-HT7 Receptor Activation against Glutamate-Induced Neurotoxicity in Human Neuroblastoma SH-SY5Y Cells via Antioxidative and Antiapoptotic Pathways. Neurotoxicol. Teratol. 2019, 72, 22–28. [Google Scholar] [CrossRef]
- Ong, Q.; Guo, S.; Duan, L.; Zhang, K.; Collier, E.A.; Cui, B. The Timing of Raf/ERK and AKT Activation in Protecting PC12 Cells against Oxidative Stress. PLoS ONE 2016, 11, e0153487. [Google Scholar] [CrossRef]
- Zheng, L.; Ishii, Y.; Tokunaga, A.; Hamashima, T.; Shen, J.; Zhao, Q.L.; Ishizawa, S.; Fujimori, T.; Nabeshima, Y.I.; Mori, H.; et al. Neuroprotective Effects of PDGF against Oxidative Stress and the Signaling Pathway Involved. J. Neurosci. Res. 2010, 88, 1273–1284. [Google Scholar] [CrossRef]
- Hartman, E.S.; Brindley, E.C.; Papoin, J.; Ciciotte, S.L.; Zhao, Y.; Peters, L.L.; Blanc, L. Increased Reactive Oxygen Species and Cell Cycle Defects Contribute to Anemia in the RASA3 Mutant Mouse Model Scat. Front. Physiol. 2018, 9, 352608. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.S.; Chang, J.H.; Hung, W.Y.; Yang, Y.C.; Chien, M.H. The Interplay of Reactive Oxygen Species and the Epidermal Growth Factor Receptor in Tumor Progression and Drug Resistance. J. Exp. Clin. Cancer Res. 2018, 37, 61. [Google Scholar] [CrossRef]
- Mak, I.T.; Kramer, J.H.; Chmielinska, J.J.; Spurney, C.F.; Weglicki, W.B. EGFR-TKI, Erlotinib, Causes Hypomagnesemia, Oxidative Stress and Cardiac Dysfunction: Attenuation by NK-1 Receptor Blockade. J. Cardiovasc. Pharmacol. 2015, 65, 54. [Google Scholar] [CrossRef]
- Coskun, M.; Salem, M.; Pedersen, J.; Nielsen, O.H. Involvement of JAK/STAT Signaling in the Pathogenesis of Inflammatory Bowel Disease. Pharmacol. Res. 2013, 76, 1–8. [Google Scholar] [CrossRef]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK–STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Manz, A.; Allenspach, K.; Kummer, S.; Richter, B.; Walter, I.; Macho-Maschler, S.; Tichy, A.; Burgener, I.A.; Luckschander-Zeller, N. Upregulation of Signal Transducer and Activator of Transcription 3 in Dogs with Chronic Inflammatory Enteropathies. J. Vet. Intern. Med. 2021, 35, 1288–1296. [Google Scholar] [CrossRef]
- Richards, C.D. The Enigmatic Cytokine Oncostatin M and Roles in Disease. ISRN Inflamm. 2013, 2013, 512103. [Google Scholar] [CrossRef]
- Werner, S.L.; Sharma, R.; Woodruff, K.; Horn, D.; Harris, S.E.; Gorin, Y.; Lee, D.Y.; Hua, R.; Gu, S.; Fajardo, R.J.; et al. CSF-1 in Osteocytes Inhibits Nox4-Mediated Oxidative Stress and Promotes Normal Bone Homeostasis. JBMR Plus 2020, 4, e10080. [Google Scholar] [CrossRef]
- Kojima, H.; Otani, A.; Oishi, A.; Makiyama, Y.; Nakagawa, S.; Yoshimura, N. Granulocyte Colony-Stimulating Factor Attenuates Oxidative Stress–Induced Apoptosis in Vascular Endothelial Cells and Exhibits Functional and Morphologic Protective Effect in Oxygen-Induced Retinopathy. Blood 2011, 117, 1091–1100. [Google Scholar] [CrossRef]
- Singh, B.; Kosuru, R.; Lakshmikanthan, S.; Sorci-Thomas, M.G.; Zhang, D.X.; Sparapani, R.; Vasquez-Vivar, J.; Chrzanowska, M. Endothelial Rap1 (Ras-Association Proximate 1) Restricts Inflammatory Signaling to Protect From the Progression of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 638–650. [Google Scholar] [CrossRef]
- Yang, Z.; Kirton, H.M.; Al-Owais, M.; Thireau, J.; Richard, S.; Peers, C.; Steele, D.S. Epac2-Rap1 Signaling Regulates Reactive Oxygen Species Production and Susceptibility to Cardiac Arrhythmias. Antioxid. Redox Signal. 2017, 27, 117. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, Y.; Shi, D.; Quilliam, L.A.; Chrzanowska-Wodnicka, M.; Wittchen, E.S.; Li, D.Y.; Hartnett, M.E. Activation of Rap1 Inhibits NADPH Oxidase-Dependent ROS Generation in Retinal Pigment Epithelium and Reduces Choroidal Neovascularization. FASEB J. 2014, 28, 265. [Google Scholar] [CrossRef] [PubMed]
- Gertzberg, N.; Neumann, P.; Rizzo, V.; Johnson, A. NAD(P)H Oxidase Mediates the Endothelial Barrier Dysfunction Induced by TNF-Alpha. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L37–L48. [Google Scholar] [CrossRef] [PubMed]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/MTOR Signaling Transduction Pathway and Targeted Therapies in Cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Chiang, J.C.; Chen, W.M.; Newman, C.; Chen, B.P.C.; Lee, H. Lysophosphatidic Acid Receptor 3 Promotes Mitochondrial Homeostasis against Oxidative Stress: Potential Therapeutic Approaches for Hutchinson-Gilford Progeria Syndrome. Antioxidants 2022, 11, 351. [Google Scholar] [CrossRef]
- Djavaheri-Mergny, M.; Javelaud, D.; Wietzerbin, J.; Besançon, F. NF-KappaB Activation Prevents Apoptotic Oxidative Stress via an Increase of Both Thioredoxin and MnSOD Levels in TNFalpha-Treated Ewing Sarcoma Cells. FEBS Lett. 2004, 578, 111–115. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 Is a Critical Regulator of the Innate Immune Response and Survival during Experimental Sepsis. J. Clin. Investig. 2016, 116, 984–995. [Google Scholar] [CrossRef]
- Greco, L.; Gobbetti, M.; Auricchio, R.; Di Mase, R.; Landolfo, F.; Paparo, F.; Di Cagno, R.; De Angelis, M.; Rizzello, C.G.; Cassone, A.; et al. Safety for Patients with Celiac Disease of Baked Goods Made of Wheat Flour Hydrolyzed During Food Processing. Clin. Gastroenterol. Hepatol. 2011, 9, 24–29. [Google Scholar] [CrossRef]
- Von Berg, A.; Koletzko, S.; Grübl, A.; Filipiak-Pittroff, B.; Wichmann, H.E.; Bauer, C.P.; Reinhardt, D.; Berdel, D. The Effect of Hydrolyzed Cow’s Milk Formula for Allergy Prevention in the First Year of Life: The German Infant Nutritional Intervention Study, a Randomized Double-Blind Trial. J. Allergy Clin. Immunol. 2003, 111, 533–540. [Google Scholar] [CrossRef]
- Mandigers, P.J.J.; Biourge, V.; Van Den Ingh, T.S.G.A.M.; Ankringa, N.; German, A.J. A Randomized, Open-Label, Positively-Controlled Field Trial of a Hydrolyzed Protein Diet in Dogs with Chronic Small Bowel Enteropathy. J. Vet. Intern. Med. 2010, 24, 1350–1357. [Google Scholar] [CrossRef]
- Cave, N.J. Hydrolyzed Protein Diets for Dogs and Cats. Vet. Clin. N. Am.-Small Anim. Pract. 2006, 36, 1251–1268. [Google Scholar] [CrossRef] [PubMed]
- Olivry, T.; Bizikova, P. A Systematic Review of the Evidence of Reduced Allergenicity and Clinical Benefit of Food Hydrolysates in Dogs with Cutaneous Adverse Food Reactions. Vet. Dermatol. 2010, 21, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, Y.M.; Neuber, S.; Borcherding, D.; Seo, Y.J.; Segarra, S.; Glanemann, B.; Garden, O.A.; Müller, U.; Adam, M.G.; Dang, V.; et al. Treatment with Hydrolyzed Diet Supplemented with Prebiotics and Glycosaminoglycans Alters Lipid Metabolism in Canine Inflammatory Bowel Disease. Front. Vet. Sci. 2020, 7, 451. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Score | Morphologic Appearance |
|---|---|
| 1 | Normal |
| 2 | Some normal ER with several distended ER |
| 3 | Few normal ER with many largely distended ER |
| 4 | Majority of ER are distended or blown out |
| Score | Morphologic Appearance |
|---|---|
| 1 | Normal cristae, size, shape |
| 2 | Some distended cristae with slight architectural voids |
| 3 | Distended cristae, larger voids with some cristae visible |
| 4 | Major distended cristae, large voids with few visible cristae |
| Pathways | Gene ID | Gene Name | Gene Description | log2 (Fold Change) | p |
|---|---|---|---|---|---|
| Ras signaling pathway, Rap1 signaling pathway | ENSCAFG00000006861 | ANGPT4 | Angiopoietin-4 | 1.458 | 0.045 |
| ENSCAFG00000005970 | FGFR1 | Fibroblast growth factor receptor-1 | 1.121 | 0.033 | |
| ENSCAFG00000006701 | FLT1 | Fms-related receptor tyrosine kinase-1 | 1.002 | 0.02 | |
| ENSCAFG00000013148 | NMDE2 (GRIN2B) | Glutamate ionotropic receptor NMDA type subunit 2B | 6.064 | 0.042 | |
| ENSCAFG00000014309 | RASGRP2 | RAS guanyl-releasing protein-2 | 1.939 | 0.035 | |
| ENSCAFG00000005870 | RASGRP3 | RAS guanyl-releasing protein-3 | 1.47 | 0.018 | |
| ENSCAFG00000029799 | RASSF5 | Ras association domain family member-5 | 1.585 | 0.02 | |
| Inflammatory bowel disease pathway, cytokine–cytokine receptor interaction | ENSCAFG00000028874 | C6L8D7 (IL-17A) | Interleukin-17A | 5.77 | 0.018 |
| ENSCAFG00000002183 | IL-17F | Interleukin-17F | 3.278 | 0.022 | |
| Oxidative phosphorylation, ROS–chemical carcinogenesis | ENSCAFG00000019536 | ATP5D (ATP5F1D) | ATP synthase F1 subunit delta | −0.834 | 0.04 |
| ENSCAFG00000016880 | ATP5G1 (ATP5MC1) | ATP-synthase membrane subunit c locus 1 | −0.981 | 0.039 | |
| ENSCAFG00000022729 | ATP6 (MT-ATP6) | Mitochondrially encoded ATP synthase membrane subunit 6 | −1.237 | 0.029 | |
| ENSCAFG00000002387 | B7ZDP5 (COX5B) | Cytochrome c oxidase subunit 5B | −1.121 | 0.016 | |
| ENSCAFG00000029155 | COX7A2 | Cytochrome c oxidase subunit 7A2 | −1.058 | 0.009 | |
| ENSCAFG00000014738 | COX8A | Cytochrome c oxidase subunit VIIIA (ubiquitous) | −0.902 | 0.047 | |
| ENSCAFG00000004842 | NDUFC2 | NADH:ubiquinone oxidoreductase subunit C2 | −0.83 | 0.033 | |
| ENSCAFG00000006913 | LOC612644 | Cytochrome c oxidase subunit 6B1 | −1.08 | 0.015 | |
| ENSCAFG00000009698 | NDUFB8 | NADH:ubiquinone oxidoreductase subunit B8 | −0.897 | 0.024 | |
| ENSCAFG00000017667 | NDUFAB1 | NADH:ubiquinone oxidoreductase subunit AB1 | −0.918 | 0.022 | |
| ENSCAFG00000025112 | NDUFA5 | NADH:ubiquinone oxidoreductase subunit A5 | −1.201 | 0.004 | |
| ENSCAFG00000031458 | LOC477508 | Cytochrome c oxidase subunit 6A1, mitochondrial | −0.963 | 0.0301 | |
| ENSCAFG00000005861 | NDUFA2 | NADH:ubiquinone oxidoreductase subunit A2 | −0.957 | 0.016 | |
| ENSCAFG00000000982 | NDUFA6 | NADH:ubiquinone oxidoreductase subunit A6 | −0.808 | 0.039 | |
| ENSCAFG00000018497 | NDUFA7 | NADH dehydrogenase [ubiquinone]-1 alpha subcomplex subunit 7 | −0.913 | 0.027 | |
| ENSCAFG00000030171 | NDUFA8 | NADH:ubiquinone oxidoreductase subunit A8 | −0.912 | 0.042 | |
| ENSCAFG00000018753 | NDUFA11 | NADH:ubiquinone oxidoreductase subunit A11 | −1.13 | 0.005 | |
| ENSCAFG00000006232 | NDUFA12 | NADH:ubiquinone oxidoreductase subunit A12 | −0.909 | 0.028 | |
| ENSCAFG00000003930 | NDUFB2 | NADH:ubiquinone oxidoreductase subunit B2 | −0.849 | 0.028 | |
| ENSCAFG00000058755 | NDUFB3 | NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 3 | −0.979 | 0.021 | |
| ENSCAFG00000011204 | NDUFB4 | NADH:ubiquinone oxidoreductase subunit B4 | −0.793 | 0.045 | |
| ENSCAFG00000004456 | NDUFB6 | NADH:ubiquinone oxidoreductase subunit B6 | −0.93 | 0.017 | |
| ENSCAFG00000052422 | NDUFS4 | NADH:ubiquinone oxidoreductase subunit S4 | −1.099 | 0.01 | |
| ENSCAFG00000030152 | NDUFS6 | NADH:ubiquinone oxidoreductase subunit S6 | −0.939 | 0.03 | |
| ENSCAFG00000019529 | NDUFS7 | NADH:ubiquinone oxidoreductase core subunit S7 | −0.956 | 0.029 | |
| ENSCAFG00000011083 | NDUFS8 | NADH:ubiquinone oxidoreductase core subunit S8 | −0.993 | 0.022 | |
| ENSCAFG00000022732 | NU3M (MT-ND3) | Mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 3 | −1.23 | 0.004 | |
| ENSCAFG00000015832 | Q0QEY4 (SDHB) | Succinate dehydrogenase complex iron sulfur subunit B | −1.01 | 0.017 | |
| ENSCAFG00000004260 | UQCRH | Cytochrome b-c1 complex subunit 6 | −0.98 | 0.019 | |
| ENSCAFG00000000919 | UQCRQ | Ubiquinol-cytochrome c reductase complex III subunit VII | −0.952 | 0.023 | |
| NF-κB signaling pathway, cytokine–cytokine receptor interaction | ENSCAFG00000000515 | TNFB (LTA) | Lymphotoxin alpha | 6.939 | 0.007 |
| ENSCAFG00000001005 | TNFRSF13C | TNF receptor superfamily member 13C | 2.822 | 0.042 | |
| ENSCAFG00000004675 | TNFSF11 | TNF superfamily member 11 | 1.798 | 0.017 | |
| NF-κB signaling pathway, PI3K/Akt signaling pathway | ENSCAFG00000002171 | B8XNP7 (SYK) | Spleen-associated tyrosine kinase | 1.634 | 0.019 |
| Rap1 signaling pathway, cGMP-PKG signaling pathway | ENSCAFG00000016413 | GNAI1 | G-protein subunit alpha i1 | 1.208 | 0.013 |
| AGE-RAGE, NF-κB signaling pathway | ENSCAFG00000031460 | ICAM-1 | Intercellular Adhesion Molecule 1 | 1.828 | 0.005 |
| TRP channels, cGMP-PKG signaling pathway | ENSCAFG00000023463 | ITPR1 | Inositol-1,4,5-trisphosphate receptor type 1 | 0.806 | 0.035 |
| ENSCAFG00000029786 | Q9BDQ4 (Bdkrb2) | B2 bradykinin receptor | 1.924 | 0.0003 | |
| PI3K/Akt signaling pathway, Rap1 signaling pathway | ENSCAFG00000004366 | MAGI2 | Membrane-associated guanylate kinase | 1.932 | 0.012 |
| JAK/STAT signaling pathway, PI3K/Akt signaling pathway | ENSCAFG00000015159 | JAK3 | Janus kinase 3 | 1.842 | 0.019 |
| PI3K/Akt signaling pathway, cGMP-PKG signaling pathway | ENSCAFG00000003005 | CREB5 | CAMP-responsive element binding protein 5 | 2.16 | 0.013 |
| ENSCAFG00000017387 | PIK3R5 | Phosphoinositide-3-kinase regulatory subunit 5 | 1.522 | 0.034 | |
| Ras signaling pathway, TRP channels | ENSCAFG00000020319 | PRKACB | Protein kinase cAMP-activated catalytic subunit beta | −0.81 | 0.048 |
| AGE-RAGE, PI3K/Akt signaling pathway | ENSCAFG00000014345 | Q28252 (FN1) | Fibronectin 1 ED-A | 2.038 | 0.0002 |
| EGFR-TKI resistance, TGF-β signaling pathway | ENSCAFG00000011590 | RPS6KB2 | Ribosomal protein S6 kinase B2 | −0.882 | 0.031 |
| cGMP-PKG signaling pathway, ROS–chemical carcinogenesis | ENSCAFG00000007596 | SLC25A4 | Solute carrier family 25 member 4 | −0.944 | 0.04 |
| AGE-RAGE, NF-κB signaling pathway | ENSCAFG00000020004 | VCAM1 | Vascular cell adhesion molecule 1 | 3.13 | 0.0002 |
| TGF-β signaling pathway, Rap1 signaling pathway | ENSCAFG00000008704 | THBS1 | Thrombospondin 1 | 1.293 | 0.026 |
| ENSCAFG00000007045 | ID1 | Inhibitor of DNA binding 1 | −0.872 | 0.049 | |
| PI3K/Akt signaling pathway, cGMP-PKG signaling pathway | ENSCAFG00000017382 | PIK3R6 | Phosphoinositide-3-kinase regulatory subunit 6 | 1.715 | 0.024 |
| TGF-β signaling pathway, cytokine–cytokine receptor interaction | ENSCAFG00000014880 | BMP4 | Bone morphogenetic protein 4 | 1.772 | 0.001 |
| ENSCAFG00000012011 | BMP7 | Bone morphogenetic protein 7 | 1.266 | 0.039 | |
| ENSCAFG00000010107 | BMPR1B | Bone morphogenetic protein receptor type 1B | 6.562 | 0.0009 | |
| NF-κB signaling pathway, cytokine–cytokine receptor interaction | ENSCAFG00000031735 | C4NZX1 (TNFSF13B) | Tumor-necrosis factor superfamily member 13b | 1.699 | 0.026 |
| ENSCAFG00000001970 | EDAR | Ectodysplasin A receptor | 3.037 | 0.033 | |
| ENSCAFG00000001954 | CCL19 | C-C motif chemokine ligand 19 | 3.286 | 0.012 | |
| JAK/STAT signaling pathway, cytokine–cytokine receptor interaction | ENSCAFG00000011034 | CRLF2 | Cytokine receptor-like factor 2 | 1.947 | 0.038 |
| ENSCAFG00000001519 | CSF2RB | Colony-stimulating factor 2 receptor subunit beta | 1.486 | 0.005 | |
| ENSCAFG00000031295 | CSF2RA | Colony-stimulating factor 2 receptor subunit alpha | 1.941 | 0.011 | |
| ENSCAFG00000018579 | GHR | Growth-hormone receptor | 1.185 | 0.0131 | |
| ENSCAFG00000018230 | IL13RA2 | Interleukin-13 receptor subunit alpha-2 | 1.774 | 0.016 | |
| ENSCAFG00000012844 | IL10RA | Interleukin-10 receptor subunit alpha | 1.83 | 0.007 | |
| ENSCAFG00000005991 | IL5RA | Interleukin-5 receptor subunit alpha | 2.389 | 0.046 | |
| ENSCAFG00000018600 | LEPR | Leptin receptor | 1.778 | 0.004 | |
| ENSCAFG00000018661 | LIFR | Leukemia inhibitory factor receptor subunit alpha | 0.928 | 0.042 | |
| ENSCAFG00000018648 | OSMR | Oncostatin M receptor | 1.436 | 0.013 | |
| ENSCAFG00000030931 | Q8SPY1 (LIF) | Leukemia inhibitory factor | 1.41 | 0.009 | |
| AGE-RAGE signaling pathway, cytokine–cytokine receptor interaction | ENSCAFG00000018349 | CCL2 | C-C motif chemokine ligand 2 | 2.134 | 0.006 |
| TRP channels, Rap1 signaling pathway and cGMP-PKG signaling pathway | ENSCAFG00000001097 | ADCY8 | Adenylate cyclase 8 | 4.351 | 0.021 |
| Ras signaling pathway, PI3K/Akt signaling pathway, Rap1 signaling pathway | ENSCAFG00000014923 | FGF7 | Fibroblast-growth factor 7 | 2.357 | 0.0074 |
| ENSCAFG00000032695 | FGF10 | Fibroblast-growth factor 10 | 2.502 | 0.0001 | |
| ENSCAFG00000002065 | KIT | Mast/stem cell growth factor receptor Kit | 1.602 | 0.0009 | |
| AGE-RAGE, TRP channels, cGMP-PKG signaling pathway | ENSCAFG00000002610 | PRKCE | Protein kinase C epsilon | 1.099 | 0.005 |
| Ras signaling pathway, Rap1 signaling pathway, EGFR-TKI resistance | ENSCAFG00000002079 | Q2XPT7 (KDR) | Vascular endothelial growth factor receptor 2 | 1.11 | 0.005 |
| ENSCAFG00000010881 | Q6QHH2 (IGF1R) | Insulin-like growth factor 1 receptor | 0.845 | 0.026 | |
| Inflammatory bowel disease, AGE-RAGE, TGF-β signaling pathway | ENSCAFG00000017388 | SMAD3 | SMAD family member 3 | 0.85 | 0.039 |
| JAK/STAT signaling pathway, PI3K/Akt signaling pathway, cytokine–cytokine receptor interaction | ENSCAFG00000016261 | CSF3 | Colony-stimulating factor 3 | 3.963 | 0.022 |
| ENSCAFG00000005210 | D2XMM7 (IL2RA) | Interleukin-2 receptor subunit alpha | 1.722 | 0.034 | |
| Inflammatory bowel disease pathway, AGE-RAGE signaling pathway, cytokine–cytokine receptor interaction | ENSCAFG00000007245 | IL1A | Interleukin-1 alpha | 1.603 | 0.022 |
| Inflammatory bowel disease pathway, JAK/STAT signaling pathway, cytokine–cytokine receptor interaction | ENSCAFG00000032590 | IL21R | Interleukin-21 receptor | 3.525 | 0.013 |
| ENSCAFG00000018542 | IL23R | Interleukin-23 receptor | 2.128 | 0.022 | |
| ENSCAFG00000000855 | IL5 | Interleukin-5 | 2.733 | 0.037 | |
| PI3K/Akt signaling pathway, cytokine–cytokine receptor interaction, JAK/STAT signaling pathway | ENSCAFG00000012468 | OSM | Oncostatin M | 7.424 | 0.0009 |
| AGE-RAGE signaling pathway, TGF-β signaling pathway, cytokine–cytokine receptor interaction | ENSCAFG00000005449 | TGFBR2 | Transforming growth factor beta receptor 2 | 1.157881378 | 0.018 |
| NF-κB signaling pathway, TRP channels, cytokine–cytokine receptor interaction | ENSCAFG00000002147 | IL1R1 | Interleukin-1 receptor type 1 | 1.169 | 0.021 |
| Ras signaling pathway, TRP channels, Rap1 signaling pathway, cGMP-PKG signaling pathway | ENSCAFG00000002646 | CALM2 | Calmodulin 2 | −1.003 | 0.011 |
| ENSCAFG00000017516 | CALM1 | Calmodulin 1 | −0.893 | 0.042 | |
| Ras signaling pathway, PI3K/Akt signaling pathway, Rap1 signaling pathway, EGFR-TKI resistance | ENSCAFG00000003996 | FGF2 | Fibroblast-growth factor 2 | 2.45 | 0.004 |
| EGFR-TKI resistance, JAK/STAT signaling pathway, NF-κB signaling pathway, AGE-RAGE | ENSCAFG00000000068 | BCL2 | BCL2 apoptosis regulator | 0.892 | 0.024 |
| AGE-RAGE, TRP channels, Rap1 signaling pathway, cGMP-PKG signaling pathway | ENSCAFG00000009036 | PLCB2 | Phospholipase C beta 2 | 1.801 | 0.0097 |
| Ras signaling pathway, PI3K/Akt signaling pathway, Rap1 signaling pathway, cytokine–cytokine receptor interaction | ENSCAFG00000019798 | CSF1 | Colony-stimulating factor 1 | 1.555 | 0.0027 |
| ENSCAFG00000018219 | CSF1R | Colony-stimulating factor 1 receptor | 1.58 | 0.034 | |
| Inflammatory bowel disease pathway, JAK/STAT signaling pathway, PI3K/Akt signaling pathway, cytokine–cytokine receptor interaction | ENSCAFG00000028978 | IL2RG | Cytokine receptor common subunit gamma | 1.627 | 0.046 |
| Inflammatory bowel disease pathway, AGE-RAGE signaling pathway, TGF-β signaling pathway, cytokine–cytokine receptor interaction | ENSCAFG00000010815 | Q19KA8 (TGFbeta2) | Transforming growth factor beta 2 | 1.128 | 0.035 |
| Ras signaling pathway, EGFR-TKI resistance, AGE-RAGE, NF-κB signaling pathway, TRP channels | ENSCAFG00000019989 | PLCG2 | Phospholipase C gamma 2 | 1.541 | 0.015 |
| Ras signaling pathway, EGFR-TKI resistance, PI3K/Akt signaling pathway, Rap1 signaling pathway, ROS–chemical carcinogenesis | ENSCAFG00000006370 | HGF | Hepatocyte growth factor | 1.854 | 0.0025 |
| Ras signaling pathway, EGFR-TKI resistance, JAK/STAT signaling pathway, PI3K/Akt signaling pathway, Rap1 signaling pathway | ENSCAFG00000002057 | PDGFRA | Platelet-derived growth factor receptor alpha | 1.472 | 0.003 |
| ENSCAFG00000018214 | PGFRB (PDGFRB) | Platelet-derived growth factor receptor beta | 1.753 | 0.0005 | |
| Ras signaling pathway, EGFR-TKI resistance, AGE-RAGE, TRP channels, Rap1 signaling pathway | ENSCAFG00000011326 | PRKCA | Protein kinase C alpha | 0.863 | 0.046 |
| Ras signaling pathway, EGFR-TKI resistance, AGE-RAGE, TRP channels, Rap1 signaling pathway, NF-κB signaling pathway | ENSCAFG00000009082 | PLCG1 | Phospholipase C gamma 1 | 0.861 | 0.046 |
| ENSCAFG00000017622 | PRKCB | Protein kinase C beta | 1.457 | 0.038 | |
| Inflammatory bowel disease pathway, AGE-RAGE signaling pathway, JAK/STAT signaling pathway, PI3K/Akt signaling pathway, cytokine–cytokine receptor interaction, EGFR-TKI resistance signaling pathway | ENSCAFG00000002733 | Q95LE4 (IL6) | Interleukin-6 | 4.433 | 0.0028 |
| Ras signaling pathway, EGFR-TKI resistance, AGE-RAGE, JAK/STAT signaling pathway, PI3K/Akt signaling pathway, Rap1 signaling pathway, ROS–chemical carcinogenesis, cGMP-PKG signaling pathway | ENSCAFG00000015806 | Q6PVW1 (AKT3) | Protein kinase B gamma-like protein | 1.727 | 0.005 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahoo, D.K.; Stewart, T.; Lindgreen, E.M.; Patel, B.; Patel, A.; Trivedi, J.N.; Parker, V.; Rudinsky, A.J.; Winston, J.A.; Bourgois-Mochel, A.; et al. Restorative Effects of Synbiotics on Colonic Ultrastructure and Oxidative Stress in Dogs with Chronic Enteropathy. Antioxidants 2025, 14, 727. https://doi.org/10.3390/antiox14060727
Sahoo DK, Stewart T, Lindgreen EM, Patel B, Patel A, Trivedi JN, Parker V, Rudinsky AJ, Winston JA, Bourgois-Mochel A, et al. Restorative Effects of Synbiotics on Colonic Ultrastructure and Oxidative Stress in Dogs with Chronic Enteropathy. Antioxidants. 2025; 14(6):727. https://doi.org/10.3390/antiox14060727
Chicago/Turabian StyleSahoo, Dipak Kumar, Tracey Stewart, Emily M. Lindgreen, Bhakti Patel, Ashish Patel, Jigneshkumar N. Trivedi, Valerie Parker, Adam J. Rudinsky, Jenessa A. Winston, Agnes Bourgois-Mochel, and et al. 2025. "Restorative Effects of Synbiotics on Colonic Ultrastructure and Oxidative Stress in Dogs with Chronic Enteropathy" Antioxidants 14, no. 6: 727. https://doi.org/10.3390/antiox14060727
APA StyleSahoo, D. K., Stewart, T., Lindgreen, E. M., Patel, B., Patel, A., Trivedi, J. N., Parker, V., Rudinsky, A. J., Winston, J. A., Bourgois-Mochel, A., Mochel, J. P., Allenspach, K., Heilmann, R. M., & Jergens, A. E. (2025). Restorative Effects of Synbiotics on Colonic Ultrastructure and Oxidative Stress in Dogs with Chronic Enteropathy. Antioxidants, 14(6), 727. https://doi.org/10.3390/antiox14060727