
Nicotinamide Adenine Dinucleotide Phosphate Oxidases and Metabolic Dysfunction-Associated Steatotic Liver Disease


Abstract
1. Introduction
2. Pathophysiology of MASLD and MASH
2.1. MASLD and MASH Development
2.2. Oxidative Stress in MASLD and MASH
2.3. NOX-Related ROS Production in the Liver
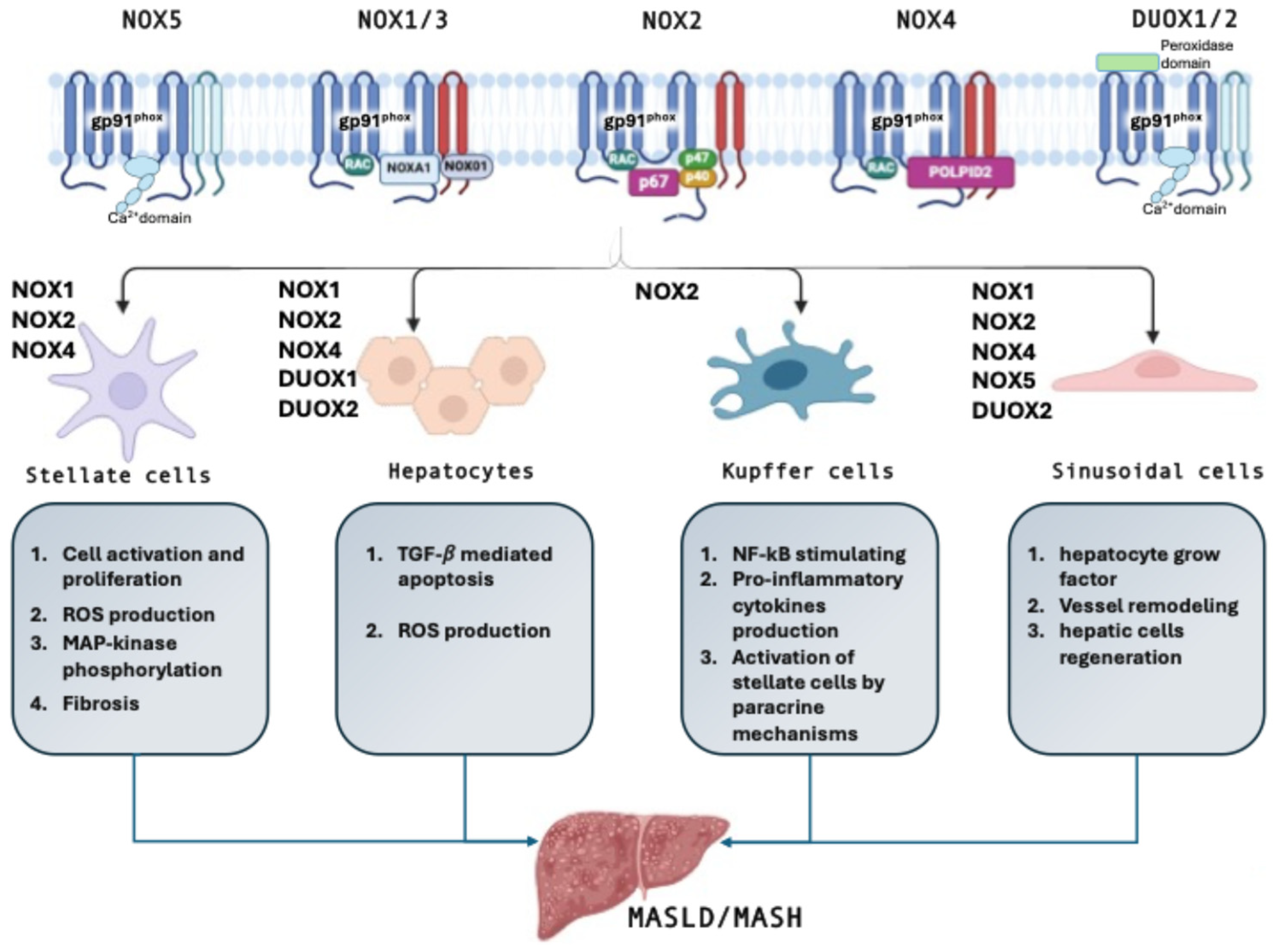
3. NOX in the Liver
3.1. Hepatic Stellate Cells
3.2. Hepatocytes
3.3. Kupffer Cells
3.4. Sinusoidal Endothelial Cells
4. NOX in MASLD Development and Progression to MASH
4.1. Experimental Studies
4.2. Human Studies
5. Perspectives and Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J. Hepatol. 2023, 79, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Khalil, M.; Mahdi, L.; Perniola, V.; Idone, V.; Graziani, A.; Baffy, G.; Di Ciaula, A. Metabolic Dysfunction-Associated Steatotic Liver Disease: From Pathogenesis to Current Therapeutic Options. Int. J. Mol. Sci. 2024, 25, 5640. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Byrne, C.D.; Tilg, H. MASLD: A systemic metabolic disorder with cardiovascular and malignant complications. Gut 2024, 73, 691–702. [Google Scholar] [CrossRef]
- Sandireddy, R.; Sakthivel, S.; Gupta, P.; Behari, J.; Tripathi, M.; Singh, B.K. Systemic impacts of metabolic dysfunction-associated steatotic liver disease (MASLD) and metabolic dysfunction-associated steatohepatitis (MASH) on heart, muscle, and kidney related diseases. Front. Cell Dev. Biol. 2024, 12, 1433857. [Google Scholar] [CrossRef]
- Ramirez-Mejia, M.M.; Qi, X.; Abenavoli, L.; Romero-Gomez, M.; Eslam, M.; Mendez-Sanchez, N. Metabolic dysfunction: The silenced connection with fatty liver disease. Ann. Hepatol. 2023, 28, 101138. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.E.; Koh, T.J.L.; Tang, A.S.P.; Quek, J.; Yong, J.N.; Tay, P.; Tan, D.J.H.; Lim, W.H.; Lin, S.Y.; Huang, D.; et al. Global Prevalence and Clinical Characteristics of Metabolic-associated Fatty Liver Disease: A Meta-Analysis and Systematic Review of 10 739 607 Individuals. J. Clin. Endocrinol. Metab. 2022, 107, 2691–2700. [Google Scholar] [CrossRef]
- Spengler, E.K.; Loomba, R. Recommendations for Diagnosis, Referral for Liver Biopsy, and Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Mayo Clin. Proc. 2015, 90, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Lekakis, V.; Papatheodoridis, G.V. Natural history of metabolic dysfunction-associated steatotic liver disease. Eur. J. Intern. Med. 2024, 122, 3–10. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e9. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Valeriani, E.; Bartimoccia, S.; Pignatelli, P.; Pastori, D. Aging and Antithrombotic Treatment. Antioxid. Redox Signal 2024, 41, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Effenberger, M. From NAFLD to MAFLD: When pathophysiology succeeds. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 387–388. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Heeren, J.; Scheja, L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef]
- Tanase, D.M.; Gosav, E.M.; Costea, C.F.; Ciocoiu, M.; Lacatusu, C.M.; Maranduca, M.A.; Ouatu, A.; Floria, M. The Intricate Relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD). J. Diabetes Res. 2020, 2020, 3920196. [Google Scholar] [CrossRef]
- Lomonaco, R.; Ortiz-Lopez, C.; Orsak, B.; Webb, A.; Hardies, J.; Darland, C.; Finch, J.; Gastaldelli, A.; Harrison, S.; Tio, F.; et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Lass, A.; Zimmermann, R.; Gorkiewicz, G.; Meyer, C.; Rozman, J.; Heldmaier, G.; Maier, R.; Theussl, C.; Eder, S.; et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006, 312, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F.; Fraterrigo, G.; Yoshino, J.; Luecking, C.; Kirbach, K.; Kelly, S.C.; de Las Fuentes, L.; He, S.; Okunade, A.L.; Patterson, B.W.; et al. Effects of Moderate and Subsequent Progressive Weight Loss on Metabolic Function and Adipose Tissue Biology in Humans with Obesity. Cell Metab. 2016, 23, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 2017, 65, 350–362. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Noworolski, S.M.; Erkin-Cakmak, A.; Korn, N.J.; Wen, M.J.; Tai, V.W.; Jones, G.M.; Palii, S.P.; Velasco-Alin, M.; Pan, K.; et al. Effects of Dietary Fructose Restriction on Liver Fat, De Novo Lipogenesis, and Insulin Kinetics in Children With Obesity. Gastroenterology 2017, 153, 743–752. [Google Scholar] [CrossRef]
- van Nierop, F.S.; Scheltema, M.J.; Eggink, H.M.; Pols, T.W.; Sonne, D.P.; Knop, F.K.; Soeters, M.R. Clinical relevance of the bile acid receptor TGR5 in metabolism. Lancet Diabetes Endocrinol. 2017, 5, 224–233. [Google Scholar] [CrossRef]
- Kajimura, S.; Spiegelman, B.M.; Seale, P. Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell Metab. 2015, 22, 546–559. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Yesilova, Z.; Yaman, H.; Oktenli, C.; Ozcan, A.; Uygun, A.; Cakir, E.; Sanisoglu, S.Y.; Erdil, A.; Ates, Y.; Aslan, M.; et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2005, 100, 850–855. [Google Scholar] [CrossRef]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression From NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef] [PubMed]
- Carpino, G.; Del Ben, M.; Pastori, D.; Carnevale, R.; Baratta, F.; Overi, D.; Francis, H.; Cardinale, V.; Onori, P.; Safarikia, S.; et al. Increased Liver Localization of Lipopolysaccharides in Human and Experimental NAFLD. Hepatology 2020, 72, 470–485. [Google Scholar] [CrossRef] [PubMed]
- Ore, A.; Akinloye, O.A. Oxidative Stress and Antioxidant Biomarkers in Clinical and Experimental Models of Non-Alcoholic Fatty Liver Disease. Medicina 2019, 55, 26. [Google Scholar] [CrossRef] [PubMed]
- Allameh, A.; Niayesh-Mehr, R.; Aliarab, A.; Sebastiani, G.; Pantopoulos, K. Oxidative Stress in Liver Pathophysiology and Disease. Antioxidants 2023, 12, 1653. [Google Scholar] [CrossRef] [PubMed]
- Koek, G.H.; Liedorp, P.R.; Bast, A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin. Chim. Acta 2011, 412, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Tariq, Z.; Green, C.J.; Hodson, L. Are oxidative stress mechanisms the common denominator in the progression from hepatic steatosis towards non-alcoholic steatohepatitis (NASH)? Liver Int. 2014, 34, e180–e190. [Google Scholar] [CrossRef] [PubMed]
- Crosas-Molist, E.; Fabregat, I. Role of NADPH oxidases in the redox biology of liver fibrosis. Redox Biol. 2015, 6, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Nazari, B.; Jaquet, V.; Krause, K.H. NOX family NADPH oxidases in mammals: Evolutionary conservation and isoform-defining sequences. Redox Biol. 2023, 66, 102851. [Google Scholar] [CrossRef]
- Bedard, K.; Jaquet, V.; Krause, K.H. NOX5: From basic biology to signaling and disease. Free Radic. Biol. Med. 2012, 52, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian. J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity During Development of Steatohepatitis in Mice. Gastroenterology 2015, 149, 468–480.e10. [Google Scholar] [CrossRef]
- Garcia-Ruiz, I.; Solis-Munoz, P.; Fernandez-Moreira, D.; Grau, M.; Munoz-Yague, T.; Solis-Herruzo, J.A. NADPH oxidase is implicated in the pathogenesis of oxidative phosphorylation dysfunction in mice fed a high-fat diet. Sci. Rep. 2016, 6, 23664. [Google Scholar] [CrossRef]
- Matsumoto, M.; Zhang, J.; Zhang, X.; Liu, J.; Jiang, J.X.; Yamaguchi, K.; Taruno, A.; Katsuyama, M.; Iwata, K.; Ibi, M.; et al. The NOX1 isoform of NADPH oxidase is involved in dysfunction of liver sinusoids in nonalcoholic fatty liver disease. Free. Radic. Biol. Med. 2018, 115, 412–420. [Google Scholar] [CrossRef]
- Paik, Y.H.; Kim, J.; Aoyama, T.; De Minicis, S.; Bataller, R.; Brenner, D.A. Role of NADPH oxidases in liver fibrosis. Antioxid. Redox Signal 2014, 20, 2854–2872. [Google Scholar] [CrossRef]
- Novo, E.; Busletta, C.; Bonzo, L.V.; Povero, D.; Paternostro, C.; Mareschi, K.; Ferrero, I.; David, E.; Bertolani, C.; Caligiuri, A.; et al. Intracellular reactive oxygen species are required for directional migration of resident and bone marrow-derived hepatic pro-fibrogenic cells. J. Hepatol. 2011, 54, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Matsuno, K.; Iwata, K.; Ibi, M.; Matsumoto, M.; Zhang, J.; Zhu, K.; Katsuyama, M.; Torok, N.J.; Yabe-Nishimura, C. NOX1/nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase promotes proliferation of stellate cells and aggravates liver fibrosis induced by bile duct ligation. Hepatology 2011, 54, 949–958. [Google Scholar] [CrossRef]
- Paik, Y.H.; Iwaisako, K.; Seki, E.; Inokuchi, S.; Schnabl, B.; Osterreicher, C.H.; Kisseleva, T.; Brenner, D.A. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 2011, 53, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Mainez, J.; Crosas-Molist, E.; Roncero, C.; Fernandez-Rodriguez, C.M.; Pinedo, F.; Huber, H.; Eferl, R.; Mikulits, W.; Fabregat, I. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS ONE 2012, 7, e45285. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Venugopal, S.; Serizawa, N.; Chen, X.; Scott, F.; Li, Y.; Adamson, R.; Devaraj, S.; Shah, V.; Gershwin, M.E.; et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology 2010, 139, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Foo, N.P.; Lin, S.H.; Lee, Y.H.; Wu, M.J.; Wang, Y.J. alpha-Lipoic acid inhibits liver fibrosis through the attenuation of ROS-triggered signaling in hepatic stellate cells activated by PDGF and TGF-beta. Toxicology 2011, 282, 39–46. [Google Scholar] [CrossRef]
- Abhilash, P.A.; Harikrishnan, R.; Indira, M. Ascorbic acid supplementation down-regulates the alcohol induced oxidative stress, hepatic stellate cell activation, cytotoxicity and mRNA levels of selected fibrotic genes in guinea pigs. Free Radic. Res. 2012, 46, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Moreau, R.; Pessayre, D.; Epperson, T.K.; Krause, K.H. NOX family NADPH oxidases in liver and in pancreatic islets: A role in the metabolic syndrome and diabetes? Biochem. Soc. Trans. 2008, 36, 920–929. [Google Scholar] [CrossRef]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free. Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Bataller, R.; Schwabe, R.F.; Choi, Y.H.; Yang, L.; Paik, Y.H.; Lindquist, J.; Qian, T.; Schoonhoven, R.; Hagedorn, C.H.; Lemasters, J.J.; et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J. Clin. Investig. 2003, 112, 1383–1394. [Google Scholar] [CrossRef]
- Novo, E.; Marra, F.; Zamara, E.; Valfre di Bonzo, L.; Caligiuri, A.; Cannito, S.; Antonaci, C.; Colombatto, S.; Pinzani, M.; Parola, M. Dose dependent and divergent effects of superoxide anion on cell death, proliferation, and migration of activated human hepatic stellate cells. Gut 2006, 55, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Paik, Y.H.; Watanabe, S.; Laleu, B.; Gaggini, F.; Fioraso-Cartier, L.; Molango, S.; Heitz, F.; Merlot, C.; Szyndralewiez, C.; et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 2012, 56, 2316–2327. [Google Scholar] [CrossRef]
- Jiang, J.X.; Chen, X.; Serizawa, N.; Szyndralewiez, C.; Page, P.; Schroder, K.; Brandes, R.P.; Devaraj, S.; Torok, N.J. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic. Biol. Med. 2012, 53, 289–296. [Google Scholar] [CrossRef]
- Lan, T.; Kisseleva, T.; Brenner, D.A. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS ONE 2015, 10, e0129743. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Togashi, H.; Suzuki, A.; Kasai, S.; Ito, J.; Sugahara, K.; Kawata, S. NAD(P)H oxidase plays a crucial role in PDGF-induced proliferation of hepatic stellate cells. Hepatology 2005, 41, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Brenner, D.A. NOX in liver fibrosis. Arch. Biochem. Biophys. 2007, 462, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.Y.; Chin, R.; Cunningham, S.C.; Habib, M.R.; Torresi, J.; Sharland, A.F.; Alexander, I.E.; Angus, P.W.; Herath, C.B. ACE2 Therapy Using Adeno-associated Viral Vector Inhibits Liver Fibrosis in Mice. Mol. Ther. 2015, 23, 1434–1443. [Google Scholar] [CrossRef]
- Jiang, F.; Liu, G.S.; Dusting, G.J.; Chan, E.C. NADPH oxidase-dependent redox signaling in TGF-beta-mediated fibrotic responses. Redox Biol. 2014, 2, 267–272. [Google Scholar] [CrossRef]
- Crosas-Molist, E.; Bertran, E.; Fabregat, I. Cross-Talk Between TGF-beta and NADPH Oxidases During Liver Fibrosis and Hepatocarcinogenesis. Curr. Pharm. Des. 2015, 21, 5964–5976. [Google Scholar] [CrossRef]
- Mortezaee, K. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) and liver fibrosis: A review. Cell Biochem. Funct. 2018, 36, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Cuenca, I.; Roncero, C.; Sancho, P.; Caja, L.; Fausto, N.; Fernandez, M.; Fabregat, I. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J. Hepatol. 2008, 49, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.; Fernandez, M.; Alvarez, A.M.; Roncero, C.; Benito, M.; Gil, J.; Fabregat, I. Activation of caspases occurs downstream from radical oxygen species production, Bcl-xL down-regulation, and early cytochrome C release in apoptosis induced by transforming growth factor beta in rat fetal hepatocytes. Hepatology 2001, 34, 548–556. [Google Scholar] [CrossRef]
- Caja, L.; Sancho, P.; Bertran, E.; Iglesias-Serret, D.; Gil, J.; Fabregat, I. Overactivation of the MEK/ERK pathway in liver tumor cells confers resistance to TGF-beta-induced cell death through impairing up-regulation of the NADPH oxidase NOX4. Cancer Res. 2009, 69, 7595–7602. [Google Scholar] [CrossRef]
- Baffy, G. Kupffer cells in non-alcoholic fatty liver disease: The emerging view. J. Hepatol. 2009, 51, 212–223. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Brenner, D.A. Oxidative stress in alcoholic liver disease: Role of NADPH oxidase complex. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. S1), S98–S103. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.; Pritchard, M.T.; McMullen, M.R.; Wang, Q.; Nagy, L.E. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: Role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J. Leukoc. Biol. 2006, 79, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Rusyn, I.; Yin, M.; Gabele, E.; Yamashina, S.; Dikalova, A.; Kadiiska, M.B.; Connor, H.D.; Mason, R.P.; Segal, B.H.; et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J. Clin. Investig. 2000, 106, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Teufelhofer, O.; Parzefall, W.; Kainzbauer, E.; Ferk, F.; Freiler, C.; Knasmuller, S.; Elbling, L.; Thurman, R.; Schulte-Hermann, R. Superoxide generation from Kupffer cells contributes to hepatocarcinogenesis: Studies on NADPH oxidase knockout mice. Carcinogenesis 2005, 26, 319–329. [Google Scholar] [CrossRef]
- Zhai, L.; Pei, H.; Yang, Y.; Zhu, Y.; Ruan, S. NOX4 promotes Kupffer cell inflammatory response via ROS-NLRP3 to aggravate liver inflammatory injury in acute liver injury. Aging 2022, 14, 6905–6916. [Google Scholar] [CrossRef]
- Minciuna, I.; Taru, M.G.; Procopet, B.; Stefanescu, H. The Interplay between Liver Sinusoidal Endothelial Cells, Platelets, and Neutrophil Extracellular Traps in the Development and Progression of Metabolic Dysfunction-Associated Steatotic Liver Disease. J. Clin. Med. 2024, 13, 1406. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Ye, T.; Sun, Y.; Ji, G.; Shido, K.; Chen, Y.; Luo, L.; Na, F.; Li, X.; Huang, Z.; et al. Targeting the vascular and perivascular niches as a regenerative therapy for lung and liver fibrosis. Sci. Transl. Med. 2017, 9, eaai8710. [Google Scholar] [CrossRef]
- Straub, A.C.; Clark, K.A.; Ross, M.A.; Chandra, A.G.; Li, S.; Gao, X.; Pagano, P.J.; Stolz, D.B.; Barchowsky, A. Arsenic-stimulated liver sinusoidal capillarization in mice requires NADPH oxidase-generated superoxide. J. Clin. Investig. 2008, 118, 3980–3989. [Google Scholar] [CrossRef]
- Nasce, A.; Gariani, K.; Jornayvaz, F.R.; Szanto, I. NADPH Oxidases Connecting Fatty Liver Disease, Insulin Resistance and Type 2 Diabetes: Current Knowledge and Therapeutic Outlook. Antioxidants 2022, 11, 1131. [Google Scholar] [CrossRef] [PubMed]
- Gabbia, D.; Cannella, L.; De Martin, S. The Role of Oxidative Stress in NAFLD-NASH-HCC Transition-Focus on NADPH Oxidases. Biomedicines 2021, 9, 687. [Google Scholar] [CrossRef] [PubMed]
- Larion, S.; Padgett, C.A.; Mintz, J.D.; Thompson, J.A.; Butcher, J.T.; Belin de Chantemele, E.J.; Haigh, S.; Khurana, S.; Fulton, D.J.; Stepp, D.W. NADPH oxidase 1 promotes hepatic steatosis in obese mice and is abrogated by augmented skeletal muscle mass. Am. J. Physiol. Gastrointest. Liver Physiol. 2024, 326, G264–G273. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Kaji, K.; Nishimura, N.; Asada, S.; Koizumi, A.; Matsuda, T.; Yorioka, N.; Tsuji, Y.; Fujinaga, Y.; Sato, S.; et al. Blockade of angiotensin II modulates insulin-like growth factor 1-mediated skeletal muscle homeostasis in experimental steatohepatitis. Biochim. Biophys. Acta Mol. Cell Res. 2024, 1871, 119649. [Google Scholar] [CrossRef]
- Greatorex, S.; Kaur, S.; Xirouchaki, C.E.; Goh, P.K.; Wiede, F.; Genders, A.J.; Tran, M.; Jia, Y.; Raajendiran, A.; Brown, W.A.; et al. Mitochondria- and NOX4-dependent antioxidant defense mitigates progression to nonalcoholic steatohepatitis in obesity. J. Clin. Investig. 2023, 134, e162533. [Google Scholar] [CrossRef]
- Ji, J.; Feng, M.; Huang, Y.; Niu, X. Liraglutide inhibits receptor for advanced glycation end products (RAGE)/reduced form of nicotinamide-adenine dinucleotide phosphate (NAPDH) signaling to ameliorate non-alcoholic fatty liver disease (NAFLD) in vivo and vitro. Bioengineered 2022, 13, 5091–5102. [Google Scholar] [CrossRef] [PubMed]
- Grossini, E.; Garhwal, D.P.; Calamita, G.; Romito, R.; Rigamonti, C.; Minisini, R.; Smirne, C.; Surico, D.; Bellan, M.; Pirisi, M. Exposure to Plasma From Non-alcoholic Fatty Liver Disease Patients Affects Hepatocyte Viability, Generates Mitochondrial Dysfunction, and Modulates Pathways Involved in Fat Accumulation and Inflammation. Front. Med. 2021, 8, 693997. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Chen, Z.; Sun, C.; Yang, D.; Zhou, Z.; Peng, X.; Zheng, L.; Tang, C. Exercise Intervention Mitigates Pathological Liver Changes in NAFLD Zebrafish by Activating SIRT1/AMPK/NRF2 Signaling. Int. J. Mol. Sci. 2021, 22, 10940. [Google Scholar] [CrossRef] [PubMed]
- Bunbupha, S.; Pakdeechote, P.; Maneesai, P.; Prasarttong, P. Nobiletin alleviates high-fat diet-induced nonalcoholic fatty liver disease by modulating AdipoR1 and gp91(phox) expression in rats. J. Nutr. Biochem. 2021, 87, 108526. [Google Scholar] [CrossRef]
- Sarkar, S.; Saha, P.; Seth, R.K.; Mondal, A.; Bose, D.; Kimono, D.; Albadrani, M.; Mukherjee, A.; Porter, D.E.; Scott, G.I.; et al. Higher intestinal and circulatory lactate associated NOX2 activation leads to an ectopic fibrotic pathology following microcystin co-exposure in murine fatty liver disease. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2020, 238, 108854. [Google Scholar] [CrossRef]
- Jiang, J.X.; Fish, S.R.; Tomilov, A.; Li, Y.; Fan, W.; Dehnad, A.; Gae, D.; Das, S.; Mozes, G.; Charville, G.W.; et al. Nonphagocytic Activation of NOX2 Is Implicated in Progressive Nonalcoholic Steatohepatitis During Aging. Hepatology 2020, 72, 1204–1218. [Google Scholar] [CrossRef] [PubMed]
- Albadrani, M.; Seth, R.K.; Sarkar, S.; Kimono, D.; Mondal, A.; Bose, D.; Porter, D.E.; Scott, G.I.; Brooks, B.; Raychoudhury, S.; et al. Exogenous PP2A inhibitor exacerbates the progression of nonalcoholic fatty liver disease via NOX2-dependent activation of miR21. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G408–G428. [Google Scholar] [CrossRef]
- Dattaroy, D.; Seth, R.K.; Das, S.; Alhasson, F.; Chandrashekaran, V.; Michelotti, G.; Fan, D.; Nagarkatti, M.; Nagarkatti, P.; Diehl, A.M.; et al. Sparstolonin B attenuates early liver inflammation in experimental NASH by modulating TLR4 trafficking in lipid rafts via NADPH oxidase activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G510–G525. [Google Scholar] [CrossRef] [PubMed]
- Dattaroy, D.; Pourhoseini, S.; Das, S.; Alhasson, F.; Seth, R.K.; Nagarkatti, M.; Michelotti, G.A.; Diehl, A.M.; Chatterjee, S. Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G298–G312. [Google Scholar] [CrossRef]
- Carmiel-Haggai, M.; Cederbaum, A.I.; Nieto, N. A high-fat diet leads to the progression of non-alcoholic fatty liver disease in obese rats. FASEB J. 2005, 19, 136–138. [Google Scholar] [CrossRef]
- Baratta, F.; Pastori, D.; Bartimoccia, S.; Cammisotto, V.; Cocomello, N.; Colantoni, A.; Nocella, C.; Carnevale, R.; Ferro, D.; Angelico, F.; et al. Poor Adherence to Mediterranean Diet and Serum Lipopolysaccharide are Associated with Oxidative Stress in Patients with Non-Alcoholic Fatty Liver Disease. Nutrients 2020, 12, 1732. [Google Scholar] [CrossRef] [PubMed]
- Rabelo, F.; Stefano, J.T.; Cavaleiro, A.M.; Lima, R.V.C.; de Campos Mazo, D.F.; Carrilho, F.J.; Correa-Giannella, M.L.; Oliveira, C.P. Association between the CYBA and NOX4 genes of NADPH oxidase and its relationship with metabolic syndrome in non-alcoholic fatty liver disease in Brazilian population. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.; Zicari, A.M.; Perri, L.; Carnevale, R.; Nocella, C.; Angelico, F.; Del Ben, M.; Mosca, A.; Zaffina, S.; Panera, N.; et al. Does Nox2 Overactivate in Children with Nonalcoholic Fatty Liver Disease? Antioxid. Redox Signal 2019, 30, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.; Baratta, F.; Ludovica, P.; Battaglia, S.; Carnevale, R.; Nocella, C.; Novo, M.; Pannitteri, G.; Ceci, F.; Angelico, F.; et al. Effects of dark chocolate on endothelial function in patients with non-alcoholic steatohepatitis. Nutr. Metab. Cardiovasc. Dis. 2017, 28, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Del Ben, M.; Polimeni, L.; Carnevale, R.; Bartimoccia, S.; Nocella, C.; Baratta, F.; Loffredo, L.; Pignatelli, P.; Violi, F.; Angelico, F. NOX2-generated oxidative stress is associated with severity of ultrasound liver steatosis in patients with non-alcoholic fatty liver disease. BMC Gastroenterol. 2014, 14, 81. [Google Scholar] [CrossRef]
- Cichoz-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, I.; Rodriguez-Juan, C.; Diaz-Sanjuan, T.; del Hoyo, P.; Colina, F.; Munoz-Yague, T.; Solis-Herruzo, J.A. Uric acid and anti-TNF antibody improve mitochondrial dysfunction in ob/ob mice. Hepatology 2006, 44, 581–591. [Google Scholar] [CrossRef]
- Garcia-Ruiz, I.; Solis-Munoz, P.; Fernandez-Moreira, D.; Grau, M.; Colina, F.; Munoz-Yague, T.; Solis-Herruzo, J.A. High-fat diet decreases activity of the oxidative phosphorylation complexes and causes nonalcoholic steatohepatitis in mice. Dis. Model. Mech. 2014, 7, 1287–1296. [Google Scholar] [CrossRef]
- Ghasemitarei, M.; Ghorbi, T.; Yusupov, M.; Zhang, Y.; Zhao, T.; Shali, P.; Bogaerts, A. Effects of Nitro-Oxidative Stress on Biomolecules: Part 1-Non-Reactive Molecular Dynamics Simulations. Biomolecules 2023, 13, 1371. [Google Scholar] [CrossRef]
- Thapaliya, S.; Wree, A.; Povero, D.; Inzaugarat, M.E.; Berk, M.; Dixon, L.; Papouchado, B.G.; Feldstein, A.E. Caspase 3 inactivation protects against hepatic cell death and ameliorates fibrogenesis in a diet-induced NASH model. Dig. Dis. Sci. 2014, 59, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Lee, G.; Heo, S.Y.; Roh, Y.S. Oxidative Stress Is a Key Modulator in the Development of Nonalcoholic Fatty Liver Disease. Antioxidants 2021, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Tiniakos, D.G. Histopathology of nonalcoholic fatty liver disease. World J. Gastroenterol. 2010, 16, 5286–5296. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tuo, Y.H.; Chen, J.W.; Wang, Q.Y.; Li, S.; Li, M.C.; Dai, G.; Wang, J.S.; Zhang, Y.L.; Feng, L.; et al. NADPH oxidase inhibitor regulates microRNAs with improved outcome after mechanical reperfusion. J. Neurointerv. Surg. 2017, 9, 702–706. [Google Scholar] [CrossRef]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Loyer, X.; Paradis, V.; Henique, C.; Vion, A.C.; Colnot, N.; Guerin, C.L.; Devue, C.; On, S.; Scetbun, J.; Romain, M.; et al. Liver microRNA-21 is overexpressed in non-alcoholic steatohepatitis and contributes to the disease in experimental models by inhibiting PPARalpha expression. Gut 2016, 65, 1882–1894. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Tang, N.; Wu, K.; Dai, W.; Ye, C.; Shi, J.; Zhang, J.; Ning, B.; Zeng, X.; Lin, Y. MiR-21 simultaneously regulates ERK1 signaling in HSC activation and hepatocyte EMT in hepatic fibrosis. PLoS ONE 2014, 9, e108005. [Google Scholar] [CrossRef] [PubMed]
- Petronio, M.S.; Zeraik, M.L.; Fonseca, L.M.; Ximenes, V.F. Apocynin: Chemical and biophysical properties of a NADPH oxidase inhibitor. Molecules 2013, 18, 2821–2839. [Google Scholar] [CrossRef]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.X.; Werstuck, G.; Chen, Y. Oxidative stress and aging diseases. Oxid. Med. Cell Longev. 2014, 2014, 569146. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Silvestri, R.; Loffredo, L.; Novo, M.; Cammisotto, V.; Castellani, V.; Bartimoccia, S.; Nocella, C.; Violi, F. Oleuropein, a component of extra virgin olive oil, lowers postprandial glycaemia in healthy subjects. Br. J. Clin. Pharmacol. 2018, 84, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [PubMed]
- El-Baz, A.M.; Shata, A.; Nouh, N.A.; Jamil, L.; Hafez, M.M.; Negm, S.; El-Kott, A.F.; AlShehri, M.A.; Khalaf, E.M. Vinpocetine and Lactobacillus improve fatty liver in rats: Role of adiponectin and gut microbiome. AMB Express 2024, 14, 89. [Google Scholar] [CrossRef]
- Sokal-Dembowska, A.; Jarmakiewicz-Czaja, S.; Ferenc, K.; Filip, R. Can Nutraceuticals Support the Treatment of MASLD/MASH, and thus Affect the Process of Liver Fibrosis? Int. J. Mol. Sci. 2024, 25, 5238. [Google Scholar] [CrossRef]
- Bode, K.; Hauri-Hohl, M.; Jaquet, V.; Weyd, H. Unlocking the power of NOX2: A comprehensive review on its role in immune regulation. Redox Biol. 2023, 64, 102795. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, G.; Szyndralewiez, C.; Molango, S.; Carnesecchi, S.; Heitz, F.; Wiesel, P.; Wood, J.M. Therapeutic potential of NADPH oxidase 1/4 inhibitors. Br. J. Pharmacol. 2017, 174, 1647–1669. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Guo, N.; Xu, H.; Li, Y.; Sun, T.; Jiang, X.; Fu, D.; You, T.; Diao, S.; Huang, Y.; et al. Caveolin-1 ameliorates hepatic injury in non-alcoholic fatty liver disease by inhibiting ferroptosis via the NOX4/ROS/GPX4 pathway. Biochem. Pharmacol. 2024, 230, 116594. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| References | NOX Isoform | Disease Status | Experimental Model | Main Outcomes in Association with NOXs |
|---|---|---|---|---|
| Experimental Studies | ||||
| Larion S. et al., 2024 [83] | NOX1 | MASLD | NOX1 knockout mice: lean and db/db | ↑ Superoxide ↑ Insulin signaling ↑ Fat accumulation in the liver |
| Tanaka M. et al., 2024 [84] | NOX2 | MASH-derived sarcopenia | MCD-fed mice steatohepatitis and skeletal muscle atrophy model | NOX2 down-regulation induces: ↓ Proinflammatory cytokines (i.e., TNFα, IL6, and IL1β) ↑ Antioxidant capacity ↑ Antioxidant enzymes ↑ Hepatic and plasma IGF-1 |
| Greatorex S. et al., 2024 [85] | NOX4 | MASLD/MASH | 1. Hepatocyte NOX4 deletion in HFD obese mice: develop steatosis, but not MASH 2. Hepatocyte NOX4 over-expression in mice fed a MASH-promoting diet | 1. ↑ Hepatic oxidative damage ↑ Inflammation ↑ T cell recruitment 2. ↓ MASH and fibrosis |
| Ji J. et al., 2022 [86] | NOX2 | MASLD | HFD-fed mice | NOX2 over- expression induces: ↓ liver function ↑ ROS levels ↑ TNF-α, IL-1β and IL-6 |
| Grossini E., et al., 2021 [87] | NOX2 | MASLD | Human hepatocellular carcinoma cells (Huh7.5) treated with plasma from: 1. 12 MAFLD patients 2. 12 Healthy subjects | Plasma of MAFLD patients induced: ↑ H2O2 ↑ Mitochondrial ROS ↓ Mitochondrial membrane potential ↑ Triglycerides ↑ NF-kB ↑ NOX2 |
| Zou Y. et al., 2021 [88] | NOX4 | MASLD | HFD-fed Zebrafish | ↑ NOX4 ↑ ROS ↓ MDA |
| Bunbupha S. et al., 2021 [89] | NOX2 | MASLD | HFD-fed rats down-regulation of liver NOX2 | In plasma and hepatic tissue ↓ MDA ↑ SOD activity |
| Sarkar S. et al., 2020 [90] | NOX2 | MASLD | p47phox knockout CD-HFD-fed mice | ↓ Collagen protein (fibrosis) in intestine |
| Jiang JX et al., 2020 [91] | NOX1 NOX2 NOX4 | MASH | Old-Fast food diet mice | ↑ NOX2 NOX4 and NOX1 not induced |
| Albadrani M. et al., 2019 [92] | NOX2 | MASLD/MASH | 1. p47phox knockout HFD-fed mice 2. Leptin-primed immortalized Kupffer cells (SV40) | 1. ↓ TNF-α ↓ Stellate cell activation ↓ NOX2-derived peroxynitrite 2. SV40 cells treated with apocynin: ↓ NOX2-derived peroxynitrite ↓ Kupffer cell activation ↓stellate cell pathology |
| Matsumoto M., 2018 [46] | NOX1 | MASLD | 1. HFD-fed and HFC-fed mice 2. NOX1-knockout mice | 1. ↑ NOX1 expression ↑ Hepatic cleaved-C3 2. ↓ Hepatic cleaved-C3 ↓ Peroxynitrite injury in hepatic sinusoids ↓ Nitrotyrosine adducts |
| García-Ruiz I. et al., 2016 [45] | NOX2 | MASH | HFD-fed NOX1- knockout mice |
|
| Dattaroy D. et al., 2015 [93] | Not specified | MASH | Steatohepatitic injury mice HFD-fed mice Kupffer Cells culture | ↑ gp91/p47phox colocalization ↑ Peroxynitrite formation |
| Dattaroy D. et al., 2014 [94] | Not specified | MASH | 1. HFD-fed mice 2. p47phox knockout CD-HFD-fed mice | 1. ↑ Oxidative stress ↑ p47phox expression ↑ NF-κB activation 2. ↓ NF-κB activation ↓ Fibrogenesis |
| Carmiel-Haggai M. et al., 2004 [95] | Not specified | MASLD | Obese HFD-fed rats | ↑ NOX activity ↑ Lipid peroxidation ↑ Protein carbonyl formation ↓ Antioxidant defense |
| Human Studies | ||||
| Baratta F. et al., 2020 [96] | NOX2 | MASLD | MASLD patients (n = 193) Cardio-metabolic patients without MASLD (n = 45) | ↑ sNOX2-dp |
| Rabelo F. et al., 2018 [97] | NOX4 | MASLD | MASLD patients (n = 207) | Association between SNPs in the NOX4 gene and alanine transferase |
| Loffredo L. et al., 2019 [98] | NOX2 | MASLD | MASLD children (n = 67) Controls (n = 73) | ↑ sNOX2-dp ↑ Isoprostanes ↑ Triglycerides ↑ HOMA-IR ↑ Fasting glucose and insulin Linear association between sNOX2-dp and degree of liver damage |
| Loffredo L. et al., 2017 [99] | NOX2 | MASH | MASH (n = 19), Fatty liver disease (n = 19), controls (n = 19) | ↑ sNOX2-dp ↑ Isoprostanes ↓ FMD ↓ NOx bioavailability |
| Matsumoto M et al., 2018 [46] | NOX1 | MASH | MASH patients | ↑ NOX1 expression in liver |
| Del Ben M. et al., 2014 [100] | NOX2 | MASLD | Steatosis patients ith or without MASLD (n = 264) | ↑ sNOX2-dp ↑ Urinary 8-iso-PGF2α Urinary 8-iso-PGF2α and sNOX2-dp increase with the liver steatosis severity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cammisotto, V.; Valeriani, E.; Pignatelli, P.; Violi, F. Nicotinamide Adenine Dinucleotide Phosphate Oxidases and Metabolic Dysfunction-Associated Steatotic Liver Disease. Antioxidants 2025, 14, 83. https://doi.org/10.3390/antiox14010083
Cammisotto V, Valeriani E, Pignatelli P, Violi F. Nicotinamide Adenine Dinucleotide Phosphate Oxidases and Metabolic Dysfunction-Associated Steatotic Liver Disease. Antioxidants. 2025; 14(1):83. https://doi.org/10.3390/antiox14010083
Chicago/Turabian StyleCammisotto, Vittoria, Emanuele Valeriani, Pasquale Pignatelli, and Francesco Violi. 2025. "Nicotinamide Adenine Dinucleotide Phosphate Oxidases and Metabolic Dysfunction-Associated Steatotic Liver Disease" Antioxidants 14, no. 1: 83. https://doi.org/10.3390/antiox14010083
APA StyleCammisotto, V., Valeriani, E., Pignatelli, P., & Violi, F. (2025). Nicotinamide Adenine Dinucleotide Phosphate Oxidases and Metabolic Dysfunction-Associated Steatotic Liver Disease. Antioxidants, 14(1), 83. https://doi.org/10.3390/antiox14010083