
The Key Role and Mechanism of Oxidative Stress in Hypertrophic Cardiomyopathy: A Systematic Exploration Based on Multi-Omics Analysis and Experimental Validation

Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Identification and Functional Annotation of Differentially Expressed Genes (DEGs)
2.3. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.4. Functional Enrichment Analysis and Protein–Protein Interaction (PPI) Network Construction
2.5. Machine Learning Model Construction and Trait Gene Screening
2.6. Construction and Validation of Diagnostic Model
2.7. Correlation Analysis Between Characteristic Genes and Immune Cells
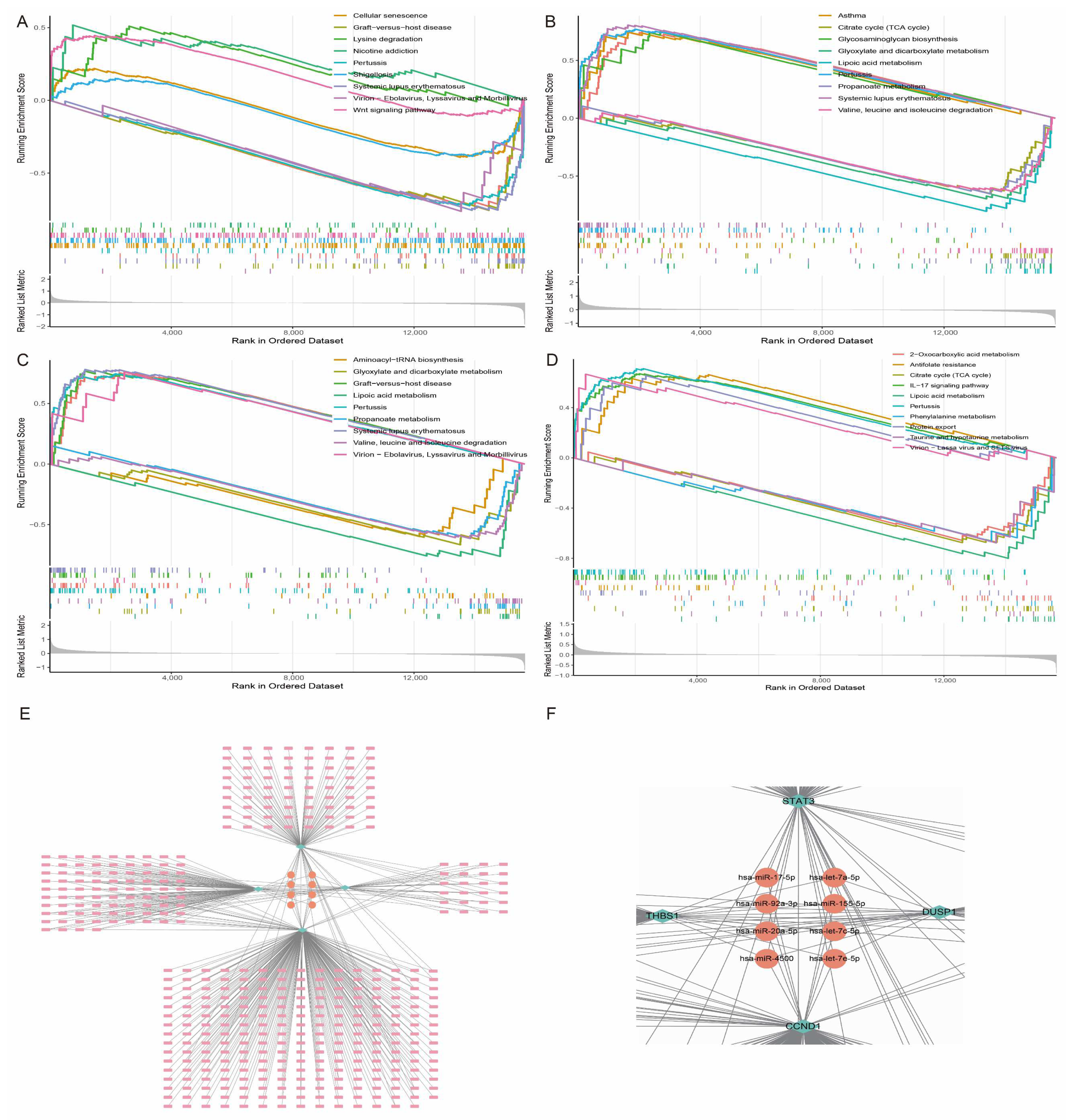
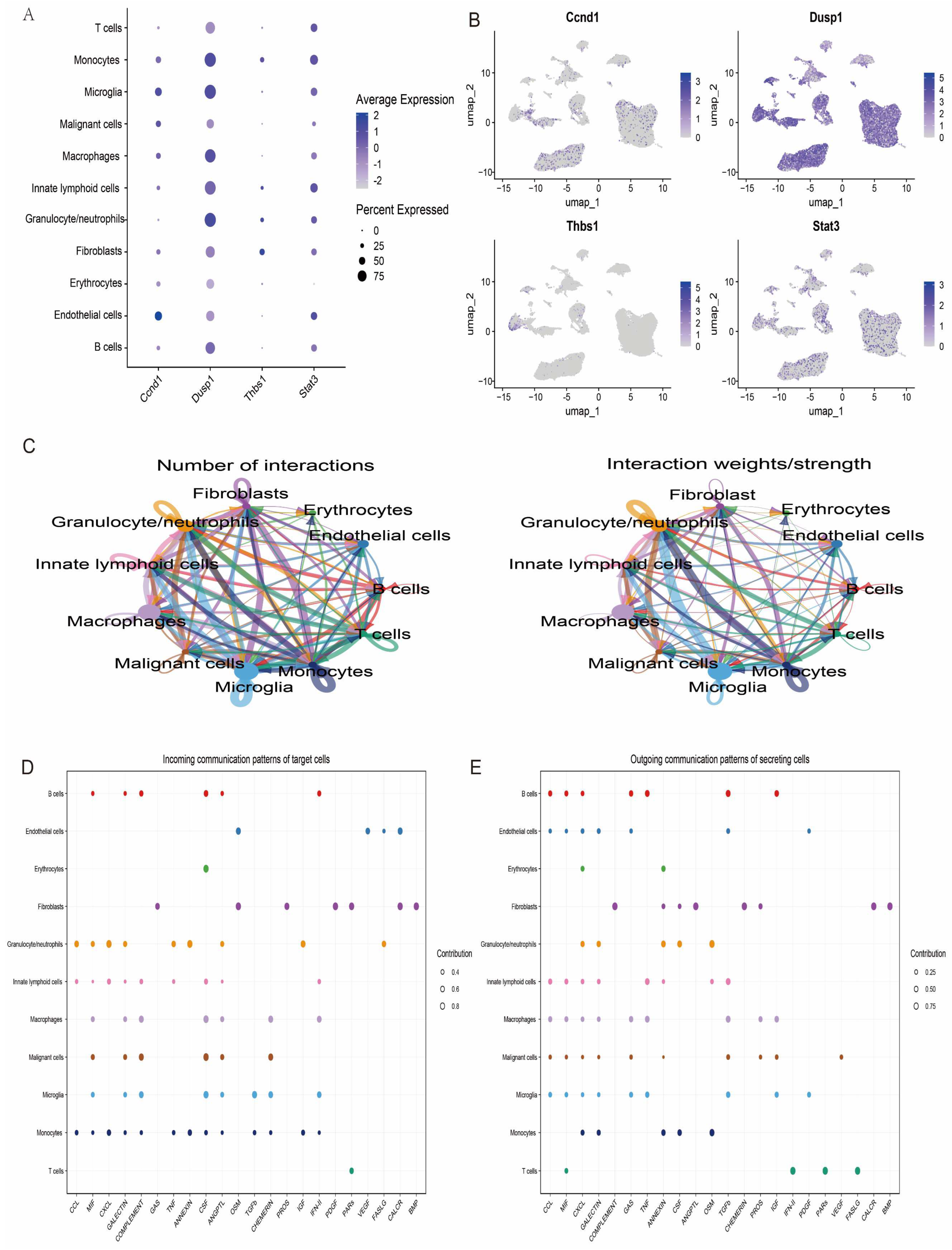
2.8. Hub Gene Enrichment and miRNA-mRNA Regulatory Network Analysis
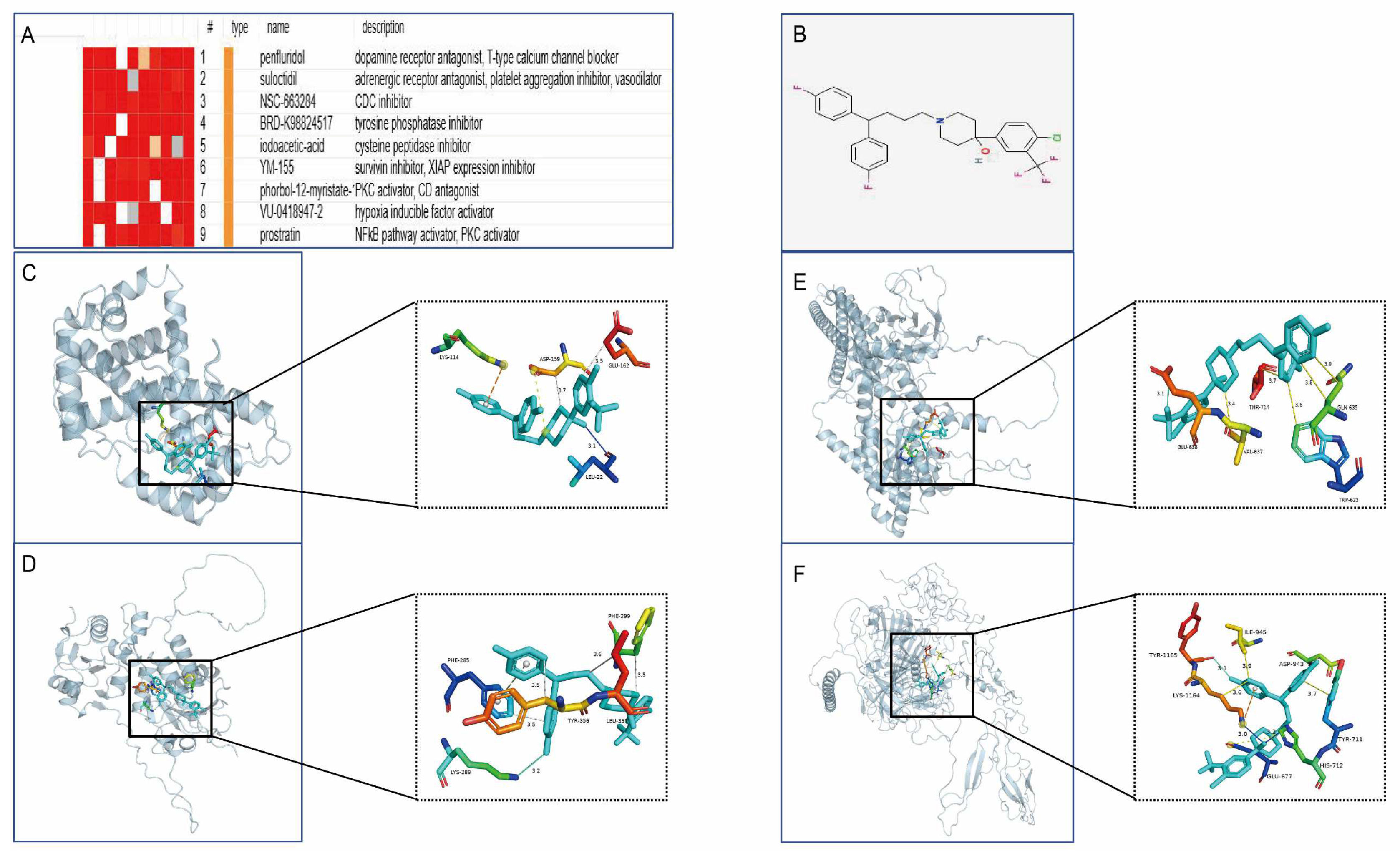
2.9. Drug Prediction and Molecular Docking Analysis
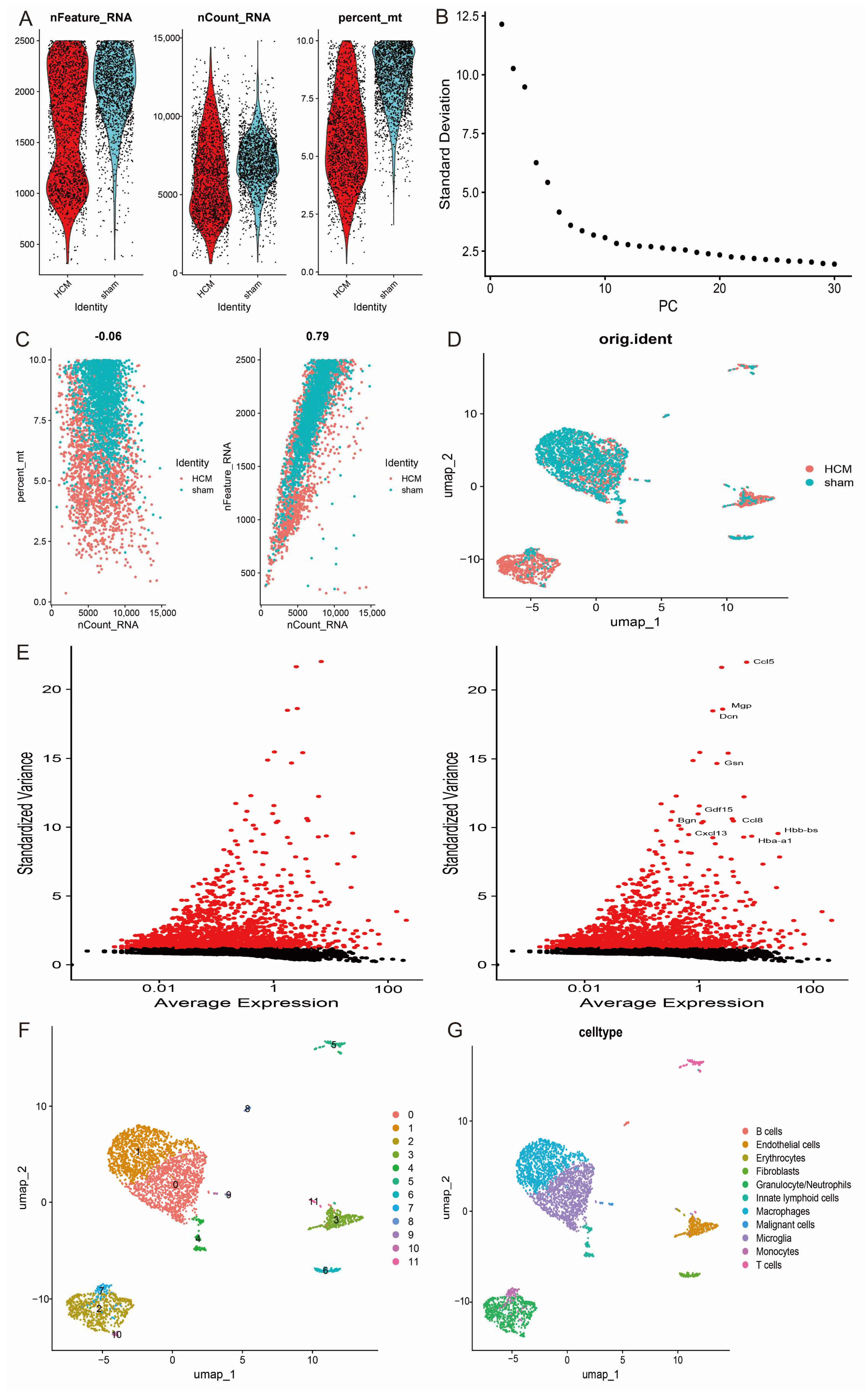
2.10. Analysis of Single-Cell RNA Sequencing Data (scRNA-seq)
2.11. Cell Culture and In Vitro Model Construction
2.12. qPCR In Vivo Validation
2.13. Statistical Analysis
3. Results
3.1. Characterisation of HCM Based on Oxidative Stress-Related Genes
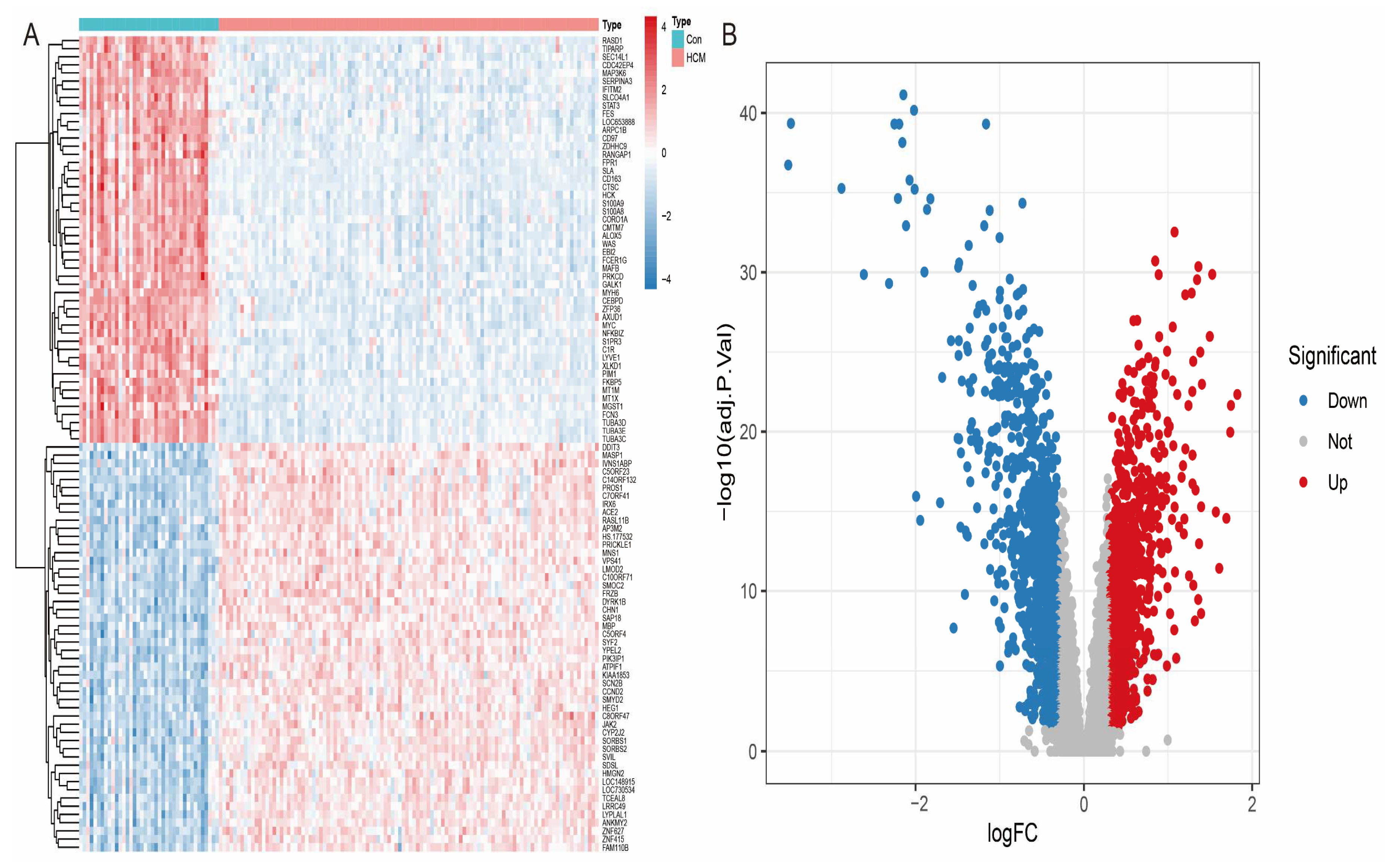
3.2. Identification of Differentially Expressed Genes (DEGs) in GSE36961 Cohort
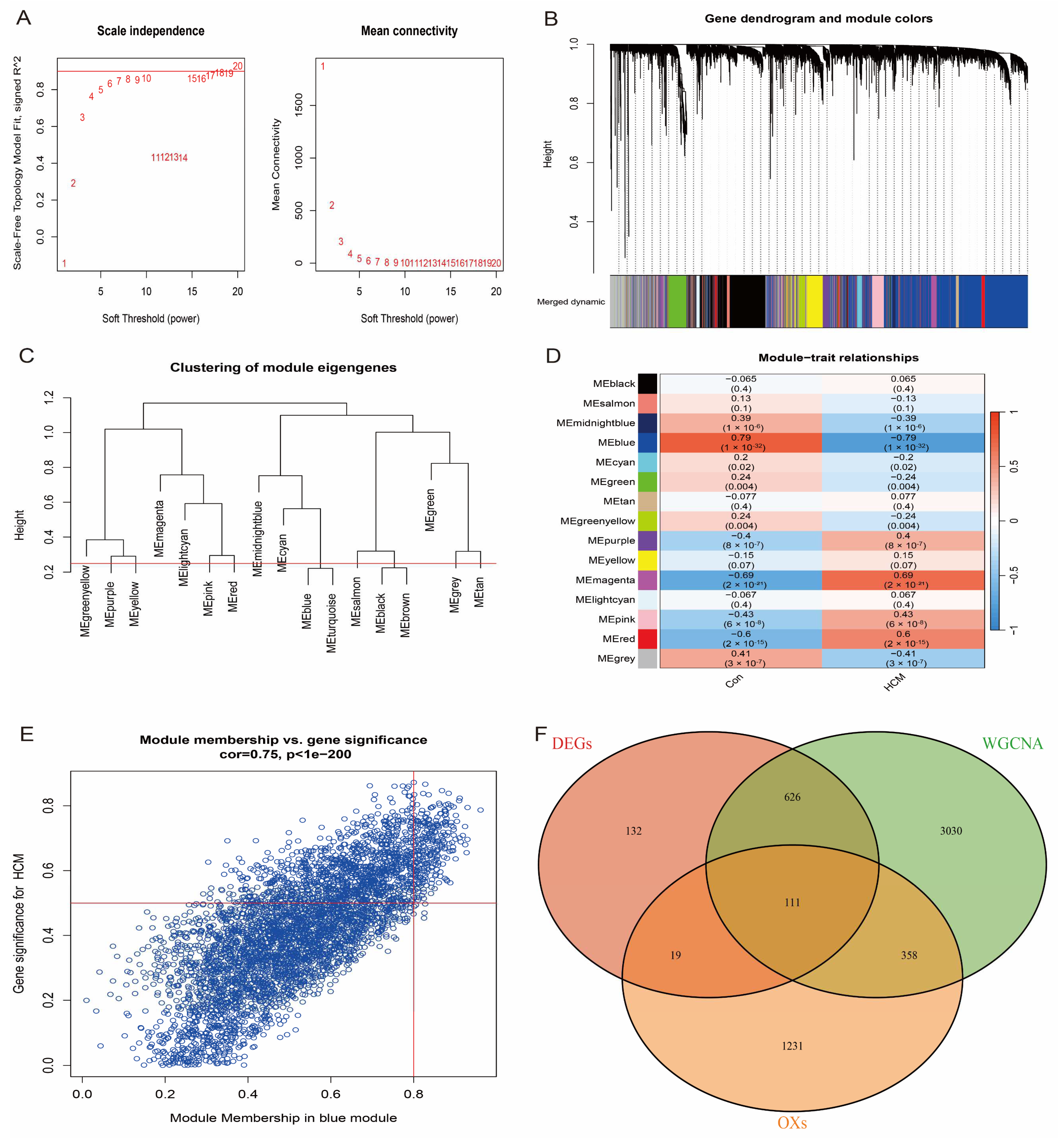
3.3. WGCNA Identifies Modular Genes Associated with HCM
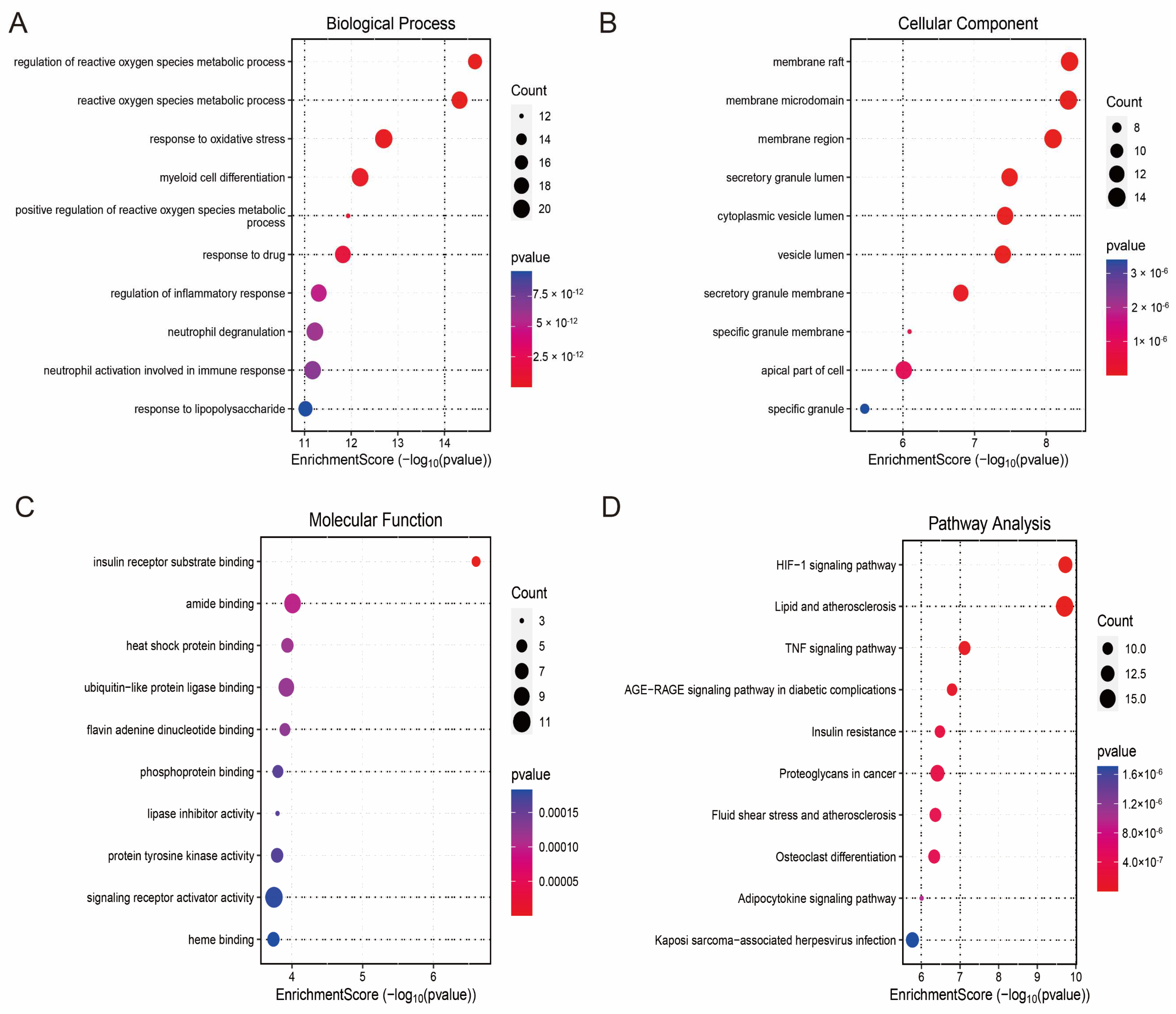
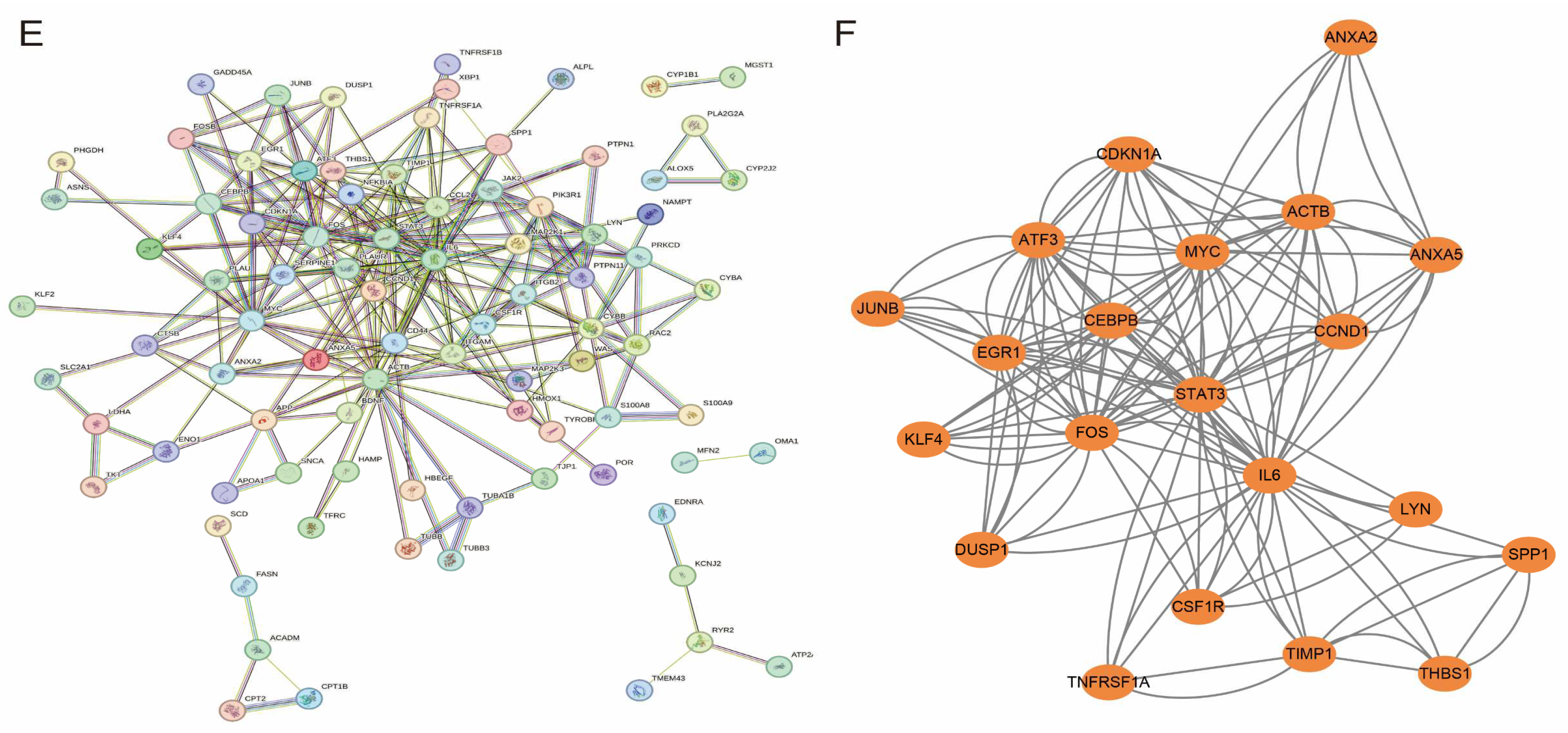
3.4. Enrichment Analysis and PPI Network
3.5. Feature Gene Selection
3.6. Evaluation of Characteristic Genes
3.7. Correlation Between Characterised Genes and Immune Cells
3.8. Characteristic Gene Enrichment Analysis and miRNA-mRNA Regulatory Network
3.9. Single-Cell Analysis
3.10. Drug Prediction and Molecular Docking
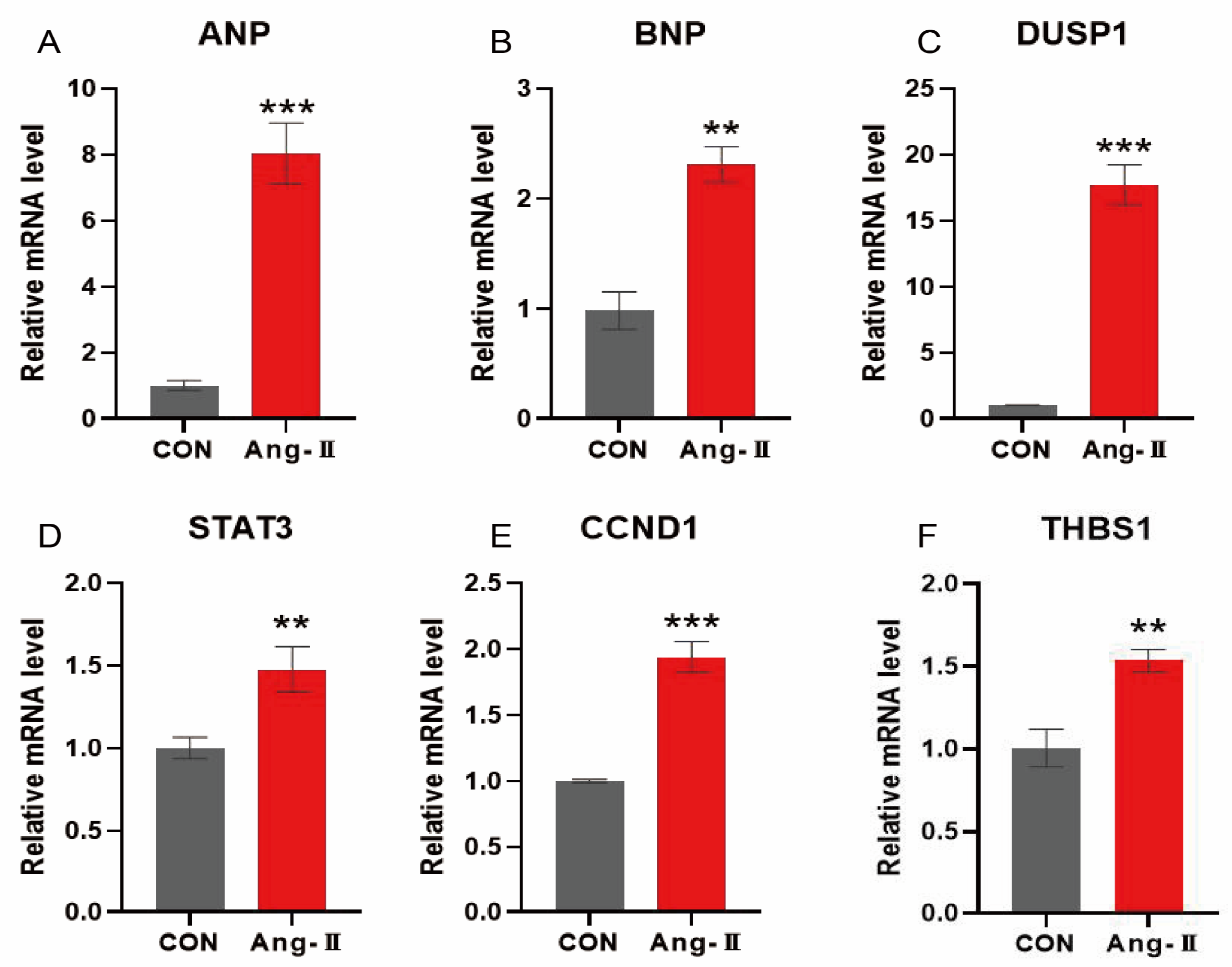
3.11. Angiotensin-Induced mRNA Expression of Hypertrophy-Characterising Genes in H9c2 Cardiomyocytes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maron, B.J.; Maron, M.S. Hypertrophic cardiomyopathy. Lancet 2013, 381, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Tuohy, C.V.; Kaul, S.; Song, H.K.; Nazer, B.; Heitner, S.B. Hypertrophic cardiomyopathy: The future of treatment. Eur. J. Heart Fail. 2020, 22, 228–240. [Google Scholar] [CrossRef]
- Wang, L.; Li, L.; Zhao, D.; Yuan, H.; Zhang, H.; Chen, J.; Pang, D.; Lu, Y.; Ouyang, H. MYH7 R453C induced cardiac remodelling via activating TGF-β/Smad2/3, ERK1/2 and Nox4/ROS/NF-κB signalling pathways. Open Biol. 2024, 14, 230427. [Google Scholar] [CrossRef]
- Reid, A.; Miller, C.; Farrant, J.P.; Polturi, R.; Clark, D.; Ray, S.; Cooper, G.; Schmitt, M. Copper chelation in patients with hypertrophic cardiomyopathy. Open Heart 2022, 9, e001803. [Google Scholar] [CrossRef]
- Valencia, A.; Abrantes, M.; Hasan, J.; Aranda, J.; Beharry, K. Reactive Oxygen Species, Biomarkers of Microvascular Maturation and Alveolarization, and Antioxidants in Oxidative Lung Injury. React. Oxyg. Species 2018, 6, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Engebretsen, S.; Bohlin, J. Statistical predictions with glmnet. Clin. Epigenet. 2019, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Sanz, H.; Valim, C.; Vegas, E.; Oller, J.M.; Reverter, F. SVM-RFE: Selection and visualization of the most relevant features through non-linear kernels. BMC Bioinform. 2018, 19, 432. [Google Scholar] [CrossRef]
- Tian, L.; Wu, W.; Yu, T. Graph Random Forest: A Graph Embedded Algorithm for Identifying Highly Connected Important Features. Biomolecules 2023, 13, 1153. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liu, Y.; Yang, J.; Liu, Q.; You, H.; Dong, Y.; Lyu, J. Development and Validation of a Nomogram for Predicting Survival in Male Patients With Breast Cancer. Front. Oncol. 2019, 9, 361. [Google Scholar] [CrossRef]
- Wolbers, M.; Koller, M.T.; Witteman, J.C.M.; Steyerberg, E.W. Prognostic Models With Competing Risks. Epidemiology 2009, 20, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.; Han, S.J.; Sughrue, M.E.; Tihan, T.; Parsa, A.T. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: Evidence of distinct immunological microenvironments that reflect tumor biology. J. Neurosurg. 2011, 115, 505–511. [Google Scholar] [CrossRef]
- Xia, J.; Gill, E.E.; Hancock, R.E.W. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844. [Google Scholar] [CrossRef]
- Yang, K.; Dinasarapu, A.R.; Reis, E.S.; Deangelis, R.A.; Ricklin, D.; Subramaniam, S.; Lambris, J.D. CMAP: Complement Map Database. Bioinformatics 2013, 29, 1832–1833. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Hou, W.; Ji, Z. Assessing GPT-4 for cell type annotation in single-cell RNA-seq analysis. Nat. Methods 2024, 21, 1462–1465. [Google Scholar] [CrossRef]
- Jin, S.; Plikus, M.V.; Nie, Q. CellChat for systematic analysis of cell–cell communication from single-cell transcriptomics. Nat. Protoc. 2025, 20, 180–219. [Google Scholar] [CrossRef] [PubMed]
- Teekakirikul, P.; Zhu, W.; Huang, H.C.; Fung, E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules 2019, 9, 878. [Google Scholar] [CrossRef]
- Chou, C.; Chin, M.T. Genetic and Molecular Mechanisms of Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 2522. [Google Scholar] [CrossRef]
- Afanas’Ev, I. ROS and RNS Signaling in Heart Disorders: Could Antioxidant Treatment Be Successful? Oxidative Med. Cell. Longev. 2011, 2011, 293769. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Kass, D.A. Role of Oxidative Stress in Cardiac Hypertrophy and Remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Biagini, E.; Olivotto, I.; Iascone, M.; Parodi, M.I.; Girolami, F.; Frisso, G.; Autore, C.; Limongelli, G.; Cecconi, M.; Maron, B.J.; et al. Significance of Sarcomere Gene Mutations Analysis in the End-Stage Phase of Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2014, 114, 769–776. [Google Scholar] [CrossRef]
- Christiansen, L.B.; Dela, F.; Koch, J.; Hansen, C.N.; Leifsson, P.S.; Yokota, T. Impaired cardiac mitochondrial oxidative phosphorylation and enhanced mitochondrial oxidative stress in feline hypertrophic cardiomyopathy. Am. J. Physiol.-Heart Circ. Physiol. 2015, 308, H1237–H1247. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Wen, Y.-C.; Ku, C.-C.; Yang, Y.-C.; Chow, J.-M.; Yang, S.-F.; Lee, W.-J.; Chien, M.-H. Penfluridol triggers cytoprotective autophagy and cellular apoptosis through ROS induction and activation of the PP2A-modulated MAPK pathway in acute myeloid leukemia with different FLT3 statuses. J. Biomed. Sci. 2019, 26, 63. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Liu, R.-H.; Cui, Y.-Z.; Hettinghouse, A.; Fu, W.-Y.; Zhang, L.; Zhang, C.; Liu, C.-J. Penfluridol targets acid sphingomyelinase to inhibit TNF signaling and is therapeutic against inflammatory autoimmune diseases. Arthritis Res. Ther. 2022, 24, 27. [Google Scholar] [CrossRef]
- Hoppstädter, J.; Ammit, A.J. Role of Dual-Specificity Phosphatase 1 in Glucocorticoid-Driven Anti-inflammatory Responses. Front. Immunol. 2019, 10, 1446. [Google Scholar] [CrossRef]
- Auger-Messier, M.; Accornero, F.; Goonasekera, S.A.; Bueno, O.F.; Lorenz, J.N.; Van Berlo, J.H.; Willette, R.N.; Molkentin, J.D. Unrestrained p38 MAPK Activation in Dusp1/4 Double-Null Mice Induces Cardiomyopathy. Circ. Res. 2013, 112, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Xing, W.; Liu, R.; Zhang, Y.; Xie, C.; Gong, F. MiR-32-5p influences high glucose-induced cardiac fibroblast proliferation and phenotypic alteration by inhibiting DUSP1. BMC Mol. Biol. 2019, 20, 21. [Google Scholar] [CrossRef]
- Montalto, F.I.; De Amicis, F. Cyclin D1 in Cancer: A Molecular Connection for Cell Cycle Control, Adhesion and Invasion in Tumor and Stroma. Cells 2020, 9, 2648. [Google Scholar] [CrossRef]
- Kang, J.Q.; Chong, Z.Z.; Maiese, K. Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J. Neurosci. Res. 2003, 74, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Adan, H.; Daniel, J.; Raptis, L. Roads to Stat3 Paved with Cadherins. Cells 2022, 11, 2537. [Google Scholar] [CrossRef]
- Camporeale, A.; Marino, F.; Papageorgiou, A.; Carai, P.; Fornero, S.; Fletcher, S.; Page, B.D.G.; Gunning, P.; Forni, M.; Chiarle, R.; et al. STAT3 activity is necessary and sufficient for the development of immune-mediated myocarditis in mice and promotes progression to dilated cardiomyopathy. EMBO Mol. Med. 2013, 5, 572–590. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, Y.; Thang, B.Q.; Ramirez, K.; Shin, S.J.; Kohata, T.; Ohata, S.; Nguyen, T.A.V.; Ohtsuki, S.; Nagayama, K.; Yanagisawa, H. Matrix mechanotransduction mediated by thrombospondin-1/integrin/YAP in the vascular remodeling. Proc. Natl. Acad. Sci. USA 2020, 117, 9896–9905. [Google Scholar] [CrossRef]
- Captur, G.; Doykov, I.; Chung, S.-C.; Field, E.; Barnes, A.; Zhang, E.; Heenan, I.; Norrish, G.; Moon, J.C.; Elliott, P.M.; et al. Novel Multiplexed Plasma Biomarker Panel Has Diagnostic and Prognostic Potential in Children With Hypertrophic Cardiomyopathy. Circ. Genom. Precis. Med. 2024, 17, e004448. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Primers (5′–3′) | |
|---|---|---|
| ANP (ID: 43595) | F | GGCACTTAGCTCCCTCTC |
| R | CCCTCAGTTTGCTTTTCA | |
| β-MHC (ID: 140781) | F | TGGATGCAGACCTCTCCC |
| R | TGCTTCTTGCCACCCTTG | |
| DUSP1 (ID: 19252) | F | GTTGTTGGATTGTCGCTCCTT |
| R | GTTGTTGGATTGTCGCTCCTT | |
| CCND1 (ID: 12443) | F | GCGTACCCTGACACCAATCTC |
| R | CTCCTCTTCGCACTTCTGCTC | |
| STAT3 (ID: 20848) | F | CAATACCATTGACCTGCCGAT |
| R | GAGCGACTCAAACTGCCCT | |
| THBS1 (ID: 21825) | F | GGGGAGATAACGGTGTGTTTG |
| R | CGGGGATCAGGTTGGCATT | |
| GAPDH (ID: 14433) | F | GAGTCAACGGATTTGGTCGT |
| R | GACAAGCTTCCCGTTCTCAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Li, T.; Sun, S.; Jiang, Y.; Sun, Y.; Meng, Y. The Key Role and Mechanism of Oxidative Stress in Hypertrophic Cardiomyopathy: A Systematic Exploration Based on Multi-Omics Analysis and Experimental Validation. Antioxidants 2025, 14, 557. https://doi.org/10.3390/antiox14050557
Zhang S, Li T, Sun S, Jiang Y, Sun Y, Meng Y. The Key Role and Mechanism of Oxidative Stress in Hypertrophic Cardiomyopathy: A Systematic Exploration Based on Multi-Omics Analysis and Experimental Validation. Antioxidants. 2025; 14(5):557. https://doi.org/10.3390/antiox14050557
Chicago/Turabian StyleZhang, Sijie, Tianzhi Li, Shiyi Sun, Yujiao Jiang, Yuxin Sun, and Yan Meng. 2025. "The Key Role and Mechanism of Oxidative Stress in Hypertrophic Cardiomyopathy: A Systematic Exploration Based on Multi-Omics Analysis and Experimental Validation" Antioxidants 14, no. 5: 557. https://doi.org/10.3390/antiox14050557
APA StyleZhang, S., Li, T., Sun, S., Jiang, Y., Sun, Y., & Meng, Y. (2025). The Key Role and Mechanism of Oxidative Stress in Hypertrophic Cardiomyopathy: A Systematic Exploration Based on Multi-Omics Analysis and Experimental Validation. Antioxidants, 14(5), 557. https://doi.org/10.3390/antiox14050557