
A Defective Circulating Mitochondrial Bioenergetics Profile Reflects the Hepatic One and Outlines Genetic MASLD

 ,
,  and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Discovery Cohort
2.2. Fibroscan-MASLD Cohort
2.3. Unrelated Liver Disease Cohort
2.4. Sample Collection, Storage and Homogenization
2.5. Frozen Liver Tissues and PBMCs Respirometry: Seahorse Assay
2.6. Statistical Analysis
3. Results
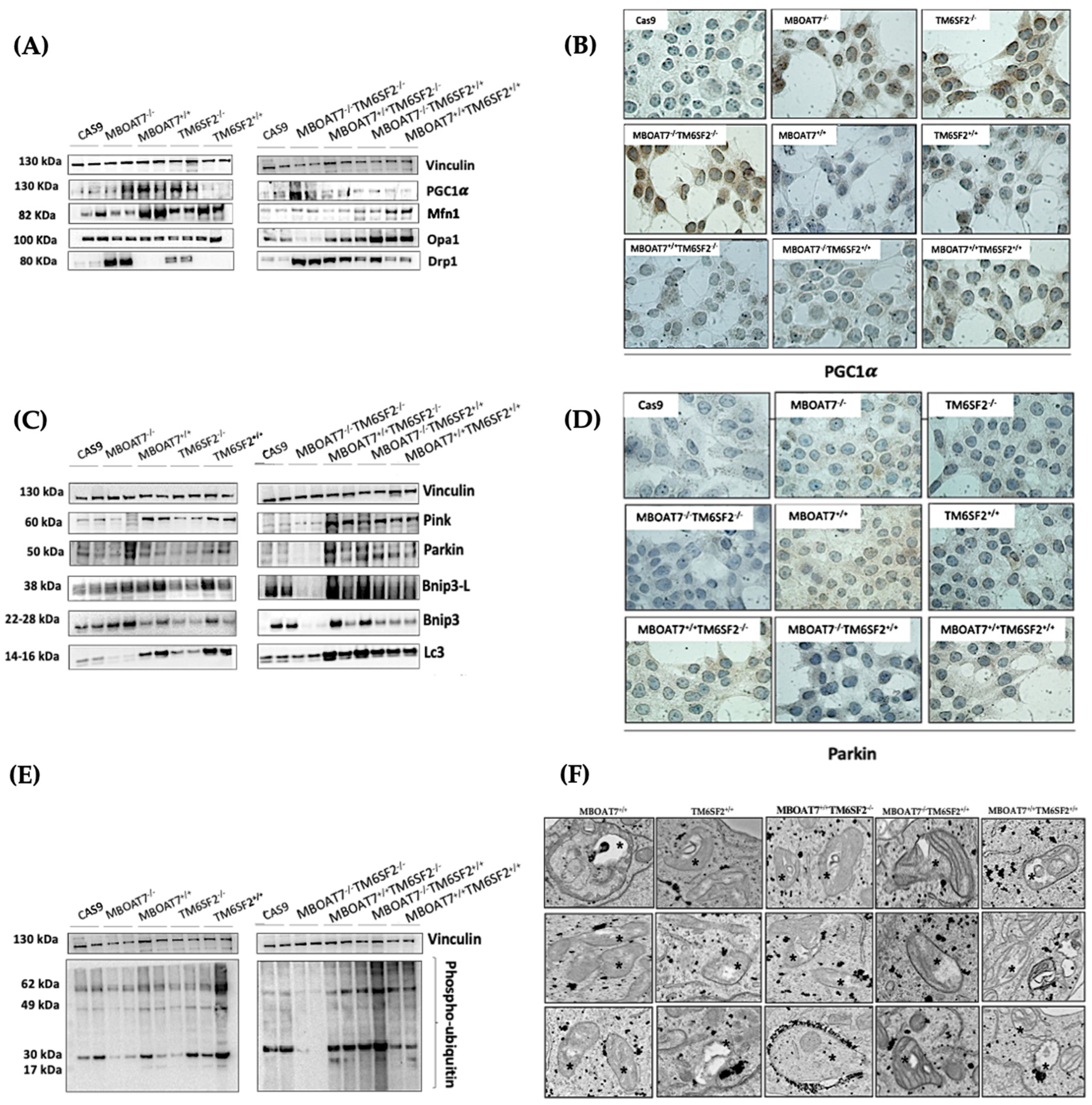
3.1. Restoring MBOAT7 and/or TM6SF2 WT Activities in KO Models Rebalances the Mitobiogenesis Process
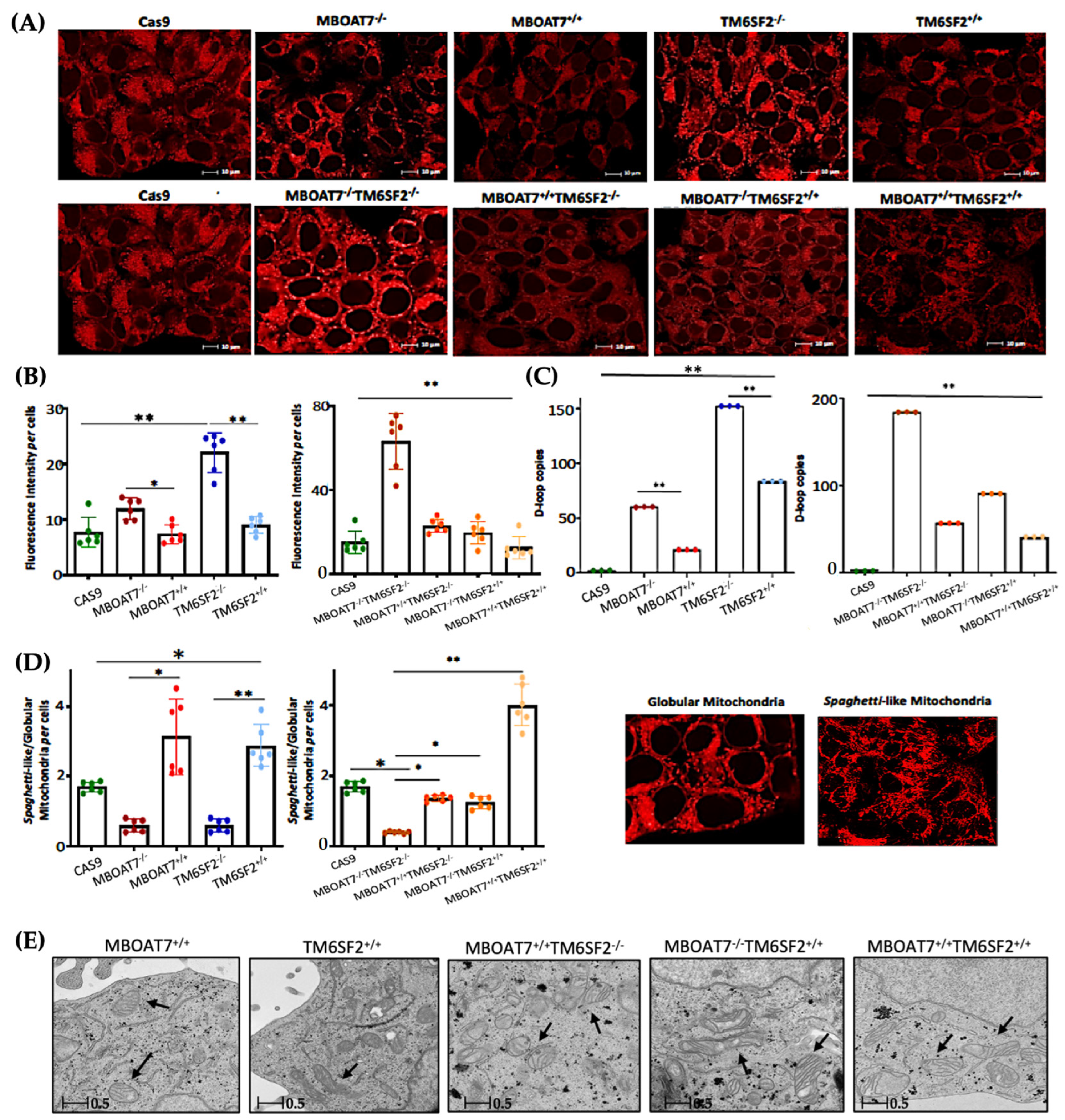
3.2. Mitochondrial Morphology Is Restored by the Expression of Wild-Type MBOAT7 and/or TM6SF2 Proteins in Knock-Out Models
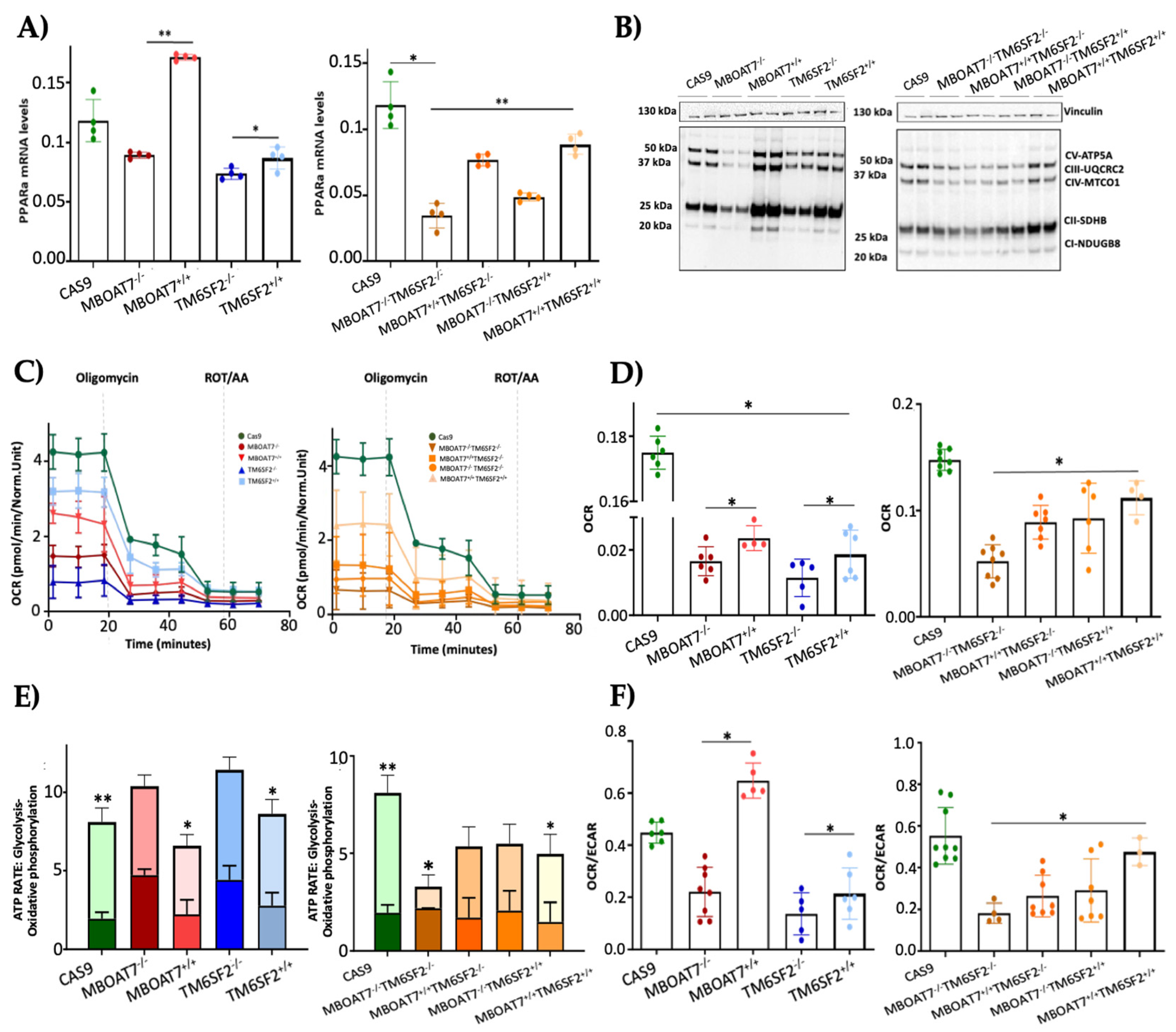
3.3. The Overexpression of WT MBOAT7 and/or TM6SF2 Proteins in KO Cells Rescues Mitochondrial Function
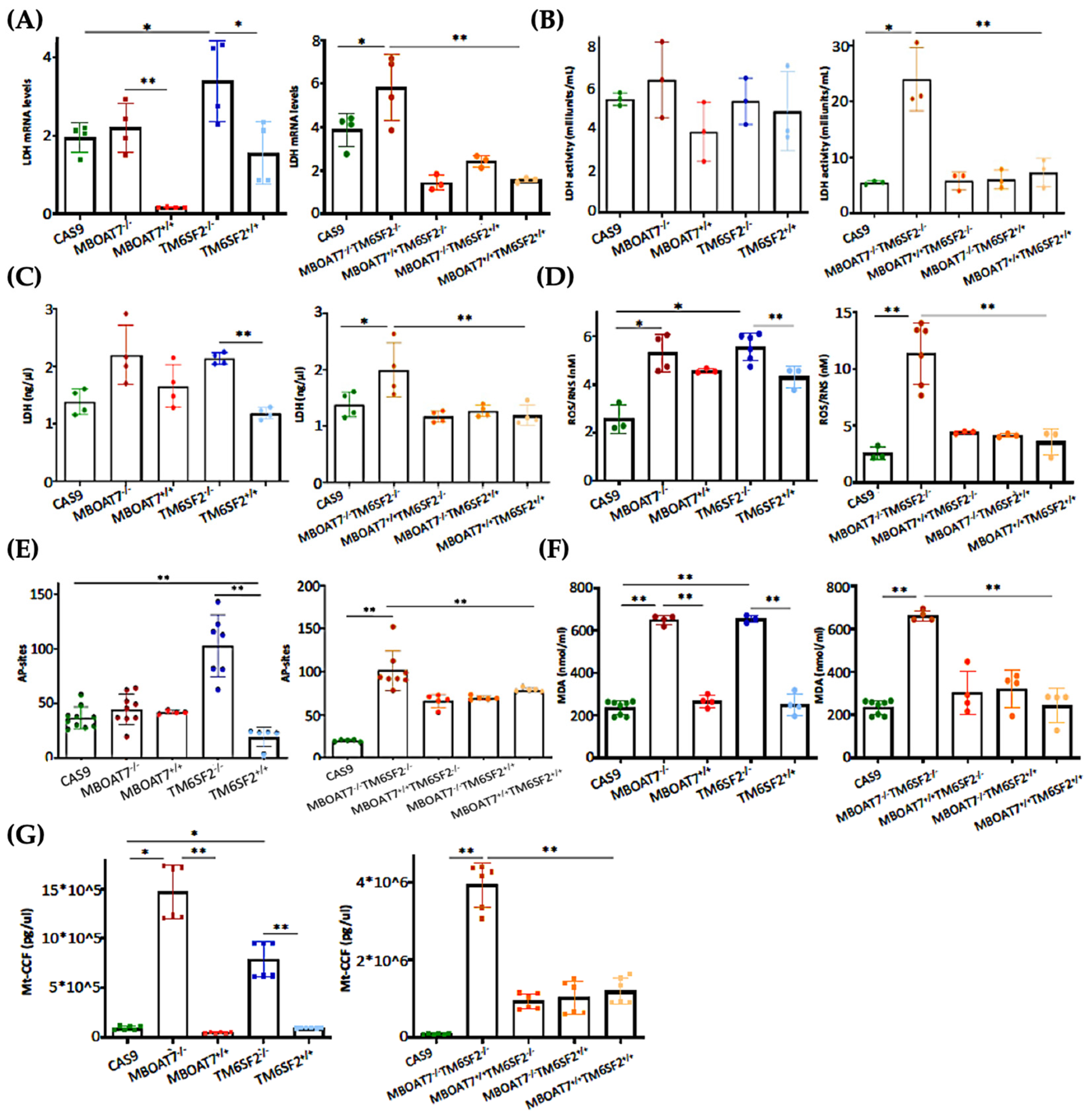
3.4. The Restoration of WT MBOAT7 and/or TM6SF2 Proteins in KO Cells Attenuates Hepatocellular Damage
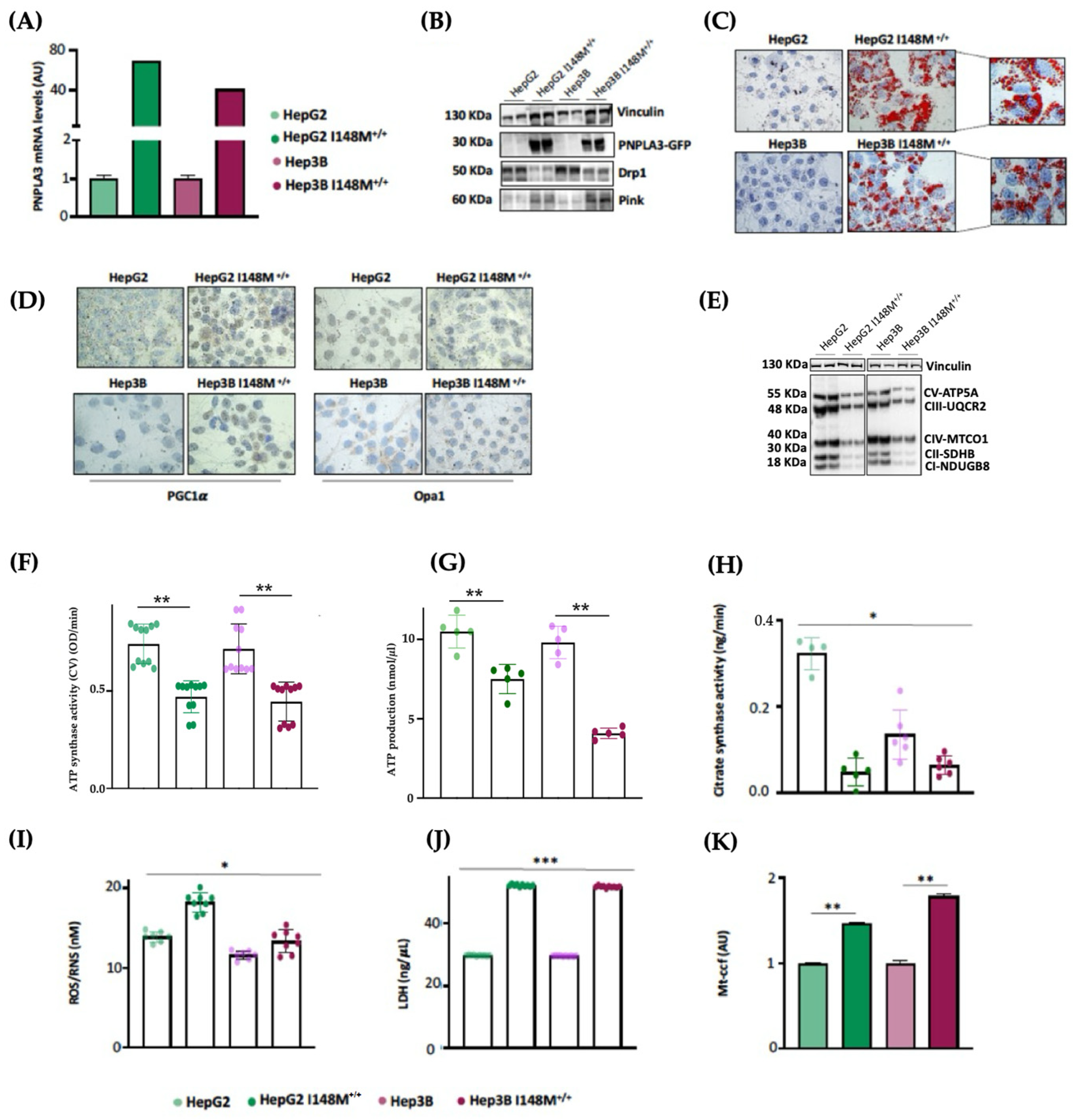
3.5. PNPLA3 I148M Overexpression in Hepatoma Cells Impairs Mitochondrial Function
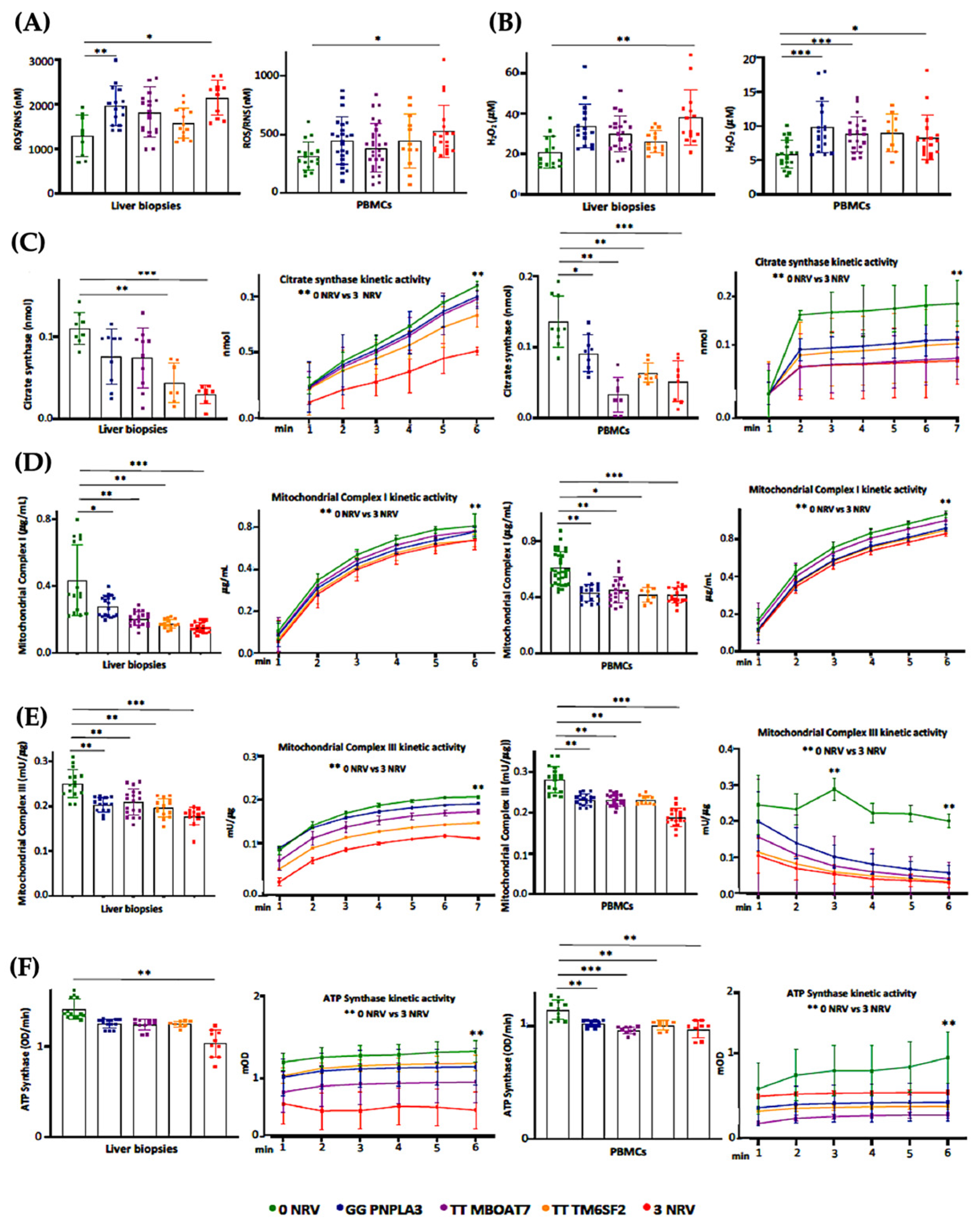
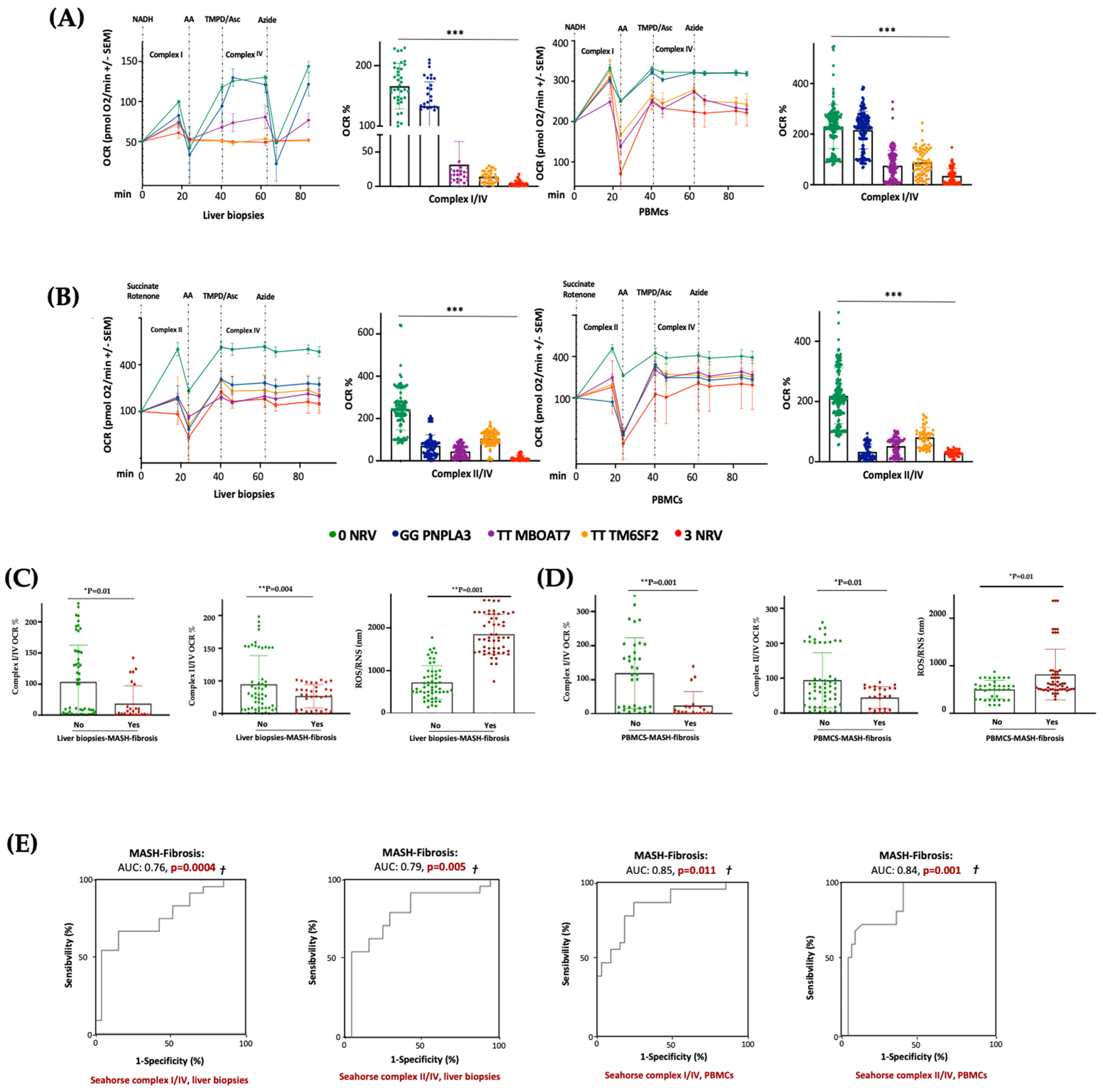
3.6. Hepatic and Circulating Mitochondrial Activity Is Impaired in MASLD Patients Carrying the Three At-Risk Variants
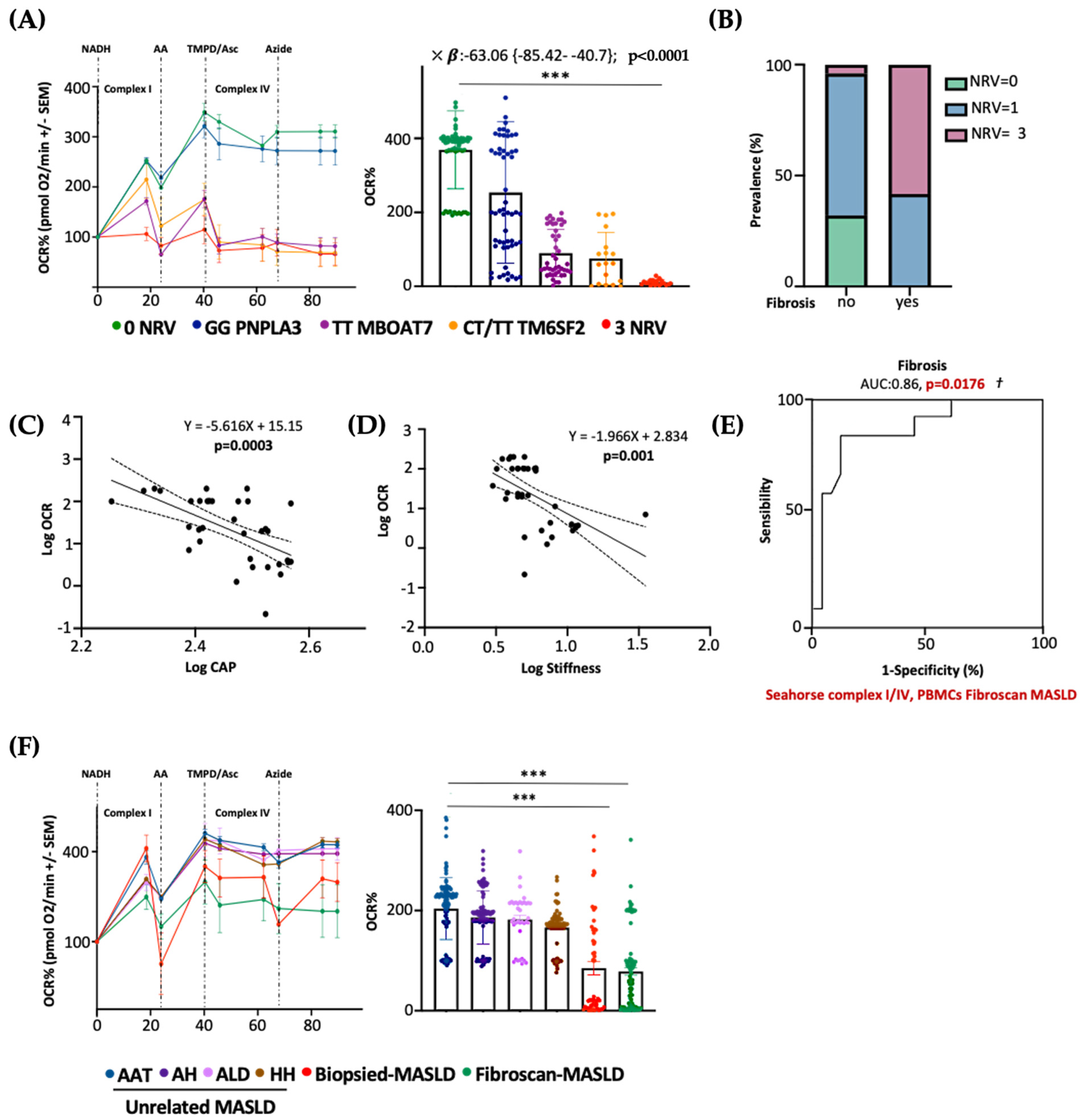
3.7. Serum Bioenergetic Profile Resembles the Hepatic One and Prognoses MASLD Severity in 3NRV Carriers
3.8. Serum Bioenergetics Predicts Fibrosis in Non-Invasively Assessed MASLD Patients Carrying 3NRV
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Abd El-Kader, S.M.; El-Den Ashmawy, E.M. Non-alcoholic fatty liver disease: The diagnosis and management. World J. Hepatol. 2015, 7, 846–858. [Google Scholar] [CrossRef]
- Protopapas, A.A.; Cholongitas, E.; Chrysavgis, L.; Tziomalos, K. Alcohol consumption in patients with nonalcoholic fatty liver disease: Yes, or no? Ann. Gastroenterol. 2021, 34, 476–486. [Google Scholar] [CrossRef]
- Marra, F.; Gastaldelli, A.; Svegliati Baroni, G.; Tell, G.; Tiribelli, C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol. Med. 2008, 14, 72–81. [Google Scholar] [CrossRef]
- Seen, T.K.; Sayed, M.; Bilal, M.; Reyes, J.V.; Bhandari, P.; Lourdusamy, V.; Al-Khazraji, A.; Syed, U.; Sattar, Y.; Bansal, R. Clinical indicators for progression of nonalcoholic steatohepatitis to cirrhosis. World J. Gastroenterol. 2021, 27, 3238–3248. [Google Scholar] [CrossRef]
- Cholankeril, G.; Patel, R.; Khurana, S.; Satapathy, S.K. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Current knowledge and implications for management. World J. Hepatol. 2017, 9, 533–543. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Blachier, M.; Leleu, H.; Peck-Radosavljevic, M.; Valla, D.C.; Roudot-Thoraval, F. The burden of liver disease in Europe: A review of available epidemiological data. J. Hepatol. 2013, 58, 593–608. [Google Scholar] [CrossRef]
- Shum, M.; Ngo, J.; Shirihai, O.S.; Liesa, M. Mitochondrial oxidative function in NAFLD: Friend or foe? Mol. Metab. 2021, 50, 101134. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef]
- Montemurro, C.; Vadrevu, S.; Gurlo, T.; Butler, A.E.; Vongbunyong, K.E.; Petcherski, A.; Shirihai, O.S.; Satin, L.S.; Braas, D.; Butler, P.C.; et al. Cell cycle-related metabolism and mitochondrial dynamics in a replication-competent pancreatic beta-cell line. Cell Cycle 2017, 16, 2086–2099. [Google Scholar] [CrossRef]
- Degli Esposti, D.; Hamelin, J.; Bosselut, N.; Saffroy, R.; Sebagh, M.; Pommier, A.; Martel, C.; Lemoine, A. Mitochondrial roles and cytoprotection in chronic liver injury. Biochem. Res. Int. 2012, 2012, 387626. [Google Scholar] [CrossRef]
- Piccinin, E.; Villani, G.; Moschetta, A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: The role of PGC1 coactivators. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 160–174. [Google Scholar] [CrossRef]
- Longo, M.; Meroni, M.; Paolini, E.; Macchi, C.; Dongiovanni, P. Mitochondrial dynamics and nonalcoholic fatty liver disease (NAFLD): New perspectives for a fairy-tale ending? Metabolism 2021, 117, 154708. [Google Scholar] [CrossRef]
- Paolini, E.; Longo, M.; Corsini, A.; Dongiovanni, P. The Non-Invasive Assessment of Circulating D-Loop and mt-ccf Levels Opens an Intriguing Spyhole into Novel Approaches for the Tricky Diagnosis of NASH. Int. J. Mol. Sci. 2023, 24, 2331. [Google Scholar] [CrossRef]
- Radosavljevic, T.; Brankovic, M.; Samardzic, J.; Djuretić, J.; Vukicevic, D.; Vucevic, D.; Jakovljevic, V. Altered Mitochondrial Function in MASLD: Key Features and Promising Therapeutic Approaches. Antioxidants 2024, 13, 906. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bedossa, P.; Guy, C.D.; Schattenberg, J.M.; Loomba, R.; Taub, R.; Labriola, D.; Moussa, S.E.; Neff, G.W.; Rinella, M.E.; et al. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. N. Engl. J. Med. 2024, 390, 497–509. [Google Scholar] [CrossRef]
- Roeb, E.; Geier, A. Nonalcoholic steatohepatitis (NASH)—Current treatment recommendations and future developments. Z. Gastroenterol. 2019, 57, 508–517. [Google Scholar] [CrossRef]
- Nalbantoglu, I.L.; Brunt, E.M. Role of liver biopsy in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 9026–9037. [Google Scholar]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Ajaz, S.; McPhail, M.J.; Gnudi, L.; Trovato, F.M.; Mujib, S.; Napoli, S.; Carey, I.; Agarwal, K. Mitochondrial dysfunction as a mechanistic biomarker in patients with non-alcoholic fatty liver disease (NAFLD). Mitochondrion 2021, 57, 119–130. [Google Scholar] [CrossRef]
- Garrafa, E.; Segala, A.; Vezzoli, M.; Bottani, E.; Zanini, B.; Vetturi, A.; Bracale, R.; Ricci, C.; Valerio, A. Mitochondrial Dysfunction in Peripheral Blood Mononuclear Cells as Novel Diagnostic Tools for Non-Alcoholic Fatty Liver Disease: Visualizing Relationships with Known and Potential Disease Biomarkers. Diagnostics 2023, 13, 2363. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Tria, G.; Dongiovanni, P. Genetics Is of the Essence to Face NAFLD. Biomedicines 2021, 9, 359. [Google Scholar] [CrossRef]
- Longo, M.; Meroni, M.; Paolini, E.; Erconi, V.; Carli, F.; Fortunato, F.; Ronchi, D.; Piciotti, R.; Sabatini, S.; Macchi, C.; et al. TM6SF2/PNPLA3/MBOAT7 Loss-of-Function Genetic Variants Impact on NAFLD Development and Progression Both in Patients and in In Vitro Models. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 759–788. [Google Scholar] [CrossRef]
- Meroni, M.; Dongiovanni, P.; Longo, M.; Carli, F.; Baselli, G.; Rametta, R.; Pelusi, S.; Badiali, S.; Maggioni, M.; Gaggini, M.; et al. Mboat7 down-regulation by hyper-insulinemia induces fat accumulation in hepatocytes. EBioMedicine 2020, 52, 102658. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. 2013, 19, 6969–6978. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Paolini, E.; Tria, G.; Ripolone, M.; Napoli, L.; Moggio, M.; Fracanzani, A.L.; Dongiovanni, P. Expanding the phenotypic spectrum of non-alcoholic fatty liver disease and hypertriglyceridemia. Front. Nutr. 2022, 9, 967899. [Google Scholar] [CrossRef]
- Min, H.K.; Sookoian, S.; Pirola, C.J.; Cheng, J.; Mirshahi, F.; Sanyal, A.J. Metabolic profiling reveals that PNPLA3 induces widespread effects on metabolism beyond triacylglycerol remodeling in Huh-7 hepatoma cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G66–G76. [Google Scholar] [CrossRef]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e6. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e1. [Google Scholar] [CrossRef]
- Ruhanen, H.; Nidhina Haridas, P.A.; Eskelinen, E.L.; Eriksson, O.; Olkkonen, V.M.; Käkelä, R. Depletion of TM6SF2 disturbs membrane lipid composition and dynamics in HuH7 hepatoma cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 676–685. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Zhou, Y.; Nidhina Haridas, P.A.; Dwivedi, O.P.; Hyötyläinen, T.; Ali, A.; Juuti, A.; Leivonen, M.; Tukiainen, T.; Ahonen, L.; et al. Impaired hepatic lipid synthesis from polyunsaturated fatty acids in TM6SF2 E167K variant carriers with NAFLD. J. Hepatol. 2017, 67, 128–136. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, E.A.; Yang, R.; Yerges-Armstrong, L.M.; Sreenivasan, U.; McFarland, R.; Leitch, C.C.; Wilson, M.H.; Narina, S.; Gorden, A.; Ryan, K.A.; et al. TM6SF2 rs58542926 impacts lipid processing in liver and small intestine. Hepatology 2017, 65, 1526–1542. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Castera, L.; Forns, X.; Alberti, A. Non-invasive evaluation of liver fibrosis using transient elastography. J. Hepatol. 2008, 48, 835–847. [Google Scholar] [CrossRef]
- Boursier, J.; Zarski, J.P.; de Ledinghen, V.; Rousselet, M.C.; Sturm, N.; Lebail, B.; Fouchard-Hubert, I.; Gallois, Y.; Oberti, F.; Bertrais, S.; et al. Determination of reliability criteria for liver stiffness evaluation by transient elastography. Hepatology 2013, 57, 1182–1191. [Google Scholar] [CrossRef]
- Walkon, L.L.; Strubbe-Rivera, J.O.; Bazil, J.N. Calcium Overload and Mitochondrial Metabolism. Biomolecules 2022, 12, 1891. [Google Scholar] [CrossRef]
- Mu, C.; Wang, S.; Wang, Z.; Tan, J.; Yin, H.; Wang, Y.; Dai, Z.; Ding, D.; Yang, F. Mechanisms and therapeutic targets of mitochondria in the progression of metabolic dysfunction-associated steatotic liver disease. Ann. Hepatol. 2024, 30, 101774. [Google Scholar] [CrossRef]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef]
- Sharpe, M.C.; Pyles, K.D.; Hallcox, T.; Kamm, D.R.; Piechowski, M.; Fisk, B.; Albert, C.J.; Carpenter, D.H.; Ulmasov, B.; Ford, D.A.; et al. Enhancing Hepatic MBOAT7 Expression in Mice With Nonalcoholic Steatohepatitis. Gastro Hep Adv. 2023, 2, 558–572. [Google Scholar] [CrossRef]
- Pant, A.; Chen, Y.; Kuppa, A.; Du, X.; Halligan, B.D.; Speliotes, E.K. Perturbation of TM6SF2 Expression Alters Lipid Metabolism in a Human Liver Cell Line. Int. J. Mol. Sci. 2021, 22, 9758. [Google Scholar] [CrossRef]
- Longo, M.; Paolini, E.; Meroni, M.; Ripolone, M.; Napoli, L.; Gentile, F.; Cespiati, A.; Maggioni, M.; Alisi, A.; Miele, L.; et al. Artificial intelligence as a ploy to delve into the intricate relationship between genetics and mitochondria in MASLD patients. bioRxiv 2024. [Google Scholar] [CrossRef]
- Gong, F.; Gao, L.; Ding, T. IDH2 protects against nonalcoholic steatohepatitis by alleviating dyslipidemia regulated by oxidative stress. Biochem. Biophys. Res. Commun. 2019, 514, 593–600. [Google Scholar] [CrossRef]
- Galloway, C.A.; Lee, H.; Brookes, P.S.; Yoon, Y. Decreasing mitochondrial fission alleviates hepatic steatosis in a murine model of nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G632–G641. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, L.; Hu, W.; Zheng, Q.; Xiang, W. Mitochondrial dysfunction during in vitro hepatocyte steatosis is reversed by omega-3 fatty acid-induced up-regulation of mitofusin 2. Metabolism 2011, 60, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, X.; Nie, J.; Zhang, J.; Kimball, S.R.; Zhang, H.; Zhang, W.J.; Jefferson, L.S.; Cheng, Z.; Ji, Q.; et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology 2015, 61, 486–496. [Google Scholar] [CrossRef]
- Boland, M.L.; Oldham, S.; Boland, B.B.; Will, S.; Lapointe, J.M.; Guionaud, S.; Rhodes, C.J.; Trevaskis, J.L. Nonalcoholic steatohepatitis severity is defined by a failure in compensatory antioxidant capacity in the setting of mitochondrial dysfunction. World J. Gastroenterol. 2018, 24, 1748–1765. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, T.E.; Chen, M.; Xu, D.; Zhu, Y.; Hu, B.Y.; Lin, Z.F.; Pan, J.J.; Wang, X.; Wu, C.; et al. MFN1-dependent alteration of mitochondrial dynamics drives hepatocellular carcinoma metastasis by glucose metabolic reprogramming. Br. J. Cancer 2020, 122, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Le, T.H.; Caldwell, S.H.; Redick, J.A.; Sheppard, B.L.; Davis, C.A.; Arseneau, K.O.; Iezzoni, J.C.; Hespenheide, E.E.; Al-Osaimi, A.; Peterson, T.C. The zonal distribution of megamitochondria with crystalline inclusions in nonalcoholic steatohepatitis. Hepatology 2004, 39, 1423–1429. [Google Scholar] [CrossRef]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef]
- García-Ruiz, I.; Rodríguez-Juan, C.; Díaz-Sanjuan, T.; del Hoyo, P.; Colina, F.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. Uric acid and anti-TNF antibody improve mitochondrial dysfunction in ob/ob mice. Hepatology 2006, 44, 581–591. [Google Scholar] [CrossRef]
- Wang, Y.; Hong, S.; Hudson, H.; Kory, N.; Kinch, L.N.; Kozlitina, J.; Cohen, J.C.; Hobbs, H.H. PNPLA3(148M) is a gain-of-function mutation that promotes hepatic steatosis by inhibiting ATGL-mediated triglyceride hydrolysis. J. Hepatol. 2025, 82, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Hubens, W.H.G.; Vallbona-Garcia, A.; de Coo, I.F.M.; van Tienen, F.H.J.; Webers, C.A.B.; Smeets, H.J.M.; Gorgels, T.G.M.F. Blood biomarkers for assessment of mitochondrial dysfunction: An expert review. Mitochondrion 2022, 62, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, R.; Nakajima, T.; Yoshimura, A.; Kawahara, Y.; Orito, C.; Yamane, M.; Handa, H.; Takada, S.; Furihata, T.; Fukushima, A.; et al. Author Correction: Enhanced mitochondrial oxidative metabolism in peripheral blood mononuclear cells is associated with fatty liver in obese young adults. Sci. Rep. 2024, 14, 6786. [Google Scholar] [CrossRef] [PubMed]
- Lindén, D.; Ahnmark, A.; Pingitore, P.; Ciociola, E.; Ahlstedt, I.; Andréasson, A.C.; Sasidharan, K.; Madeyski-Bengtson, K.; Zurek, M.; Mancina, R.M.; et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol. Metab. 2019, 22, 49–61. [Google Scholar] [CrossRef]
- BasuRay, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017, 66, 1111–1124. [Google Scholar] [CrossRef]
- Weber, M.; Mera, P.; Casas, J.; Salvador, J.; Rodríguez, A.; Alonso, S.; Sebastián, D.; Soler-Vázquez, M.C.; Montironi, C.; Recalde, S.; et al. Liver CPT1A gene therapy reduces diet-induced hepatic steatosis in mice and highlights potential lipid biomarkers for human NAFLD. FASEB J. 2020, 34, 11816–11837. [Google Scholar] [CrossRef]
- Vilà, L.; Elias, I.; Roca, C.; Ribera, A.; Ferré, T.; Casellas, A.; Lage, R.; Franckhauser, S.; Bosch, F. AAV8-mediated Sirt1 gene transfer to the liver prevents high carbohydrate diet-induced nonalcoholic fatty liver disease. Mol. Ther. Methods Clin. Dev. 2014, 1, 14039. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paolini, E.; Longo, M.; Meroni, M.; Podini, P.; Maggioni, M.; Quattrini, A.; Fracanzani, A.L.; Dongiovanni, P. A Defective Circulating Mitochondrial Bioenergetics Profile Reflects the Hepatic One and Outlines Genetic MASLD. Antioxidants 2025, 14, 618. https://doi.org/10.3390/antiox14060618
Paolini E, Longo M, Meroni M, Podini P, Maggioni M, Quattrini A, Fracanzani AL, Dongiovanni P. A Defective Circulating Mitochondrial Bioenergetics Profile Reflects the Hepatic One and Outlines Genetic MASLD. Antioxidants. 2025; 14(6):618. https://doi.org/10.3390/antiox14060618
Chicago/Turabian StylePaolini, Erika, Miriam Longo, Marica Meroni, Paola Podini, Marco Maggioni, Angelo Quattrini, Anna Ludovica Fracanzani, and Paola Dongiovanni. 2025. "A Defective Circulating Mitochondrial Bioenergetics Profile Reflects the Hepatic One and Outlines Genetic MASLD" Antioxidants 14, no. 6: 618. https://doi.org/10.3390/antiox14060618
APA StylePaolini, E., Longo, M., Meroni, M., Podini, P., Maggioni, M., Quattrini, A., Fracanzani, A. L., & Dongiovanni, P. (2025). A Defective Circulating Mitochondrial Bioenergetics Profile Reflects the Hepatic One and Outlines Genetic MASLD. Antioxidants, 14(6), 618. https://doi.org/10.3390/antiox14060618