1. Background
Pulmonary fibrosis (PF) is a progressive disorder characterized by alveolar architecture destruction and interstitial fibrosis, ultimately leading to respiratory failure and death. No definitive cure is currently available for PF. Cigarette smoking-induced pulmonary inflammation and oxidative stress leading to structural lung damage form the basis of PF development [
1]. Two FDA-approved drugs—nintedanib and pirfenidone—which inhibit fibroblast proliferation and collagen production, are available for the treatment of PF; however, they only slow the disease progression and cannot cure it. Moreover, severe side effects are observed in patients treated with nintedanib and pirfenidone [
2,
3]. Therefore, there is an urgent need to develop new, more effective drugs for PF.
TGF-β1, a key pro-fibrotic factor, is exacerbated by pulmonary inflammation and oxidative stress, leading to PF [
4]. The downstream signaling pathway, TGF-β1-driven PF, primarily involves Smad signaling (Smad2 and Smad3) and non-Smad signaling (MAPK and ERK1/2) pathways [
5]. Activation of the TGF-β1 signaling pathway induces fibroblast proliferation, migration, and fibroblast-to-myofibroblast transition (FMT). TGF-β1 induces myofibroblasts to excessively secrete extracellular matrix (ECM) components, including collagen I and fibronectin, ultimately leading to collagen deposition [
6,
7]. It also upregulates matrix metalloproteinases (MMPs) that selectively degrade ECM in lung tissue, further exacerbating PF [
8]. Thus, inhibiting myofibroblast activation by targeting TGF-β1 may be an effective strategy for preserving normal lung structure in the context of PF.
Traditional Chinese medicine has always been a valuable source of drugs, whether for the discovery of leading compounds or direct use as therapeutic agents.
Artemisia rupestris L. is widely distributed in Xinjiang, China. As part of traditional Chinese medicine (TCM),
Artemisia rupestris L. contains multiple chemical components, including flavonoids, organic acids, and sesquiterpenes [
9,
10]. These active constituents have demonstrated significant anti-inflammatory, antioxidant, and immunomodulatory activities [
11,
12]. Studies have shown that flavonoid compounds can treat PF [
13], and suppressing TGF-β1 induces myofibroblast activation and reduces the production of ECM, thereby alleviating PF [
14,
15]. Therefore, investigating whether
Artemisia rupestris L. has antifibrotic properties and identifying its potential treatment components has important significance for the discovery of PF treatments.
In the present research, we investigated the therapeutic effects and mechanisms of action of Artemisia rupestris L. ethanol extract (ER) against cigarette smoke (CS)-induced PF and isolated the active flavonoid compounds that treat PF by targeting TGF-β1. Using in vitro and in vivo experiments, we confirmed that the ER suppressed fibroblast activation and FMT via the TGF-β1 signaling pathway and decreased collagen production, thus treating PF. We used network pharmacology and molecular docking to screen potential active components and, combined with LC–MS and pull-down assays, we identified the active ingredient in ER that binds to TGF-β1 as Luteolin 7-glucuronide (LG). LG inhibited fibroblast activation and FMT, while increasing E-cadherin expression to enhance fibroblast adhesion via the TGF-β1 signaling pathway. Furthermore, LG alleviated PF by decreasing collagen I production, thus inhibiting collagen deposition.
2. Materials and Methods
2.1. Reagents
Luteolin 7-glucuronide (LG, C21H18O12, MW: 462.36, >99.8% purity) was purchased from Derick Biotechnology company (Chengdu, China). Dexamethasone (DEX, C22H29FO5, MW: 392.46, >97% purity) was purchased from Sigma-Aldrich (Cat# HY-14648; Saint Louis, MO, USA). Mouse TNF-α ELISA Kit (Cat#E-EL-M0044, Elabscience, Wuhan, China), Mouse IL-6 ELISA Kit (Cat#E-EL-M3063, Elabscience, China), and Mouse IL-10 ELISA Kit (Cat#E-EL-M0046, Elabscience, China) were purchased from Elabscience; TGF-β1 proteins were purchased from SinoBiological (Cat#10804-HNAC; Beijing, China); superoxide dismutase (SOD), catalase (CAT), and glutathione (GSH) kits were purchased from Solarbio (Cat#BC5165, Cat#BC0205, Cat#BC1175; Beijing, China); and rProtein A/G kit was purchased from Yeasen (Cat#36421ES40; Shanghai, China).
2.2. Drug Preparation and Extraction
Artemisia rupestris L. was collected at Balikun prairie in Xinjiang province, China, and identified by Prof. Li Baoli. A voucher specimen (No. 2018-17) was deposited in the Institute of Medicinal Plant Development, Beijing, China. A 100 g mass of Artemisia rupestris L., except roots, was extracted using 1 L of hot 80% ethanol under reflux for 2 h at 80 °C. This was repeated three times. The 3 L of extract was concentrated to 100 mL (1 g/mL). Subsequently, freeze-drying was performed using a lyophilizer, and the ER was stored at −80 °C.
2.3. Animals
Animal experiments were performed according to ethical policies and procedures approved by the Institutional Animal Care and Use Committee of China Agriculture University, Beijing, China (Approval number NO.AW72403202-2-1). Male C57BL/6 mice (weight: 20–22 g; age: 6–8 weeks) were purchased from SPF biotechnology company (Beijing, China) and raised under standard specific pathogen-free conditions of 23 ± 2 °C and 12/12 h light/dark cycles, and given standard animal chow and water. The mice were randomly assigned into seven groups (
n = 7 per group): (a) control group, (b) CS group, (c) CS + ER-L (125 mg/kg) group, (d) CS + ER-H (500 mg/kg) group, and (e) CS + DEX (2.5 mg/kg) group. Mice in the CS and CS + ER groups were placed in a homemade smoke box (40 × 30 × 25 cm) with a three-way valve to draw the smoke and guide it into the box. Commercial cigarette-removed filters, comprising 10 mg of tar, 11 mg of carbon monoxide, and 1.0 mg of nicotine per cigarette, were used in the study. The animals stimulated by CS were placed in one box, received CS from 12 cigarettes per day, 6 days/week, for a total of 10 weeks. The methods were adapted from a previous study [
16]. ER was dissolved in normal saline and administered to the mice 1 h before CS exposure.
2.4. Histopathological Observation
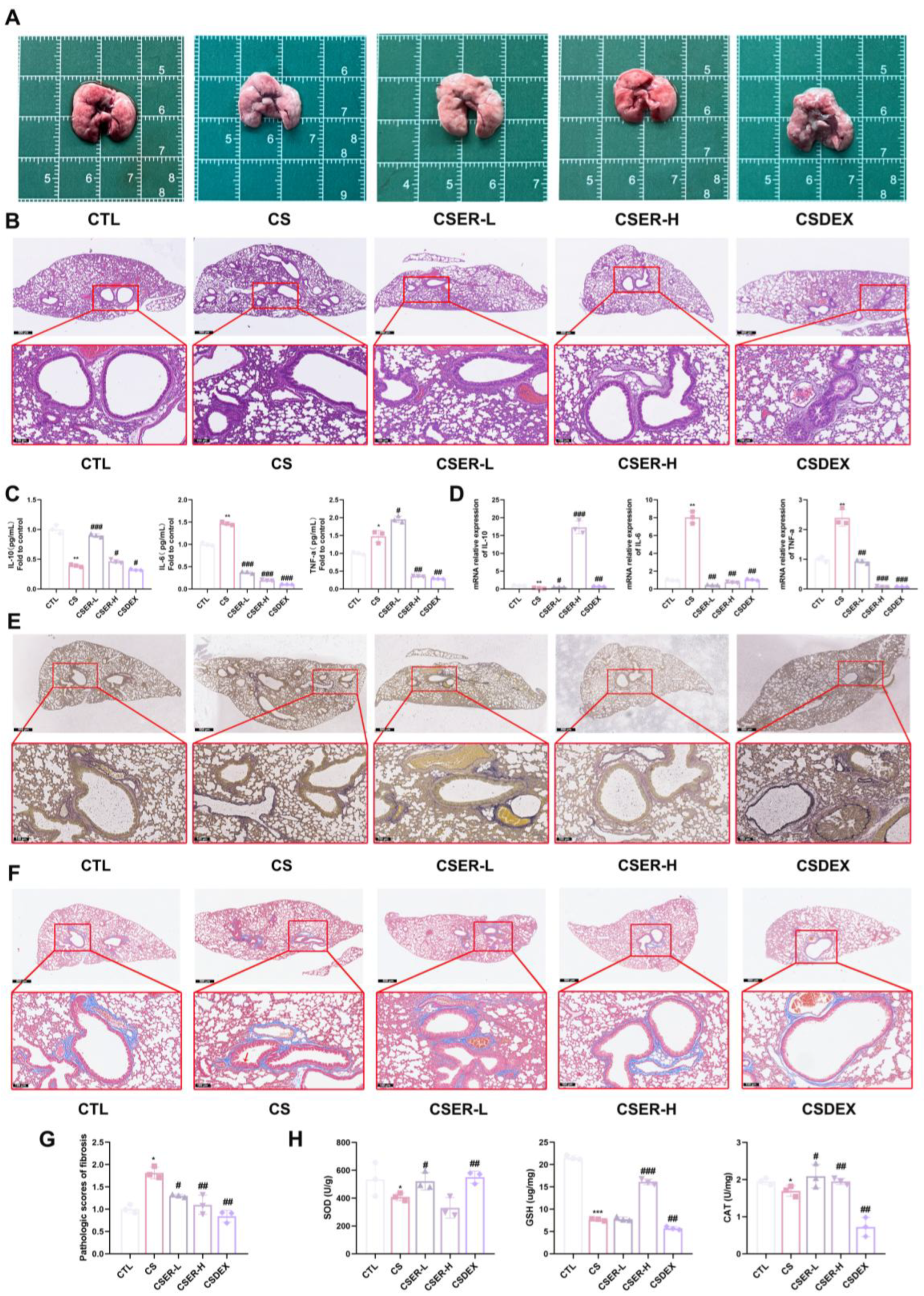
Lung tissues from identical regions in the mice were fixed in 4% formaldehyde, paraffin-embedded, and sectioned into 4 μm slices. To evaluate pulmonary histoarchitecture, tissue sections were stained with hematoxylin and eosin (H&E). Collagen and elastic fibers were visualized using Masson’s trichrome (Masson) and Elastica van Gieson (EVG) staining, respectively. The blue-stained regions surrounding the airway were designated as positive areas and subsequently outlined for further analysis. The proportion of the positive area within the bronchial wall was calculated to quantify the extent of subepithelial fibrosis using the Image J software 1.54p.
2.5. Preparation of Cigarette Smoke Extract
Cigarette smoke extract (CSE) solution was prepared as previously described [
17], but with some modifications. Briefly, CSE was prepared by burning commercial cigarettes without filters. The smoke was bubbled into 10 mL of PBS for 5 min, and CSE was standardized by measuring the absorbance (OD = 0.9–1.0) at a wavelength of 320 nm. The pH was adjusted to 7.0–7.5 and the extract filtered using a 0.22 μm syringe filter to remove bacteria and large particles. The concentration of CSE in the PBS was defined as 100%, and the prepared CSE was used within 30 min of collection.
2.6. Cell Culture and Treatment
L929 cells were obtained from Pricella Biotechnology (Cat#CL-0137; Wuhan, China). and cultured in Dulbecco’s modified Eagle medium (DMEM, Cat#11965092; Thermo Fisher, Waltham, MA, USA) containing 10% fetal bovine serum (FBS, Cat#SE100-B; WISENT, Nanjing, China) and 1% penicillin–streptomycin. All cells were free from mycoplasma contamination and were grown at 37 °C in an atmosphere with 5% CO2. The freeze-dried ER powder was dissolved in DMEM to prepare a 40 mg/mL drug solution. This solution was filtered through a 0.22 μm filter (Cat#BS-PES-22; Biosharp, Hefei, China) to obtain the stock solution, which was diluted with DMEM to obtain a working solution. The drug treatment groups were pre-incubated with ER or LG for 1 h, and then co-incubated with 5% CSE or TGF-β1 (10 ng/mL) for 24 h. The experimental groups were grouped as follows: control group (0 mg/mL ER and 0% CSE), ER-H group (5 mg/mL ER and 0% CSE), CSE group (0 mg/mL ER and 5% CSE), CSEER-L (1.25 mg/mL ER and 5% CSE), CSEER-M (2.5 mg/mL and 5% CSE), CSEER-H (5 mg/mL and 5% CSE), LG-H (50 ug/mL LG and 0 ng/mL TGF-β1), TGF (0 ug/mL LG and 10 ng/mL TGF-β1), TGFLG-L (12.5 ug/mL LG and 10 ng/mL TGF-β1), TGFLG-M (25 ug/mL LG and 10 ng/mL TGF-β1), and TGFLG-H (50 ug/mL LG and 10 ng/mL TGF-β1).
2.7. Total RNA Extraction and Real-Time Quantitative PCR
Total RNA was isolated using TRIzol (Cat#R1100; Solarbio, Beijing, China), and the RNA concentrations were measured using a Nanodrop spectrophotometer (Thermo Fisher, Waltham, MA, USA). cDNA synthesis was performed using RevertAid First Strand cDNA Synthesis Kit (Cat#K1622; Thermo Fisher, Waltham, MA, USA). RT-qPCR was carried out on the CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA) using SYBR Green Master Mix (Cat#Q712-02; Vazyme Biotech, Nanjing, China). The primers used are presented in
Table S1.
2.8. Biochemical Index
Lung tissue samples were analyzed to measure TNF-a, IL-6, IL-10, superoxide dismutase (SOD), catalase (CAT), and glutathione (GSH). TNF-α, IL-6, and IL-10 detection was performed using enzyme-linked immunosorbent assay (ELISA). All biochemical assays were performed according to the kit instructions provided by the supplier.
2.9. Immunofluorescence (IF) Staining
The L929 cells were seeded in a 24-well plate as previously described [
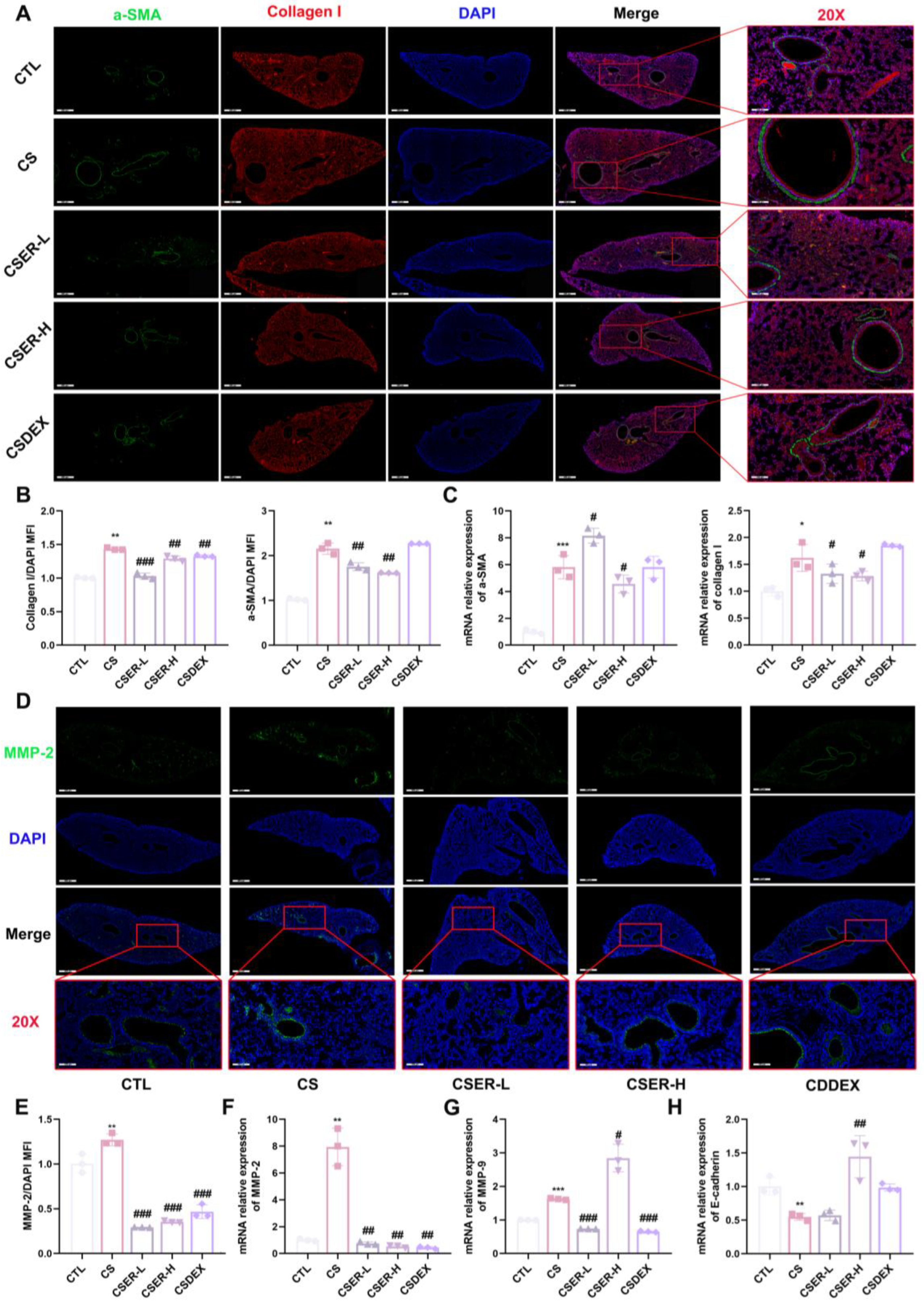
18]. At the end of the treatment, cells were fixed with paraformaldehyde (Cat#P1110; Solarbio, Beijing, China) for 15 min at room temperature and washed three times with PBS. Antigen retrieval from the lung sections was conducted using 5% bovine serum albumin (BSA) blocking. The cells and sections were cultured overnight with the primary antibody. Primary antibodies against α-SMA (1:100; Cat#67735-1-Ig; Proteintech, Wuhan, China), collagen I (1:200; Cat#bs-0578R; Bioss, Beijing, China), and MMP-2 (1:100; Cat#ab92536; Abcam, Cambridge, MA, USA) were prepared in advance. Finally, observations were made under a fluorescence microscope for image acquisition. The mean fluorescence intensity (MFI) of the images was determined using Image J 1.54p.
2.10. Cell Viability Assay
An MTT assay was used to evaluate drug cytotoxicity. Briefly, L929 cells were added to 96-well plates at a concentration of 5 × 103 cells per well and maintained under culture conditions at 37 °C overnight. After treatment with drugs for 24 h, the cells were exposed to MTT solution for an extra 4 h at 37 °C. Afterward, the supernatant was discarded, and 150 μL of DMSO was dispensed into each well. A microplate reader (Tecan, Mannedorf, Switzerland) was used to measure absorbance at 490 nm. The average absorbance of three independent experiments, each consisting of three replicates, was calculated.
2.11. Wound Healing Assay
L929 cells were seeded in a 6-well plate and cultured for 24 h. A scratch wound assay was performed using a 200 μL pipette tip. The scratches were observed under a microscope (Leica, Solms, Germany) after the cells were treated with drugs. Image J software was used to determine the widths of the scratches.
2.12. Western Blot
Protein lysis buffer containing inhibitors was added to the sample in each group, and then the sample was incubated for 10 min at 4 °C. The supernatant was collected after centrifugation (10 min, 12,000 rpm, 4 °C). BCA kits (Cat#CW0014S; Cowin Biotech, TaiZhou, China) were used to determine protein concentration. Protein samples were separated using SDS-PAGE and transferred onto a PVDF membrane. The membrane was incubated with the homologous antibodies and tested using enhanced ECL reagents (Cat#PK10003; Proteintech, Wuhan, China). The corresponding primary antibodies were diluted with the blocking solution as follows: GAPDH (Cat#60004-1-Ig; Proteintech, Wuhan, China; 1:5000), Collagen I (Cat#66761-1-Ig; Proteintech, Wuhan, China; 1:1000), α-SMA (Cat#67735-1-Ig; Proteintech, Wuhan, China; 1:2000), MMP-2 (Cat#66366-1-Ig; Proteintech, Wuhan, China; 1:1000), p-Smad2/3 (Cat#8828; Cell Signaling Technology, Danvers, MA, USA; 1:1000), Smad2/3 (Cat#ab202445; Abcam, Cambridge, MA, USA; 1:2000), MMP-9 (Cat#ab76003; Abcam, Cambridge, MA, USA; 1:1000), TGF-β1 (Cat#ab315254; Abcam, Cambridge, MA, USA; 1:2000), ERK1/2 (Cat#11257-1-AP; Proteintech, Wuhan, China; 1:1000), and P38 (Cat#14064-1-AP; Proteintech, Wuhan, China; 1:1000). The proteins were then incubated with the corresponding HRP-labeled secondary antibody (Cat#A21020; Abbkine scientific, Wuhan, China; 1:5000). All antibodies were diluted with 5% BSA. Quantitative analysis of protein bands was conducted using Image J software.
2.13. Flow Cytometry
L929 cells were grown in 6-well plates for 24 h at a density of 10 × 10
4 cells/well as previously described [
19] to quantify DNA in nuclei stained with propidium iodide (PI). After drug treatment, a 0.25% trypsin–EDTA solution was used to trypsinize the cell pellets. Centrifugation was used to harvest the cell pellets, which were then washed with PBS solution. The cell pellets were incubated in ice-cold 70% ethanol at 4 °C for 24 h and then washed three times with PBS solution. Subsequently, the cells were incubated with RNase A and PI (Solarbio, Beijing, China) for thirty minutes. Cell cycle distribution analysis was carried out using a CytoFLEX flow cytometer (Beckman Coulter, Brea, CA, USA).
2.14. Detection of Cell Proliferation Using EdU
Cell proliferation was assessed using the BeyoClick EdU-488 kit (Cat#C0071S; Beyotime, Shanghai, China) according to the manufacturer’s protocol, as previously described [
20]. In brief, the L929 cells were pre-treated for 1 h using medium containing various drug concentrations, followed by co-incubation with TGF-β1 or CSE for 24 h. EdU-488 was added to the medium, resulting in a final concentration of 10 mM. The cells were then incubated for 2 h. The cells were incubated with the click reaction solution in the dark at room temperature for 30 min, then washed three times with TBST. The cells were then incubated with Hoechst 33,342 (diluted 1:1000) in the dark at room temperature for 10 min. After washing, they were observed under a fluorescence microscope for image acquisition. The MFIs of the images were determined using Image J 1.54p.
2.15. LC–MS Analysis
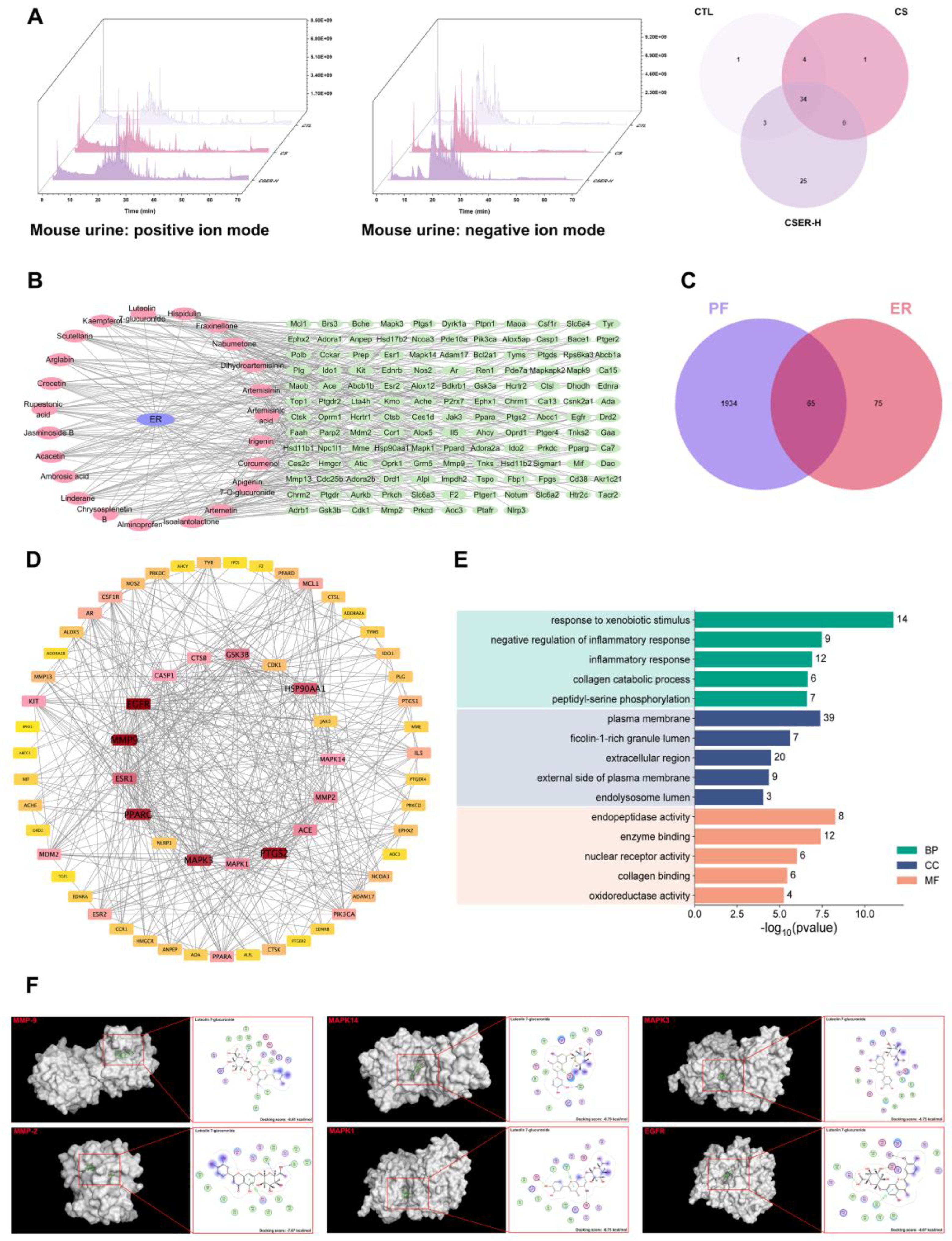
One hour after administering ER to the mice, their urine was collected. The samples were redissolved in 1 mL of 80% methanol, and then sonicated and centrifuged at 14,000 g for 10 min. Chromatographic separation was performed using the Q Exactive Plus Orbitrap system (Thermo) with an Acquity UPLC HSS T3 column (2.1 × 100 mm, 1.8 μm). The elution process occurred at a flow rate of 0.3 mL/min using 0.1% formic acid in water and 0.1% formic acid in acetonitrile. Data analysis was performed using the Compound Discoverer v3.3 software, and compounds were identified using local and online mzCloud databases.
2.16. Network Pharmacology
Potential active ingredients in ER were identified based on LC–MS detection of natural components in urine. The Traditional Chinese Medicine Database and Analysis Platform (TCMSP) (
https://old.tcmsp-e.com/tcmsp.php; accessed on 12 November 2024) was used to identify the targets of active compounds. PF targets were searched in Drugbank, GeneCards, and Online Mendelian Inheritance in Man (OMIM) databases. Finally, a network of drug genes was constructed to extract the IDs of the overlapping genes in the databases. The overlapping targets were input into the online Database for Annotation, Visualization, and Integrated Discovery (DAVID), and enriched candidate pathways were further analyzed to identify associated biological processes.
2.17. Molecular Docking
The 3D structures of proteins (MAPK14, PDB ID: 7BE5; EGFR, PDB ID: 1XKK; MAPK1, PDB ID: 1TVO; MAPK3, PDB ID: 2ZOQ; MMP-2, PDB ID: 7XGJ; MMP-9, PDB ID: 1GKC; TGF-β1, PDB ID: 1KLA) were obtained from the Protein Data Bank (
https://www.rcsb.org/, accessed on 15 December 2024). The 3D structure of the small molecule was downloaded from the PubChem database (
https://pubchem.ncbi.nlm.nih.gov/, accessed on 15 December 2024). Prior to docking, the receptor protein was pre-processed using the Molecular Operating Environment (MOE, Version 2022.02) software, and water molecules, salt ions, and small molecules were removed. Docking of chemical constituents with protein was conducted using the MOE software, and a docking score was calculated.
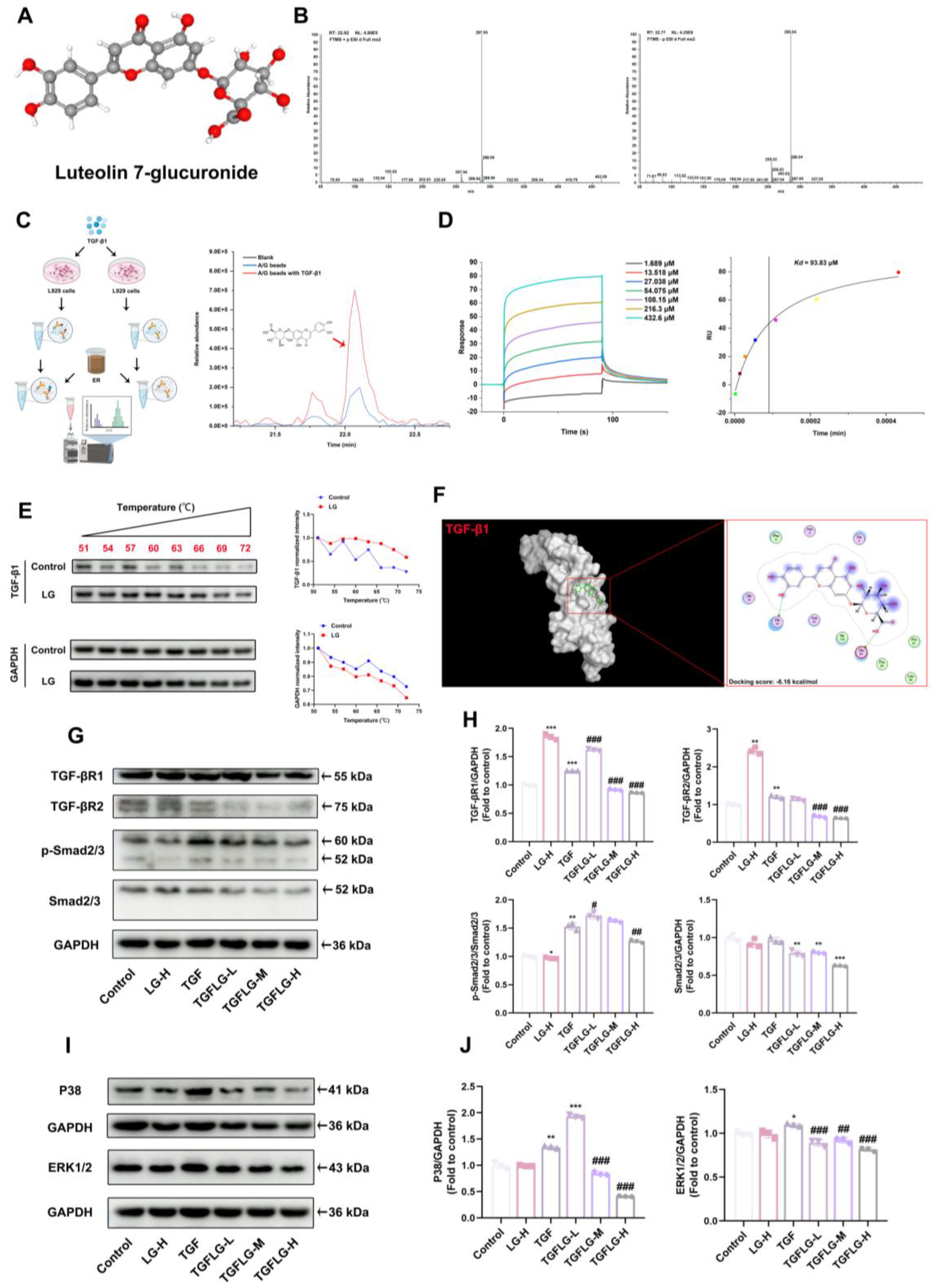
2.18. Pull-Down Assay
To perform the pull-down assay, L929 cells were pre-treated with 10 ng/mL of TGF-β1 for 24 h, and then collected and lysed using RIPA buffer containing protease and phosphatase inhibitors. The primary antibody targeting TGF-β1 (1:30; Cat#bsm-33287M; Bioss, Beijing, China) was reacted with protein A/G magnetic beads and incubated overnight at 4 °C. The antibody–bead complexes were then mixed with whole-cell lysates and incubated overnight at 4 °C. Subsequently, the protein mixture was separated using a magnetic rack, and the precipitates were washed three times with TBS solution. The precipitates were then incubated with 5 mg/mL of ER for 1 h to isolate target compounds. After incubation, the precipitates were washed three times with TBS solution. The active target components were eluted from Protein A/G magnetic beads using 0.1% (v/v) aqueous formic acid and then subjected to LC–MS analysis.
2.19. Surface Plasmon Resonance (SPR) Analysis
The binding affinity of LG to TGF-β1 was measured using Biacore T200 (Cytiva, Marlborough, MA, USA) with a CM5 sensor chip (GE Healthcare, Marlborough, MA, USA). Solutions of compounds were prepared in a running buffer by serial dilution of stock solutions. Subsequently, different concentrations of LG (3.125–200 μg/mL) were diluted in running buffer and injected into the system as the analyte. The parameters for SPR were set as follows: flow rate, 30 μL/min; association time, 90 s; dissociation time, 90 s; temperature, 25 °C. The interaction parameters were obtained using the Biacore evaluation software (Version 1.0).
2.20. CETSA Assay
L929 cells were cultured overnight in 100 mm dishes. RIPA buffer (Cat#R0010; Solarbio, Beijing, China) was used to obtain cellular proteins, and BCA kits were used to quantify protein content. The proteins were diluted to 5 μg/μL before drug treatment. The proteins were mixed with LG (50 μg/mL) and rotated for 1 h at room temperature. The proteins were incubated at specific temperatures (51 °C, 54 °C, 57 °C, 60 °C, 63 °C, 66 °C, 69 °C, 72 °C) for 3 min. Western blot analysis was performed on each sample.
4. Discussion
PF causes irreversible lung damage and severely threatens health. Chronic exposure to cigarette smoke is a major contributing factor to the pathogenesis of PF. Cigarette smoke induces sustained inflammatory and oxidative stress responses in the lungs, serving as an initiating factor for fibrosis development. Currently approved PF therapies can only slow the progression of the disease, failing to halt its advancement. Although numerous small molecule agents are under development, their clinical translation faces a protracted timeline. Artemisia rupestris L., a traditional Chinese herb with lung-clearing, lung-moistening, and anti-inflammatory properties, has never been investigated for its antifibrotic potential. In this study, we investigated the therapeutic effects of ER against CS-induced pulmonary inflammation, oxidative stress, and fibrosis.
PF usually occurs as the final stage of lung function damage, such as chronic obstructive pulmonary disease (COPD), lung inflammation, and so on. Persistent lung injury due to oxidative stress and chronic inflammation is a central feature of lung fibrosis [
26,
27]. In this study, we established a CS-induced mouse model to investigate the antifibrotic activity of ER and its potential as a new therapeutic for PF. The results showed that mice models of PF induced by CS have characteristics similar to human PF, such as airway remodeling, lung inflammation, fiber imbalance, and oxidative stress. ER significantly prevented inflammation, airway remodeling, oxidative stress, and collagen deposition, improving lung structure. However, DEX—a common treatment for lung inflammation and PF—had no significant protective effect on airway remodeling in the CS-exposed mice and exacerbated CS-induced pulmonary emphysema in the mice. Oxidative stress and chronic inflammation persistently activate myofibroblasts, promoting the secretion of collagen. Inhibiting the proliferation and activation of myofibroblasts is an important factor in the treatment of PF. This leads to the reduced secretion of collagen (such as collagen I) and MMPs, thereby attenuating collagen deposition in lung tissue [
28,
29]. EMT and FMT are critical factors driving the increase in myofibroblasts. Preventing the reduction in E-cadherin expression in cells, suppressing the upregulation of MMPs, and alleviating the imbalance in the ECM composition serve as key strategies for inhibiting EMT and FMT [
30,
31]. The in vivo experimental results demonstrated that CS induced a significant decrease in
E-cadherin mRNA expression, suggesting that EMT could also contribute to CS-induced PF. The α-SMA protein expression as a marker of myofibroblasts was examined using an IF assay [
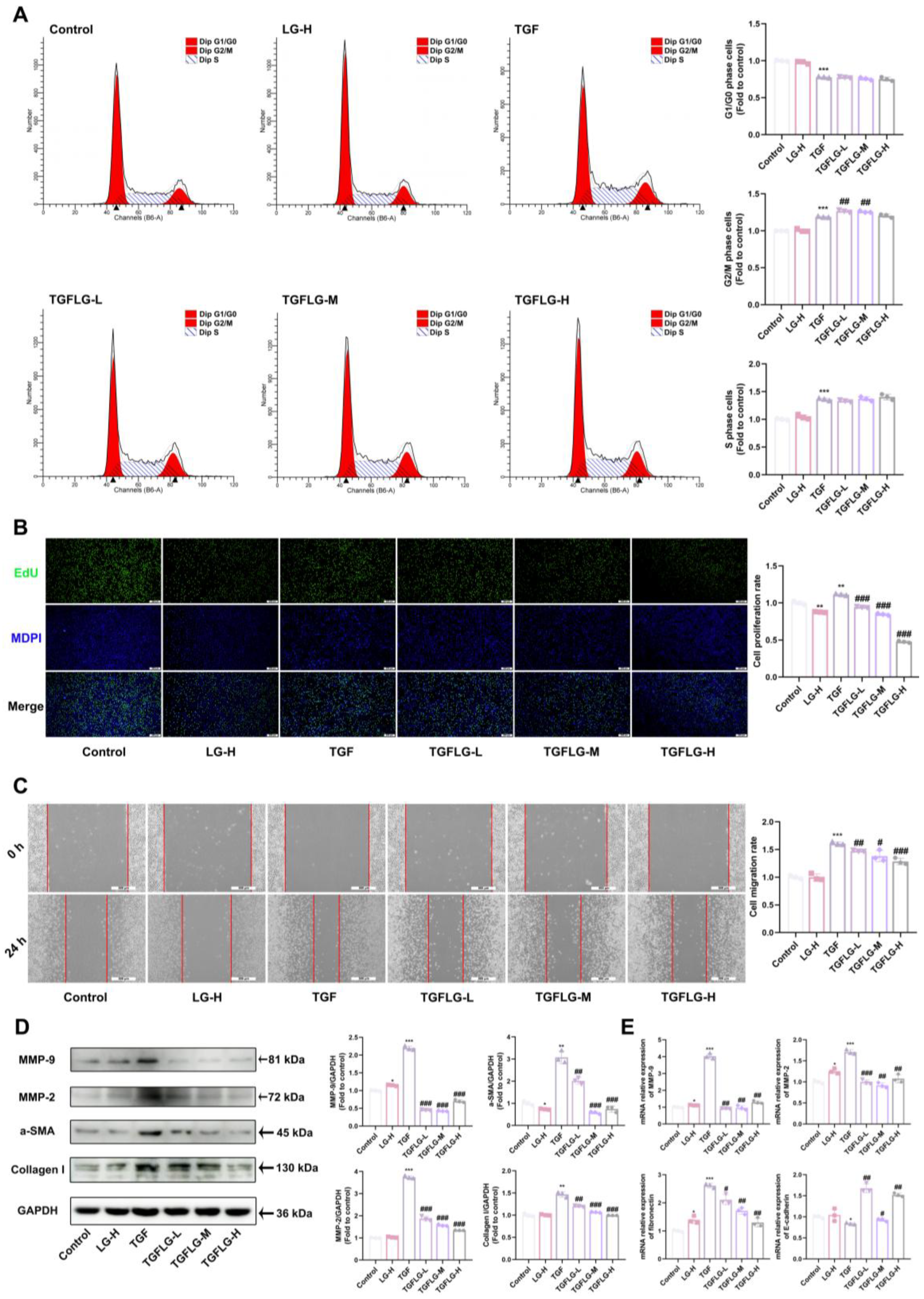
32]. The results showed that ER inhibited the CS-induced increase in α-SMA protein expression around the airway, suppressing myofibroblast accumulation. Concurrently, the secretion of collagen I and MMP-2 decreased, along with a decrease in fibronectin mRNA, which alleviated collagen deposition in lung tissue. To further validate these findings, an in vitro model of L929 cells was used to demonstrate that ER significantly inhibited the CSE-induced activation and proliferation of L929 cells via the TGF-β1 signaling pathway, suppressed FMT and collagen deposition, and increased E-cadherin expression to enhance adhesion, thus confirming its antifibrotic effects.
To date, only two therapeutic agents—pirfenidone (targeting inflammasome pathways) and nintedanib (a tyrosine kinase inhibitor)—have been approved by international clinical regulators for the treatment of PF; thus, identifying and screening TCM extracts for chemical constituents that can be used for the treatment of PF is essential for single-chemical drug development.
Artemisia rupestris L. mainly contains flavonoids, organic acids, and sesquiterpenes [
33]. We used LC–MS to identify 25 active natural compounds in mouse urine. The potential targets of these active compounds against PF were predicted using network pharmacology and molecular docking. Based on network pharmacology, MMP-9, MMP-2, MAPK14, MAPK1, MAPK3, and EGFR were identified as key targets of active ingredients in ER for treating PF. The molecular docking simulations of the binding of the 25 active compounds with these key targets revealed LG as the compound exhibiting favorable binding to all key targets, consistent with the targets identified in the in vitro ER experimental results. LG was screened as a potential active compound for treating PF.
TGF-β1 is one of the most researched profibrotic cytokines and plays a central regulatory role in the pathogenesis of PF [
34]. It regulates the activation of Smad and non-Smad downstream signaling pathways, inducing FMT and the activation of fibroblasts [
35]. Targeting and binding the TGF-β1 protein to affect its function is an effective method for preserving normal lung structure in PF. Therefore, we performed pull-down and LC–MS testing to screen and identify compounds that bind to TGF-β1 in ER. We identified the active ingredient in ER that binds to TGF-β1 as LG. SPR and CETSA analyses were performed to quantify the interaction between LG and TGF-β1. In addition, a molecular docking analysis was carried out to elucidate the binding mode of LG and TGF-β1. LG interacted with His40 and Glu84 in TGF-β1. Finally, LG inhibits the activation of both Smad and non-Smad downstream signaling pathways by binding to TGF-β1; reverses the upregulation of α-SMA, collagen I, MMP-2, and MMP-9 protein expression; suppresses
fibronectin expression; and increases
E-cadherin expression to enhance fibroblast adhesion. Thus, it can suppress fibroblast activation and FMT to inhibit collagen deposition, hence treating PF.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}