

Hyperphosphataemia and NADPH Oxidase Regulation in Pathophysiological Processes: Implications for Oxidative Stress and Disease Progression

Abstract
1. Introduction
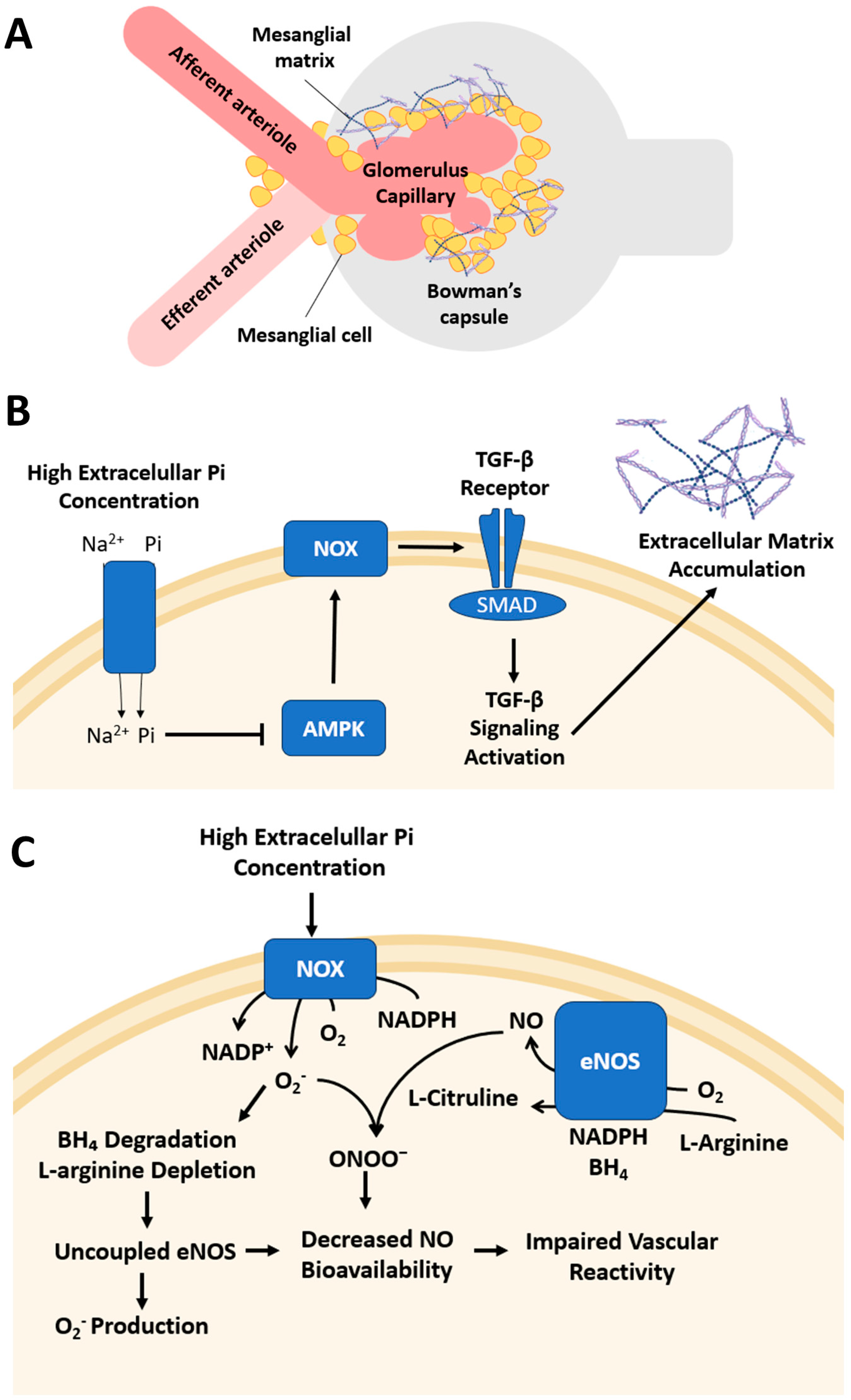
2. Chronic Kidney Disease
3. Vascular Calcification
3.1. Dextromethorphan as an NADPH Oxidase Inhibitor
3.2. Role of Macrophages Activated in Vascular Calcification
4. Ageing-Related Vascular Dysfunction
5. Renal Osteodystrophy
6. Atrial Fibrillation
7. Cancer
- (1)
- Short-term exposure (1 h): Pi hyperpolarises the mitochondrial membrane, increases mitochondrial ROS production, inhibits O2 consumption, and enhances PKC activity [25].
- (2)
- Long-term exposure (24 h): The source of Pi-induced H2O2 production shifts from mitochondria to NADPH oxidase. Using the NOX inhibitor VAS2870, Lacerda-Abreu et al. [63] demonstrated that this compound effectively inhibited H2O2 production only during prolonged Pi exposure but not in the short term, confirming the role of NOX as the primary ROS source under sustained Pi elevation [25].
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations:
| AF | Atrial fibrillation |
| ALP | Alkaline phosphatase |
| AMPK | 5′ Adenosine monophosphate-activated protein kinase |
| ANG II | Angiotensin II |
| ATP | Adenosine triphosphate |
| BMDMs | Bone marrow-derived macrophages |
| BMP2 | Bone morphogenetic protein 2 |
| CBFA1 | Core-binding factor alpha 1 (Runx2) |
| CVD | Cardiovascular disease |
| CKD | Chronic kidney disease |
| CKD-MBD | Chronic kidney disease–mineral and bone disorder |
| cAMP | Cyclic Adenosine Monophosphate |
| DHA | Dihydroxyadenine |
| DUOX | Dual oxidase |
| DUOXA | Dual oxidase activator |
| DXM | Dextromethorphan |
| ECM | Extracellular matrix |
| ET-1 | Endothelin-1 |
| ETC | Electron transport chain |
| Gpx1 | Glutathione peroxidase 1 |
| HAoSMCs | Human aortic smooth muscle cells |
| IFN-γ | Interferon gamma |
| iHMCs | Immortalised human mesangial cells |
| IL | Interleukin |
| M0φs | Nonpolarised macrophages |
| M1φs | Classically activated macrophages |
| M2φs | Alternatively activated macrophages |
| MPiφs | Phosphate-activated macrophages |
| mRNA | Messenger ribonucleic acid |
| MSX2 | Msh homeobox 2 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-κB | Nuclear factor kappa B |
| NO | Nitric oxide |
| NOX | NADPH oxidase |
| NOXA1 | NADPH oxidase activator 1 |
| NOXO1 | NADPH oxidase organiser 1 |
| O2•− | Superoxide anion |
| OS | Oxidative Stress |
| •OH | Hydroxyl radical |
| OSX | Osterix (SP7 transcription factor) |
| Pi | Inorganic phosphate |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PLC | Phospholipase C |
| PMA | Phorbol 12-myristate 13-acetate |
| Poldip2 | Polymerase (DNA-directed), delta interacting protein 2 |
| PTDlns(4,5)P2 | Phosphatidylinositol 4,5-bisphosphate |
| PTH | Parathyroid hormone |
| RAC1 | Ras-related C3 botulinum toxin substrate 1 |
| ROD | Renal osteodystrophy |
| ROS | Reactive oxygen species |
| RUNX2 | Runt-related transcription factor 2 |
| SMA | Smooth muscle actin |
| SM22α | Smooth muscle 22 alpha |
| SMAD | Mothers against decapentaplegic homolog |
| SOD | Superoxide dismutase |
| SOX9 | SRY-box transcription factor 9 |
| STAT3 | Signal transducer activator of transcription 3 |
| TGF-β | Transforming growth factor beta |
| TNBC | Triple-negative breast cancer |
| TNF-α | Tumour necrosis factor alpha |
| VC | Vascular calcification |
| VSMCs | Vascular smooth muscle cells |
References
- Lacerda-Abreu, M.A.; Russo-Abrahão, T.; Monteiro, R.Q.; Rumjanek, F.D.; Meyer-Fernandes, J.R. Inorganic phosphate transporters in cancer: Functions, molecular mechanisms and possible clinical applications. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Lacerda-Abreu, M.A.; Dick, C.F.; Meyer-Fernandes, J.R. The Role of Inorganic Phosphate Transporters in Highly Proliferative Cells: From Protozoan Parasites to Cancer Cells. Membranes 2022, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Lacerda-Abreu, M.A.; Russo-Abrahão, T.; Meyer-Fernandes, J.R. The Roles of Sodium-Independent Inorganic Phosphate Transporters in Inorganic Phosphate Homeostasis and in Cancer and Other Diseases. Int. J. Mol. Sci. 2021, 21, 9298. [Google Scholar] [CrossRef]
- Lacerda-Abreu, M.A.; Meyer-Fernandes, J.R. Extracellular Inorganic Phosphate-Induced Release of Reactive Oxygen Species: Roles in Physiological Processes and Disease Development. Int. J. Mol. Sci. 2021, 22, 7768. [Google Scholar] [CrossRef]
- Williams, M.J.; Patel, H.M.; Halling, C.B.; Hruska, K.A. The impact of a Western diet high in phosphate on the CKD-MBD in an Alport syndrome model. bioRxiv 2025. bioRxiv: 2025.01.17.633378. [Google Scholar] [CrossRef]
- Rudolph, E.H.; Gonin, J.M. Disorders of Phosphorus Metabolism. In Nephrology Secrets; Elsevier: Amsterdam, The Netherlands, 2012; pp. 551–559. [Google Scholar]
- Lacerda-Abreu, M.A.; Meyer-Fernandes, J.R. Inorganic Phosphate (Pi) in the Breast Cancer Microenvironment: Production, Transport and Signal Transduction as Potential Targets for Anticancer Strategies. Curr. Cancer Drug Targets 2023, 23, 187–198. [Google Scholar] [CrossRef]
- Francisqueti, F.V.; Chiaverini, L.C.; Santos, K.C.; Minatel, I.O.; Ronchi, C.B.; Ferron, A.J.; Ferreira, A.L.A.; Corrêa, C.R.; Unesp, B. The role of oxidative stress on the pathophysiology of metabolic syndrome. Rev. Assoc. Med. Bras. 2017, 63, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Rohman, M.M.; Islam, M.R.; Habib, S.H.; Choudhury, D.A.; Mohi-Ud-Din, M. NADPH oxidase-mediated reactive oxygen species, antioxidant isozymes, and redox homeostasis regulate salt sensitivity in maize genotypes. Heliyon 2024, 10, e26920. [Google Scholar] [CrossRef]
- Kim, J.; Moon, J.S. Molecular Roles of NADPH Oxidase-Mediated Oxidative Stress in Alzheimer’s Disease: Isoform-Specific Contributions. Int. J. Mol. Sci. 2024, 25, 12299. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. Tissue distribution and physiological functions of NADPH oxidases. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Paletta-Silva, R.; Rocco-Machado, N.; Meyer-Fernandes, J.R. NADPH oxidase biology and the regulation of tyrosine kinase receptor signaling and cancer drug cytotoxicity. Int. J. Mol. Sci. 2013, 14, 3683–3704. [Google Scholar] [CrossRef]
- Ganguly, U.; Kaur, U.; Chakrabarti, S.S.; Sharma, P.; Agrawal, B.K.; Saso, L.; Chakrabarti, S. Oxidative Stress, Neuroinflammation, and NADPH Oxidase: Implications in the Pathogenesis and Treatment of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2021, 2021, 7086512. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, A.; Peixoto, E.B.; Silva, K.C.; Lopes de Faria, J.M.; Lopes de Faria, J.B. Increase in AMPK brought about by cocoa is renoprotective in experimental diabetes mellitus by reducing NOX4/TGFβ-1 signalling. J. Nutr. Biochem. 2014, 25, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Taniguchi, M.; Tokumoto, M.; Toyonaga, J.; Fujisaki, K.; Suehiro, T.; Noguchi, H.; Iida, M.; Tsuruya, K.; Kitazono, T. The antioxidant tempol ameliorates arterial medial calcification in uremic rats: Important role of oxidative stress in the pathogenesis of vascular calcification in chronic kidney disease. J. Bone Miner. Res. 2012, 27, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Ryu, H.M.; Oh, E.J.; Choi, J.Y.; Cho, J.H.; Kim, C.D.; Kim, Y.-L.; Park, S.-H. Dipeptidyl peptidase-4 inhibitor gemigliptin protects against vascular calcification in an experimental chronic kidney disease and vascular smooth muscle cells. PLoS ONE 2017, 12, e0180393. [Google Scholar] [CrossRef]
- Luong, T.T.D.; Schelski, N.; Boehme, B.; Makridakis, M.; Vlahou, A.; Lang, F.; Pieske, B.; Alesutan, I.; Voelkl, J. Fibulin-3 Attenuates Phosphate-Induced Vascular Smooth Muscle Cell Calcification by Inhibition of Oxidative Stress. Cell. Physiol. Biochem. 2018, 46, 1305–1316. [Google Scholar] [CrossRef]
- Liu, E.S.; Chen, N.C.; Jao, T.M.; Chen, C.L. Dextromethorphan Reduces Oxidative Stress and Inhibits Uremic Artery Calcification. Int. J. Mol. Sci. 2021, 22, 12277. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Hamczyk, M.R.; Andrés, V. Novel phosphate-activated macrophages prevent ectopic calcification by increasing extracellular ATP and pyrophosphate. PLoS ONE 2017, 12, e0174998. [Google Scholar] [CrossRef]
- Van, T.V.; Watari, E.; Taketani, Y.; Kitamura, T.; Shiota, A.; Tanaka, T.; Tanimura, A.; Harada, N.; Nakaya, Y.; Yamamoto, H.; et al. Dietary phosphate restriction ameliorates endothelial dysfunction in adenine-induced kidney disease rats. J. Clin. Biochem. Nutr. 2012, 51, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Taguchi, M.; Osaki, T.; Fukumoto, S.; Fujita, T. Phosphate enhances reactive oxygen species production and suppresses osteoblastic differentiation. J. Bone Miner. Metab. 2014, 32, 393–399. [Google Scholar] [CrossRef]
- Hsu, Y.J.; Chang, G.J.; Lai, Y.J.; Chan, Y.H.; Chen, W.J.; Kuo, C.T.; Yeh, Y. High-phosphate diet causes atrial remodeling and increases atrial fibrillation vulnerability via STAT3/NF-κB signaling and oxidative stress. Acta Physiol. 2023, 238, e13964. [Google Scholar] [CrossRef]
- Lacerda-Abreu, M.A.; Russo-Abrahão, T.; Rocco-Machado, N.; Cosentino-Gomes, D.; Dick, C.F.; Carvalho-Kelly, L.F.; Nascimento, M.T.C.; Rocha-Vieira, T.C.; Meyer-Fernandes, J.R. Hydrogen Peroxide Generation as an Underlying Response to High Extracellular Inorganic Phosphate (Pi) in Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 10096. [Google Scholar] [CrossRef]
- Chatziantoniou, C.; Dussaule, J.C. Insights into the mechanisms of renal fibrosis: Is it possible to achieve regression? Am. J. Physiol. Renal Physiol. 2005, 289, F227–F234. [Google Scholar] [CrossRef]
- Abboud, H.E. Mesangial cell biology. Exp. Cell Res. 2012, 318, 979–985. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, D.; McCarthy, T.L.; Centrella, M.; Zhang, Y.; Moeckel, G.W. Inorganic phosphate stimulates fibronectin expression in renal fibroblasts. Cell. Physiol. Biochem. 2012, 30, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Ladilov, Y.; Aslam, M. New insights into the basic and translational aspects of AMPK signaling. Cells 2023, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Hallows, K.R.; Mount, P.F.; Pastor-Soler, N.M.; Power, D.A. Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. Am. J. Physiol. Renal Physiol. 2010, 298, F1067–F1077. [Google Scholar] [CrossRef]
- Papadimitriou, A.; Peixoto, E.B.; Silva, K.C.; Lopes de Faria, J.M.; Lopes de Faria, J.B. Inactivation of AMPK mediates high phosphate-induced extracellular matrix accumulation via NOX4/TGFβ-1 signaling in human mesangial cells. Cell. Physiol. Biochem. 2014, 34, 1260–1272. [Google Scholar] [CrossRef]
- Johnson, R.C.; Leopold, J.A.; Loscalzo, J. Vascular calcification: Pathobiological mechanisms and clinical implications. Am. J. Nephrol. 2011, 34, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Niepolski, L.; Malinowska-Loba, K. Association of Circulating Endothelial Nitric Oxide Synthase Levels with Phosphataemia in Patients on Haemodialysis. Biomedicines 2024, 12, 687. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R. Vascular Calcification: Key Roles of Phosphate and Pyrophosphate. Int. J. Mol. Sci. 2021, 22, 13536. [Google Scholar] [CrossRef]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef]
- Giachelli, C.M. Vascular calcification: In vitro evidence for the role of inorganic phosphate. J. Am. Soc. Nephrol. 2003, 14, S300–S304. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zheng, L.; Xu, H.; Tang, D.; Lin, L.; Zhang, J.; Li, C.; Wang, W.; Yuan, Q.; Tao, L.; et al. Oxidative stress contributes to vascular calcification in patients with chronic kidney disease. J. Mol. Cell. Cardiol. 2020, 138, 256–268. [Google Scholar] [CrossRef]
- Hu, C.T.; Shao, Y.D.; Liu, Y.Z.; Xiao, X.; Cheng, Z.B.; Qu, S.L.; Huang, L.; Zhang, C. Oxidative stress in vascular calcification. Clin. Chim. Acta 2021, 519, 101–110. [Google Scholar] [CrossRef]
- Brendel, H.; Shahid, A.; Hofmann, A.; Mittag, J.; Bornstein, S.R.; Morawietz, H.; Brunssen, C.; Langbein, H. NADPH oxidase 4 mediates the protective effects of physical activity against obesity-induced vascular dysfunction. Cardiovasc. Res. 2020, 116, 1767–1778. [Google Scholar] [CrossRef]
- Tamagaki, K.; Yuan, Q.; Ohkawa, H.; Imazeki, I.; Moriguchi, Y.; Imai, N.; Sasaki, S.; Takeda, K.; Fukagawa, M. Severe hyperparathyroidism with bone abnormalities and metastatic calcification in rats with adenine-induced uraemia. Nephrol. Dial. Transplant. 2006, 21, 651–659. [Google Scholar] [CrossRef]
- McClure, E.W.; Daniels, R.N. Classics in Chemical Neuroscience: Dextromethorphan (DXM). ACS Chem. Neurosci. 2023, 14, 2256–2270. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.C.; Chao, C.Y.; Lin, S.J.; Chen, J.W. Low-Dose Dextromethorphan, a NADPH Oxidase Inhibitor, Reduces Blood Pressure and Enhances Vascular Protection in Experimental Hypertension. PLoS ONE 2012, 7, e46067. [Google Scholar] [CrossRef] [PubMed]
- Cannata-Andía, J.B.; Carrillo-López, N.; Messina, O.D.; Hamdy, N.A.T.; Panizo, S.; Ferrari, S.L.; on behalf of the International Osteoporosis Foundation (IOF) Working Group on Bone and Cardiovascular Diseases. Pathophysiology of Vascular Calcification and Bone Loss: Linked Disorders of Ageing? Nutrients 2021, 13, 3835. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Q.; Xu, D. New insights into macrophage subsets in atherosclerosis. J. Mol. Med. 2022, 100, 1239–1251. [Google Scholar] [CrossRef]
- Pello, O.M.; Silvestre, C.; De Pizzol, M.; Andrés, V. A glimpse on the phenomenon of macrophage polarization during atherosclerosis. Immunobiology 2011, 216, 1172–1176. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Hamczyk, M.R.; Andrés, V. Alternatively activated macrophages exhibit an anticalcifying activity dependent on extracellular ATP/pyrophosphate metabolism. Am. J. Physiol. Cell Physiol. 2016, 310, C788–C799. [Google Scholar] [CrossRef]
- Yang, D.R.; Wang, M.Y.; Zhang, C.L.; Wang, Y. Endothelial dysfunction in vascular complications of diabetes: A comprehensive review of mechanisms and implications. Front. Endocrinol. 2024, 15, 1359255. [Google Scholar] [CrossRef]
- Bloom, S.I.; Islam, M.T.; Lesniewski, L.A.; Donato, A.J. Mechanisms and consequences of endothelial cell senescence. Nat. Rev. Cardiol. 2023, 20, 38–51. [Google Scholar] [CrossRef]
- Vgontzas, A.N.; Zoumakis, M.; Bixler, E.O.; Lin, H.M.; Prolo, P.; Vela-Bueno, A.; Kales, A.; Chrousos, G.P. Impaired nighttime sleep in healthy old versus young adults is associated with elevated plasma interleukin-6 and cortisol levels: Physiologic and therapeutic implications. J. Clin. Endocrinol. Metab. 2003, 88, 2087–2095. [Google Scholar] [CrossRef]
- Pitocco, D.; Tesauro, M.; Alessandro, R.; Ghirlanda, G.; Cardillo, C. Oxidative stress in diabetes: Implications for vascular and other complications. Int. J. Mol. Sci. 2013, 14, 21525–21550. [Google Scholar] [CrossRef]
- Aguilar, A.; Gifre, L.; Ureña-Torres, P.; Carrillo-López, N.; Rodriguez-García, M.; Massó, E.; da Silva, I.; López-Báez, V.; Sánchez-Bayá, M.; Prior-Español, Á.; et al. Pathophysiology of bone disease in chronic kidney disease: From basics to renal osteodystrophy and osteoporosis. Front. Physiol. 2023, 14, 1177829. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.C. On a form of late rickets associated with albuminuria, rickets of adolescents. Lancet 1883, 121, 993–994. [Google Scholar] [CrossRef]
- Zhang, X.; Li, T.; Wang, L.; Li, Y.; Ruan, T.; Guo, X.; Wang, Q.; Meng, X. Relative comparison of chronic kidney disease-mineral and bone disorder rat models. Front. Physiol. 2023, 14, 1083725. [Google Scholar] [CrossRef]
- Brundel, B.J.J.M.; Ai, X.; Hills, M.T.; Kuipers, M.F.; Lip, G.Y.H.; de Groot, N.M.S. Atrial fibrillation. Nat. Rev. Dis. Primers 2022, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Kong, X.J.; Ji, Y.Y.; Fan, J.; Ji, C.C.; Chen, X.; Ma, Y.-D.; Tang, A.-L.; Cheng, Y.-J.; Wu, S.-H. Serum electrolyte concentrations and risk of atrial fibrillation: An observational and mendelian randomization study. BMC Genomics 2024, 25, 280. [Google Scholar] [CrossRef]
- Gao, G.; Dudley, S.C., Jr. Redox regulation, NF-kappaB, and atrial fibrillation. Antioxid. Redox Signal. 2009, 11, 2265–2277. [Google Scholar] [CrossRef]
- Tsai, C.T.; Lin, J.L.; Lai, L.P.; Lin, C.S.; Huang, S.K. Membrane translocation of small GTPase Rac1 and activation of STAT1 and STAT3 in pacing-induced sustained atrial fibrillation. Heart Rhythm 2008, 5, 1285–1293. [Google Scholar] [CrossRef]
- Brown, R.B.; Razzaque, M.S. Phosphate toxicity and tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Elser, J.J.; Bracken, M.E.; Cleland, E.E.; Gruner, D.S.; Harpole, W.S.; Hillebrand, H.; Ngai, J.T.; Seabloom, E.W.; Shurin, J.B.; Smith, J.E. Global analysis of nitrogen and phosphorus limitation of primary producers in freshwater, marine and terrestrial ecosystems. Ecol. Lett. 2007, 10, 1135–1142. [Google Scholar] [CrossRef]
- Papaloucas, C.D.; Papaloucas, M.D.; Kouloulias, V.; Neanidis, K.; Pistevou-Gompaki, K.; Kouvaris, J.; Zygogianni, A.; Mystakidou, K.; Papaloucas, A. Measurement of blood phosphorus: A quick and inexpensive method for detection of the existence of cancer in the body. Too good to be true, or forgotten knowledge of the past? Med. Hypotheses 2014, 82, 24–25. [Google Scholar] [CrossRef]
- Jin, H.; Xu, C.X.; Lim, H.T.; Park, S.J.; Shin, J.Y.; Chung, Y.S.; Park, S.-C.; Chang, S.-H.; Youn, H.-J.; Lee, K.-H.; et al. High dietary inorganic phosphate increases lung tumorigenesis and alters Akt signaling. Am. J. Respir. Crit. Care Med. 2009, 179, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Camalier, C.E.; Young, M.R.; Bobe, G.; Perella, C.M.; Colburn, N.H.; Beck, G.R., Jr. Elevated phosphate activates N-ras and promotes cell transformation and skin tumorigenesis. Cancer Prev. Res. 2010, 3, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Lacerda-Abreu, M.A.; Meyer-Fernandes, J.R. Elevated extracellular inorganic phosphate inhibits ecto-phosphatase activity in breast cancer cells: Regulation by hydrogen peroxide. Cell Biol. Int. 2024, 48, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento-Salinas, F.L.; Delgado-Magallón, A.; Montes-Alvarado, J.B.; Ramírez-Ramírez, D.; Flores-Alonso, J.C.; Cortés-Hernández, P.; Reyes-Leyva, J.; Herrera-Camacho, I.; Anaya-Ruiz, M.; Pelayo, R.; et al. Breast Cancer Subtypes Present a Differential Production of Reactive Oxygen Species (ROS) and Susceptibility to Antioxidant Treatment. Front. Oncol. 2019, 9, 480. [Google Scholar] [CrossRef]
- Cosentino-Gomes, D.; Rocco-Machado, N.; Meyer-Fernandes, J.R. Cell signaling through protein kinase C oxidation and activation. Int. J. Mol. Sci. 2012, 13, 10697–10721. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Isoforms | Subunits | Regulators | Expression Sites |
|---|---|---|---|
| NOX1 | p22phox, NOXA1, NOXO1, RAC1 | ANG II, PDGF | Colon epithelial cells, vascular smooth cells, endothelial cells, uterus, placenta, osteoclasts, retinal pericytes, and macrophages. |
| NOX2 | gp91phox, p22phox, p40phox, p47phox, p67phox, RAC1 | PKC, TNF-α, phosphatidic acid | Phagocytes, vascular cells, endothelium, fibroblasts, cardiomyocytes, skeletal muscle, hepatocytes, and haematopoietic stem cells. |
| NOX3 | p22phox, NOXO1, NOXA1, RAC1 | Unknown | Inner ear, lung endothelial cells, foetal spleen, kidney, lung, and skull. |
| NOX4 | P22phox | Poldip2 | Kidney, smooth muscle cells, endothelial cells, fibroblasts, keratinocytes, osteoclasts, neurons, and hepatocytes. |
| NOX5 | none | Ca2+, ptdlns(4,5)p2 | Lymphoid tissues, testes, spleen and endothelial cells. |
| DUOX1 | DUOXA1, DUOXA2 | IL-4, IL-3, cAMP, PKA | Thyroid gland, airway epithelia, placenta, prostate, testis, pancreas, and heart. |
| DUOX2 | DUOXA1, DUOXA2 | IFN-γ, PLC, PKC | Thyroid gland, airway epithelia, epithelial cells in salivary excretory ducts and rectal glands. |
| Disease | Model Used | Pi Concentration | Pi Exposure | Correlation with NOX | Ref. |
|---|---|---|---|---|---|
| Chronic Kidney Disease | Immortalised human mesangial cells (iHMCs) | 5 mM | 24 h | High Pi level induced OS by NOX4 activation following AMPK inhibition. | [16] |
| Vascular calcification | Male Sprague Dawley (SD) rats | 1.2% (diet) | 6 weeks | NOX4 was time-dependently upregulated in the aortic media of uraemic rats. | [17] |
| Vascular calcification | Rat model of adenine-induced CKD and cultured VSMCs | 3 mM | 14 days | High Pi level increased expression of NOX4 and p22phox, enhancing ROS generation. | [18] |
| Vascular calcification | Primary human aortic smooth muscle cells (HAoSMCs) | 2 mM β-glycerophosphate | 24 h | Pi treatment upregulated NOX4 and CYBA, key components of NADPH oxidase. | [19] |
| Vascular calcification | Wistar rat model with adenine-induced CKD and HASMCs | 2.5 mM Pi | 14 days | Hyperphosphataemia induced ROS via NOX, leading to vascular calcification. | [20] |
| Vascular calcification | Mouse bone marrow-derived macrophages (BMDMs) | 2.5 mM | 7 days | High Pi level led to downregulation of NOX1 in macrophages. | [21] |
| Vascular dysfunction related to ageing | C57BL6 mice (young: 5 months, old: 24 months). | 0.6% (diet) | 3 months | Hyperphosphataemia increased NOX4 expression and ROS production. | [22] |
| Osteodystrophy | Osteoblastic murine MC3T3-E1 cells | 5 mM | 42 h | Pi increased ROS production through NOX1 and NOX4. | [23] |
| Atrial Fibrillation | 8-week-old male C57BL/6 mice | 2% (diet) | 10 weeks | Elevated Pi level increased NOX4 expression and ROS production. | [24] |
| Breast cancer | MDA-MB-231 cells. | 8 mM | 24 h | Pi induced ROS production through PKC-mediated NOX activation. | [25] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lacerda-Abreu, M.A.; Meyer-Fernandes, J.R. Hyperphosphataemia and NADPH Oxidase Regulation in Pathophysiological Processes: Implications for Oxidative Stress and Disease Progression. Antioxidants 2025, 14, 461. https://doi.org/10.3390/antiox14040461
Lacerda-Abreu MA, Meyer-Fernandes JR. Hyperphosphataemia and NADPH Oxidase Regulation in Pathophysiological Processes: Implications for Oxidative Stress and Disease Progression. Antioxidants. 2025; 14(4):461. https://doi.org/10.3390/antiox14040461
Chicago/Turabian StyleLacerda-Abreu, Marco Antonio, and José Roberto Meyer-Fernandes. 2025. "Hyperphosphataemia and NADPH Oxidase Regulation in Pathophysiological Processes: Implications for Oxidative Stress and Disease Progression" Antioxidants 14, no. 4: 461. https://doi.org/10.3390/antiox14040461
APA StyleLacerda-Abreu, M. A., & Meyer-Fernandes, J. R. (2025). Hyperphosphataemia and NADPH Oxidase Regulation in Pathophysiological Processes: Implications for Oxidative Stress and Disease Progression. Antioxidants, 14(4), 461. https://doi.org/10.3390/antiox14040461