

Environmental Toxins and Oxidative Stress: The Link to Cardiovascular Diseases

 ,
, 
Abstract
1. Introduction
2. Methodology
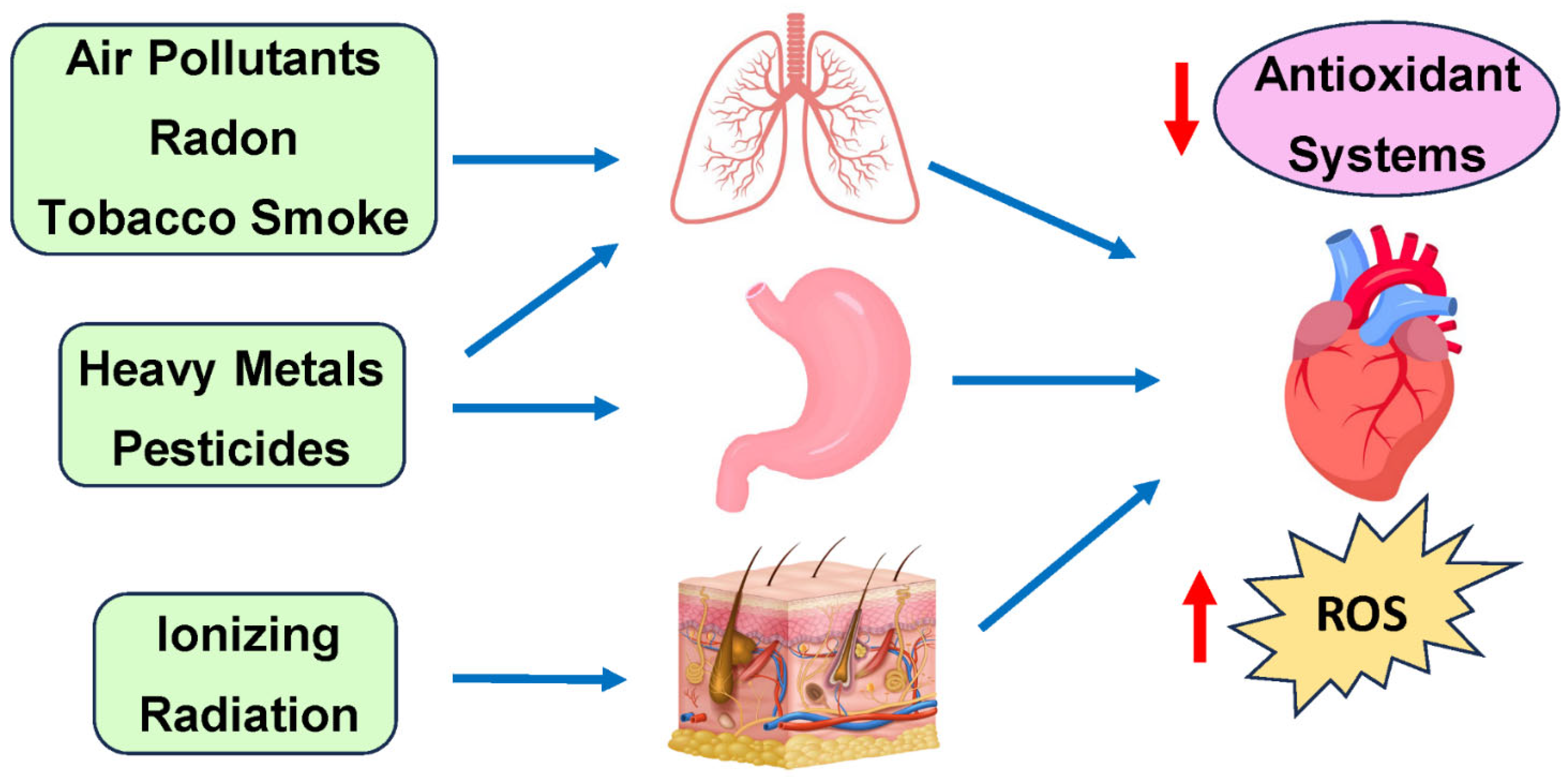
3. Common Environmental Toxins
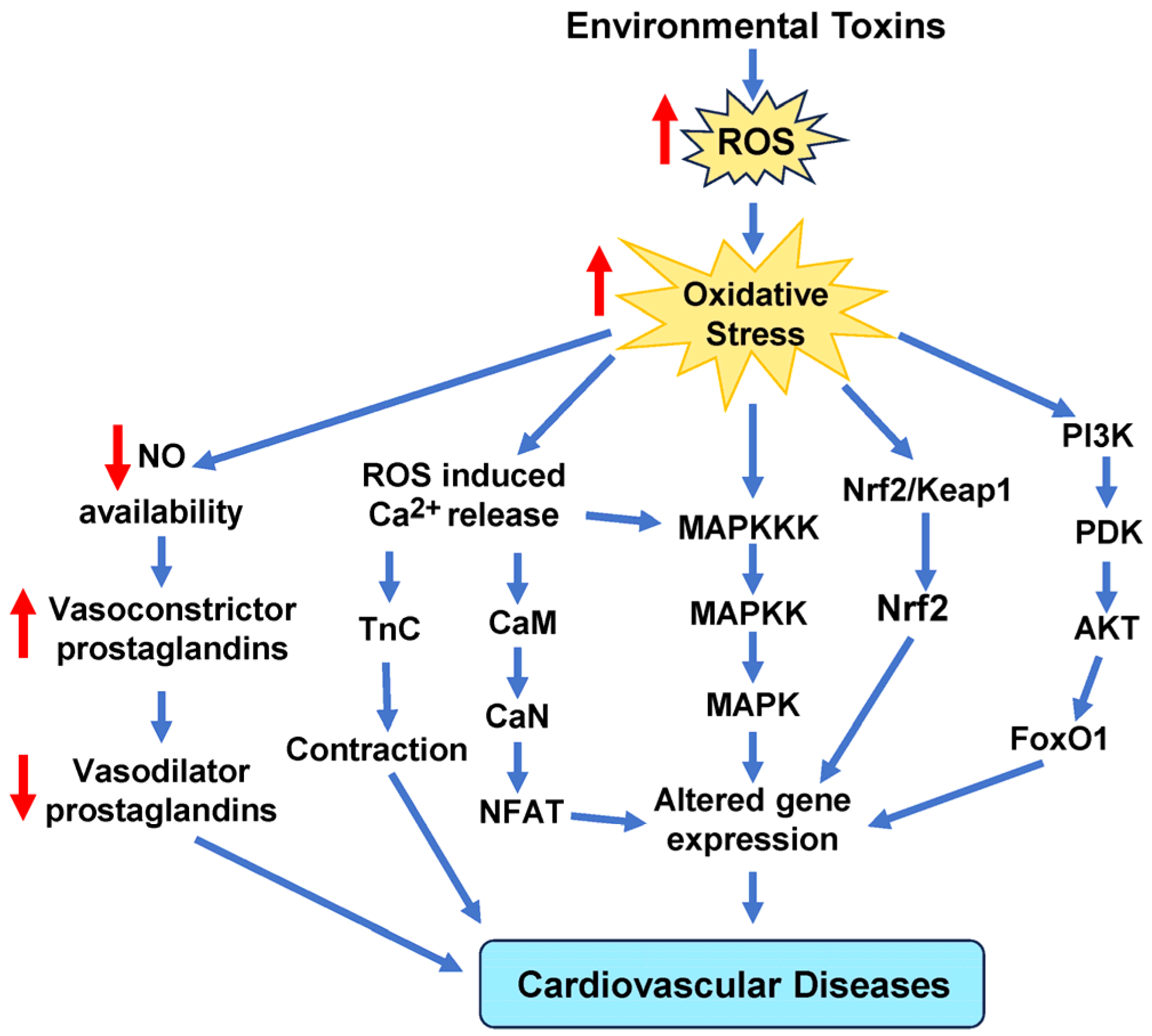
4. Oxidative and Antioxidative Stress
5. Air Pollutants—Particulate Matter (PM)
6. Common Heavy Metal Pollutants
6.1. Lead
6.2. Cadmium
7. Household Toxins
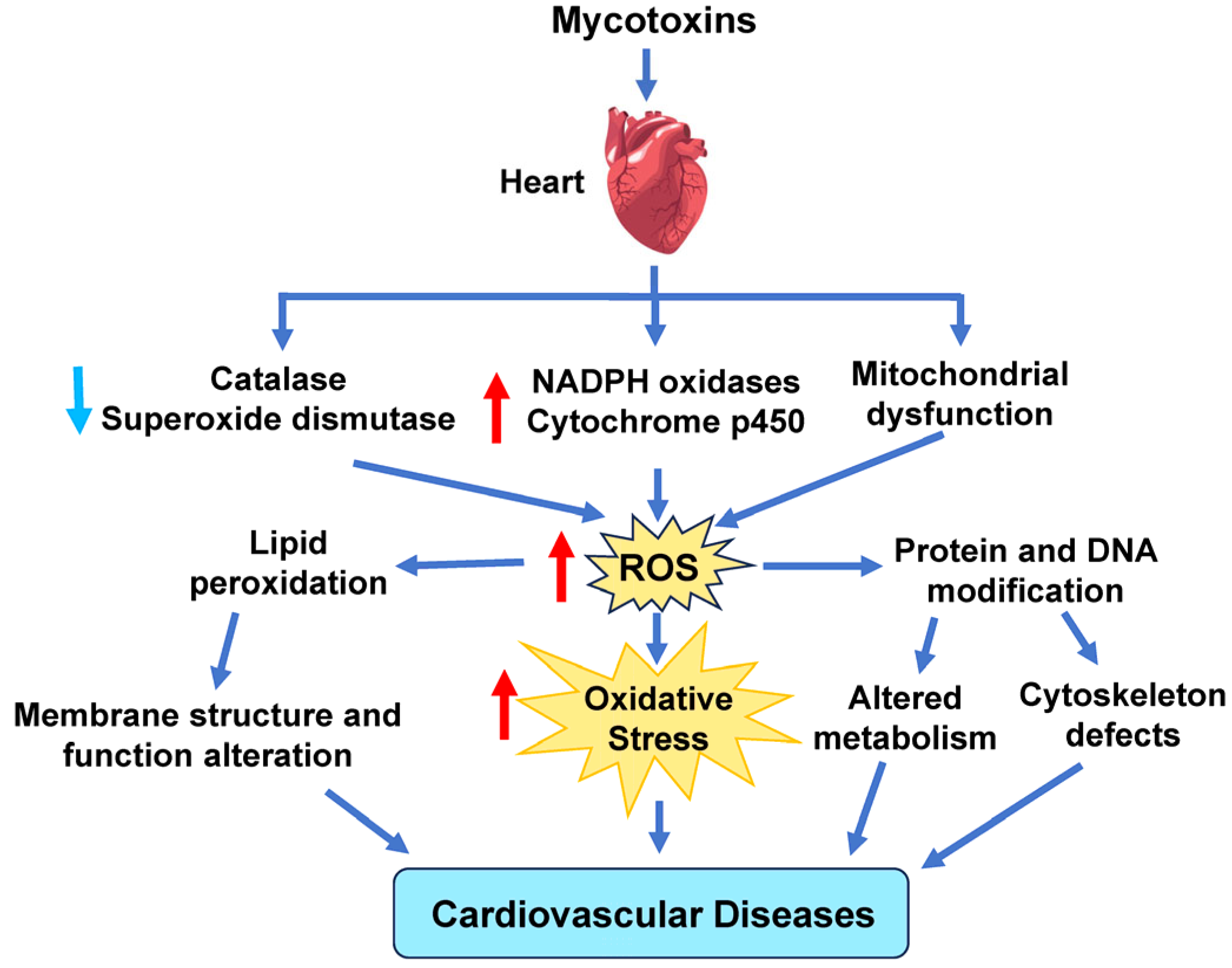
8. Mycotoxins
9. Pharmaceutical Products
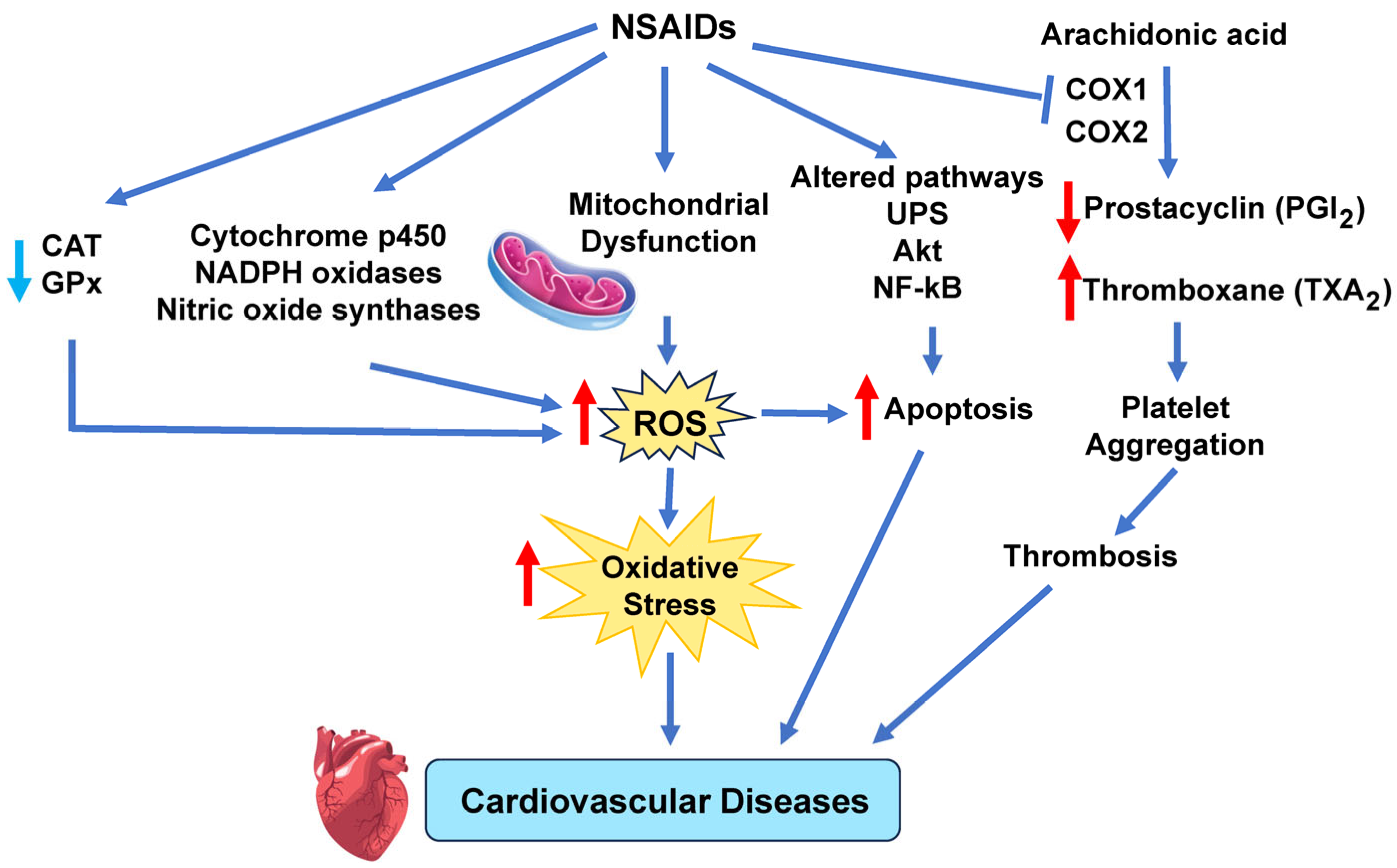
9.1. Ibuprofen
9.2. Mechanism and Signaling Pathways by Which NSAIDs Increase Cardiovascular Disease Risk
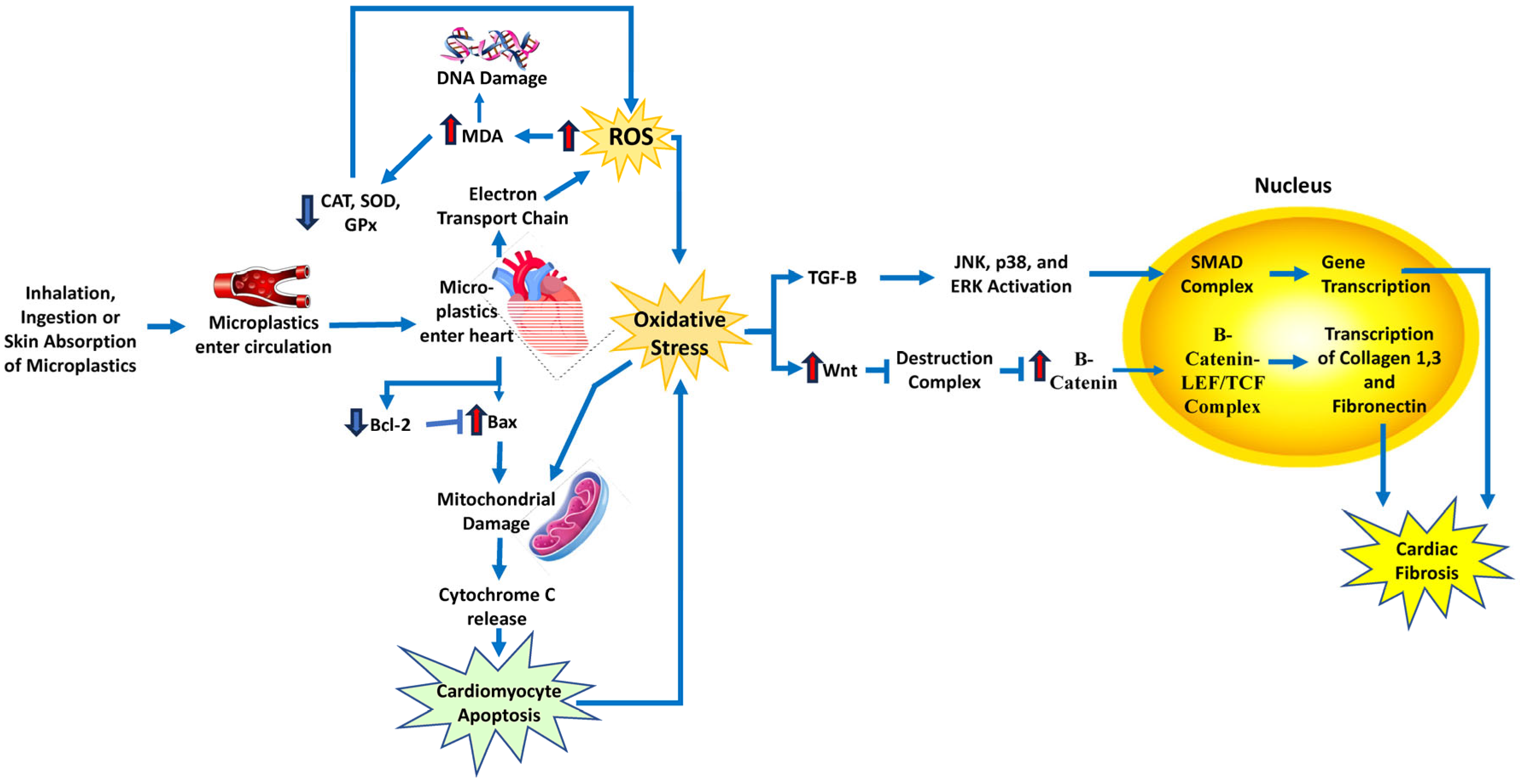
10. Microplastics
11. Clinical Implications of Oxidative Stress
12. Conclusions
13. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BPA | Bisphenol A |
| CAT | Catalase |
| CD | Cadmium |
| CHD | Coronary heart disease |
| CVD | Cardiovascular disease |
| EDCs | Endocrine-disrupting chemicals |
| ETC | Mitochondrial electron transport chain |
| GPx | Glutathione peroxidase |
| GSH | Glutathione |
| Gst | Glutathione S-transferase |
| NO | Nitric oxide |
| NOX | NADPH oxidase |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| PCBs | Polychlorinated biphenyls |
| PM | Particulate matter |
| PFCs | Perfluorinated compounds |
| PFOS | Perfluorinated compound perfluorooctane sulfonate |
| SOD | Superoxide dismutase |
| TrxR | Thioredoxin reductase |
| XO | Xanthine oxidase |
References
- Burroughs Peña, M.S.; Rollins, A. Environmental Exposures and Cardiovascular Disease: A Challenge for Health and Development in Low-and Middle-Income Countries. Cardiol. Clin. 2017, 35, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Naidu, R.; Biswas, B.; Willett, I.R.; Cribb, J.; Kumar Singh, B.; Paul Nathanail, C.; Coulon, F.; Semple, K.T.; Jones, K.C.; Barclay, A.; et al. Chemical pollution: A growing peril and potential catastrophic risk to humanity. Environ. Int. 2021, 156, 106616. [Google Scholar] [CrossRef]
- Landrigan, P.J.; Fuller, R.; Acosta, N.J.R.; Adeyi, O.; Arnold, R.; Basu, N.; Baldé, A.B.; Bertollini, R.; Bose-O’Reilly, S.; Boufford, J.I.; et al. The Lancet Commission on pollution and health. Lancet 2018, 391, 462–512. [Google Scholar] [CrossRef] [PubMed]
- Cosselman, K.E.; Navas-Acien, A.; Kaufman, J.D. Environmental factors in cardiovascular disease. Nat. Rev. Cardiol. 2015, 12, 627–642. [Google Scholar] [CrossRef]
- Aggarwal, V.; Mehndiratta, M.M.; Wasay, M.; Garg, D. Environmental Toxins and Brain: Life on Earth is in Danger. Ann. Indian Acad. Neurol. 2022, 25, S15–S21. [Google Scholar] [CrossRef]
- Yang, A.-M.; Lo, K.; Zheng, T.-Z.; Yang, J.-L.; Bai, Y.-N.; Feng, Y.-Q.; Cheng, N.; Liu, S.-M.; Cui, Y. Environmental heavy metals and cardiovascular diseases: Status and future direction. Chronic Dis. Transl. Med. 2020, 6, 251–259. [Google Scholar] [CrossRef]
- Alissa, E.M.; Ferns, G.A. Heavy Metal Poisoning and Cardiovascular Disease. J. Toxicol. 2011, 2011, 870125. [Google Scholar] [CrossRef]
- Lind, L.; Araujo, J.A.; Barchowsky, A.; Belcher, S.; Berridge, B.R.; Chiamvimonvat, N.; Chiu, W.A.; Cogliano, V.J.; Elmore, S.; Farraj, A.K.; et al. Key Characteristics of Cardiovascular Toxicants. Environ. Health Perspect. 2021, 129, 095001. [Google Scholar] [CrossRef] [PubMed]
- Scimeca, M.; Palumbo, V.; Giacobbi, E.; Servadei, F.; Casciardi, S.; Cornella, E.; Cerbara, F.; Rotondaro, G.; Seghetti, C.; Scioli, M.P.; et al. Impact of the environmental pollution on cardiovascular diseases: From epidemiological to molecular evidence. Heliyon 2024, 10, e38047. [Google Scholar] [CrossRef]
- Niu, X.; Yu, J.; Sun, J.; Zhang, X.; Zhou, L.; Liu, X.; He, K.; Peng, Z.; Niu, X.; Xu, H.; et al. New mechanisms of PM2.5 induced atherosclerosis: Source dependent toxicity and pathogenesis. Environ. Res. 2025, 266, 120535. [Google Scholar] [CrossRef]
- Santibanez-Andrade, M.; Quezada-Maldonado, E.M.; Rivera-Pineda, A.; Chirino, Y.I.; Garcia-Cuellar, C.M.; Sanchez-Perez, Y. The Road to Malignant Cell Transformation after Particulate Matter Exposure: From Oxidative Stress to Genotoxicity. Int. J. Mol. Sci. 2023, 24, 1782. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.Y.; Kim, G.D. Particulate Matter-Induced Emerging Health Effects Associated with Oxidative Stress and Inflammation. Antioxidants 2024, 13, 1256. [Google Scholar] [CrossRef]
- Hatzis, C.; Godleski, J.J.; Gonzalez-Flecha, B.; Wolfson, J.M.; Koutrakis, P. Ambient particulate matter exhibits direct inhibitory effects on oxidative stress enzymes. Environ. Sci. Technol. 2006, 40, 2805–2811. [Google Scholar] [CrossRef] [PubMed]
- Galuszka-Bulaga, A.; Tkacz, K.; Weglarczyk, K.; Siedlar, M.; Baran, J. Air pollution induces pyroptosis of human monocytes through activation of inflammasomes and Caspase-3-dependent pathways. J. Inflamm. 2023, 20, 26. [Google Scholar] [CrossRef]
- Bhol, N.K.; Bhanjadeo, M.M.; Singh, A.K.; Dash, U.C.; Ojha, R.R.; Majhi, S.; Duttaroy, A.K.; Jena, A.B. The interplay between cytokines, inflammation, and antioxidants: Mechanistic insights and therapeutic potentials of various antioxidants and anti-cytokine compounds. Biomed. Pharmacother. 2024, 178, 117177. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Jiang, S.; Zeng, X.; Zhang, J.; Pan, K.; Song, L.; Zhou, J.; Kan, H.; Sun, Q.; Zhao, J.; et al. Fine particulate matter-induced cardiovascular injury is associated with NLRP3 inflammasome activation in Apo E−/− mice. Ecotoxicol. Environ. Saf. 2019, 174, 92–99. [Google Scholar] [CrossRef]
- Ying, Z.; Xie, X.; Bai, Y.; Chen, M.; Wang, X.; Zhang, X.; Morishita, M.; Sun, Q.; Rajagopalan, S. Exposure to concentrated ambient particulate matter induces reversible increase of heart weight in spontaneously hypertensive rats. Part. Fibre Toxicol. 2015, 12, 15. [Google Scholar] [CrossRef]
- Su, X.; Tian, J.; Li, B.; Zhou, L.; Kang, H.; Pei, Z.; Zhang, M.; Li, C.; Wu, M.; Wang, Q.; et al. Ambient PM2.5 caused cardiac dysfunction through FoxO1-targeted cardiac hypertrophy and macrophage-activated fibrosis in mice. Chemosphere 2020, 247, 125881. [Google Scholar] [CrossRef]
- Qin, G.; Xia, J.; Zhang, Y.; Guo, L.; Chen, R.; Sang, N. Ambient fine particulate matter exposure induces reversible cardiac dysfunction and fibrosis in juvenile and older female mice. Part. Fibre Toxicol. 2018, 15, 27. [Google Scholar] [CrossRef]
- Chen, J.J.; Ma, W.M.; Yuan, J.L.; Cui, L.Q. PM2.5 exposure aggravates left heart failure induced pulmonary hypertension. Acta Cardiol. 2019, 74, 238–244. [Google Scholar] [CrossRef]
- Zhang, Y.; Ji, X.; Ku, T.; Li, B.; Li, G.; Sang, N. Ambient fine particulate matter exposure induces cardiac functional injury and metabolite alterations in middle-aged female mice. Environ. Pollut. 2019, 248, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, V.; Adelstein, J.M.; Grimmer, J.A.; Youtz, D.J.; Sugar, B.P.; Wold, L.E. PM2.5 exposure in utero contributes to neonatal cardiac dysfunction in mice. Environ. Pollut. 2017, 230, 116–124. [Google Scholar] [CrossRef]
- Liu, X.; Liu, H.; Nie, H.; Tian, L.; Shi, Y.; Lai, W.; Xi, Z.; Lin, B. Oil mist particulate matter induces myocardial tissue injury by impairing fatty acid metabolism and mitochondrial bioenergetics function via inhibiting the PPAR alpha signaling pathway in rats. Environ. Pollut. 2025, 365, 125340. [Google Scholar] [CrossRef]
- Marchini, T.; Magnani, N.; Garces, M.; Kelly, J.; Paz, M.; Caceres, L.; Calabro, V.; Lasagni Vitar, R.; Caltana, L.; Contin, M.; et al. Chronic exposure to polluted urban air aggravates myocardial infarction by impaired cardiac mitochondrial function and dynamics. Environ. Pollut. 2022, 295, 118677. [Google Scholar] [CrossRef] [PubMed]
- Lodovici, M.; Bigagli, E. Oxidative stress and air pollution exposure. J. Toxicol. 2011, 2011, 487074. [Google Scholar] [CrossRef]
- Gangwar, R.S.; Bevan, G.H.; Palanivel, R.; Das, L.; Rajagopalan, S. Oxidative stress pathways of air pollution mediated toxicity: Recent insights. Redox Biol. 2020, 34, 101545. [Google Scholar] [CrossRef] [PubMed]
- Kelly, F.J.; Fussell, J.C. Role of oxidative stress in cardiovascular disease outcomes following exposure to ambient air pollution. Free. Radic. Biol. Med. 2017, 110, 345–367. [Google Scholar] [CrossRef]
- Bocci, V.; Valacchi, G.; Corradeschi, F.; Aldinucci, C.; Silvestri, S.; Paccagnini, E.; Gerli, R. Studies on the biological effects of ozone: 7. Generation of reactive oxygen species (ROS) after exposure of human blood to ozone. J. Biol. Regul. Homeost. Agents 1998, 12, 67–75. [Google Scholar]
- Hazari, M.S.; Stratford, K.M.; Krantz, Q.T.; King, C.; Krug, J.; Farraj, A.K.; Gilmour, M.I. Comparative Cardiopulmonary Effects of Particulate Matter-and Ozone-Enhanced Smog Atmospheres in Mice. Environ. Sci. Technol. 2018, 52, 3071–3080. [Google Scholar] [CrossRef]
- Singh, N.; Kumar, A.; Gupta, V.K.; Sharma, B. Biochemical and Molecular Bases of Lead-Induced Toxicity in Mammalian Systems and Possible Mitigations. Chem. Res. Toxicol. 2018, 31, 1009–1021. [Google Scholar] [CrossRef]
- Patra, R.C.; Rautray, A.K.; Swarup, D. Oxidative stress in lead and cadmium toxicity and its amelioration. Vet. Med. Int. 2011, 2011, 457327. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. Mechanisms of lead-induced hypertension and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H454–H465. [Google Scholar] [CrossRef] [PubMed]
- Navas-Acien, A.; Guallar, E.; Silbergeld, E.K.; Rothenberg, S.J. Lead exposure and cardiovascular Disease—A systematic review. Environ. Health Perspect. 2007, 115, 472–482. [Google Scholar] [CrossRef]
- Cheng, Y.; Schwartz, J.; Vokonas, P.S.; Weiss, S.T.; Aro, A.; Hu, H. Electrocardiographic conduction disturbances in association with low-level lead exposure (the Normative Aging Study). Am. J. Cardiol. 1998, 82, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J. Lead, blood pressure, and cardiovascular disease in men and women. Environ. Health Perspect. 1991, 91, 71–75. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Lin, C.Y.; Farmand, F.; Sindhu, R.K. Superoxide dismutase, catalase, glutathione peroxidase and NADPH oxidase in lead-induced hypertension. Kidney Int. 2003, 63, 186–194. [Google Scholar] [CrossRef]
- Okoye, C.N.; Koren, S.A.; Wojtovich, A.P. Mitochondrial complex I ROS production and redox signaling in hypoxia. Redox Biol. 2023, 67, 102926. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.L.; Posser, T.; Dunkley, P.R.; Dickson, P.W.; Mattos, J.J.; Martins, R.; Bainy, A.C.; Marques, M.R.; Dafre, A.L.; Farina, M. Methylmercury neurotoxicity is associated with inhibition of the antioxidant enzyme glutathione peroxidase. Free. Radic. Biol. Med. 2009, 47, 449–457. [Google Scholar] [CrossRef]
- Lemos, N.B.; Angeli, J.K.; Faria Tde, O.; Ribeiro Junior, R.F.; Vassallo, D.V.; Padilha, A.S.; Stefanon, I. Low mercury concentration produces vasoconstriction, decreases nitric oxide bioavailability and increases oxidative stress in rat conductance artery. PLoS ONE 2012, 7, e49005. [Google Scholar] [CrossRef]
- Antunes Dos Santos, A.; Ferrer, B.; Marques Goncalves, F.; Tsatsakis, A.M.; Renieri, E.A.; Skalny, A.V.; Farina, M.; Rocha, J.B.T.; Aschner, M. Oxidative Stress in Methylmercury-Induced Cell Toxicity. Toxics 2018, 6, 47. [Google Scholar] [CrossRef]
- Omanwar, S.; Fahim, M. Mercury Exposure and Endothelial Dysfunction: An Interplay Between Nitric Oxide and Oxidative Stress. Int. J. Toxicol. 2015, 34, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.H.; Huang, Y.L.; Huang, S.F. Lipid peroxidation in liver of rats administrated with methyl mercuric chloride. Biol. Trace Elem. Res. 1996, 54, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kobal, A.B.; Horvat, M.; Prezelj, M.; Briski, A.S.; Krsnik, M.; Dizdarevic, T.; Mazej, D.; Falnoga, I.; Stibilj, V.; Arneric, N.; et al. The impact of long-term past exposure to elemental mercury on antioxidative capacity and lipid peroxidation in mercury miners. J. Trace Elem. Med. Biol. 2004, 17, 261–274. [Google Scholar] [CrossRef]
- Genchi, G.; Sinicropi, M.S.; Carocci, A.; Lauria, G.; Catalano, A. Mercury Exposure and Heart Diseases. Int. J. Environ. Res. Public Health 2017, 14, 74. [Google Scholar] [CrossRef] [PubMed]
- Houston, M.C. Role of mercury toxicity in hypertension, cardiovascular disease, and stroke. J. Clin. Hypertens. 2011, 13, 621–627. [Google Scholar] [CrossRef]
- Hu, X.F.; Lowe, M.; Chan, H.M. Mercury exposure, cardiovascular disease, and mortality: A systematic review and dose-response meta-analysis. Environ. Res. 2021, 193, 110538. [Google Scholar] [CrossRef]
- Branca, J.J.V.; Pacini, A.; Gulisano, M.; Taddei, N.; Fiorillo, C.; Becatti, M. Cadmium-Induced Cytotoxicity: Effects on Mitochondrial Electron Transport Chain. Front. Cell Dev. Biol. 2020, 8, 604377. [Google Scholar] [CrossRef]
- Chen, L.; Xu, B.; Liu, L.; Luo, Y.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Kontos, C.D.; Huang, S. Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free. Radic. Biol. Med. 2011, 50, 624–632. [Google Scholar] [CrossRef]
- Dabas, A.; Nagpure, N.S.; Kumar, R.; Kushwaha, B.; Kumar, P.; Lakra, W.S. Assessment of tissue-specific effect of cadmium on antioxidant defense system and lipid peroxidation in freshwater murrel, Channa punctatus. Fish Physiol. Biochem. 2012, 38, 469–482. [Google Scholar] [CrossRef]
- Xu, B.; Chen, S.; Luo, Y.; Chen, Z.; Liu, L.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Chen, L.; et al. Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS ONE 2011, 6, e19052. [Google Scholar] [CrossRef]
- Vallée, A.; Gabet, A.; Grave, C.; Blacher, J.; Olié, V. Associations between urinary cadmium levels, blood pressure, and hypertension: The ESTEBAN survey. Environ. Sci. Pollut. Res. 2020, 27, 10748–10756. [Google Scholar] [CrossRef] [PubMed]
- Tellez-Plaza, M.; Navas-Acien, A.; Crainiceanu, C.M.; Guallar, E. Cadmium Exposure and Hypertension in the 1999–2004 National Health and Nutrition Examination Survey (NHANES). Environ. Health Perspect. 2008, 116, 51–56. [Google Scholar] [CrossRef]
- Everett, C.J.; Frithsen, I.L. Association of urinary cadmium and myocardial infarction. Environ. Res. 2008, 106, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, B.; Bergström, G.; Borén, J.; Barregard, L. Cadmium exposure is accompanied by increased prevalence and future growth of atherosclerotic plaques in 64-year-old women. J. Intern. Med. 2012, 272, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.N.; Patel, M. The role of oxidative stress in organophosphate and nerve agent toxicity. Ann. N. Y. Acad. Sci. 2016, 1378, 17–24. [Google Scholar] [CrossRef]
- Lorke, D.E.; Oz, M. A review on oxidative stress in organophosphate-induced neurotoxicity. Int. J. Biochem. Cell Biol. 2025, 180, 106735. [Google Scholar] [CrossRef]
- Sule, R.O.; Condon, L.; Gomes, A.V. A Common Feature of Pesticides: Oxidative Stress—The Role of Oxidative Stress in Pesticide-Induced Toxicity. Oxidative Med. Cell. Longev. 2022, 2022, 5563759. [Google Scholar] [CrossRef]
- Hung, D.Z.; Yang, H.J.; Li, Y.F.; Lin, C.L.; Chang, S.Y.; Sung, F.C.; Tai, S.C. The Long-Term Effects of Organophosphates Poisoning as a Risk Factor of CVDs: A Nationwide Population-Based Cohort Study. PLoS ONE 2015, 10, e0137632. [Google Scholar] [CrossRef]
- Xia, T.; Guo, J.; Zhang, B.; Song, C.; Zhao, Q.; Cui, B.; Liu, Y. Bisphenol A Promotes the Progression of Colon Cancer Through Dual-Targeting of NADPH Oxidase and Mitochondrial Electron-Transport Chain to Produce ROS and Activating HIF-1alpha/VEGF/PI3K/AKT Axis. Front. Endocrinol. 2022, 13, 933051. [Google Scholar] [CrossRef]
- Kim, K.; Kwon, J.S.; Ahn, C.; Jeung, E.B. Endocrine-Disrupting Chemicals and Their Adverse Effects on the Endoplasmic Reticulum. Int. J. Mol. Sci. 2022, 23, 1581. [Google Scholar] [CrossRef]
- Molinari, F.; Franco, G.A.; Tranchida, N.; Di Paola, R.; Cordaro, M. Molecular Mechanism of Action of Endocrine-Disrupting Chemicals on the Respiratory System. Int. J. Mol. Sci. 2024, 25, 12540. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, B.; Yan, Y.; Zhang, N.; Shao, S.; Yang, L.; Ouyang, L.; Wu, P.; Duan, H.; Zhou, K.; et al. Maternal exposure to bisphenol A induces congenital heart disease through mitochondrial dysfunction. FASEB J. 2025, 39, e70351. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, L.; Wu, X.; Hou, L.; Li, Z.; Ju, J.; Li, Q.; Qin, W.; Li, J.; Zhang, Q.; et al. Bisphenol A, an environmental estrogen-like toxic chemical, induces cardiac fibrosis by activating the ERK1/2 pathway. Toxicol. Lett. 2016, 250–251, 1–9. [Google Scholar] [CrossRef]
- Saura, M.; Marquez, S.; Reventun, P.; Olea-Herrero, N.; Arenas, M.I.; Moreno-Gómez-Toledano, R.; Gómez-Parrizas, M.; Muñóz-Moreno, C.; González-Santander, M.; Zaragoza, C.; et al. Oral administration of bisphenol A induces high blood pressure through angiotensin II/CaMKII-dependent uncoupling of eNOS. FASEB J. 2014, 28, 4719–4728. [Google Scholar] [CrossRef]
- Chaudhuri, L.; Sarsour, E.H.; Kalen, A.L.; Aykin-Burns, N.; Spitz, D.R.; Goswami, P.C. Polychlorinated biphenyl induced ROS signaling delays the entry of quiescent human breast epithelial cells into the proliferative cycle. Free. Radic. Biol. Med. 2010, 49, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tan, Y.; Song, E.; Song, Y. A Critical Review of Polychlorinated Biphenyls Metabolism, Metabolites, and Their Correlation with Oxidative Stress. Chem. Res. Toxicol. 2020, 33, 2022–2042. [Google Scholar] [CrossRef]
- Perkins, J.T.; Petriello, M.C.; Newsome, B.J.; Hennig, B. Polychlorinated biphenyls and links to cardiovascular disease. Environ. Sci. Pollut. Res. Int. 2016, 23, 2160–2172. [Google Scholar] [CrossRef]
- Deen, L.; Clark, A.; Hougaard, K.S.; Meyer, H.W.; Frederiksen, M.; Pedersen, E.B.; Petersen, K.U.; Flachs, E.M.; Bonde, J.P.E.; Tøttenborg, S.S. Risk of cardiovascular diseases following residential exposure to airborne polychlorinated biphenyls: A register-based cohort study. Environ. Res. 2023, 222, 115354. [Google Scholar] [CrossRef]
- Åkesson, A.; Donat-Vargas, C.; Berglund, M.; Glynn, A.; Wolk, A.; Kippler, M. Dietary exposure to polychlorinated biphenyls and risk of heart failure—A population-based prospective cohort study. Environ. Int. 2019, 126, 1–6. [Google Scholar] [CrossRef]
- Carpenter, D. Exposure to Polychlorinated Biphenyls Is Associated With an Increased Risk of Hypertension and Cardiovascular Disease. Epidemiology 2011, 22, S147. [Google Scholar] [CrossRef]
- Wang, C.; Liu, X.; Zhai, J.; Zhong, C.; Zeng, H.; Feng, L.; Yang, Y.; Li, X.; Ma, M.; Luan, T.; et al. Effect of oxidative stress induced by 2,3,7,8- tetrachlorodibenzo-p-dioxin on DNA damage. J. Hazard. Mater. 2024, 472, 134485. [Google Scholar] [CrossRef] [PubMed]
- Senft, A.P.; Dalton, T.P.; Nebert, D.W.; Genter, M.B.; Hutchinson, R.J.; Shertzer, H.G. Dioxin increases reactive oxygen production in mouse liver mitochondria. Toxicol. Appl. Pharmacol. 2002, 178, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.H.; Sutter, C.H.; Leon Carrion, S.; Tran, Q.T.; Bodreddigari, S.; Kensicki, E.; Mohney, R.P.; Sutter, T.R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated production of reactive oxygen species is an essential step in the mechanism of action to accelerate human keratinocyte differentiation. Toxicol. Sci. 2013, 132, 235–249. [Google Scholar] [CrossRef]
- Jokinen, M.P.; Walker, N.J.; Brix, A.E.; Sells, D.M.; Haseman, J.K.; Nyska, A. Increase in cardiovascular pathology in female Sprague-Dawley rats following chronic treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin and 3,3′,4,4′,5-pentachlorobiphenyl. Cardiovasc. Toxicol. 2003, 3, 299–310. [Google Scholar] [CrossRef]
- Kopf, P.G.; Huwe, J.K.; Walker, M.K. Hypertension, cardiac hypertrophy, and impaired vascular relaxation induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin are associated with increased superoxide. Cardiovasc. Toxicol. 2008, 8, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Dalton, T.P.; Kerzee, J.K.; Wang, B.; Miller, M.; Dieter, M.Z.; Lorenz, J.N.; Shertzer, H.G.; Nerbert, D.W.; Puga, A. Dioxin exposure is an environmental risk factor for ischemic heart disease. Cardiovasc. Toxicol. 2001, 1, 285–298. [Google Scholar] [CrossRef]
- Humblet, O.; Birnbaum, L.; Rimm, E.; Mittleman, M.A.; Hauser, R. Dioxins and cardiovascular disease mortality. Environ. Health Perspect. 2008, 116, 1443–1448. [Google Scholar] [CrossRef]
- Ivnitski-Steele, I.D.; Friggens, M.; Chavez, M.; Walker, M.K. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) inhibition of coronary vasculogenesis is mediated, in part, by reduced responsiveness to endogenous angiogenic stimuli, including vascular endothelial growth factor A (VEGF-A). Birth Defects Res. Part A Clin. Mol. Teratol. 2005, 73, 440–446. [Google Scholar] [CrossRef]
- Puga, A. Perspectives on the Potential Involvement of the Ah Receptor-Dioxin Axis in Cardiovascular Disease. Toxicol. Sci. 2010, 120, 256–261. [Google Scholar] [CrossRef]
- Aragon, A.C.; Kopf, P.G.; Campen, M.J.; Huwe, J.K.; Walker, M.K. In utero and lactational 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure: Effects on fetal and adult cardiac gene expression and adult cardiac and renal morphology. Toxicol. Sci. 2008, 101, 321–330. [Google Scholar] [CrossRef]
- Thackaberry, E.A.; Nunez, B.A.; Ivnitski-Steele, I.D.; Friggins, M.; Walker, M.K. Effect of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin on Murine Heart Development: Alteration in Fetal and Postnatal Cardiac Growth, and Postnatal Cardiac Chronotropy. Toxicol. Sci. 2005, 88, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Kamp, D.W.; Graceffa, P.; Pryor, W.A.; Weitzman, S.A. The role of free radicals in asbestos-induced diseases. Free. Radic. Biol. Med. 1992, 12, 293–315. [Google Scholar] [CrossRef]
- Shukla, A.; Jung, M.; Stern, M.; Fukagawa, N.K.; Taatjes, D.J.; Sawyer, D.; Van Houten, B.; Mossman, B.T. Asbestos induces mitochondrial DNA damage and dysfunction linked to the development of apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L1018–L1025. [Google Scholar] [CrossRef]
- Rong, Y.; Luo, X.; Zhang, Z.; Cui, X.; Liu, Y.; Chen, W. Occupational exposure to asbestos and cardiovascular related diseases: A meta-analysis. Prev. Med. Rep. 2015, 2, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, Y.; Chen, C.; Field, R.W.; Kahe, K. Radon exposure and risk of cerebrovascular disease: A systematic review and meta-analysis in occupational and general population studies. Environ. Sci. Pollut. Res. 2022, 29, 45031–45043. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Sun, J.; Zhai, X.; Chen, X.; Wan, J.; Tian, H. Repeated radon exposure induced lung damage via oxidative stress-mediated mitophagy in human bronchial epithelial cells and mice. Environ. Toxicol. Pharmacol. 2022, 90, 103812. [Google Scholar] [CrossRef]
- Nobari, H.; Nejad, H.A.; Kargarfard, M.; Mohseni, S.; Suzuki, K.; Carmelo Adsuar, J.; Perez-Gomez, J. The Effect of Acute Intense Exercise on Activity of Antioxidant Enzymes in Smokers and Non-Smokers. Biomolecules 2021, 11, 171. [Google Scholar] [CrossRef]
- Seo, Y.S.; Park, J.M.; Kim, J.H.; Lee, M.Y. Cigarette Smoke-Induced Reactive Oxygen Species Formation: A Concise Review. Antioxidants 2023, 12, 1732. [Google Scholar] [CrossRef]
- Emma, R.; Caruso, M.; Campagna, D.; Pulvirenti, R.; Li Volti, G. The Impact of Tobacco Cigarettes, Vaping Products and Tobacco Heating Products on Oxidative Stress. Antioxidants 2022, 11, 1829. [Google Scholar] [CrossRef]
- El-Mahdy, M.A.; Abdelghany, T.M.; Hemann, C.; Ewees, M.G.; Mahgoup, E.M.; Eid, M.S.; Shalaan, M.T.; Alzarie, Y.A.; Zweier, J.L. Chronic cigarette smoke exposure triggers a vicious cycle of leukocyte and endothelial-mediated oxidant stress that results in vascular dysfunction. Am. J. Physiol.-Heart Circ. Physiol. 2020, 319, H51–H65. [Google Scholar] [CrossRef]
- Andersson, E.M.; Fagerberg, B.; Sallsten, G.; Borné, Y.; Hedblad, B.; Engström, G.; Barregard, L. Partial Mediation by Cadmium Exposure of the Association Between Tobacco Smoking and Atherosclerotic Plaques in the Carotid Artery. Am. J. Epidemiol. 2017, 187, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Ockene, I.S.; Miller, N.H. Cigarette Smoking, Cardiovascular Disease, and Stroke. Circulation 1997, 96, 3243–3247. [Google Scholar] [CrossRef]
- Yin, H.; Han, S.; Chen, Y.; Wang, Y.; Li, D.; Zhu, Q. T-2 Toxin Induces Oxidative Stress, Apoptosis and Cytoprotective Autophagy in Chicken Hepatocytes. Toxins 2020, 12, 90. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Huang, T.; Chen, Y.; Song, W.; Chen, F.; Jiang, Y.; Zhang, C.; Yang, X. Nrf2: A Main Responsive Element of the Toxicity Effect Caused by Trichothecene (T-2) Mycotoxin. Toxics 2023, 11, 393. [Google Scholar] [CrossRef]
- Doi, K.; Uetsuka, K. Mechanisms of mycotoxin-induced neurotoxicity through oxidative stress-associated pathways. Int. J. Mol. Sci. 2011, 12, 5213–5237. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Lin, X.; Liu, H.; Xiang, R.; Zhan, J.; Deng, F.; Bao, M.; He, H.; Wen, X.; Deng, H.; et al. T-2 toxin induces cardiac fibrosis by causing metabolic disorders and up-regulating Sirt3/FoxO3alpha/MnSOD signaling pathway-mediated oxidative stress. J. Environ. Sci. 2025, 150, 532–544. [Google Scholar] [CrossRef]
- Ghosh, R.; Alajbegovic, A.; Gomes, A.V. NSAIDs and Cardiovascular Diseases: Role of Reactive Oxygen Species. Oxidative Med. Cell. Longev. 2015, 2015, 536962. [Google Scholar] [CrossRef]
- Fan, Z.; Yang, Y.; Hu, P.; Huang, Y.; He, L.; Hu, R.; Zhao, K.; Zhang, H.; Liu, C. Molecular mechanism of ethylparaben on zebrafish embryo cardiotoxicity based on transcriptome analyses. Sci. Total Environ. 2022, 842, 156785. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wang, B.; Wang, H.; Meng, L.; Zhao, Q.; Li, X.; Xin, Y.; Jiang, X. Radiation-Induced Normal Tissue Damage: Oxidative Stress and Epigenetic Mechanisms. Oxidative Med. Cell. Longev. 2019, 2019, 3010342. [Google Scholar] [CrossRef]
- Kim, W.; Lee, S.; Seo, D.; Kim, D.; Kim, K.; Kim, E.; Kang, J.; Seong, K.M.; Youn, H.; Youn, B. Cellular Stress Responses in Radiotherapy. Cells 2019, 8, 1105. [Google Scholar] [CrossRef]
- Belzile-Dugas, E.; Eisenberg, M.J. Radiation-Induced Cardiovascular Disease: Review of an Underrecognized Pathology. J. Am. Heart Assoc. 2021, 10, e021686. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wei, J.; Zheng, Q.; Meng, L.; Xin, Y.; Yin, X.; Jiang, X. Radiation-induced heart disease: A review of classification, mechanism and prevention. Int. J. Biol. Sci. 2019, 15, 2128–2138. [Google Scholar] [CrossRef]
- Brown, K.N.; Hussain, K.; Richards, J.R. Radiation-Induced Coronary Artery Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Ellahham, S.; Khalouf, A.; Elkhazendar, M.; Dababo, N.; Manla, Y. An overview of radiation-induced heart disease. Radiat. Oncol. J. 2022, 40, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef] [PubMed]
- Uzma, N.; Kumar, B.S.; Hazari, M.A. Exposure to benzene induces oxidative stress, alters the immune response and expression of p53 in gasoline filling workers. Am. J. Ind. Med. 2010, 53, 1264–1270. [Google Scholar] [CrossRef]
- Martinez-Alfaro, M.; Alcaraz-Contreras, Y.; Carabez-Trejo, A.; Leo-Amador, G.E. Oxidative stress effects of thinner inhalation. Indian J. Occup. Environ. Med. 2011, 15, 87–92. [Google Scholar] [CrossRef]
- Cordiano, R.; Papa, V.; Cicero, N.; Spatari, G.; Allegra, A.; Gangemi, S. Effects of Benzene: Hematological and Hypersensitivity Manifestations in Resident Living in Oil Refinery Areas. Toxics 2022, 10, 678. [Google Scholar] [CrossRef]
- Abplanalp, W.; DeJarnett, N.; Riggs, D.W.; Conklin, D.J.; McCracken, J.P.; Srivastava, S.; Xie, Z.; Rai, S.; Bhatnagar, A.; O’Toole, T.E. Benzene exposure is associated with cardiovascular disease risk. PLoS ONE 2017, 12, e0183602. [Google Scholar] [CrossRef]
- Kadac-Czapska, K.; Osko, J.; Knez, E.; Grembecka, M. Microplastics and Oxidative Stress-Current Problems and Prospects. Antioxidants 2024, 13, 579. [Google Scholar] [CrossRef]
- Mahmud, F.; Sarker, D.B.; Jocelyn, J.A.; Sang, Q.A. Molecular and Cellular Effects of Microplastics and Nanoplastics: Focus on Inflammation and Senescence. Cells 2024, 13, 1788. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, S.; Liu, Q.; Wei, J.; Jin, Y.; Wang, X.; Zhang, L. Polystyrene microplastics cause cardiac fibrosis by activating Wnt/β-catenin signaling pathway and promoting cardiomyocyte apoptosis in rats. Environ. Pollut. 2020, 265, 115025. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wang, X.; Liu, Q.; Zhou, N.; Zhu, S.; Li, Z.; Li, X.; Yao, J.; Zhang, L. The impact of polystyrene microplastics on cardiomyocytes pyroptosis through NLRP3/Caspase-1 signaling pathway and oxidative stress in Wistar rats. Environ. Toxicol. 2021, 36, 935–944. [Google Scholar] [CrossRef]
- Persiani, E.; Cecchettini, A.; Ceccherini, E.; Gisone, I.; Morales, M.A.; Vozzi, F. Microplastics: A Matter of the Heart (and Vascular System). Biomedicines 2023, 11, 264. [Google Scholar] [CrossRef]
- Jones, D.P.; Sies, H.; Klaunig, J.E. Redox biology: The old and the new. Arch. Toxicol. 2019, 93, 1–6. [Google Scholar]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Hu, Y.; Huang, Y.; Zong, L.; Lin, J.; Liu, X.; Ning, S. Emerging roles of ferroptosis in pulmonary fibrosis: Current perspectives, opportunities and challenges. Cell Death Discov. 2024, 10, 301. [Google Scholar] [CrossRef]
- Werbner, B.; Tavakoli-Rouzbehani, O.M.; Fatahian, A.N.; Boudina, S. The dynamic interplay between cardiac mitochondrial health and myocardial structural remodeling in metabolic heart disease, aging, and heart failure. J. Cardiovasc. Aging 2023, 3, 9. [Google Scholar] [CrossRef]
- Wang, L.L.; Wang, C.F.; Liu, T.; Xuan, H.C.; Li, X.Q.; Shi, X.X.; Dai, F.; Chen, J.H.; Li, D.Y.; Xu, T.D. Association of low-level lead exposure with all-cause and cardiovascular disease mortality in US adults with hypertension: Evidence from the National Health and Nutrition Examination Survey 2003–2010. Arch. Public Health 2023, 81, 146. [Google Scholar] [CrossRef]
- Jain, N.B.; Potula, V.; Schwartz, J.; Vokonas, P.S.; Sparrow, D.; Wright, R.O.; Nie, H.; Hu, H. Lead Levels and Ischemic Heart Disease in a Prospective Study of Middle-Aged and Elderly Men: The VA Normative Aging Study. Environ. Health Perspect. 2007, 115, 871–875. [Google Scholar] [CrossRef]
- Weisskopf, M.G.; Jain, N.; Nie, H.; Sparrow, D.; Vokonas, P.; Schwartz, J.; Hu, H. A Prospective Study of Bone Lead Concentration and Death From All Causes, Cardiovascular Diseases, and Cancer in the Department of Veterans Affairs Normative Aging Study. Circulation 2009, 120, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Navas-Acien, A.; Selvin, E.; Sharrett, A.R.; Calderon-Aranda, E.; Silbergeld, E.; Guallar, E. Lead, Cadmium, Smoking, and Increased Risk of Peripheral Arterial Disease. Circulation 2004, 109, 3196–3201. [Google Scholar] [CrossRef]
- Lee, M.S.; Park, S.K.; Hu, H.; Lee, S. Cadmium exposure and cardiovascular disease in the 2005 Korea National Health and Nutrition Examination Survey. Environ Res. 2011, 111, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Boyd, R.; McMullen, H.; Beqaj, H.; Kalfa, D. Environmental Exposures and Congenital Heart Disease. Pediatrics 2021, 149, e2021052151. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, M.F.; Lo, C.W.H.; Cheung, T.T.; Cheung, B.M.Y. Blood lead level and risk of hypertension in the United States National Health and Nutrition Examination Survey 1999–2016. Sci. Rep. 2021, 11, 3010. [Google Scholar] [CrossRef]
- MacMahon, S.; Rodgers, A. Blood pressure, antihypertensive treatment and stroke risk. J. Hypertens. 1994, 12, S5–S14. [Google Scholar]
- Hertz-Picciotto, I.; Croft, J. Review of the Relation between Blood Lead and Blood Pressure. Epidemiol. Rev. 1993, 15, 352–373. [Google Scholar] [CrossRef]
- Nawrot, T.S.; Thijs, L.; Den Hond, E.M.; Roels, H.A.; Staessen, J.A. An epidemiological re-appraisal of the association between blood pressure and blood lead: A meta-analysis. J. Hum. Hypertens. 2002, 16, 123–131. [Google Scholar] [CrossRef]
- Schwartz, J. Lead, Blood Pressure, and Cardiovascular Disease in Men. Arch. Environ. Health Int. J. 1995, 50, 31–37. [Google Scholar] [CrossRef]
- Staessen, J.A.; Bulpitt, C.J.; Fagard, R.; Lauwerys, R.R.; Roels, H.; Thijs, L.; Amery, A. Hypertension Caused by Low-Level Lead Exposure: Myth or Fact? Eur. J. Cardiovasc. Prev. Rehabil. 1994, 1, 87–97. [Google Scholar] [CrossRef]
- Staessen, J.A.; Roels, H.; Lauwerys, R.R.; Amery, A. Low-level lead exposure and blood pressure. J. Hum. Hypertens. 1995, 9, 303–328. [Google Scholar]
- Ahmad, S.A.; Khan, M.H.; Khandker, S.; Sarwar, A.F.M.; Yasmin, N.; Faruquee, M.H.; Yasmin, R. Blood Lead Levels and Health Problems of Lead Acid Battery Workers in Bangladesh. Sci. World J. 2014, 2014, 974104. [Google Scholar] [CrossRef] [PubMed]
- Shiue, I. Higher urinary heavy metal, arsenic, and phthalate concentrations in people with high blood pressure: US NHANES, 2009–2010. Blood Press. 2014, 23, 363–369. [Google Scholar] [CrossRef]
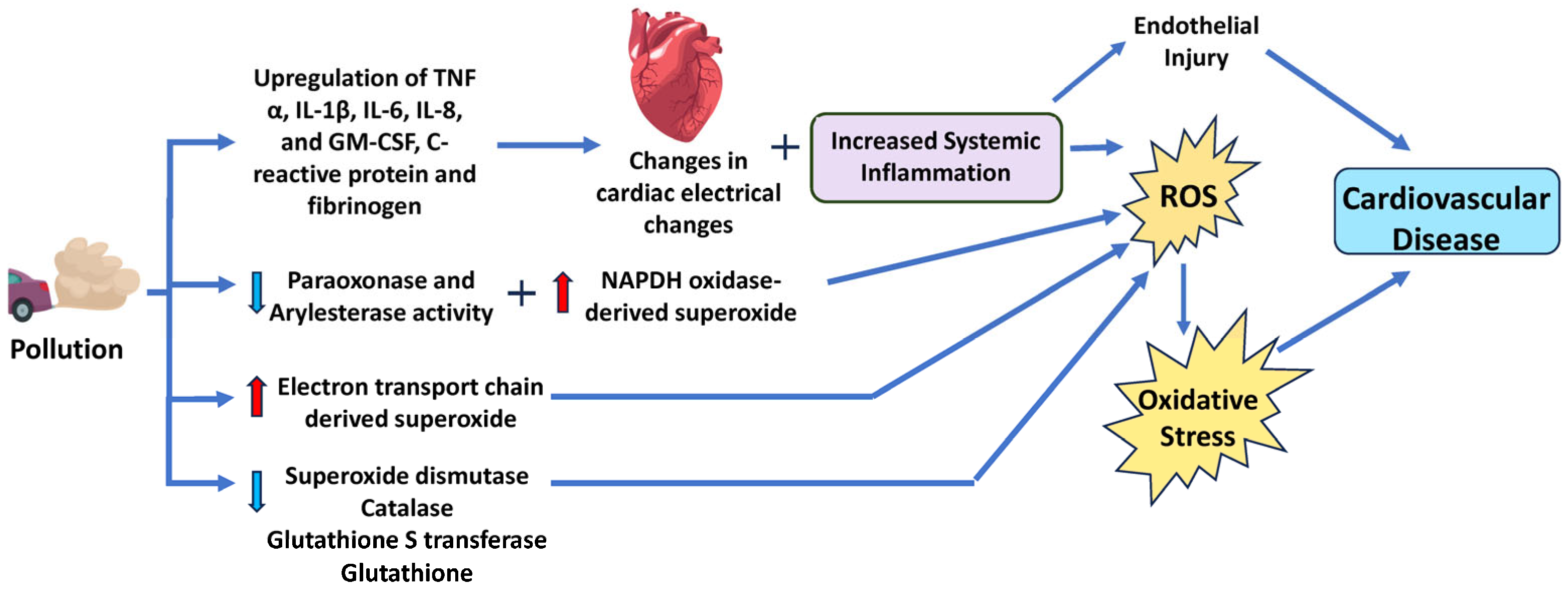
- Cai, H.; Harrison, D.G. Endothelial Dysfunction in Cardiovascular Diseases: The Role of Oxidant Stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation, Atherosclerosis, and Coronary Artery Disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef]
- Shah, P.K. Mechanisms of plaque vulnerability and rupture. J. Am. Coll. Cardiol. 2003, 41, S15–S22. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Rodríguez-Iturbe, B. Mechanisms of Disease: Oxidative stress and inflammation in the pathogenesis of hypertension. Nat. Clin. Pract. Nephrol. 2006, 2, 582–593. [Google Scholar] [CrossRef]
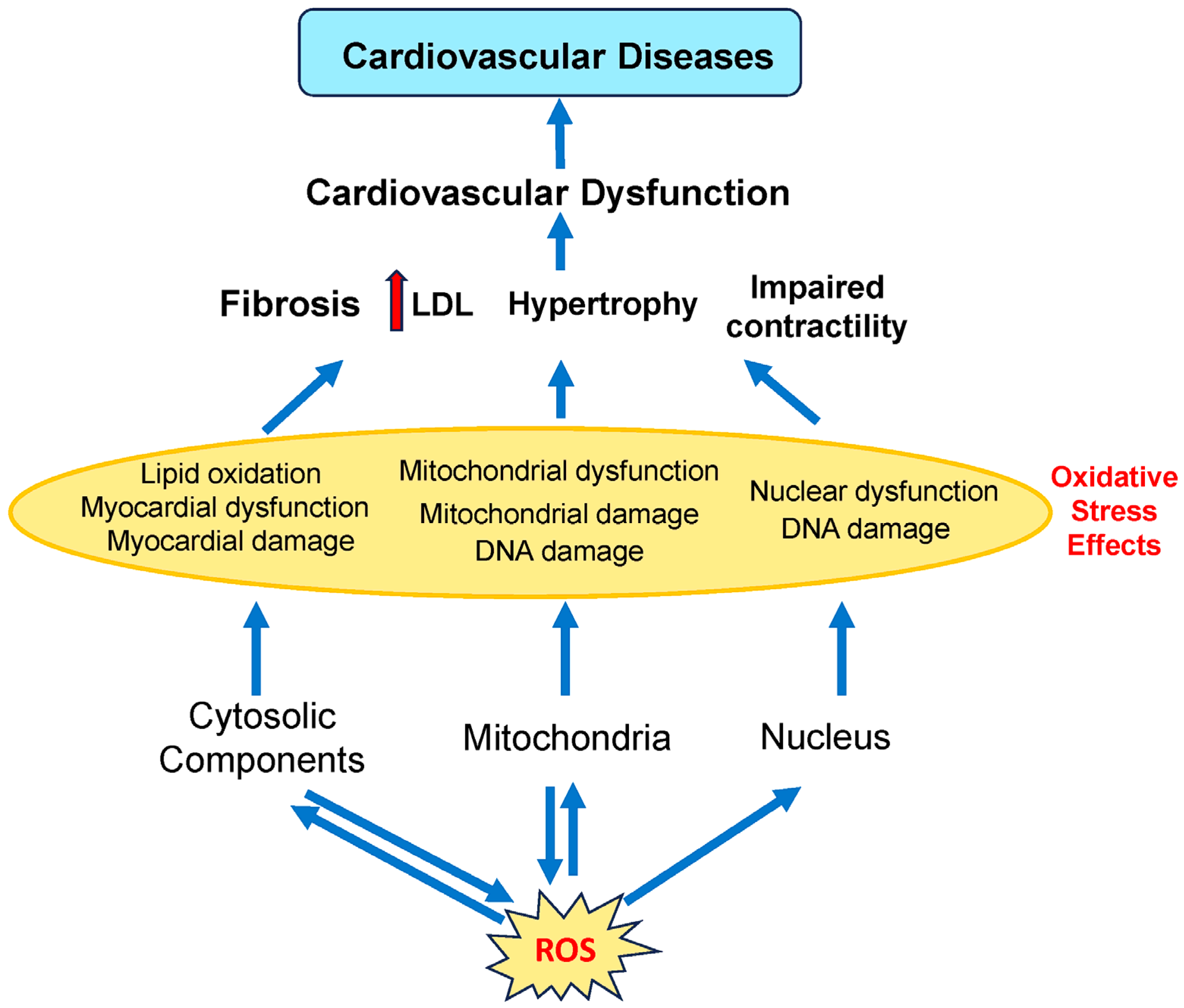
- Moris, D.; Spartalis, M.; Spartalis, E.; Karachaliou, G.S.; Karaolanis, G.I.; Tsourouflis, G.; Tsilimigras, D.I.; Tzatzaki, E.; Theocharis, S. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann. Transl. Med. 2017, 5, 326. [Google Scholar] [CrossRef]
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv. Med. 2016, 2016, 9152732. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Taverne, Y.J.H.J.; Bogers, A.J.J.C.; Duncker, D.J.; Merkus, D. Reactive Oxygen Species and the Cardiovascular System. Oxidative Med. Cell. Longev. 2013, 2013, 862423. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Förstermann, U.; Münzel, T. Endothelial Nitric Oxide Synthase in Vascular Disease. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef]
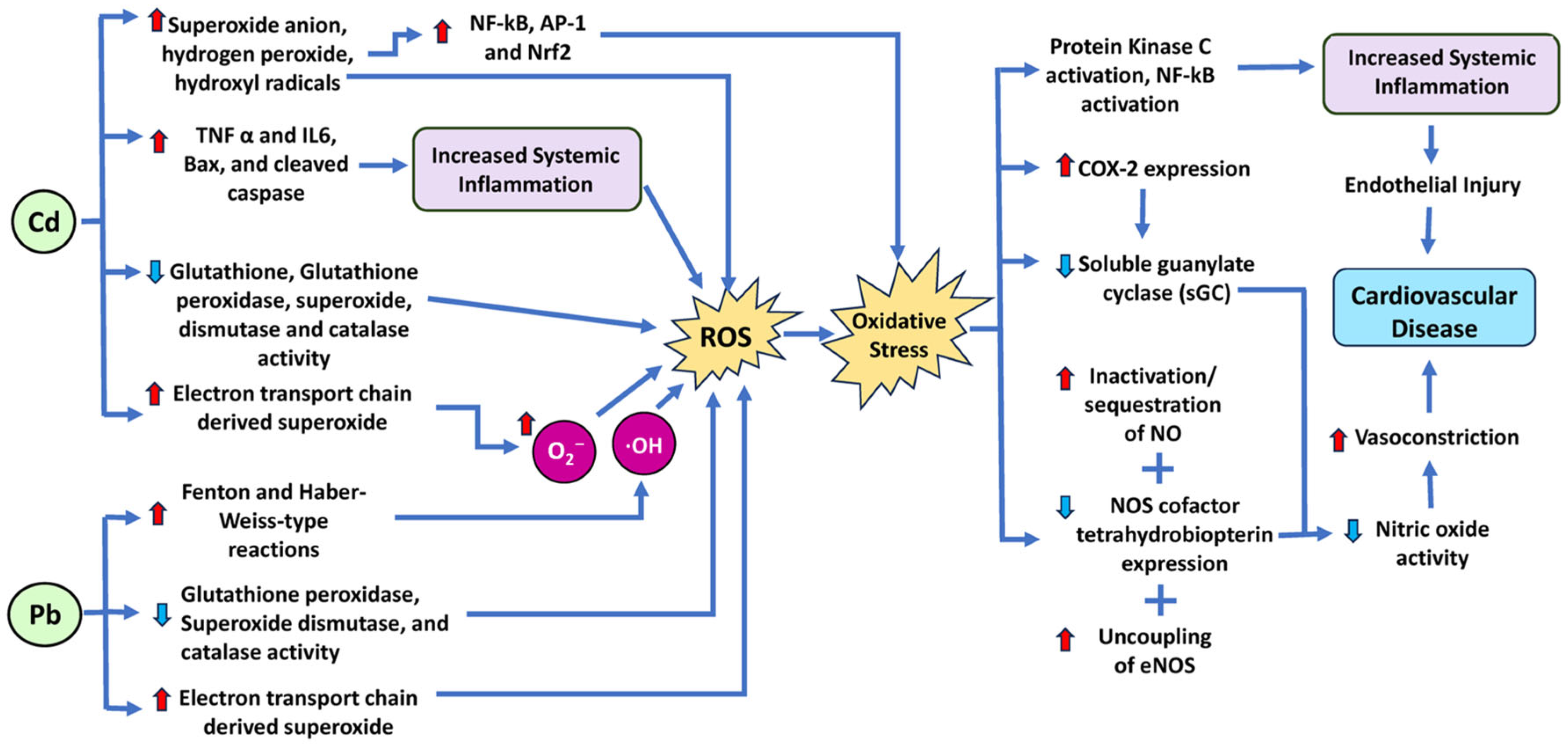
- Courtois, E.; Marques, M.; Barrientos, A.; Casado, S.; Lopez-Farre, A. Lead-Induced Downregulation of Soluble Guanylate Cyclase in Isolated Rat Aortic Segments Mediated by Reactive Oxygen Species and Cyclooxygenase-2. J. Am. Soc. Nephrol. 2003, 14, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Rafati Rahimzadeh, M.; Rafati Rahimzadeh, M.; Kazemi, S.; Moghadamnia, A.A. Cadmium toxicity and treatment: An update. Casp. J. Intern. Med. 2017, 8, 135–145. [Google Scholar] [CrossRef]
- Satarug, S. Dietary Cadmium Intake and Its Effects on Kidneys. Toxics 2018, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-C.; Hao, W.-M.; Chu, P.-H. Cadmium and cardiovascular disease: An overview of pathophysiology, epidemiology, therapy, and predictive value. Rev. Port. Cardiol. 2021, 40, 611–617. [Google Scholar] [CrossRef]
- Menke, A.; Muntner, P.; Silbergeld, E.K.; Platz, E.A.; Guallar, E. Cadmium Levels in Urine and Mortality among U.S. Adults. Environ. Health Perspect. 2009, 117, 190–196. [Google Scholar] [CrossRef]
- Deering, K.E.; Callan, A.C.; Prince, R.L.; Lim, W.H.; Thompson, P.L.; Lewis, J.R.; Hinwood, A.L.; Devine, A. Low-level cadmium exposure and cardiovascular outcomes in elderly Australian women: A cohort study. Int. J. Hyg. Environ. Health 2018, 221, 347–354. [Google Scholar] [CrossRef]
- Hsu, C.W.; Weng, C.H.; Lee, C.C.; Lin-Tan, D.T.; Chu, P.H.; Chen, K.H.; Yen, T.H.; Huang, W.H. Urinary cadmium levels predict mortality of patients with acute heart failure. Ther. Clin. Risk Manag. 2017, 13, 379–386. [Google Scholar] [CrossRef]
- Tellez-Plaza, M.; Guallar, E.; Howard, B.V.; Umans, J.G.; Francesconi, K.A.; Goessler, W.; Silbergeld, E.K.; Devereux, R.B.; Navas-Acien, A. Cadmium exposure and incident cardiovascular disease. Epidemiology 2013, 24, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, C.; Ferreira de Oliveira, J.M.P.; Pinho, F.; Bastos, V.; Oliveira, H.; Peixoto, F.; Santos, C. Biochemical and transcriptional analyses of cadmium-induced mitochondrial dysfunction and oxidative stress in human osteoblasts. J. Toxicol. Environ. Health Part A 2018, 81, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Alkharashi, N.A.O.; Periasamy, V.S.; Athinarayanan, J.; Alshatwi, A.A. Cadmium triggers mitochondrial oxidative stress in human peripheral blood lymphocytes and monocytes: Analysis using in vitro and system toxicology approaches. J. Trace Elem. Med. Biol. 2017, 42, 117–128. [Google Scholar] [CrossRef]
- Vicente-Sánchez, C.; Egido, J.; Sánchez-González, P.D.; Pérez-Barriocanal, F.; López-Novoa, J.M.; Morales, A.I. Effect of the flavonoid quercetin on cadmium-induced hepatotoxicity. Food Chem. Toxicol. 2008, 46, 2279–2287. [Google Scholar] [CrossRef]
- Wang, J.; Wang, K.; Ding, L.; Zhao, P.; Zhang, C.; Wang, H.; Yang, Z.; Liu, Z. Alleviating effect of quercetin on cadmium-induced oxidative damage and apoptosis by activating the Nrf2-keap1 pathway in BRL-3A cells. Front. Pharmacol. 2022, 13, 969892. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, E.E.; Jacob, M.; Sanadi, D.R.; Bradley, L.B. Uncoupling of Oxidative Phosphorylation by Cadmium Ion. J. Biol. Chem. 1956, 223, 147–156. [Google Scholar] [CrossRef]
- Hirst, J.; King, M.S.; Pryde, K.R. The production of reactive oxygen species by complex I. Biochem. Soc. Trans. 2008, 36, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular Mechanisms of Angiotensin II–Mediated Mitochondrial Dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef]
- Sevim, Ç.; Doğan, E.; Comakli, S. Cardiovascular disease and toxic metals. Curr. Opin. Toxicol. 2020, 19, 88–92. [Google Scholar] [CrossRef]
- Banik, S.; Akter, M.; Corpus Bondad, S.E.; Saito, T.; Hosokawa, T.; Kurasaki, M. Carvacrol inhibits cadmium toxicity through combating against caspase dependent/independent apoptosis in PC12 cells. Food Chem. Toxicol. 2019, 134, 110835. [Google Scholar] [CrossRef]
- Roberts, J.A.; Rainbow, R.D.; Sharma, P. Mitigation of Cardiovascular Disease and Toxicity through NRF2 Signalling. Int. J. Mol. Sci. 2023, 24, 6723. [Google Scholar] [CrossRef] [PubMed]
- Cracowski, J.L.; Durand, T.; Bessard, G. Isoprostanes and their products as biomarkers of oxidative stress in humans. Br. J. Pharmacol. 2012, 165, 1084–1096. [Google Scholar] [CrossRef]
- Pan, Q.; Qiu, W.Y.; Huo, Y.N.; Yao, Y.F.; Lou, M.F. Low levels of hydrogen peroxide stimulate corneal epithelial cell adhesion, migration, and wound healing. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1723–1734. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Lu, H.; Zhang, X.; Shi, X.; Jiang, S.; Wang, L.; Lu, Q. Paraben exposures and their interactions with ESR1/2 genetic polymorphisms on hypertension. Environ. Res. 2022, 213, 113651. [Google Scholar] [CrossRef]
- Pollack, A.Z.; Mumford, S.L.; Krall, J.R.; Carmichael, A.; Andriessen, V.C.; Kannan, K.; Schisterman, E.F. Urinary levels of environmental phenols and parabens and antioxidant enzyme activity in the blood of women. Environ. Res 2020, 186, 109507. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, I.L.; Dirven, H.; Couderq, S.; David, A.; D’Cruz, S.C.; Fernández, M.F.; Mustieles, V.; Rodríguez-Carrillo, A.; Hofer, T. Bisphenols and Oxidative Stress Biomarkers-Associations Found in Human Studies, Evaluation of Methods Used, and Strengths and Weaknesses of the Biomarkers. Int. J. Environ. Res. Public Health 2020, 17, 3609. [Google Scholar] [CrossRef]
- Moon, S.; Yu, S.H.; Lee, C.B.; Park, Y.J.; Yoo, H.J.; Kim, D.S. Effects of bisphenol A on cardiovascular disease: An epidemiological study using National Health and Nutrition Examination Survey 2003-2016 and meta-analysis. Sci. Total Environ. 2021, 763, 142941. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, X.; Xiao, Q.; Han, L.; Yang, J.; Li, X.; Xu, J.; Zheng, Q.; Ma, J.; Chen, J.; et al. Co-Exposure to Bisphenols, Parabens, and Antimicrobials and Association with Coronary Heart Disease: Oxidative Stress as a Potential Mediating Factor? Environ. Sci. Technol. 2023, 57, 531–538. [Google Scholar] [CrossRef]
- Simpson, K.L.; Hayes, K.P.J.W.R. Drinking water disinfection by-products: An Australian perspective. Water Res. 1998, 32, 1522–1528. [Google Scholar] [CrossRef]
- Rooney, M.R.; Lutsey, P.L.; Bhatti, P.; Prizment, A. Urinary 2,5-dicholorophenol and 2,4-dichlorophenol concentrations and prevalent disease among adults in the National Health and Nutrition Examination Survey (NHANES). Occup. Environ. Med. 2019, 76, 181–188. [Google Scholar] [CrossRef]
- Li, E.; Bolser, D.G.; Kroll, K.J.; Brockmeier, E.K.; Falciani, F.; Denslow, N.D. Comparative toxicity of three phenolic compounds on the embryo of fathead minnow, Pimephales promelas. Aquat. Toxicol. 2018, 201, 66–72. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. 2022. Available online: https://www.fda.gov/cosmetics/cosmetic-ingredients/phthalates-cosmetics (accessed on 11 May 2025).
- Cui, J.-G.; Zhao, Y.; Zhang, H.; Li, X.-N.; Li, J.-L. Lycopene regulates the mitochondrial unfolded protein response to prevent DEHP-induced cardiac mitochondrial damage in mice. Food Funct. 2022, 13, 4527–4536. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, L.; Li, M.Z.; Wang, H.R.; Zhao, Y.; Li, J.L. Lycopene prevents Di-(2-ethylhexyl) phthalate-induced mitophagy and oxidative stress in mice heart via modulating mitochondrial homeostasis. J. Nutr. Biochem. 2023, 115, 109285. [Google Scholar] [CrossRef]
- Wen, X.; Wang, M.; Xu, X.; Li, T. Exposure to Per- and Polyfluoroalkyl Substances and Mortality in U.S. Adults: A Population-Based Cohort Study. Environ. Health Perspect. 2022, 130, 67007. [Google Scholar] [CrossRef] [PubMed]
- Rubio, L.; Marcos, R.; Hernández, A. Potential adverse health effects of ingested micro- and nanoplastics on humans. Lessons learned from in vivo and in vitro mammalian models. J. Toxicol. Environ. Health Part B 2020, 23, 51–68. [Google Scholar] [CrossRef]
- Wang, D.; Tan, Z.; Yang, J.; Li, L.; Li, H.; Zhang, H.; Liu, H.; Liu, Y.; Wang, L.; Li, Q.; et al. Perfluorooctane sulfonate promotes atherosclerosis by modulating M1 polarization of macrophages through the NF-κB pathway. Ecotoxicol. Environ. Saf. 2023, 249, 114384. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Li, L.; Tang, L.; Guo, M.; Yang, J. Perfluorooctane sulfonate induces heart toxicity involving cardiac apoptosis and inflammation in rats. Exp. Ther. Med. 2022, 23, 14. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.L.; Wang, J.D.; Xu, T.T.; Zhao, Z.; Zheng, J.J.; Ge, R.S.; Zhu, D.Y. Mitochondrial toxicity of perfluorooctane sulfonate in mouse embryonic stem cell-derived cardiomyocytes. Toxicology 2017, 382, 108–116. [Google Scholar] [CrossRef]
- Patel, N.; Ivantsova, E.; Konig, I.; Souders, C.L., 2nd; Martyniuk, C.J. Perfluorotetradecanoic Acid (PFTeDA) Induces Mitochondrial Damage and Oxidative Stress in Zebrafish (Danio rerio) Embryos/Larvae. Toxics 2022, 10, 776. [Google Scholar] [CrossRef]
- Liu, H.; Sheng, N.; Zhang, W.; Dai, J. Toxic effects of perfluorononanoic acid on the development of Zebrafish (Danio rerio) embryos. J. Environ. Sci. 2015, 32, 26–34. [Google Scholar] [CrossRef]
- Hagenaars, A.; Vergauwen, L.; De Coen, W.; Knapen, D. Structure-activity relationship assessment of four perfluorinated chemicals using a prolonged zebrafish early life stage test. Chemosphere 2011, 82, 764–772. [Google Scholar] [CrossRef]
- Qian, Y.; Ducatman, A.; Ward, R.; Leonard, S.; Bukowski, V.; Lan Guo, N.; Shi, X.; Vallyathan, V.; Castranova, V. Perfluorooctane sulfonate (PFOS) induces reactive oxygen species (ROS) production in human microvascular endothelial cells: Role in endothelial permeability. J. Toxicol. Environ. Health Part A 2010, 73, 819–836. [Google Scholar] [CrossRef] [PubMed]
- Malvandi, A.M.; Shahba, S.; Mehrzad, J.; Lombardi, G. Metabolic Disruption by Naturally Occurring Mycotoxins in Circulation: A Focus on Vascular and Bone Homeostasis Dysfunction. Front. Nutr. 2022, 9, 915681. [Google Scholar] [CrossRef]
- Hope, J. A review of the mechanism of injury and treatment approaches for illness resulting from exposure to water-damaged buildings, mold, and mycotoxins. Sci. World J. 2013, 2013, 767482. [Google Scholar] [CrossRef]
- Alshannaq, A.; Yu, J.H. Occurrence, Toxicity, and Analysis of Major Mycotoxins in Food. Int. J. Environ. Res. Public Health 2017, 14, 632. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Xu, Z.L.; Yu, C.; Xu, X.H. Effects of aflatoxin B1 on mitochondrial respiration, ROS generation and apoptosis in broiler cardiomyocytes. Anim. Sci. J. 2017, 88, 1561–1568. [Google Scholar] [CrossRef]
- Adeleye, A.S.; Xue, J.; Zhao, Y.; Taylor, A.A.; Zenobio, J.E.; Sun, Y.; Han, Z.; Salawu, O.A.; Zhu, Y. Abundance, fate, and effects of pharmaceuticals and personal care products in aquatic environments. J. Hazard. Mater. 2022, 424, 127284. [Google Scholar] [CrossRef]
- Chopra, S.; Kumar, D. Ibuprofen as an emerging organic contaminant in environment, distribution and remediation. Heliyon 2020, 6, e04087. [Google Scholar] [CrossRef] [PubMed]
- Schjerning, A.-M.; McGettigan, P.; Gislason, G. Cardiovascular effects and safety of (non-aspirin) NSAIDs. Nat. Rev. Cardiol. 2020, 17, 574–584. [Google Scholar] [CrossRef]
- Santos, J.L.; Aparicio, I.; Callejón, M.; Alonso, E. Occurrence of pharmaceutically active compounds during 1-year period in wastewaters from four wastewater treatment plants in Seville (Spain). J. Hazard. Mater. 2009, 164, 1509–1516. [Google Scholar] [CrossRef]
- Han, E.J.; Lee, D.S. Significance of metabolites in the environmental risk assessment of pharmaceuticals consumed by human. Sci. Total Environ. 2017, 592, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Maric, F. Mitigating the environmental impact of NSAIDs—Physiotherapy as a contribution to One Health and the SDGs. Eur. J. Physiother. 2021, 25, 51–55. [Google Scholar] [CrossRef]
- Tambosi, J.; Yamanaka, L.; Humberto, J.; José, H.; De, R.; Moreira, R.; Moreira, M.; Schröder, H. Recent research data on the removal of pharmaceuticals from sewage treatment plants (STP). Quim. Nova 2010, 33, 411–420. [Google Scholar] [CrossRef]
- Tyumina, E.A.; Bazhutin, G.A.; Cartagena Gómez, A.d.P.; Ivshina, I.B.J.M. Nonsteroidal Anti-inflammatory Drugs as Emerging Contaminants. Microbiology 2020, 89, 148–163. [Google Scholar] [CrossRef]
- Bouissou-Schurtz, C.; Houeto, P.; Guerbet, M.; Bachelot, M.; Casellas, C.; Mauclaire, A.-C.; Panetier, P.; Delval, C.; Masset, D. Ecological risk assessment of the presence of pharmaceutical residues in a French national water survey. Regul. Toxicol. Pharmacol. 2014, 69, 296–303. [Google Scholar] [CrossRef]
- Tiwari, S.; Mishra, M.; Salemi, M.R.; Phinney, B.S.; Newens, J.L.; Gomes, A.V. Gender-specific changes in energy metabolism and protein degradation as major pathways affected in livers of mice treated with ibuprofen. Sci. Rep. 2020, 10, 3386. [Google Scholar] [CrossRef]
- Arfè, A.; Scotti, L.; Varas-Lorenzo, C.; Nicotra, F.; Zambon, A.; Kollhorst, B.; Schink, T.; Garbe, E.; Herings, R.; Straatman, H.; et al. Non-steroidal anti-inflammatory drugs and risk of heart failure in four European countries: Nested case-control study. BMJ 2016, 354, i4857. [Google Scholar] [CrossRef]
- Kohli, P.; Steg, P.G.; Cannon, C.P.; Smith, S.C., Jr.; Eagle, K.A.; Ohman, E.M.; Alberts, M.J.; Hoffman, E.; Guo, J.; Simon, T.; et al. NSAID use and association with cardiovascular outcomes in outpatients with stable atherothrombotic disease. Am. J. Med. 2014, 127, 53–60.e51. [Google Scholar] [CrossRef]
- Aw, T.J.; Haas, S.J.; Liew, D.; Krum, H. Meta-analysis of cyclooxygenase-2 inhibitors and their effects on blood pressure. Arch. Intern. Med. 2005, 165, 490–496. [Google Scholar] [CrossRef]
- White, W.B.; Kent, J.; Taylor, A.; Verburg, K.M.; Lefkowith, J.B.; Whelton, A. Effects of celecoxib on ambulatory blood pressure in hypertensive patients on ACE inhibitors. Hypertension 2002, 39, 929–934. [Google Scholar] [CrossRef]
- Kumar, B.; Swee, M.L. Nonsteroidal Anti-inflammatory Drug Use in a Patient With Hypertension: A Teachable Moment. JAMA Intern. Med. 2015, 175, 892–893. [Google Scholar] [CrossRef] [PubMed]
- Kömhoff, M.; Grone, H.J.; Klein, T.; Seyberth, H.W.; Nüsing, R.M. Localization of cyclooxygenase-1 and -2 in adult and fetal human kidney: Implication for renal function. Am. J. Physiol. 1997, 272, F460–F468. [Google Scholar] [CrossRef]
- Breyer, M.D.; Hao, C.; Qi, Z. Cyclooxygenase-2 selective inhibitors and the kidney. Curr. Opin. Crit. Care 2001, 7, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hortmann, M.; Daiber, A.; Oelze, M.; Ostad, M.A.; Schwarz, P.M.; Xu, H.; Xia, N.; Kleschyov, A.L.; Mang, C.; et al. Cyclooxygenase 2-selective and nonselective nonsteroidal anti-inflammatory drugs induce oxidative stress by up-regulating vascular NADPH oxidases. J. Pharmacol. Exp. Ther. 2008, 326, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Hwang, S.M.; Cui, Z.; Gilda, J.E.; Gomes, A.V. Different effects of the nonsteroidal anti-inflammatory drugs meclofenamate sodium and naproxen sodium on proteasome activity in cardiac cells. J. Mol. Cell. Cardiol. 2016, 94, 131–144. [Google Scholar] [CrossRef]
- Husain, M.A.; Sarwar, T.; Rehman, S.U.; Ishqi, H.M.; Tabish, M. Ibuprofen causes photocleavage through ROS generation and intercalates with DNA: A combined biophysical and molecular docking approach. Phys. Chem. Chem. Phys. 2015, 17, 13837–13850. [Google Scholar] [CrossRef]
- Kim, M.; Lee, E.J.; Lim, K.M. Ibuprofen Increases the Hepatotoxicity of Ethanol through Potentiating Oxidative Stress. Biomol. Ther. 2021, 29, 205–210. [Google Scholar] [CrossRef]
- Sule, R.O.; Phinney, B.S.; Salemi, M.R.; Gomes, A.V. Mitochondrial and Proteasome Dysfunction Occurs in the Hearts of Mice Treated with Triazine Herbicide Prometryn. Int. J. Mol. Sci. 2023, 24, 15266. [Google Scholar] [CrossRef]
- Gilda, J.E.; Gomes, A.V. Proteasome dysfunction in cardiomyopathies. J. Physiol. 2017, 595, 4051–4071. [Google Scholar] [CrossRef]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef]
- Liou, J.Y.; Wu, C.C.; Chen, B.R.; Yen, L.B.; Wu, K.K. Nonsteroidal anti-inflammatory drugs induced endothelial apoptosis by perturbing peroxisome proliferator-activated receptor-delta transcriptional pathway. Mol. Pharmacol. 2008, 74, 1399–1406. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, C.; Duan, X.; Liang, B.; Genbo Xu, E.; Huang, Z. Micro- and nanoplastics: A new cardiovascular risk factor? Environ. Int. 2023, 171, 107662. [Google Scholar] [CrossRef] [PubMed]
- Roshanzadeh, A.; Oyunbaatar, N.E.; Ganjbakhsh, S.E.; Park, S.; Kim, D.S.; Kanade, P.P.; Lee, S.; Lee, D.W.; Kim, E.S. Exposure to nanoplastics impairs collective contractility of neonatal cardiomyocytes under electrical synchronization. Biomaterials 2021, 278, 121175. [Google Scholar] [CrossRef]
- Lee, W.S.; Cho, H.J.; Kim, E.; Huh, Y.H.; Kim, H.J.; Kim, B.; Kang, T.; Lee, J.S.; Jeong, J. Bioaccumulation of polystyrene nanoplastics and their effect on the toxicity of Au ions in zebrafish embryos. Nanoscale 2019, 11, 3173–3185. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y.; Lu, L.; Zheng, M.; Zhang, X.; Tian, H.; Wang, W.; Ru, S. Polystyrene microplastics cause tissue damages, sex-specific reproductive disruption and transgenerational effects in marine medaka (Oryzias melastigma). Environ. Pollut. 2019, 254, 113024. [Google Scholar] [CrossRef]
- Lonn, E.; Bosch, J.; Yusuf, S.; Sheridan, P.; Pogue, J.; Arnold, J.M.; Ross, C.; Arnold, A.; Sleight, P.; Probstfield, J.; et al. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: A randomized controlled trial. JAMA 2005, 293, 1338–1347. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Elfowiris, A.; Banigesh, A. Evaluation of Antioxidant Therapeutic Value of Ace Inhibitor as Adjunct Therapy on T2dm Patients with Cardiovascular Disease. Int. J. Stroke 2023, 18, 310. [Google Scholar]
- Das, S.K.; Nerune, S.M.; Das, K.K. Antioxidant therapy for hepatic diseases: A double-edged sword. J. Basic Clin. Physiol. Pharmacol. 2024, 35, 7–14. [Google Scholar] [CrossRef]
- Stocker, R.; Keaney, J.F., Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef]
- Kukreja, R.C.; Salloum, F.N. Pharmacological preconditioning with natural compounds. Vasc. Pharmacol. 2009, 51, 228–243. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Graham, D.; Huynh, N.N.; Hamilton, C.A.; Beattie, E.; Smith, R.A.; Cocheme, H.M.; Murphy, M.P.; Dominiczak, A.F. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension 2009, 54, 322–328. [Google Scholar] [CrossRef]
- Su, X.; Wang, S.; Zhang, H.; Yang, G.; Bai, Y.; Liu, P.; Meng, L.; Jiang, X.; Xin, Y. Sulforaphane prevents angiotensin II-induced cardiomyopathy by activation of Nrf2 through epigenetic modification. J. Cell. Mol. Med. 2021, 25, 4408–4419. [Google Scholar] [CrossRef]
- Rana, A.; Rera, M.; Walker, D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 8638–8643. [Google Scholar] [CrossRef]
- Li, J.; Ichikawa, T.; Janicki, J.S.; Cui, T. Targeting the Nrf2 pathway against cardiovascular disease. Expert Opin. Ther. Targets. 2009, 13, 785–794. [Google Scholar] [CrossRef]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyürek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308. [Google Scholar] [CrossRef]
- Zou, Z.V.; Le, G.K.; El Zowalaty, A.E.; Pehlivanoglu, L.E.; Garellick, V.; Gul, N.; Ibrahim, M.X.; Bergh, P.O.; Henricsson, M.; Wiel, C.; et al. Antioxidants Promote Intestinal Tumor Progression in Mice. Antioxidants 2021, 10, 241. [Google Scholar] [CrossRef]
- Kashino, I.; Mizoue, T.; Serafini, M.; Akter, S.; Sawada, N.; Ishihara, J.; Kotemori, A.; Inoue, M.; Yamaji, T.; Goto, A.; et al. Higher Dietary Non-enzymatic Antioxidant Capacity Is Associated with Decreased Risk of All-Cause and Cardiovascular Disease Mortality in Japanese Adults. J. Nutr. 2019, 149, 1967–1976. [Google Scholar] [CrossRef]
- Ma, J.; Li, H. The Role of Gut Microbiota in Atherosclerosis and Hypertension. Front. Pharmacol. 2018, 9, 1082. [Google Scholar] [CrossRef]
- Jonsson, A.L.; Backhed, F. Role of gut microbiota in atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 79–87. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environmental Toxin | Enzymes and Mechanisms Involved in ROS Production | Effect on Cardiovascular Disease |
|---|---|---|
| Air Pollutants | ||
| Particulate Matter (air pollutant) | Fine particulate matter in polluted air, such as Particulate Matter 2.5 (PM2.5) and PM10, can generate ROS in the respiratory tract when inhaled, by increased NADPH oxidases (NOX), increased leaking of electrons from the mitochondrial electron transport chains (ETCs), stimulation of immune cells that produce ROS, decrease of glutathione (GSH) levels, and decrease of catalase (CAT), glutathione S transferase (GST), and superoxide dismutase (SOD) activity [11,12,13]. Particulate matter can induce inflammation, which increases ROS production via cytokine signaling (e.g., tumor necrosis factor-alpha (TNF-α)) [14,15]. | Induces cardiac dysfunction, increased cardiac injury, and acute myocardial infarction in Apolipoprotein E−/− (ApoE−/−) mice [16]. |
| Increased blood expression, heart weight, cardiac expression of hypertrophic markers (ACTA1 (α-actin) and MYH7 (β-myosin heavy chain)), and decreased cardiac stroke volume and output in rats [17]. | ||
| Cardiac hypertrophy and fibrosis led to a decrease in cardiac systolic function in mice [18]. | ||
| Cardiac fibrosis in 4-week and 10-month-old mice. 10-month-old mice demonstrated cardiac diastolic dysfunction, elevated heart rate/blood pressure, and cardiac systolic dysfunction [19]. | ||
| Aggravates left ventricle failure-induced pulmonary hypertension in mice [20]. | ||
| Decreased cardiac growth, cardiac dysfunction, abnormal mitochondrial structure and function, and cardiac metabolic disorder [21]. | ||
| Neonatal and adult cardiac dysfunction [22]. | ||
| Oil Mist PM induces oxidative stress, inflammation, and mitochondrial dysfunction, inducing myocardial tissue injury [23]. | ||
| Polluted urban air aggravates myocardial infarction by enhancing mitochondrial H2O2 production and altering mitochondrial ultrastructure and function [24]. | ||
| Ozone (O3) (air pollutant) | Ground-level ozone, a component of smog, can induce ROS in the heart by increasing the activity of NOX, increased leaking of electrons from the ETC and reducing SOD and CAT activity [25,26,27]. | Platelet aggregation [28]. |
| Increased heart rate variability. Significant increase in the number of cardiac arrhythmias [29]. | ||
| Heavy Metals | ||
| Lead (heavy metal) | Lead stimulates ROS production through various mechanisms, including ETC complexes, NOX, and xanthine oxidase (XO) [30,31]. Lead reduces SOD, CAT, and glutathione peroxidase (GPx) activities. Lead elevates cytosolic Ca2+, activating protein kinase C (PKC) and phospholipases that amplify ROS generation [30]. | Elevated arterial pressure [32]. |
| Increased systolic blood pressure, increased prevalence of left ventricular hypertrophy in humans [33]. | ||
| Left ventricular hypertrophy and alterations in cardiac rhythm [34,35]. | ||
| Hypertension—marked rise in systolic blood pressure [36]. | ||
| Mercury (heavy metal) | Mercury exposure, especially in its methylmercury form found in contaminated fish, can lead to ROS production through various mechanisms, including ETC complexes and NOX. Mercury reduces GPx and thioredoxin reductase (TrxR) activity [37,38,39,40]. It stimulates Nitric oxide synthase (NOS) to produce nitric oxide (NO), which can form peroxynitrite, a potent oxidant [39,41]. | Increased platelet aggregation and thrombosis [42]. |
| Increased systolic blood pressure [43]. | ||
| Increased arterial blood pressure [44]. | ||
| Hypertension, generalized atherosclerosis, coronary heart disease (CHD), myocardial infarction (MI), cardiac arrhythmias, heart rate variability, sudden death, cerebrovascular accidents (CVA), and carotid artery disease [45]. | ||
| Increased nonfatal ischemic heart disease (IHD), CVD mortality, and mortality due to other heart diseases [46]. | ||
| Cadmium (heavy metal) | Cadmium (Cd) stimulates ROS production via ETC complexes and NOX. SOD, CAT, GPx, and GST activities are suppressed [47,48,49]. Cd increases cytosolic Ca2+, activating phospholipases and ROS overproduction [50]. | Atherosclerosis and impaired cardiac function [51]. |
| Elevated blood pressure [52]. | ||
| Acute myocardial infarction and increased CHD mortality [53]. | ||
| Increased formation of atherosclerotic plaques [54]. | ||
| Pesticides | ||
| Organophosphates (pesticide) | Organophosphates can promote ROS production through various mechanisms, including ETC complexes, NOX, XO, cytochrome P450 (CYP)-induced ROS, and reduced SOD, GSH, CAT, and GPx activity [55,56,57]. | High rates of developing arrhythmia, CAD, and congestive heart failure (CHF) [58]. |
| Endocrine-Disrupting Chemicals (EDCs) | ||
| Endocrine-Disrupting Chemicals | EDCs increase ROS by NOX and ETC complexes [59]. Changes in the unfolded protein response (UPR) due to endoplasmic reticulum (ER) stress increase ROS through Ca2+ release and disrupted protein folding. ROS levels are further elevated by lower GSH amounts and lower SOD, CAT, and GPx enzyme activities [60,61]. | High-dose BPA exposure in fetal mouse heart development causes oxidative stress, mitochondrial dysfunction, and congenital heart defects [62]. |
| Bisphenol-A induced arrhythmia and triggered atherosclerosis in rats [63,64]. | ||
| Polychlorinated Biphenyls (PCBs) | ||
| Polychlorinated Biphenyls (PCBs) | PCBs induce ROS production by NOX, CYP, and ETC complexes [65]. Decreased SOD and GSH activities increase ROS levels [66]. | Increased risk of hypertension [67]. |
| Elevated risk of hospitalization for CHD, acute myocardial infarction (AMI), and stroke [68]. | ||
| Increased heart failure risk [69]. | ||
| Increased rate of hypertension [70]. | ||
| Dioxins | ||
| Dioxin | Dioxins found in certain environmental pollutants, like the byproducts of waste incineration, can stimulate ROS production, leading to oxidative stress and various health issues. ROS levels are increased by CYP-derived ROS, decreased CAT and SOD activities, and lower GSH levels [71,72,73]. | Increased incidence of degenerative cardiovascular lesions, including cardiomyopathy and chronic active arteritis in rats [74]. |
| Increased blood pressure and heart weight in mice [75]. | ||
| Increased blood pressure and development of severe atherosclerotic lesions in ApoE−/− mice [76]. | ||
| Associated with mortality from ischemic heart disease (IHD); reduced blood supply to the heart [77]. | ||
| Induced dilated cardiomyopathy and myocardial hypoxia in avian embryos [78]. | ||
| Several forms of congenital cardiovascular malformations in humans [79]. | ||
| Cardiac hypertrophy and bradycardia in mice [80,81]. | ||
| Asbestos | ||
| Asbestos | Asbestos fibers can induce ROS production when inhaled, contributing to oxidative stress in lung tissues. ROS levels are increased by ETC complexes and decreased GSH levels [82,83]. | Increased risk of cardiovascular-related diseases in exposed workers [84]. |
| Radon | ||
| Radon | Inhalation of radon causes increased ROS generation via radiation-induced ionization [85]. Radon disrupts ETC complexes and depletes SOD, resulting in increased ROS levels [86]. | Radon accelerates low-density lipoprotein (LDL) accumulation, foam cell formation, and arterial thickening or fibrosis [85]. |
| Tobacco Smoke | ||
| Cigarette Smoke | Tobacco smoke contains a mixture of toxic chemicals, including ROS-generating compounds like free radicals and reactive carbonyls, which contribute to oxidative stress and are linked to smoking-related diseases. Increased ROS production comes from NOX, CYP, NOS, and ETC complexes. Lower levels of GSH, SOD, and CAT weaken antioxidant defenses, leading to increased ROS accumulation [87,88,89,90]. | Atherosclerotic plaques in the carotid artery [91]. |
| Increases the risk of ischemic stroke [92]. | ||
| Mold and Indoor Air Contaminants | ||
| Mold Byproduct | T-2 mycotoxin increases higher ROS levels via NOX, CYP, and ETC complexes. Reduced GSH levels and SOD and CAT activity further raise ROS levels [93,94,95]. | T-2 mycotoxin induces interstitial hemorrhage, capillary dilation, mitochondrial impairment, and fibrotic damage [96]. |
| Pharmaceuticals and Personal Care Products | ||
| Pharmaceuticals and Personal Care Products | NSAIDs such as naproxen and ibuprofen are associated with increased ROS production. This occurs by NOX, CYP, ETC complexes, and NOS, which contribute to peroxynitrite. ROS levels are further elevated due to decreased CAT and GPx activities [97]. | Increased risk of stroke, hypertension, and coronary artery disease [97]. |
| Induced abnormal cardiac function and morphology (pericardial effusion) and abnormal heart rate in zebrafish embryos [98]. | ||
| Radiation | ||
| Radiation | Exposure to ionizing radiation, such as from nuclear accidents or medical procedures, can lead to upregulation of several enzymes, including NOX and NOS, that generate ROS [99]. ROS is also induced by ETC complexes, and decreased SOD activity contributes to higher ROS levels [99,100]. | Fibrosis significantly increases the risk of coronary artery disease, cardiomyopathy, valvulopathy, arrhythmias, and pericardial disease [101,102,103,104]. |
| Chemical Waste and Spills | ||
| Chemical Waste and Spills | Some organic solvents used in industrial processes, like benzene and toluene, can produce ROS when metabolized in the body, contributing to oxidative stress and potential toxicity. Chemical waste-induced ROS is often produced by NOX, CYP, ETC complexes, peroxisomal oxidases, and XO. ROS also increases due to reduced SOD and CAT activity [105,106,107,108]. | Mice inhaling volatile benzene had significantly reduced levels of circulating angiogenic cells (Flk-1+/Sca-1+) and increased levels of plasma LDL [109]. |
| Microplastics | ||
| Microplastics | Microplastics induce ROS production via ETC complexes. ROS also increases due to reduced SOD, GPx, and CAT activity [110,111]. | Cardiac fibrosis and damage in Wistar rats [112]. |
| Damaged cardiac structure and function [113]. | ||
| Cardiotoxicity, pericardial edema, and impaired heart rate in fish cardiac tissue [114]. | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sule, R.O.; Rivera, G.D.T.; Vaidya, T.; Gartrell, E.; Gomes, A.V. Environmental Toxins and Oxidative Stress: The Link to Cardiovascular Diseases. Antioxidants 2025, 14, 604. https://doi.org/10.3390/antiox14050604
Sule RO, Rivera GDT, Vaidya T, Gartrell E, Gomes AV. Environmental Toxins and Oxidative Stress: The Link to Cardiovascular Diseases. Antioxidants. 2025; 14(5):604. https://doi.org/10.3390/antiox14050604
Chicago/Turabian StyleSule, Rasheed O., Gabriela Del Toro Rivera, Tanishq Vaidya, Emily Gartrell, and Aldrin V. Gomes. 2025. "Environmental Toxins and Oxidative Stress: The Link to Cardiovascular Diseases" Antioxidants 14, no. 5: 604. https://doi.org/10.3390/antiox14050604
APA StyleSule, R. O., Rivera, G. D. T., Vaidya, T., Gartrell, E., & Gomes, A. V. (2025). Environmental Toxins and Oxidative Stress: The Link to Cardiovascular Diseases. Antioxidants, 14(5), 604. https://doi.org/10.3390/antiox14050604