



Deciphering Oxidative Stress in Cardiovascular Disease Progression: A Blueprint for Mechanistic Understanding and Therapeutic Innovation


Abstract
1. Introduction
2. The Dynamic Role of Oxidative Stress in the Progression of Cardiovascular Disease
2.1. Molecular Mechanisms Underlying Oxidative Stress
2.2. Sex-Based Differences in Redox Status and Their Implications for CVD
2.3. Oxidative Stress Profile at Various Stages of CVDs
2.3.1. Early Triggering Mechanisms of Oxidative Stress in Endothelial Injury
2.3.2. Oxidative Stress in the Formation of Atherosclerotic Lesions
2.3.3. Oxidative Stress and Reperfusion Injury in Acute Ischemic Events
2.3.4. Oxidative Stress Mechanisms in Cardiac Remodeling and Chronic Heart Failure
2.3.5. Protective Roles of ROS in CVDs
3. The Protective Effects of Antioxidants and the Underlying Redox Signaling Mechanisms in the Heart
3.1. Redox Signaling Pathways
3.1.1. Nrf2/Keap1/ARE Signaling Pathway
3.1.2. PI3K/Akt Pathway
3.1.3. AMPK Pathway
3.1.4. SIRT1 Pathway
3.1.5. MAPK Pathway
3.1.6. CVDs Associated with Oxidative Stress and Their Underlying Signaling Pathways

{kind=link}
{kind=link}
| CVDs | Core Signaling Pathways and Their Mechanisms | Role of ROS in CVDs | Reference | |
|---|---|---|---|---|
| AS | NF-κB Nrf2 MAPK PI3K/Akt SIRT1 | NF-κB drives inflammation, Nrf2 promotes antioxidant defense, SIRT1 regulates metabolism, and MAPK/Akt promotes proliferation. | Increases lipid oxidation promotes plaque formation and progression. | [193,194,195] |
| HTN | RAAS ROS ET-1 NOX AMPK | RAAS increases vascular tension, NOX generates ROS, AMPK regulates energy metabolism and improves vascular function, and ET-1 promotes vasoconstriction. | Leads to vascular stiffening, promoting increased vascular resistance and endothelial dysfunction. | [196,197] |
| CAD | NF-κB p38 MAPK PI3K/Akt SIRT3 | p38 MAPK regulates inflammation, SIRT3 promotes antioxidant defense, and NF-κB induces inflammation. | Induces endothelial cell apoptosis, promoting coronary plaque instability. | [198,199] |
| AMI | JAK/STAT NF-κB p38 MAPK HIF-1α | HIF-1α activates protective genes, JAK/STAT regulates inflammation, and p38 MAPK promotes cardiac repair. | Results in cardiomyocyte apoptosis and necrosis, leading to aggravated myocardial injury. | [200,201] |
| DCM | TGF-β PI3K/Akt JNK ERK | TGF-β promotes fibrosis, JNK/ERK induces injury, and PI3K/Akt prevents apoptosis. | Exacerbates cardiac fibrosis, leading to ventricular remodeling and functional failure. | [202,203] |
| HCM | IGF-1 mTOR ERK GATA4 | GATA4 induces hypertrophic genes and IGF-1/mTOR promotes growth. | Exacerbates cardiac hypertrophy and oxidative stress, leading to cardiac dysfunction. | [204] |
| PH | ET-1 TGF-β NFAT PI3K/Akt mTOR | NFAT and TGF-β induce vascular smooth muscle cell proliferation and remodeling and mTOR regulates cell proliferation. | Induces vascular smooth muscle cell proliferation and increases pulmonary arterial pressure. | [205] |
| MIRI |
ROS JAK/STAT ERK1/2 NOX | NOX induces oxidative damage and ERK1/2 regulates cell growth and repair. | Induces cardiomyocyte apoptosis and necrosis, leading to reperfusion injury. | [206,207] |
| CVI | NOX NF-κB VEGF MMP | MMPs degrade the extracellular matrix, facilitating vascular remodeling, and VEGF promotes angiogenesis. | Increases inflammation and vascular permeability, leading to chronic vascular wall injury. | [208,209] |
| VTE | P-selectin TGF-β NF-κB COX-2 | COX-2 promotes inflammation and P-selectin mediates leukocyte and platelet adhesion. | Promotes thrombus formation and exacerbates vascular occlusion. | [210,211] |
| RHD | JAK/STAT NF-κB IL-17 | IL-17 drives chronic inflammation and induces immune responses. | Induces chronic inflammation and fibrosis, impairing cardiac valve function. | [212] |
| CAV | NF-κB mTOR JAK/STAT PD-1/PD-L1 | PD-1/PD-L1 regulates immune suppression and mTOR regulates cell proliferation. | Promotes chronic rejection and vascular remodeling, leading to allograft heart injury. | [213,214] |
| Afib | CaMKII NF-κB TGF-β | TGF-β induces fibrosis and atrial remodeling and CaMKII modulates myocardial electrical remodeling. | Leads to atrial remodeling, increasing the incidence of atrial fibrillation. | [215] |
| HF | β-AR PI3K/Akt NF-κB Notch | Notch signaling modulates cell proliferation and apoptosis and β-AR potentiates cardiac contractility. | Increases apoptosis and fibrosis, leading to impaired cardiac function. | [216,217] |
| Heart inflammation | TLR NF-κB MAPK IL-1β | IL-1β mediates the inflammatory response and TLRs mediate the recognition of pathogens and trigger immune responses. | Induces oxidative stress and immune responses, thereby exacerbating cardiac injury. | [218,219] |
| Rheumatic carditis | NF-κB TGF-β JAK/STAT IL-6 | IL-6 drives chronic inflammation and JAK/STAT signaling modulates immune cell activation. | Induces chronic myocardial inflammation, thereby exacerbating cardiac injury. | [220,221] |
| MVP | TGF-β MMP NF-κB SMAD | Smad signaling contributes to valvular fibrosis by promoting extracellular matrix protein synthesis. | Increases valvular fibrosis, thereby impairing valve function. | [222,223] |
| HOCM | CaMKII ROS PI3K/Akt ERK | ERK mediates cardiac hypertrophy and PI3K/Akt regulates cell survival and proliferation. | Exacerbates myocardial hypertrophy and dysfunction. | [224,225] |
| CHD | Wnt/β-catenin Notch NF-κB SHH | Wnt and SHH signaling regulate cardiac development and Notch regulates cell differentiation. | Causes developmental abnormalities, thereby exacerbating congenital heart disease. | [226,227] |
| Coronary artery spasm angina | PKC eNOS ET-1 CaMKII | CaMKII affects myocardial contractility and eNOS regulates nitric oxide production. | Causes coronary artery constriction, leading to ischemic events. | [228] |
| PSVT | CaMKII PKA RyR SERCA | SERCA regulates calcium ion reuptake and RyR regulates calcium ion release. | Causes arrhythmia, thereby increasing myocardial workload. | [229] |
| CHF | β-AR ROS NF-κB SIRT3 | SIRT3 protects mitochondrial function and reduces oxidative stress. | Promotes myocardial fibrosis and apoptosis, leading to aggravated heart failure. | [230] |
| Left ventricular insufficiency | TGF-β NF-κB PI3K/Akt IL-1β | IL-1β induces cardiac fibrosis and TGF-β triggers structural remodeling. | Causes myocardial fibrosis. | [231] |
3.2. Antioxidants
| Antioxidant | Main Mechanism of Action | CVDs Applied | Animal Model | Reference |
|---|---|---|---|---|
| Vitamin C | Antioxidant scavenging of free radicals, reduction of lipid peroxidation, protection of endothelial function, and inhibition of inflammatory response. | AS CHD HTN | LDLr−/− mice | [232] |
| Vitamin E | Inhibits lipid peroxidation, reduces oxidative damage, and inhibits inflammation and cell proliferation. | AS MIRI | CETP transgenic rats | [233] |
| Glutathione | Increases antioxidant reserves, regulates redox balance, and attenuates mitochondrial damage. | HTN CHD Myocardial fibrosis | Aldosterone-induced hypertension in C57BL/6 mice | [234] |
| lipoic acid | Inhibits oxidative stress and inflammatory responses and improves mitochondrial function. | DbCM HTN HF | RAS-activated mice | [235] |
| CoQ10 | Enhances mitochondrial respiratory chain function, reduces ROS production, and improves myocardial tolerance. | HF MIRI AS | ApoE−/− mice | [236] |
| Resveratrol | Activates SIRT1, reduces oxidative stress, and inhibits inflammation. | MIRI AS | ApoE−/− mice | [237] |
| N-Acetylcysteine | Provides GSH precursors, reduces ROS, and enhances antioxidant capacity. | CHD MIRI | MIRI in C57BL/6 Mice | [238] |
| Astaxanthin | A potent antioxidant that reduces lipid peroxidation and inhibits inflammatory responses. | MIRI HF AS | SOD2-deficient mice | [239] |
| Quercetin | Inhibits oxidative stress and lipid peroxidation and enhances anti-inflammatory capacity. | HTN AS CHD | AT1 transgenic mice | [240] |
| Tea polyphenols | Inhibits endothelial inflammation and lipid peroxidation and protects the myocardium and blood vessels. | Diabetic AS CMP | Leprdb/db mice | [241] |
| Sodium thiosulfate | Scavenges ROS, reduces mitochondrial oxidative stress, and inhibits calcium overload and apoptosis. | MIRI HF | SIRT3 gene-deficient mice | [242] |
| Statin drugs | Reduces cholesterol, inhibits ROS production, and attenuates vascular endothelial damage. | AS CHD HTN | LDLr−/− and ApoE−/− mice | [243] |
| Vitamin D | Regulates calcium metabolism, reduces vascular smooth muscle cell proliferation, and protects the endothelium. | HTN AS Myocardial Fibrosis | HTN-induced ApoE−/− mice | [244] |
| Gallic acid | Inhibits oxidative stress and smooth muscle cell proliferation and regulates endothelial cell activity. | AS DbCM | Cholesterol-hypercholesterolemic rat model | [245] |
| Hydrogen sulfide | Acts as an endogenous antioxidant molecule, regulates mitochondrial function, and inhibits apoptosis. | MIRI HF DbCM | Diabetes-induced ZDF in rats | [246] |
| Thioredoxin | Inhibits ROS generation, protects endothelial cells and mitochondria, and regulates calcium ion balance. | AS HTN HF | Angiotensin II-induced hypertension in C57BL/6 mice | [247] |
4. Emerging Paradigms in Cardiovascular Protection
4.1. Drug Therapy
| Category | Drug | Mechanism | Advantages and Innovation | Reference |
|---|---|---|---|---|
| Mitochondria-Targeted Antioxidants | MitoTEMPO | Targets mitochondria to reduce ROS generation and improve mitochondrial function. | Targeting mitochondria with high specificity, directly acting on major ROS production sites, and reducing side effects. | [273] |
| SkQ1 | Penetrates the mitochondrial membrane to reduce oxidative damage and delay cell apoptosis. | Unique mitochondrial penetration mechanism for effective myocardial cell protection. | [274] | |
| Nrf2 Activators | Bardoxolone methyl | Activates the Nrf2/ARE pathway to enhance antioxidant enzyme (e.g., HO-1) expression. | Enhances endogenous antioxidant capacity via transcriptional regulation, providing stable long-term effects. | [275] |
| Small Molecule Scavengers | Edaravone derivatives | Scavenges free radicals and reduces acute ROS levels. | Optimized molecular structure for higher efficiency and rapid action. | |
| Tempol | Mimics superoxide dismutase (SOD) to inhibit superoxide production. | Simple synthetic pathway, providing an economical antioxidant treatment option. | ||
| Enzyme-Based Therapies | PEG-SOD/PEG-Catalase | Modifies enzymes to extend circulation time and improve ROS scavenging capacity. | Enhanced stability and bioavailability, reducing the need for frequent administration. | |
| ROS Metabolism Inhibitors | GKT137831 | Inhibits NOX to reduce ROS production. | High-specificity NOX inhibition with minimal side effects. | |
| p66Shc Inhibitor | Targets upstream regulators of mitochondrial ROS production, delaying cardiovascular aging. | Innovative target directly addressing core mitochondrial oxidative stress mechanisms. | ||
| Multifunctional Antioxidant Molecules | RTA 408 | Simultaneously scavenges ROS and inhibits inflammation. | Integrated mechanism combining anti-inflammatory and antioxidant functions, broadening indications. |
4.2. Gene Therapy
| Typology | Advantages | Disadvantages | Related Research | Reference |
|---|---|---|---|---|
| Gene editing | High-precision targeted gene editing enabling permanent repair and reducing recurrence risk in monogenic diseases. | The potential risks of this technology include off-target mutations, immune responses, and challenges in precise delivery, which are coupled with high technical complexity and cost. | CRISPR-mediated gene editing ameliorates cardiac hypertrophy and improves cardiac function in a murine model of MYBPC3-related cardiomyopathy. | [277] |
| Gene replacement therapy | The precise targeting and repair of defective genes, leading to the restoration of normal cellular physiology, highlighting its broad clinical potential. | The limitations of this technology, including low insertion efficiency, off-target effects, and immunogenicity, necessitate further investigations into its long-term safety profile. | DMD gene replacement significantly improves cardiac function in a mouse model of Duchenne muscular dystrophy. | [278] |
| RNA interference | Highly specific targeting of specific gene expression to inhibit pathological effects of disease-causing genes; applicable to reversible regulation without affecting gene ontology; and short duration of action to modulate efficacy and safety. | Shorter expression time, requiring repeated administration; lower stability in vivo, difficult to resist enzymatic breakdown; complex drug delivery and dose regulation, frequent dosing cycles; and may trigger cytotoxic and immune responses. | RNAi inhibition of PCSK9 significantly reduces LDL-C levels and inhibits the course of atherosclerosis in ApoE−/− mice. | [279] |
| Gene enhancement therapy | Can upregulate protective gene expression and enhance antioxidant, anti-inflammatory and metabolic regulation functions; suitable for chronic diseases lacking protective mechanisms; and can delay lesions by modulating multiple protective pathways. | Higher dosage requirements, overexpression may trigger toxic effects; dependence on the level of gene expression makes it difficult to control the side effects of gene overexpression; the immune system may reject exogenous genes; and long-term monitoring and observation of safety is required. | SIRT1 overexpression protects against myocardial ischemia-reperfusion injury. | [280] |
| AAV-mediated gene delivery | Persistent gene expression, which facilitates long-term treatment; low immunogenicity, which reduces the risk of immune rejection; cardiac-specific delivery potential, which enhances therapeutic efficacy; and suitability for long-acting treatments for chronic diseases and genetic defects. | The limited capacity of AAV vectors restricts large-scale gene delivery; it is difficult to completely avoid immune recognition, so we need to be vigilant about potential immune reactions; the uneven integration of vectors may trigger gene insertion mutations; and the risk of long-term expression needs to be adequately verified. | AAV delivery of SERCA2a restores calcium homeostasis, reduces myocardial fibrosis, and restores cardiac function in mouse and porcine models of heart failure. | [281] |
| Genetic vaccine | Stimulates immune system regulation and reduces arterial inflammation and lipid accumulation; intervenes well in the early stages of disease; is easy to administer multiple times at relatively low cost; and can be personalised to improve vaccine specificity. | The immune response is difficult to control accurately and may trigger an autoimmune response; there are individual differences in vaccine tolerance and durability; the vaccine development and evaluation cycle is long; and some of the vaccine components may cause toxicity or side effects. | Gene vaccine targeting; CD68 and LDLR reduce arterial plaque formation and enhance anti-inflammation and lipid metabolism in a mouse model of atherosclerosis. | [282] |
4.3. Cellular and Regenerative Medicine
4.3.1. Cardiac Cell Therapy
4.3.2. Exocrine Therapy
4.4. Inflammation Regulation and Immunotherapy
4.5. Metabolic Regulator
5. Outlook and Future Perspective
Funding
Conflicts of Interest
References
- Tomii, D.; Pilgrim, T.; Borger, M.A.; De Backer, O.; Lanz, J.; Reineke, D.; Siepe, M.; Windecker, S. Aortic Stenosis and Coronary Artery Disease: Decision-Making Between Surgical and Transcatheter Management. Circulation 2024, 150, 2046–2069. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.C.; Chan, W.; Dart, A.; Shaw, J.A. Novel therapeutic targets and emerging treatments for atherosclerotic cardiovascular disease. Eur. Heart J. Cardiovasc. Pharmacother. 2024, 10, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Hummelgaard, S.; Vilstrup, J.P.; Gustafsen, C.; Glerup, S.; Weyer, K. Targeting PCSK9 to tackle cardiovascular disease. Pharmacol. Ther. 2023, 249, 108480. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, A.; Minhas, A.S.; Kazzi, B.; Varma, B.; Choi, E.; Thakkar, A.; Michos, E.D. Sex-specific differences in cardiovascular risk factors and implications for cardiovascular disease prevention in women. Atherosclerosis 2023, 384, 117269. [Google Scholar] [CrossRef]
- Ariza Corbo, M.J.; Muñiz-Grijalvo, O.; Blanco Echevarría, A.; Díaz-Díaz, J.L. Genetic basis of hypertriglyceridemia. Clin. Investig. Arterioscler. 2024, 36 (Suppl. 2), S3–S12. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Bayo Jimenez, M.T.; Vujacic-Mirski, K.; Helmstädter, J.; Kröller-Schön, S.; Münzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxidative Med. Cell. Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, S.; Marino, A.; Morabito, R.; Remigante, A. Interplay Between Metabolic Pathways and Increased Oxidative Stress in Human Red Blood Cells. Cells 2024, 13, 2026. [Google Scholar] [CrossRef] [PubMed]
- Shahidi, F.; Zhong, Y. Lipid oxidation and improving the oxidative stability. Chem. Soc. Rev. 2010, 39, 4067–4079. [Google Scholar] [CrossRef] [PubMed]
- Hauck, A.K.; Huang, Y.; Hertzel, A.V.; Bernlohr, D.A. Adipose oxidative stress and protein carbonylation. J. Biol. Chem. 2019, 294, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Elimban, V.; Bartekova, M.; Adameova, A. Involvement of Oxidative Stress in the Development of Subcellular Defects and Heart Disease. Biomedicines 2022, 10, 393. [Google Scholar] [CrossRef]
- Evans, C.E.; Cober, N.D.; Dai, Z.; Stewart, D.J.; Zhao, Y.Y. Endothelial cells in the pathogenesis of pulmonary arterial hypertension. Eur. Respir. J. 2021, 58, 2003957. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xu, X.D.; Ma, M.Q.; Liang, Y.; Cai, Y.B.; Zhu, Z.X.; Xu, T.; Zhu, L.; Ren, K. The mechanisms of ferroptosis and its role in atherosclerosis. Biomed. Pharmacother. 2024, 171, 116112. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Pecchillo Cimmino, T.; Ammendola, R.; Cattaneo, F.; Esposito, G. NOX Dependent ROS Generation and Cell Metabolism. Int. J. Mol. Sci. 2023, 24, 2086. [Google Scholar] [CrossRef]
- Bortolotti, M.; Polito, L.; Battelli, M.G.; Bolognesi, A. Xanthine oxidoreductase: One enzyme for multiple physiological tasks. Redox Biol. 2021, 41, 101882. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Chi, R.; Peng, Y.; Sun, K.; Liu, H.; Guo, F.; Guo, J. The Role and Interactive Mechanism of Endoplasmic Reticulum Stress and Ferroptosis in Musculoskeletal Disorders. Biomolecules 2024, 14, 1369. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Guo, R.; Zong, S.; Wu, M.; Gu, J.; Yang, M. Architecture of Human Mitochondrial Respiratory Megacomplex I2III2IV2. Cell 2017, 170, 1247–1257.e12. [Google Scholar] [CrossRef]
- Panov, A.V.; Mayorov, V.I.; Dikalov, S.I. Role of Fatty Acids β-Oxidation in the Metabolic Interactions Between Organs. Int. J. Mol. Sci. 2024, 25, 12740. [Google Scholar] [CrossRef] [PubMed]
- Hunte, C.; Zickermann, V.; Brandt, U. Functional modules and structural basis of conformational coupling in mitochondrial complex I. Science 2010, 329, 448–451. [Google Scholar] [CrossRef]
- Esteban-Amo, M.J.; Jiménez-Cuadrado, P.; Serrano-Lorenzo, P.; de la Fuente, M.; Simarro, M. Succinate Dehydrogenase and Human Disease: Novel Insights into a Well-Known Enzyme. Biomedicines 2024, 12, 2050. [Google Scholar] [CrossRef]
- Čunátová, K.; Fernández-Vizarra, E. Pathological variants in nuclear genes causing mitochondrial complex III deficiency: An update. J. Inherit. Metab. Dis. 2024, 47, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B.; Hüttemann, M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015, 24, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Zotta, A.; O’Neill, L.A.J.; Yin, M. Unlocking potential: The role of the electron transport chain in immunometabolism. Trends Immunol. 2024, 45, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Pham, L.; Arroum, T.; Wan, J.; Pavelich, L.; Bell, J.; Morse, P.T.; Lee, I.; Grossman, L.I.; Sanderson, T.H.; Malek, M.H.; et al. Regulation of mitochondrial oxidative phosphorylation through tight control of cytochrome c oxidase in health and disease—Implications for ischemia/reperfusion injury, inflammatory diseases, diabetes, and cancer. Redox Biol. 2024, 78, 103426. [Google Scholar] [CrossRef]
- Le Bras, M.; Clément, M.V.; Pervaiz, S.; Brenner, C. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol. Histopathol. 2005, 20, 205–219. [Google Scholar] [CrossRef]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updates 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Brand, M.D.; Pakay, J.L.; Ocloo, A.; Kokoszka, J.; Wallace, D.C.; Brookes, P.S.; Cornwall, E.J. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem. J. 2005, 392, 353–362. [Google Scholar] [CrossRef]
- Roussel, D.; Harding, M.; Runswick, M.J.; Walker, J.E.; Brand, M.D. Does any yeast mitochondrial carrier have a native uncoupling protein function? J. Bioenerg. Biomembr. 2002, 34, 165–176. [Google Scholar] [CrossRef]
- O’Reilly, A.; Zhao, W.; Wickström, S.; Arnér, E.S.J.; Kiessling, R. Reactive oxygen species: Janus-faced molecules in the era of modern cancer therapy. J. ImmunoTher. Cancer 2024, 12, e009409. [Google Scholar] [CrossRef]
- Gan, Z.; van der Stelt, I.; Li, W.; Hu, L.; Song, J.; Grefte, S.; van de Westerlo, E.; Zhang, D.; van Schothorst, E.M.; Claahsen-van der Grinten, H.L.; et al. Mitochondrial Nicotinamide Nucleotide Transhydrogenase: Role in Energy Metabolism, Redox Homeostasis, and Cancer. Antioxid. Redox Signal. 2024, 41, 927–956. [Google Scholar] [CrossRef]
- Kamal, R.; Awasthi, A.; Pundir, M.; Thakur, S. Healing the diabetic wound: Unlocking the secrets of genes and pathways. Eur. J. Pharmacol. 2024, 975, 176645. [Google Scholar] [CrossRef]
- Akhigbe, R.; Ajayi, A. The impact of reactive oxygen species in the development of cardiometabolic disorders: A review. Lipids Health Dis. 2021, 20, 23. [Google Scholar] [CrossRef]
- Silveira, T.H.R.; Silva, F.H.; Hill, W.G.; Antunes, E.; de Oliveira, M.G. Targeting NADPH Oxidase as an Approach for Diabetic Bladder Dysfunction. Antioxidants 2024, 13, 1155. [Google Scholar] [CrossRef]
- Yan, H.; Yin, Y.; Zhou, Y.; Li, Z.; Li, Y.; Ren, L.; Wen, J.; Wang, W. Regulation of cardiovascular diseases by histone deacetylases and NADPH oxidases. Redox Biol. 2024, 77, 103379. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Tang, B.; Zhang, C. Signaling pathways of chronic kidney diseases, implications for therapeutics. Signal Transduct. Target. Ther. 2022, 7, 182. [Google Scholar] [CrossRef]
- García, J.G.; Ansorena, E.; Izal, I.; Zalba, G.; de Miguel, C.; Milagro, F.I. Structure, regulation, and physiological functions of NADPH oxidase 5 (NOX5). J. Physiol. Biochem. 2023, 79, 383–395. [Google Scholar] [CrossRef]
- Sylvester, A.L.; Zhang, D.X.; Ran, S.; Zinkevich, N.S. Inhibiting NADPH Oxidases to Target Vascular and Other Pathologies: An Update on Recent Experimental and Clinical Studies. Biomolecules 2022, 12, 823. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Mondragón, R.; Lozhkin, A.; Vendrov, A.E.; Runge, M.S.; Isom, L.L.; Madamanchi, N.R. NADPH Oxidases and Oxidative Stress in the Pathogenesis of Atrial Fibrillation. Antioxidants 2023, 12, 1833. [Google Scholar] [CrossRef]
- Furuhashi, M. New insights into purine metabolism in metabolic diseases: Role of xanthine oxidoreductase activity. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E827–E834. [Google Scholar] [CrossRef]
- Mokhosoev, I.M.; Astakhov, D.V.; Terentiev, A.A.; Moldogazieva, N.T. Human Cytochrome P450 Cancer-Related Metabolic Activities and Gene Polymorphisms: A Review. Cells 2024, 13, 1958. [Google Scholar] [CrossRef]
- Knaus, U.G. Oxidants in Physiological Processes. Handb. Exp. Pharmacol. 2021, 264, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Ullah, Z.; Yue, P.; Mao, G.; Zhang, M.; Liu, P.; Wu, X.; Zhao, T.; Yang, L. A comprehensive review on recent xanthine oxidase inhibitors of dietary based bioactive substances for the treatment of hyperuricemia and gout: Molecular mechanisms and perspective. Int. J. Biol. Macromol. 2024, 278, 134832. [Google Scholar] [CrossRef]
- Engin, A. Protein Kinases in Obesity, and the Kinase-Targeted Therapy. Adv. Exp. Med. Biol. 2024, 1460, 199–229. [Google Scholar] [CrossRef]
- Benítez-King, G.; Argueta, J.; Miranda-Riestra, A.; Muñoz-Delgado, J.; Estrada-Reyes, R. Interaction of the Melatonin/Ca2+-CaM Complex with Calmodulin Kinase II: Physiological Importance. Mol. Pharmacol. 2024, 106, 3–12. [Google Scholar] [CrossRef]
- Mirzavi, F.; Rajabian, A.; Hosseini, H. The potential protective role of carotenoids from saffron: A focus on endoplasmic reticulum stress-related organ damage. Food Sci. Nutr. 2024, 12, 6108–6122. [Google Scholar] [CrossRef]
- Engin, A. Nonalcoholic Fatty Liver Disease and Staging of Hepatic Fibrosis. Adv. Exp. Med. Biol. 2024, 1460, 539–574. [Google Scholar] [CrossRef]
- Laurindo, F.R.; Pescatore, L.A.; Fernandes Dde, C. Protein disulfide isomerase in redox cell signaling and homeostasis. Free Radic. Biol. Med. 2012, 52, 1954–1969. [Google Scholar] [CrossRef] [PubMed]
- Meneses-Valdés, R.; Gallero, S.; Henríquez-Olguín, C.; Jensen, T.E. Exploring NADPH oxidases 2 and 4 in cardiac and skeletal muscle adaptations—A cross-tissue comparison. Free Radic. Biol. Med. 2024, 223, 296–305. [Google Scholar] [CrossRef]
- Nkhumeleni, Z.; Phoswa, W.N.; Mokgalaboni, K. Purslane Ameliorates Inflammation and Oxidative Stress in Diabetes Mellitus: A Systematic Review. Int. J. Mol. Sci. 2024, 25, 12276. [Google Scholar] [CrossRef] [PubMed]
- Massart, J.; Begriche, K.; Hartman, J.H.; Fromenty, B. Role of Mitochondrial Cytochrome P450 2E1 in Healthy and Diseased Liver. Cells 2022, 11, 288. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Das Gupta, S.; Wang, Z.; Jiang, H.; Velkov, T.; Shen, J. T-2 toxin and its cardiotoxicity: New insights on the molecular mechanisms and therapeutic implications. Food Chem. Toxicol. 2022, 167, 113262. [Google Scholar] [CrossRef] [PubMed]
- Antelo-Cea, D.A.; Martínez-Rojas, L.; Cabrerizo-Ibáñez, I.; Roudi Rashtabady, A.; Hernández-Alvarez, M.I. Regulation of Mitochondrial and Peroxisomal Metabolism in Female Obesity and Type 2 Diabetes. Int. J. Mol. Sci. 2024, 25, 11237. [Google Scholar] [CrossRef]
- Lan, R.; Zhang, M.J.; Liu, K.; Meng, F.F.; Xu, X.H.; Wang, C.C.; Zhang, M.Q.; Yan, Y.; Kou, J.J.; Zhao, L.L.; et al. Cytochrome P450-derived Epoxyeicosatrienoic Acid, the Regulation of Cardiovascular-related Diseases, and the Implication for Pulmonary Hypertension. Cardiovasc. Drugs Ther. 2024, 38, 1–17. [Google Scholar] [CrossRef]
- Blagov, A.V.; Summerhill, V.I.; Sukhorukov, V.N.; Zhigmitova, E.B.; Postnov, A.Y.; Orekhov, A.N. Potential use of antioxidants for the treatment of chronic inflammatory diseases. Front. Pharmacol. 2024, 15, 1378335. [Google Scholar] [CrossRef]
- Lu, Y.; George, J. Interaction between fatty acid oxidation and ethanol metabolism in liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2024, 326, G483–G494. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Lin, C.C.; Lett, C.; Karpinska, B.; Wright, M.H.; Foyer, C.H. Catalase: A critical node in the regulation of cell fate. Free Radic. Biol. Med. 2023, 199, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Rius-Pérez, S. p53 at the crossroad between mitochondrial reactive oxygen species and necroptosis. Free Radic. Biol. Med. 2023, 207, 183–193. [Google Scholar] [CrossRef]
- Di Florio, D.N.; Sin, J.; Coronado, M.J.; Atwal, P.S.; Fairweather, D. Sex differences in inflammation, redox biology, mitochondria and autoimmunity. Redox Biol. 2020, 31, 101482. [Google Scholar] [CrossRef] [PubMed]
- Olié, V.; Grave, C.; Helft, G.; Nguyen-Thanh, V.; Andler, R.; Quatremere, G.; Pasquereau, A.; Lahaie, E.; Lailler, G.; Verdot, C.; et al. Epidemiology of cardiovascular risk factors: Behavioural risk factors. Arch. Cardiovasc. Dis. 2024, 117, 770–784. [Google Scholar] [CrossRef]
- Lailler, G.; Gabet, A.; Grave, C.; Boudet-Berquier, J.; El Rafei, R.; Regnault, N.; Acar, P.; Thomas-Chabaneix, J.; Tuppin, P.; Béjot, Y.; et al. Cardiovascular hospitalizations and deaths in adults, children and pregnant women. Arch. Cardiovasc. Dis. 2024, 117, 751–760. [Google Scholar] [CrossRef]
- Vona, R.; Ascione, B.; Malorni, W.; Straface, E. Mitochondria and Sex-Specific Cardiac Function. Adv. Exp. Med. Biol. 2018, 1065, 241–256. [Google Scholar] [CrossRef]
- Robert, J. Sex differences in vascular endothelial cells. Atherosclerosis 2023, 384, 117278. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Xu, W.; Yang, F.; Li, J.; Wei, W.; Chen, X.; Liu, Y.; Zhang, Z. Protective and Detoxifying Effects of Resveratrol on Zearalenone-Mediated Toxicity: A Review. Int. J. Mol. Sci. 2024, 25, 11003. [Google Scholar] [CrossRef] [PubMed]
- Krzyżewska, A.; Kurakula, K. Sex Dimorphism in Pulmonary Arterial Hypertension Associated with Autoimmune Diseases. Arterioscler. Thromb. Vasc. Biol. 2024, 44, 2169–2190. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Safabakhsh, S.; Palumbo, A.; Fiset, C.; Shen, C.; Parker, J.; Foster, L.J.; Laksman, Z. Sex-Based Mechanisms of Cardiac Development and Function: Applications for Induced-Pluripotent Stem Cell Derived-Cardiomyocytes. Int. J. Mol. Sci. 2024, 25, 5964. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kang, P.M. Oxidative Stress and Antioxidant Treatments in Cardiovascular Diseases. Antioxidants 2020, 9, 1292. [Google Scholar] [CrossRef] [PubMed]
- Padmanaban, A.M.; Ganesan, K.; Ramkumar, K.M. A Co-Culture System for Studying Cellular Interactions in Vascular Disease. Bioengineering 2024, 11, 1090. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Chen, J.; Zhang, X.; Ma, Z.; Wang, J.; Wu, Q. Role of Neutrophil Extracellular Traps in Hypertension and Their Impact on Target Organs. J. Clin. Hypertens. 2024, 26, 1323–1534. [Google Scholar] [CrossRef]
- Manoj, H.; Gomes, S.M.; Thimmappa, P.Y.; Nagareddy, P.R.; Jamora, C.; Joshi, M.B. Cytokine signalling in formation of neutrophil extracellular traps: Implications for health and diseases. Cytokine Growth Factor Rev. 2024; in press. [Google Scholar] [CrossRef]
- Latif, F.; Mubbashir, A.; Khan, M.S.; Shaikh, Z.; Memon, A.; Alvares, J.; Azhar, A.; Jain, H.; Ahmed, R.; Kanagala, S.G. Trimethylamine N-oxide in cardiovascular disease: Pathophysiology and the potential role of statins. Life Sci. 2024, 361, 123304. [Google Scholar] [CrossRef]
- Wang, L.; Cao, Y.; Gorshkov, B.; Zhou, Y.; Yang, Q.; Xu, J.; Ma, Q.; Zhang, X.; Wang, J.; Mao, X.; et al. Ablation of endothelial Pfkfb3 protects mice from acute lung injury in LPS-induced endotoxemia. Pharmacol. Res. 2019, 146, 104292. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef] [PubMed]
- Alvandi, Z.; Bischoff, J. Endothelial-Mesenchymal Transition in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2357–2369. [Google Scholar] [CrossRef]
- Eity, T.A.; Bhuia, M.S.; Chowdhury, R.; Ahmmed, S.; Salehin, S.; Akter, R.; Islam, M.T. Therapeutic Efficacy of Quercetin and Its Nanoformulation Both the Mono- or Combination Therapies in the Management of Cancer: An Update with Molecular Mechanisms. J. Trop. Med. 2024, 2024, 5594462. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, K.; Wu, B.; Lu, X.; Sun, L.; Privratsky, J.R.; Xing, C.; Robson, M.J.; Mao, H.; Blakely, R.D.; et al. Divergent Actions of Renal Tubular and Endothelial Type 1 IL-1 Receptor Signaling in Toxin-Induced AKI. J. Am. Soc. Nephrol. 2023, 34, 1629–1646. [Google Scholar] [CrossRef]
- Ge, Y.L.; Li, P.J.; Bu, Y.R.; Zhang, B.; Xu, J.; He, S.Y.; Cao, Q.L.; Bai, Y.G.; Ma, J.; Zhang, L.; et al. TNF-α and RPLP0 drive the apoptosis of endothelial cells and increase susceptibility to high-altitude pulmonary edema. Apoptosis 2024, 29, 1600–1618. [Google Scholar] [CrossRef]
- Palikuqi, B.; Nguyen, D.T.; Li, G.; Schreiner, R.; Pellegata, A.F.; Liu, Y.; Redmond, D.; Geng, F.; Lin, Y.; Gómez-Salinero, J.M.; et al. Adaptable haemodynamic endothelial cells for organogenesis and tumorigenesis. Nature 2020, 585, 426–432. [Google Scholar] [CrossRef]
- Chiu, J.J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef]
- Tokarz-Deptuła, B.; Baraniecki, Ł.; Palma, J.; Stosik, M.; Deptuła, W. Characterization of Platelet Receptors and Their Involvement in Immune Activation of These Cells. Int. J. Mol. Sci. 2024, 25, 12611. [Google Scholar] [CrossRef]
- Steinberg, D. The LDL modification hypothesis of atherogenesis: An update. J. Lipid Res. 2009, 50, S376–S381. [Google Scholar] [CrossRef]
- Rajpoot, A.; Aggarwal, T.; Sharma, V. Unraveling the enigma of cardiac damage caused by lead: Understanding the intricate relationship between oxidative stress and other multifactorial mechanisms. Toxicology 2024, 509, 153984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, J.; Duan, H.; Li, R.; Peng, W.; Wu, C. Activation of Nrf2/HO-1 signaling: An important molecular mechanism of herbal medicine in the treatment of atherosclerosis via the protection of vascular endothelial cells from oxidative stress. J. Adv. Res. 2021, 34, 43–63. [Google Scholar] [CrossRef]
- Jacob, R.; Khan, M. Cardiac Biomarkers: What Is and What Can Be. Indian J. Cardiovasc. Dis. Women WINCARS 2018, 3, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Kirichenko, T.V.; Sobenin, I.A.; Nikolic, D.; Rizzo, M.; Orekhov, A.N. Anti-cytokine therapy for prevention of atherosclerosis. Phytomedicine 2016, 23, 1198–1210. [Google Scholar] [CrossRef] [PubMed]
- Ouweneel, A.B.; Van Eck, M. Lipoproteins as modulators of atherothrombosis: From endothelial function to primary and secondary coagulation. Vascul. Pharmacol. 2016, 82, 1–10. [Google Scholar] [CrossRef]
- Engin, A. Endothelial Dysfunction in Obesity and Therapeutic Targets. Adv. Exp. Med. Biol. 2024, 1460, 489–538. [Google Scholar] [CrossRef] [PubMed]
- Kardassis, D.; Vindis, C.; Stancu, C.S.; Toma, L.; Gafencu, A.V.; Georgescu, A.; Alexandru-Moise, N.; Molica, F.; Kwak, B.R.; Burlacu, A.; et al. Unravelling molecular mechanisms in atherosclerosis using cellular models and omics technologies. Vascul. Pharmacol. 2024, 158, 107452. [Google Scholar] [CrossRef]
- Yang, B.; Hang, S.; Xu, S.; Gao, Y.; Yu, W.; Zang, G.; Zhang, L.; Wang, Z. Macrophage polarisation and inflammatory mechanisms in atherosclerosis: Implications for prevention and treatment. Heliyon 2024, 10, e32073. [Google Scholar] [CrossRef]
- Song, J.; Cao, C.; Wang, Z.; Li, H.; Yang, L.; Kang, J.; Meng, H.; Li, L.; Liu, J. Mechanistic insights into the regression of atherosclerotic plaques. Front. Physiol. 2024, 15, 1473709. [Google Scholar] [CrossRef]
- Saki, N.; Haybar, H.; Maniati, M.; Davari, N.; Javan, M.; Moghimian-Boroujeni, B. Modification macrophage to foam cells in atherosclerosis disease: Some factors stimulate or inhibit this process. J. Diabetes Metab. Disord. 2024, 23, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Brown, O.I.; Bridge, K.I.; Kearney, M.T. Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Glucose Homeostasis and Diabetes-Related Endothelial Cell Dysfunction. Cells 2021, 10, 2315. [Google Scholar] [CrossRef]
- Yu, X.H.; Fu, Y.C.; Zhang, D.W.; Yin, K.; Tang, C.K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wu, X.; Hu, J.; Liu, X.; Guo, Z.; Wu, J.; Shao, Y.; Hao, M.; Zhang, S.; Hu, W.; et al. The translational potential of miR-26 in atherosclerosis and development of agents for its target genes ACC1/2, COL1A1, CPT1A, FBP1, DGAT2, and SMAD7. Cardiovasc. Diabetol. 2024, 23, 21. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, H.; Chen, Y.; Liu, X.; Tian, J.; Shen, W. The Role of Macrophage Iron Overload and Ferroptosis in Atherosclerosis. Biomolecules 2022, 12, 1702. [Google Scholar] [CrossRef]
- Obare, L.M.; Temu, T.; Mallal, S.A.; Wanjalla, C.N. Inflammation in HIV and Its Impact on Atherosclerotic Cardiovascular Disease. Circ. Res. 2024, 134, 1515–1545. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Adameová, A.; Barile, L.; Cabrera-Fuentes, H.A.; Lazou, A.; Pagliaro, P.; Stensløkken, K.O.; Garcia-Dorado, D. Mitochondrial and mitochondrial-independent pathways of myocardial cell death during ischaemia and reperfusion injury. J. Cell. Mol. Med. 2020, 24, 3795–3806. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Li, Z.; Gautam, M.; Ghosh, A.; Man, S.M. Molecular mechanisms of emerging inflammasome complexes and their activation and signaling in inflammation and pyroptosis. Immunol. Rev. 2024, 38, 1–18. [Google Scholar] [CrossRef]
- Jiang, Y.; Xing, W.; Li, Z.; Zhao, D.; Xiu, B.; Xi, Y.; Bai, S.; Li, X.; Zhang, Z.; Zhang, W.; et al. The calcium-sensing receptor alleviates endothelial inflammation in atherosclerosis through regulation of integrin β1-NLRP3 inflammasome. FEBS J. 2024, 291, 1–18. [Google Scholar] [CrossRef]
- Minutoli, L.; Puzzolo, D.; Rinaldi, M.; Irrera, N.; Marini, H.; Arcoraci, V.; Bitto, A.; Crea, G.; Pisani, A.; Squadrito, F.; et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxidative Med. Cell. Longev. 2016, 2016, 2183026. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Feng, C. Research progress on pyroptosis and its effect on the central nervous system. Neurobiol. Dis. 2023, 188, 106333. [Google Scholar] [CrossRef]
- Gupta, S.; Cassel, S.L.; Sutterwala, F.S.; Dagvadorj, J. Regulation of the NLRP3 inflammasome by autophagy and mitophagy. Immunol. Rev. 2024. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xu, X.; Zhang, X. Targeting different phenotypes of macrophages: A potential strategy for natural products to treat inflammatory bone and joint diseases. Phytomedicine 2023, 118, 154952. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, M.; Peters, A.S.; Erhart, P.; Körfer, D.; Böckler, D.; Dihlmann, S. Inflammasomes in the Pathophysiology of Aortic Disease. Cells 2021, 10, 2433. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Yurdagul, A., Jr.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef]
- Prouse, T.; Majumder, S.; Majumder, R. Functions of TAM Receptors and Ligands Protein S and Gas6 in Atherosclerosis and Cardiovascular Disease. Int. J. Mol. Sci. 2024, 25, 12736. [Google Scholar] [CrossRef]
- Zhou, D.; Yang, Y.; Han, R.; He, J.; Liu, D.; Xia, W.; Cai, Y.; Perek, B.; Xia, Z. Ferroptosis and its Potential Determinant Role in Myocardial Susceptibility to Ischemia/Reperfusion Injury in Diabetes. Rev. Cardiovasc. Med. 2024, 25, 360. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Zhang, Z.; Zhang, Y.; Yang, M.; Wang, C.; Xu, J.; Zhu, Y.; Mi, Y.; Jiang, J.; Sun, Z. Ferroptosis and myocardial ischemia-reperfusion: Mechanistic insights and new therapeutic perspectives. Front. Pharmacol. 2024, 15, 1482986. [Google Scholar] [CrossRef]
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- Chen, M.; Wang, R.; Liao, L.; Li, Y.; Sun, X.; Wu, H.; Lan, Q.; Deng, Z.; Liu, P.; Xu, T.; et al. DanShen Decoction targets miR-93-5p to provide protection against MI/RI by regulating the TXNIP/NLRP3/Caspase-1 signaling pathway. Phytomedicine 2024, 135, 156225. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.H.; Chen, J.; Wu, X.F.; Zhang, Q.; Xia, G.Y.; Chu, X.Y.; Xia, H.; Lin, S.; Shang, H.C. Salvianolic acid B alleviated myocardial ischemia-reperfusion injury via modulating SIRT3-mediated crosstalk between mitochondrial ROS and NLRP3. Phytomedicine 2024, 136, 156260. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, D.H.; Zhang, Y.; Zheng, F.; Gao, F.; Sun, J.; Shi, G. Geniposide suppresses NLRP3 inflammasome-mediated pyroptosis via the AMPK signaling pathway to mitigate myocardial ischemia/reperfusion injury. Chin. Med. 2022, 17, 73. [Google Scholar] [CrossRef]
- Pagliaro, P.; Penna, C. Inhibitors of NLRP3 Inflammasome in Ischemic Heart Disease: Focus on Functional and Redox Aspects. Antioxidants 2023, 12, 1396. [Google Scholar] [CrossRef] [PubMed]
- Alloatti, G.; Penna, C.; Comità, S.; Tullio, F.; Aragno, M.; Biasi, F.; Pagliaro, P. Aging, sex and NLRP3 inflammasome in cardiac ischaemic disease. Vascul. Pharmacol. 2022, 145, 107001. [Google Scholar] [CrossRef]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhang, Z.; Zhang, W.; Liu, X. Mitochondrial dysfunction and mitochondrial therapies in heart failure. Pharmacol. Res. 2022, 175, 106038. [Google Scholar] [CrossRef] [PubMed]
- Atici, A.E.; Crother, T.R.; Noval Rivas, M. Mitochondrial quality control in health and cardiovascular diseases. Front. Cell Dev. Biol. 2023, 11, 1290046. [Google Scholar] [CrossRef]
- Chen, T.H.; Wang, H.C.; Chang, C.J.; Lee, S.Y. Mitochondrial Glutathione in Cellular Redox Homeostasis and Disease Manifestation. Int. J. Mol. Sci. 2024, 25, 1314. [Google Scholar] [CrossRef]
- Peng, Z.; Liang, Y.; Liu, X.; Shao, J.; Hu, N.; Zhang, X. New insights into the mechanisms of diabetic kidney disease: Role of circadian rhythm and Bmal1. Biomed. Pharmacother. 2023, 166, 115422. [Google Scholar] [CrossRef]
- Fu, L.; Zhang, L. Physiological functions of CKIP-1: From molecular mechanisms to therapy implications. Ageing Res. Rev. 2019, 53, 100908. [Google Scholar] [CrossRef]
- Oduro, P.K.; Zheng, X.; Wei, J.; Yang, Y.; Wang, Y.; Zhang, H.; Liu, E.; Gao, X.; Du, M.; Wang, Q. The cGAS-STING signaling in cardiovascular and metabolic diseases: Future novel target option for pharmacotherapy. Acta Pharm. Sin. B 2022, 12, 50–75. [Google Scholar] [CrossRef]
- Takada, S.; Maekawa, S.; Furihata, T.; Kakutani, N.; Setoyama, D.; Ueda, K.; Nambu, H.; Hagiwara, H.; Handa, H.; Fumoto, Y.; et al. Succinyl-CoA-based energy metabolism dysfunction in chronic heart failure. Proc. Natl. Acad. Sci. USA 2022, 119, e2203628119. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Shao, Y.; Guo, H.C.; Zhi, Y.; Qiao, B.; Ma, K.; Du, J.; Lai, Y.Q.; Li, Y. MicroRNA-27b-3p down-regulates FGF1 and aggravates pathological cardiac remodelling. Cardiovasc. Res. 2022, 118, 2139–2151. [Google Scholar] [CrossRef]
- Mongirdienė, A.; Skrodenis, L.; Varoneckaitė, L.; Mierkytė, G.; Gerulis, J. Reactive Oxygen Species Induced Pathways in Heart Failure Pathogenesis and Potential Therapeutic Strategies. Biomedicines 2022, 10, 602. [Google Scholar] [CrossRef] [PubMed]
- Noctor, G.; Foyer, C.H. Intracellular Redox Compartmentation and ROS-Related Communication in Regulation and Signaling. Plant Physiol. 2016, 171, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Cortassa, S.; Juhaszova, M.; Aon, M.A.; Zorov, D.B.; Sollott, S.J. Mitochondrial Ca2+, redox environment and ROS emission in heart failure: Two sides of the same coin? J. Mol. Cell. Cardiol. 2021, 151, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef]
- Bej, E.; Cesare, P.; d’Angelo, M.; Volpe, A.R.; Castelli, V. Neuronal Cell Rearrangement During Aging: Antioxidant Compounds as a Potential Therapeutic Approach. Cells 2024, 13, 1945. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Kulshreshtha, A.; Lall, R.; Gupta, S.C. Inflammation and ROS in arthritis: Management by Ayurvedic medicinal plants. Food Funct. 2021, 12, 8227–8247. [Google Scholar] [CrossRef]
- Li, D.; Rui, Y.X.; Guo, S.D.; Luan, F.; Liu, R.; Zeng, N. Ferulic acid: A review of its pharmacology, pharmacokinetics and derivatives. Life Sci. 2021, 284, 119921. [Google Scholar] [CrossRef] [PubMed]
- Downey, J.M.; Cohen, M.V. A really radical observation—A comment on Penna et al. in Basic Res Cardiol (2006) 101:180–189. Basic. Res. Cardiol. 2006, 101, 190–191. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.Z.; Chen, W.; Liu, X.; Kou, X.; Khan, A.; Khan, R.U.; Zahoor, M.; Wang, C. An Overview of Bioactive Compounds’ Role in Modulating the Nrf2/Keap1/NF-κB Pathway to Alleviate Lipopolysaccharide-Induced Endometritis. Int. J. Mol. Sci. 2024, 25, 10319. [Google Scholar] [CrossRef]
- Penna, C.; Rastaldo, R.; Mancardi, D.; Raimondo, S.; Cappello, S.; Gattullo, D.; Losano, G.; Pagliaro, P. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic. Res. Cardiol. 2006, 101, 180–189. [Google Scholar] [CrossRef]
- Tsutsumi, Y.M.; Yokoyama, T.; Horikawa, Y.; Roth, D.M.; Patel, H.H. Reactive oxygen species trigger ischemic and pharmacological postconditioning: In vivo and in vitro characterization. Life Sci. 2007, 81, 1223–1227. [Google Scholar] [CrossRef]
- Voronkov, N.S.; Popov, S.V.; Naryzhnaya, N.V.; Prasad, N.R.; Petrov, I.M.; Kolpakov, V.V.; Tomilova, E.A.; Sapozhenkova, E.V.; Maslov, L.N. Effect of Cold Adaptation on the State of Cardiovascular System and Cardiac Tolerance to Ischemia/Reperfusion Injury. Iran. Biomed. J. 2024, 28, 59–70. [Google Scholar] [CrossRef]
- Xu, M.; Wu, G.; You, Q.; Chen, X. The Landscape of Smart Biomaterial-Based Hydrogen Therapy. Adv. Sci. 2024, 11, e2401310. [Google Scholar] [CrossRef]
- Di Pietro, M.; Filardo, S.; Falasca, F.; Turriziani, O.; Sessa, R. Infectious Agents in Atherosclerotic Cardiovascular Diseases through Oxidative Stress. Int. J. Mol. Sci. 2017, 18, 2459. [Google Scholar] [CrossRef]
- Ju, S.; Singh, M.K.; Han, S.; Ranbhise, J.; Ha, J.; Choe, W.; Yoon, K.S.; Yeo, S.G.; Kim, S.S.; Kang, I. Oxidative Stress and Cancer Therapy: Controlling Cancer Cells Using Reactive Oxygen Species. Int. J. Mol. Sci. 2024, 25, 12387. [Google Scholar] [CrossRef] [PubMed]
- Procaccio, V.; Bris, C.; Chao de la Barca, J.M.; Oca, F.; Chevrollier, A.; Amati-Bonneau, P.; Bonneau, D.; Reynier, P. Perspectives of drug-based neuroprotection targeting mitochondria. Rev. Neurol. 2014, 170, 390–400. [Google Scholar] [CrossRef]
- Sommers, O.; Tomsine, R.A.; Khacho, M. Mitochondrial Dynamics Drive Muscle Stem Cell Progression from Quiescence to Myogenic Differentiation. Cells 2024, 13, 1773. [Google Scholar] [CrossRef]
- Maqoud, F.; Scala, R.; Hoxha, M.; Zappacosta, B.; Tricarico, D. ATP-sensitive Potassium Channel Subunits in Neuroinflammation: Novel Drug Targets in Neurodegenerative Disorders. CNS Neurol. Disord. Drug Targets 2022, 21, 130–149. [Google Scholar] [CrossRef]
- Macrì, R.; Musolino, V.; Gliozzi, M.; Carresi, C.; Maiuolo, J.; Nucera, S.; Scicchitano, M.; Bosco, F.; Scarano, F.; Ruga, S.; et al. Ferula L. Plant Extracts and Dose-Dependent Activity of Natural Sesquiterpene Ferutinin: From Antioxidant Potential to Cytotoxic Effects. Molecules 2020, 25, 5768. [Google Scholar] [CrossRef]
- Wang, L.; Apel, K. Dose-dependent effects of 1O2 in chloroplasts are determined by its timing and localization of production. J. Exp. Bot. 2019, 70, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Kopacz, A.; Kloska, D.; Forman, H.J.; Jozkowicz, A.; Grochot-Przeczek, A. Beyond repression of Nrf2: An update on Keap1. Free Radic. Biol. Med. 2020, 157, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Tanase, D.M.; Gosav, E.M.; Anton, M.I.; Floria, M.; Seritean Isac, P.N.; Hurjui, L.L.; Tarniceriu, C.C.; Costea, C.F.; Ciocoiu, M.; Rezus, C. Oxidative Stress and NRF2/KEAP1/ARE Pathway in Diabetic Kidney Disease (DKD): New Perspectives. Biomolecules 2022, 12, 1227. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Chen, Y.; Luo, Z.; Nie, G.; Dai, Y. Role of oxidative stress and inflammation-related signaling pathways in doxorubicin-induced cardiomyopathy. Cell Commun. Signal. 2023, 21, 61. [Google Scholar] [CrossRef]
- Chauhan, W.; Zennadi, R. Keap1-Nrf2 Heterodimer: A Therapeutic Target to Ameliorate Sickle Cell Disease. Antioxidants 2023, 12, 740. [Google Scholar] [CrossRef]
- Wu, L.; Wang, L.; Du, Y.; Zhang, Y.; Ren, J. Mitochondrial quality control mechanisms as therapeutic targets in doxorubicin-induced cardiotoxicity. Trends Pharmacol. Sci. 2023, 44, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Ubaid, S. Role of Silent Information Regulator 1 (SIRT1) in Regulating Oxidative Stress and Inflammation. Inflammation 2020, 43, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Qi, Y.; Xu, L.; Tao, X.; Han, X.; Yin, L.; Peng, J. MicroRNA-140-5p aggravates doxorubicin-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biol. 2018, 15, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Tao, X.; Qi, Y.; Xu, L.; Yin, L.; Peng, J. Protective effect of dioscin against doxorubicin-induced cardiotoxicity via adjusting microRNA-140-5p-mediated myocardial oxidative stress. Redox Biol. 2018, 16, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, C.; Abodi, M.; D’Oria, V.; Milani, G.P.; Agostoni, C.; Mazzocchi, A. Alpha-Linolenic Acid and Cardiovascular Events: A Narrative Review. Int. J. Mol. Sci. 2023, 24, 14319. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Mamun, A.A.; Jakaria, M.; Thangapandiyan, S.; Ahmad, J.; Rahman, M.A.; Mathew, B.; Abdel-Daim, M.M.; Aleya, L. Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci. Total Environ. 2020, 707, 135624. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.M.; Coleman, R.L.; Holman, R.R. Prognostic significance of silent myocardial infarction in newly diagnosed type 2 diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS) 79. Circulation 2013, 127, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Cenci, A.; Macchia, I.; La Sorsa, V.; Sbarigia, C.; Di Donna, V.; Pietraforte, D. Mechanisms of Action of Ozone Therapy in Emerging Viral Diseases: Immunomodulatory Effects and Therapeutic Advantages with Reference to SARS-CoV-2. Front. Microbiol. 2022, 13, 871645. [Google Scholar] [CrossRef] [PubMed]
- Karan, A.; Bhakkiyalakshmi, E.; Jayasuriya, R.; Sarada, D.V.L.; Ramkumar, K.M. The pivotal role of nuclear factor erythroid 2-related factor 2 in diabetes-induced endothelial dysfunction. Pharmacol. Res. 2020, 153, 104601. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, R.; Zou, J.; Yin, H.; Zhao, M.; Zhao, L. The impact of chitooligosaccharides with a certain degree of polymerization on diabetic nephropathic mice and high glucose-damaged HK-2 cells. Food Sci. Nutr. 2024, 12, 4173–4184. [Google Scholar] [CrossRef]
- Nie, C.; He, T.; Zhang, W.; Zhang, G.; Ma, X. Branched Chain Amino Acids: Beyond Nutrition Metabolism. Int. J. Mol. Sci. 2018, 19, 954. [Google Scholar] [CrossRef]
- Ramasubbu, K.; Devi Rajeswari, V. Impairment of insulin signaling pathway PI3K/Akt/mTOR and insulin resistance induced AGEs on diabetes mellitus and neurodegenerative diseases: A perspective review. Mol. Cell. Biochem. 2023, 478, 1307–1324. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Zhang, Y.; An, S.T.; Chen, Y. Annexin A3 gene silencing promotes myocardial cell repair through activation of the PI3K/Akt signaling pathway in rats with acute myocardial infarction. J. Cell. Physiol. 2019, 234, 10535–10546. [Google Scholar] [CrossRef]
- Chen, X.; Zhabyeyev, P.; Azad, A.K.; Vanhaesebroeck, B.; Grueter, C.E.; Murray, A.G.; Kassiri, Z.; Oudit, G.Y. Pharmacological and cell-specific genetic PI3Kα inhibition worsens cardiac remodeling after myocardial infarction. J. Mol. Cell. Cardiol. 2021, 157, 17–30. [Google Scholar] [CrossRef]
- Qin, W.; Cao, L.; Massey, I.Y. Role of PI3K/Akt signaling pathway in cardiac fibrosis. Mol. Cell. Biochem. 2021, 476, 4045–4059. [Google Scholar] [CrossRef] [PubMed]
- Puthusseri, B.; Marudamuthu, A.; Tiwari, N.; Fu, J.; Idell, S.; Shetty, S. Regulation of p53-mediated changes in the uPA-fibrinolytic system and in lung injury by loss of surfactant protein C expression in alveolar epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L783–L796. [Google Scholar] [CrossRef] [PubMed]
- Cameselle-García, S.; Abdulkader-Nallib, I.; Sánchez-Ares, M.; Cameselle-Teijeiro, J.M. Cribriform morular thyroid carcinoma: Clinicopathological and molecular basis for both a preventive and therapeutic approach for a rare tumor (Review). Oncol. Rep. 2024, 52, 119. [Google Scholar] [CrossRef] [PubMed]
- Alfadda, A.A.; Sallam, R.M. Reactive oxygen species in health and disease. J. Biomed. Biotechnol. 2012, 2012, 936486. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Imanaka, S. Mitochondrial DNA Damage and Its Repair Mechanisms in Aging Oocytes. Int. J. Mol. Sci. 2024, 25, 13144. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, C.; You, J.; Xie, Z. The interplay between autophagy and apoptosis in the diabetic heart. J. Mol. Cell. Cardiol. 2014, 71, 71–80. [Google Scholar] [CrossRef]
- Penugurti, V.; Manne, R.K.; Bai, L.; Kant, R.; Lin, H.K. AMPK: The energy sensor at the crossroads of aging and cancer. Semin. Cancer Biol. 2024, 106–107, 15–27. [Google Scholar] [CrossRef]
- Abdul Khaliq, H.; Alhouayek, M.; Quetin-Leclercq, J.; Muccioli, G.G. 5’AMP-activated protein kinase: An emerging target of phytochemicals to treat chronic inflammatory diseases. Crit. Rev. Food Sci. Nutr. 2024, 64, 4763–4788. [Google Scholar] [CrossRef]
- Zhou, R.; Barnes, K.; Gibson, S.; Fillmore, N. Dual-edged role of SIRT1 in energy metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2024, 327, H1162–H1173. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Koya, D. The protective role of Sirt1 in vascular tissue: Its relationship to vascular aging and atherosclerosis. Aging 2016, 8, 2290–2307. [Google Scholar] [CrossRef] [PubMed]
- Campagna, R.; Mazzanti, L.; Pompei, V.; Alia, S.; Vignini, A.; Emanuelli, M. The Multifaceted Role of Endothelial Sirt1 in Vascular Aging: An Update. Cells 2024, 13, 1469. [Google Scholar] [CrossRef] [PubMed]
- Sazdova, I.; Hadzi-Petrushev, N.; Keremidarska-Markova, M.; Stojchevski, R.; Sopi, R.; Shileiko, S.; Mitrokhin, V.; Gagov, H.; Avtanski, D.; Lubomirov, L.T.; et al. SIRT-associated attenuation of cellular senescence in vascular wall. Mech. Ageing Dev. 2024, 220, 111943. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Wang, Z.; Chen, H.Z.; Zhou, S.; Zheng, W.; Liu, G.; Wei, Y.S.; Cai, H.; Liu, D.P.; Liang, C.C. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc. Res. 2008, 80, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Balestrieri, M.L.; Sica, V.; Lerman, L.O.; Crimi, E.; De Rosa, G.; Schiano, C.; Servillo, L.; D’Armiento, F.P. Beneficial effects of low doses of red wine consumption on perturbed shear stress-induced atherogenesis. Heart Vessel. 2008, 23, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chen, H.Z. Histone Deacetylase SIRT1, Smooth Muscle Cell Function, and Vascular Diseases. Front. Pharmacol. 2020, 11, 537519. [Google Scholar] [CrossRef]
- Law, M.; Wang, P.C.; Zhou, Z.Y.; Wang, Y. From Microcirculation to Aging-Related Diseases: A Focus on Endothelial SIRT1. Pharmaceuticals 2024, 17, 1495. [Google Scholar] [CrossRef]
- Donato, A.J.; Magerko, K.A.; Lawson, B.R.; Durrant, J.R.; Lesniewski, L.A.; Seals, D.R. SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J. Physiol. 2011, 589, 4545–4554. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Kumari, A.; Verma, A.; Chaudhary, V.; Agrawal, V.; Yadav, H.N. Silent Information Regulator 1/Peroxisome Proliferator-Activated Receptor-γ Coactivator-1α Axis: A Promising Target for Parkinson’s and Alzheimer’s Disease Therapies. J. Biochem. Mol. Toxicol. 2024, 38, e70078. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 2013, 25, 1939–1948. [Google Scholar] [CrossRef]
- Xue, H.Z.; Chen, Y.; Wang, S.D.; Yang, Y.M.; Cai, L.Q.; Zhao, J.X.; Huang, W.J.; Xiao, Y.H. Radix Astragali and Its Representative Extracts for Diabetic Nephropathy: Efficacy and Molecular Mechanism. J. Diabetes Res. 2024, 2024, 5216113. [Google Scholar] [CrossRef] [PubMed]
- Xian, Y.; Ye, J.; Tang, Y.; Zhang, N.; Peng, C.; Huang, W.; He, G. Deubiquitinases as novel therapeutic targets for diseases. MedComm 2024, 5, e70036. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Calzetta, L.; Rogliani, P.; Matera, M.G. Emerging Anti-Inflammatory COPD Treatments: Potential Cardiovascular Impacts. Int. J. Chronic Obstr. Pulm. Dis. 2024, 19, 2481–2495. [Google Scholar] [CrossRef]
- Khanahmadi, M.; Ebrahimi Fard, M.; Baghani, M.; Shayan, M.; Baghani, M. Exploring STK3 in melanoma: A systematic review of signaling networks and therapeutic opportunities. Mol. Biol. Rep. 2024, 52, 8. [Google Scholar] [CrossRef] [PubMed]
- Puranik, N.; Jung, H.; Song, M. SPROUTY2, a Negative Feedback Regulator of Receptor Tyrosine Kinase Signaling, Associated with Neurodevelopmental Disorders: Current Knowledge and Future Perspectives. Int. J. Mol. Sci. 2024, 25, 11043. [Google Scholar] [CrossRef]
- Ramli, F.F.; Ali, A.; Ibrahim, N. Molecular-Signaling Pathways of Ginsenosides Rb in Myocardial Ischemia-Reperfusion Injury: A Mini Review. Int. J. Med. Sci. 2022, 19, 65–73. [Google Scholar] [CrossRef]
- Spirrison, A.N.; Lannigan, D.A. RSK1 and RSK2 as therapeutic targets: An up-to-date snapshot of emerging data. Expert Opin. Ther. Targets 2024, 27, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Bueno, O.F.; Wilkins, B.J.; Kuan, C.Y.; Xia, Y.; Molkentin, J.D. c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling. EMBO J. 2003, 22, 5079–5089. [Google Scholar] [CrossRef] [PubMed]
- Attachaipanich, T.; Chattipakorn, S.C.; Chattipakorn, N. Cardiovascular toxicities by calcineurin inhibitors: Cellular mechanisms behind clinical manifestations. Acta Physiol. 2024, 240, e14199. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.M.; Jin, X.; Ren, J.; Avery, J.; DeBosch, B.J.; Treskov, I.; Lupu, T.S.; Kovacs, A.; Weinheimer, C.; Muslin, A.J. The 14-3-3τ phosphoserine-binding protein is required for cardiomyocyte survival. Mol. Cell. Biol. 2007, 27, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Muslin, A.J. MAPK signalling in cardiovascular health and disease: Molecular mechanisms and therapeutic targets. Clin. Sci. 2008, 115, 203–218. [Google Scholar] [CrossRef]
- Feng, X.; Du, M.; Li, S.; Zhang, Y.; Ding, J.; Wang, J.; Wang, Y.; Liu, P. Hydroxysafflor yellow A regulates lymphangiogenesis and inflammation via the inhibition of PI3K on regulating AKT/mTOR and NF-κB pathway in macrophages to reduce atherosclerosis in ApoE-/- mice. Phytomedicine 2023, 112, 154684. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Wan, X.; Zou, X.; Sun, S.; Hao, X.; Liang, C.; Zhang, Z.; Zhang, F.; Sun, B.; Li, H.; et al. Arsenic trioxide induces macrophage autophagy and atheroprotection by regulating ROS-dependent TFEB nuclear translocation and AKT/mTOR pathway. Cell Death Dis. 2021, 12, 88. [Google Scholar] [CrossRef]
- D’Onofrio, N.; Servillo, L.; Balestrieri, M.L. SIRT1 and SIRT6 Signaling Pathways in Cardiovascular Disease Protection. Antioxid. Redox Signal. 2018, 28, 711–732. [Google Scholar] [CrossRef] [PubMed]
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.H. Hypertension: Renin-angiotensin-aldosterone system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Motte, S.; McEntee, K.; Naeije, R. Endothelin receptor antagonists. Pharmacol. Ther. 2006, 110, 386–414. [Google Scholar] [CrossRef]
- Valanti, E.K.; Dalakoura-Karagkouni, K.; Fotakis, P.; Vafiadaki, E.; Mantzoros, C.S.; Chroni, A.; Zannis, V.; Kardassis, D.; Sanoudou, D. Reconstituted HDL-apoE3 promotes endothelial cell migration through ID1 and its downstream kinases ERK1/2, AKT and p38 MAPK. Metabolism 2022, 127, 154954. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, Y.; Ran, X.; Wang, D.; Zheng, X.; Zhang, M.; Yu, B.; Sun, Y.; Wu, J. Mettl14 mediates the inflammatory response of macrophages in atherosclerosis through the NF-κB/IL-6 signaling pathway. Cell. Mol. Life Sci. 2022, 79, 311. [Google Scholar] [CrossRef]
- Li, X.; Liu, R.; Liu, W.; Liu, X.; Fan, Z.; Cui, J.; Wu, Y.; Yin, H.; Lin, Q. Panax quinquefolium L. and Salvia miltiorrhiza Bunge. Enhances Angiogenesis by Regulating the miR-155-5p/HIF-1α/VEGF Axis in Acute Myocardial Infarction. Drug Des. Dev. Ther. 2023, 17, 3249–3267. [Google Scholar] [CrossRef] [PubMed]
- Negoro, S.; Kunisada, K.; Tone, E.; Funamoto, M.; Oh, H.; Kishimoto, T.; Yamauchi-Takihara, K. Activation of JAK/STAT pathway transduces cytoprotective signal in rat acute myocardial infarction. Cardiovasc. Res. 2000, 47, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Guo, X.; Chen, Y.; Zeng, Y.; Mo, X.; Hong, S.; He, H.; Li, J.; Steinmetz, R.; Liu, Q. TAB2 deficiency induces dilated cardiomyopathy by promoting RIPK1-dependent apoptosis and necroptosis. J. Clin. Investig. 2022, 132, e152297. [Google Scholar] [CrossRef] [PubMed]
- Shahzadi, S.K.; Marzook, H.; Qaisar, R.; Ahmad, F. Nicotinamide riboside kinase-2 inhibits JNK pathway and limits dilated cardiomyopathy in mice with chronic pressure overload. Clin. Sci. 2022, 136, 181–196. [Google Scholar] [CrossRef]
- Khalilimeybodi, A.; Saucerman, J.J.; Rangamani, P. Modeling cardiomyocyte signaling and metabolism predicts genotype-to-phenotype mechanisms in hypertrophic cardiomyopathy. Comput. Biol. Med. 2024, 175, 108499. [Google Scholar] [CrossRef] [PubMed]
- El Chami, H.; Hassoun, P.M. Inflammatory mechanisms in the pathogenesis of pulmonary arterial hypertension. Compr. Physiol. 2011, 1, 1929–1941. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shao, M.; Yao, J.; Yang, S.; Cheng, W.; Ma, L.; Li, W.; Cao, J.; Zhang, Y.; Hu, Y.; et al. Neocryptotanshinone protects against myocardial ischemia-reperfusion injury by promoting autolysosome degradation of protein aggregates via the ERK1/2-Nrf2-LAMP2 pathway. Phytomedicine 2023, 110, 154625. [Google Scholar] [CrossRef]
- Xu, M.; Li, X.; Song, L. Baicalin regulates macrophages polarization and alleviates myocardial ischaemia/reperfusion injury via inhibiting JAK/STAT pathway. Pharm. Biol. 2020, 58, 655–663. [Google Scholar] [CrossRef]
- Slonková, V.; Slonková, V., Jr.; Vašků, A.; Vašků, V. Genetic predisposition for chronic venous insufficiency in several genes for matrix metalloproteinases (MMP-2, MMP-9, MMP-12) and their inhibitor TIMP-2. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1746–1752. [Google Scholar] [CrossRef]
- Bharath, V.; Kahn, S.R.; Lazo-Langner, A. Genetic polymorphisms of vein wall remodeling in chronic venous disease: A narrative and systematic review. Blood 2014, 124, 1242–1250. [Google Scholar] [CrossRef]
- Swamy, S.; Ueland, T.; Hansen, J.B.; Snir, O.; Brækkan, S.K. Plasma levels of P-selectin and future risk of incident venous thromboembolism. J. Thromb. Haemost. 2023, 21, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C. The Multifaceted Clinical Readouts of Platelet Inhibition by Low-Dose Aspirin. J. Am. Coll. Cardiol. 2015, 66, 74–85. [Google Scholar] [CrossRef]
- Ji, E.; Lee, S. Antibody-Based Therapeutics for Atherosclerosis and Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 5770. [Google Scholar] [CrossRef] [PubMed]
- Gremese, E.; Alivernini, S.; Ferraccioli, E.S.; Ferraccioli, G. Checkpoint inhibitors (CPI) and autoimmune chronic inflammatory diseases (ACIDs): Tolerance and loss of tolerance in the occurrence of immuno-rheumatologic manifestations. Clin. Immunol. 2020, 214, 108395. [Google Scholar] [CrossRef]
- Sperry, B.W.; Zein, R.E.; Fendler, T.J.; Sauer, A.J.; Khumri, T.M.; Magalski, A.; Austin, B.A.; Safley, D.; Kao, A.C. Stabilization of Rapidly Progressive Cardiac Allograft Vasculopathy Using mTOR Inhibition After Heart Transplantation. J. Card. Fail. 2024, 30, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Heijman, J.; Zhou, L.; Dobrev, D. Molecular Basis of Atrial Fibrillation Pathophysiology and Therapy: A Translational Perspective. Circ. Res. 2020, 127, 51–72. [Google Scholar] [CrossRef]
- Trac, D.; Maxwell, J.T.; Brown, M.E.; Xu, C.; Davis, M.E. Aggregation of Child Cardiac Progenitor Cells Into Spheres Activates Notch Signaling and Improves Treatment of Right Ventricular Heart Failure. Circ. Res. 2019, 124, 526–538. [Google Scholar] [CrossRef]
- Qian, J.F.; Liang, S.Q.; Wang, Q.Y.; Xu, J.C.; Luo, W.; Huang, W.J.; Wu, G.J.; Liang, G. Isoproterenol induces MD2 activation by β-AR-cAMP-PKA-ROS signalling axis in cardiomyocytes and macrophages drives inflammatory heart failure. Acta Pharmacol. Sin. 2024, 45, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Higashikuni, Y.; Liu, W.; Numata, G.; Tanaka, K.; Fukuda, D.; Tanaka, Y.; Hirata, Y.; Imamura, T.; Takimoto, E.; Komuro, I.; et al. NLRP3 Inflammasome Activation Through Heart-Brain Interaction Initiates Cardiac Inflammation and Hypertrophy During Pressure Overload. Circulation 2023, 147, 338–355. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Kerschbaumer, A.; Kastrati, K.; Dejaco, C.; Dougados, M.; McInnes, I.B.; Sattar, N.; Stamm, T.A.; Takeuchi, T.; Trauner, M.; et al. Consensus statement on blocking interleukin-6 receptor and interleukin-6 in inflammatory conditions: An update. Ann. Rheum. Dis. 2023, 82, 773–787. [Google Scholar] [CrossRef]
- Kumar, R.K.; Antunes, M.J.; Beaton, A.; Mirabel, M.; Nkomo, V.T.; Okello, E.; Regmi, P.R.; Reményi, B.; Sliwa-Hähnle, K.; Zühlke, L.J.; et al. Contemporary Diagnosis and Management of Rheumatic Heart Disease: Implications for Closing the Gap: A Scientific Statement from the American Heart Association. Circulation 2020, 142, e337–e357. [Google Scholar] [CrossRef]
- Kruithof, B.P.T.; Paardekooper, L.; Hiemstra, Y.L.; Goumans, M.J.; Palmen, M.; Delgado, V.; Klautz, R.J.M.; Ajmone Marsan, N. Stress-induced remodelling of the mitral valve: A model for leaflet thickening and superimposed tissue formation in mitral valve disease. Cardiovasc. Res. 2020, 116, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Hagler, M.A.; Hadley, T.M.; Zhang, H.; Mehra, K.; Roos, C.M.; Schaff, H.V.; Suri, R.M.; Miller, J.D. TGF-β signalling and reactive oxygen species drive fibrosis and matrix remodelling in myxomatous mitral valves. Cardiovasc. Res. 2013, 99, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, L.; Zhao, D.; Yuan, H.; Zhang, H.; Chen, J.; Pang, D.; Lu, Y.; Ouyang, H. MYH7 R453C induced cardiac remodelling via activating TGF-β/Smad2/3, ERK1/2 and Nox4/ROS/NF-κB signalling pathways. Open Biol. 2024, 14, 230427. [Google Scholar] [CrossRef]
- Robinson, P.; Liu, X.; Sparrow, A.; Patel, S.; Zhang, Y.H.; Casadei, B.; Watkins, H.; Redwood, C. Hypertrophic cardiomyopathy mutations increase myofilament Ca(2+) buffering, alter intracellular Ca2+ handling, and stimulate Ca2+-dependent signaling. J. Biol. Chem. 2018, 293, 10487–10499. [Google Scholar] [CrossRef] [PubMed]
- Moncla, L.M.; Briend, M.; Bossé, Y.; Mathieu, P. Calcific aortic valve disease: Mechanisms, prevention and treatment. Nat. Rev. Cardiol. 2023, 20, 546–559. [Google Scholar] [CrossRef]
- Miao, Y.; Tian, L.; Martin, M.; Paige, S.L.; Galdos, F.X.; Li, J.; Klein, A.; Zhang, H.; Ma, N.; Wei, Y.; et al. Intrinsic Endocardial Defects Contribute to Hypoplastic Left Heart Syndrome. Cell Stem Cell 2020, 27, 574–589.e8. [Google Scholar] [CrossRef]
- Yoshimura, T.; Hisatomi, A.; Kajihara, S.; Yasutake, T.; Ogawa, Y.; Mizuta, T.; Ozaki, I.; Utsunomiyai, T.; Yamamoto, K. The relationship between insulin resistance and polymorphisms of the endothelial nitric oxide synthase gene in patients with coronary artery disease. J. Atheroscler. Thromb. 2003, 10, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Motloch, L.J.; Cacheux, M.; Ishikawa, K.; Xie, C.; Hu, J.; Aguero, J.; Fish, K.M.; Hajjar, R.J.; Akar, F.G. Primary Effect of SERCA 2a Gene Transfer on Conduction Reserve in Chronic Myocardial Infarction. J. Am. Heart Assoc. 2018, 7, e009598. [Google Scholar] [CrossRef]
- Hu, J.; Liu, T.; Fu, F.; Cui, Z.; Lai, Q.; Zhang, Y.; Yu, B.; Liu, F.; Kou, J.; Li, F. Omentin1 ameliorates myocardial ischemia-induced heart failure via SIRT3/FOXO3a-dependent mitochondrial dynamical homeostasis and mitophagy. J. Transl. Med. 2022, 20, 447. [Google Scholar] [CrossRef]
- Kundu, S.; Gairola, S.; Verma, S.; Mugale, M.N.; Sahu, B.D. Chronic kidney disease activates the HDAC6-inflammatory axis in the heart and contributes to myocardial remodeling in mice: Inhibition of HDAC6 alleviates chronic kidney disease-induced myocardial remodeling. Basic Res. Cardiol. 2024, 119, 831–852. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Zheng, J.S.; Mason, A.M.; Burgess, S.; Larsson, S.C. Genetically predicted circulating vitamin C in relation to cardiovascular disease. Eur. J. Prev. Cardiol. 2022, 28, 1829–1837. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Nocella, C.; Loffredo, L.; Carnevale, R.; Pignatelli, P. Interventional study with vitamin E in cardiovascular disease and meta-analysis. Free Radic. Biol. Med. 2022, 178, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Ghrayeb, A.; Finney, A.C.; Agranovich, B.; Peled, D.; Anand, S.K.; McKinney, M.P.; Sarji, M.; Yang, D.; Weissman, N.; Drucker, S.; et al. Serine synthesis via reversed SHMT2 activity drives glycine depletion and acetaminophen hepatotoxicity in MASLD. Cell Metab. 2024, 36, 116–129.e7. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, Y.; Qi, B.; Liu, Y.; Cheng, X.; Feng, J.; Gao, W.; Li, T. α-Lipoic acid alleviates myocardial injury and induces M2b macrophage polarization after myocardial infarction via HMGB1/NF-kB signaling pathway. Int. Immunopharmacol. 2023, 121, 110435. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Mariscal, F.M.; de la Cruz-Ares, S.; Torres-Peña, J.D.; Alcalá-Diaz, J.F.; Yubero-Serrano, E.M.; López-Miranda, J. Coenzyme Q(10) and Cardiovascular Diseases. Antioxidants 2021, 10, 906. [Google Scholar] [CrossRef]
- Dyck, G.J.B.; Raj, P.; Zieroth, S.; Dyck, J.R.B.; Ezekowitz, J.A. The Effects of Resveratrol in Patients with Cardiovascular Disease and Heart Failure: A Narrative Review. Int. J. Mol. Sci. 2019, 20, 904. [Google Scholar] [CrossRef]
- Martinez de Lizarrondo, S.; Gakuba, C.; Herbig, B.A.; Repessé, Y.; Ali, C.; Denis, C.V.; Lenting, P.J.; Touzé, E.; Diamond, S.L.; Vivien, D.; et al. Potent Thrombolytic Effect of N-Acetylcysteine on Arterial Thrombi. Circulation 2017, 136, 646–660. [Google Scholar] [CrossRef]
- Ciaraldi, T.P.; Boeder, S.C.; Mudaliar, S.R.; Giovannetti, E.R.; Henry, R.R.; Pettus, J.H. Astaxanthin, a natural antioxidant, lowers cholesterol and markers of cardiovascular risk in individuals with prediabetes and dyslipidaemia. Diabetes Obes. Metab. 2023, 25, 1985–1994. [Google Scholar] [CrossRef]
- Ferenczyova, K.; Kalocayova, B.; Bartekova, M. Potential Implications of Quercetin and its Derivatives in Cardioprotection. Int. J. Mol. Sci. 2020, 21, 1585. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Li, K.; Lin, Y.; Liu, Y. Protective effects and molecular mechanisms of tea polyphenols on cardiovascular diseases. Front. Nutr. 2023, 10, 1202378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Dugbartey, G.J.; Juriasingani, S.; Sener, A. Hydrogen Sulfide Metabolite, Sodium Thiosulfate: Clinical Applications and Underlying Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 6452. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Laufs, U.; Ray, K.K.; Leiter, L.A.; Bays, H.E.; Goldberg, A.C.; Stroes, E.S.; MacDougall, D.; Zhao, X.; Catapano, A.L. Bempedoic acid plus ezetimibe fixed-dose combination in patients with hypercholesterolemia and high CVD risk treated with maximally tolerated statin therapy. Eur. J. Prev. Cardiol. 2020, 27, 593–603. [Google Scholar] [CrossRef] [PubMed]
- de la Guía-Galipienso, F.; Martínez-Ferran, M.; Vallecillo, N.; Lavie, C.J.; Sanchis-Gomar, F.; Pareja-Galeano, H. Vitamin D and cardiovascular health. Clin. Nutr. 2021, 40, 2946–2957. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.; Centner, A.M.; Ukhanov, V.; Nagpal, R.; Salazar, G. Gallic acid ameliorates atherosclerosis and vascular senescence and remodels the microbiome in a sex-dependent manner in ApoE(-/-) mice. J. Nutr. Biochem. 2022, 110, 109132. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.H.; Taha, F.M.; Omar, H.S.; Elwi, H.M.; Abdelnasser, M. Hydrogen sulfide modulates SIRT1 and suppresses oxidative stress in diabetic nephropathy. Mol. Cell. Biochem. 2019, 457, 1–9. [Google Scholar] [CrossRef]
- El Hadri, K.; Smith, R.; Duplus, E.; El Amri, C. Inflammation, Oxidative Stress, Senescence in Atherosclerosis: Thioredoxine-1 as an Emerging Therapeutic Target. Int. J. Mol. Sci. 2021, 23, 77. [Google Scholar] [CrossRef]
- Liu, B.; Chen, L.; Gao, M.; Dai, M.; Zheng, Y.; Qu, L.; Zhang, J.; Gong, G. A comparative study of the efficiency of mitochondria-targeted antioxidants MitoTEMPO and SKQ1 under oxidative stress. Free Radic. Biol. Med. 2024, 224, 117–129. [Google Scholar] [CrossRef]
- Chang, B.; Su, Y.; Li, T.; Zheng, Y.; Yang, R.; Lu, H.; Wang, H.; Ding, Y. Mito-TEMPO Ameliorates Sodium Palmitate Induced Ferroptosis in MIN6 Cells through PINK1/Parkin-Mediated Mitophagy. Biomed. Environ. Sci. 2024, 37, 1128–1141. [Google Scholar] [CrossRef]
- Zinovkina, L.A.; Makievskaya, C.I.; Galkin, I.I.; Zinovkin, R.A. Mitochondria-targeted Uncouplers Decrease Inflammatory Reactions in Endothelial Cells by Enhancing Methylation of the ICAM1 Gene Promoter. Curr. Mol. Pharmacol. 2024, 17, e150823219723. [Google Scholar] [CrossRef]
- Liu, S.Q.; Xie, S.Y.; Zhang, T.; Zhang, H.; Chen, M.Y.; Xing, Y.; Zhao, N.; Li, L.; Chen, S.; Wang, S.S.; et al. Impeding Nucleotide-Binding Oligomerization Domain-Like Receptor 3 Inflammasome Ameliorates Cardiac Remodeling and Dysfunction in Obesity-Associated Cardiomyopathy. J. Am. Heart Assoc. 2024, 13, e035234. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Sheng, J.; Lei, J.; Gan, W.; Yang, Y. Mitochondrial Targeted Antioxidant SKQ1 Ameliorates Acute Kidney Injury by Inhibiting Ferroptosis. Oxidative Med. Cell. Longev. 2022, 2022, 2223957. [Google Scholar] [CrossRef]
- Jabůrek, M.; Klöppel, E.; Průchová, P.; Mozheitova, O.; Tauber, J.; Engstová, H.; Ježek, P. Mitochondria to plasma membrane redox signaling is essential for fatty acid β-oxidation-driven insulin secretion. Redox Biol. 2024, 75, 103283. [Google Scholar] [CrossRef]
- Park, C.; Cha, H.J.; Kim, M.Y.; Bang, E.; Moon, S.K.; Yun, S.J.; Kim, W.J.; Noh, J.S.; Kim, G.Y.; Cho, S.; et al. Phloroglucinol Attenuates DNA Damage and Apoptosis Induced by Oxidative Stress in Human Retinal Pigment Epithelium ARPE-19 Cells by Blocking the Production of Mitochondrial ROS. Antioxidants 2022, 11, 2353. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhao, L.; Liu, Y.; Chen, M.; Guo, X.; Wang, J. N-acetylcysteine, a small molecule scavenger of reactive oxygen species, alleviates cardiomyocyte damage by regulating OPA1-mediated mitochondrial quality control and apoptosis in response to oxidative stress. J. Thorac. Dis. 2024, 16, 5323–5336. [Google Scholar] [CrossRef]
- Quiles, J.M.; Pepin, M.E.; Sunny, S.; Shelar, S.B.; Challa, A.K.; Dalley, B.; Hoidal, J.R.; Pogwizd, S.M.; Wende, A.R.; Rajasekaran, N.S. Identification of Nrf2-responsive microRNA networks as putative mediators of myocardial reductive stress. Sci. Rep. 2021, 11, 11977. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, W.; Song, Y. Impairing Ferroptosis Through the PI3K/Akt/Nrf2 Pathway: The Way for Nerve Growth Factor to Mitigate Hypoxia-induced Cardiomyocyte Damage. Cell Biochem. Biophys. 2024, 1–11. [Google Scholar] [CrossRef]
- Dong, S.; Zhang, C.; Wang, Y.; Liu, S.; Yang, J.; Li, L.; Ma, Y.; Liu, J. The protective effect of rutin on sciatic nerve injury in acrylamide-exposed rats and its mechanisms. Food Chem. Toxicol. 2024, 195, 115106. [Google Scholar] [CrossRef] [PubMed]
- Asle-Rousta, M.; Peirovy, Y. Neuroprotective Effects of Thymol and p-Cymene in Immobilized Male rats through Alterations in Molecular, Biochemical, Histological, and Behavioral Parameters. Neurochem. Res. 2024, 50, 5. [Google Scholar] [CrossRef]
- Zheng, X.B.; Wang, C.; Zhang, M.; Yao, B.Q.; Wu, H.Y.; Hou, S.X. Exogenous H2S targeting PI3K/AKT/mTOR pathway alleviates chronic intermittent hypoxia-induced myocardial damage through inhibiting oxidative stress and enhancing autophagy. Sleep Breath. 2024, 29, 43. [Google Scholar] [CrossRef]
- Sahasrabudhe, S.A.; Terluk, M.R.; Kartha, R.V. N-acetylcysteine Pharmacology and Applications in Rare Diseases-Repurposing an Old Antioxidant. Antioxidants 2023, 12, 1316. [Google Scholar] [CrossRef]
- Song, G.; Wang, J.; Liu, J.; Ruan, Y. Dimethyl fumarate ameliorates erectile dysfunction in bilateral cavernous nerve injury rats by inhibiting oxidative stress and NLRP3 inflammasome-mediated pyroptosis of nerve via activation of Nrf2/HO-1 signaling pathway. Redox Biol. 2023, 68, 102938. [Google Scholar] [CrossRef] [PubMed]
- Carlström, M.; Weitzberg, E.; Lundberg, J.O. Nitric Oxide Signaling and Regulation in the Cardiovascular System: Recent Advances. Pharmacol. Rev. 2024, 76, 1038–1062. [Google Scholar] [CrossRef]
- Tomar, A.; Sahoo, S.; Aathi, M.; Kuila, S.; Khan, M.A.; Ravi, G.R.R.; Jeyaraman, J.; Mehta, J.L.; Varughese, K.I.; Arockiasamy, A. Exploring the druggability of oxidized low-density lipoprotein (ox-LDL) receptor, LOX-1, a proatherogenic drug target involved in atherosclerosis. Biochem. Biophys. Res. Commun. 2022, 623, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Sun, J.; Cavalera, M.; Al-Sharify, D.; Matthes, F.; Barghouth, M.; Tengryd, C.; Dunér, P.; Persson, A.; Sundius, L.; et al. Dysregulation of MMP2-dependent TGF-ß2 activation impairs fibrous cap formation in type 2 diabetes-associated atherosclerosis. Nat. Commun. 2024, 15, 10464. [Google Scholar] [CrossRef]
- Zeng, L.; Zhang, X.; Huang, Z.; Song, S.; Li, M.; Wang, T.; Sun, A.; Ge, J. Ubiquitin proteasome system in cardiac fibrosis. J. Adv. Res. 2024; in press. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H.; Tanaka, T.; Koury, M.J. Hypoxia-inducible factor activators: A novel class of oral drugs for the treatment of anemia of chronic kidney disease. Hematol. Am. Soc. Hematol. Educ. Program. 2024, 2024, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Zhang, M.; Li, S.; Liu, W.; Xu, C.; Li, Y.; Xie, R. Controlled Stimulus-Responsive Delivery Systems for Osteoarthritis Treatment. Int. J. Mol. Sci. 2024, 25, 11799. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhang, Y.; Shi, W.; Lu, L.; Wei, J.; Wang, J.; Zhang, H.; Pu, Y.; Yin, L. Angiotensin II Induces Vascular Endothelial Dysfunction by Promoting Lipid Peroxidation-Mediated Ferroptosis via CD36. Biomolecules 2024, 14, 1456. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Huang, X.; Li, M.; Lou, P.; Ma, H.; Jiang, X.; Zhou, Y.; Liu, Y. Ferroptosis in diabetic cardiomyopathy: From its mechanisms to therapeutic strategies. Front. Endocrinol. 2024, 15, 1421838. [Google Scholar] [CrossRef] [PubMed]
- Alsereidi, F.R.; Khashim, Z.; Marzook, H.; Al-Rawi, A.M.; Salomon, T.; Almansoori, M.K.; Madkour, M.M.; Hamam, A.M.; Ramadan, M.M.; Peterson, Q.P.; et al. Dapagliflozin mitigates cellular stress and inflammation through PI3K/AKT pathway modulation in cardiomyocytes, aortic endothelial cells, and stem cell-derived β cells. Cardiovasc. Diabetol. 2024, 23, 388. [Google Scholar] [CrossRef] [PubMed]
- Drakontaeidi, A.; Papanotas, I.; Pontiki, E. Multitarget Pharmacology of Sulfur-Nitrogen Heterocycles: Anticancer and Antioxidant Perspectives. Antioxidants 2024, 13, 898. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Barros-Becker, F.; Ogelman, R.; Camci, E.D.; Linbo, T.H.; Simon, J.A.; Rubel, E.W.; Raible, D.W. Multiple mechanisms of aminoglycoside ototoxicity are distinguished by subcellular localization of action. Front. Neurol. 2024, 15, 1480435. [Google Scholar] [CrossRef]
- Lyamzaev, K.G.; Huan, H.; Panteleeva, A.A.; Simonyan, R.A.; Avetisyan, A.V.; Chernyak, B.V. Exogenous Iron Induces Mitochondrial Lipid Peroxidation, Lipofuscin Accumulation, and Ferroptosis in H9c2 Cardiomyocytes. Biomolecules 2024, 14, 730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shi, C.; Yao, Z.; Qian, S. Bardoxolone methyl attenuates doxorubicin-induced cardiotoxicity by inhibiting the TXNIP-NLRP3 pathway through Nrf2 activation. Environ. Toxicol. 2024, 39, 1936–1950. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, M.; Nakagami, H.; Sanada, F.; Morishita, R. Progress of Gene Therapy in Cardiovascular Disease. Hypertension 2020, 76, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Han, Y.; Jin, Z.; Hang, W.; Shu, H.; Wen, Z.; Ni, L.; Wang, D.W. Homology-directed repair of an MYBPC3 gene mutation in a rat model of hypertrophic cardiomyopathy. Gene Ther. 2023, 30, 520–527. [Google Scholar] [CrossRef]
- Argiro, A.; Bui, Q.; Hong, K.N.; Ammirati, E.; Olivotto, I.; Adler, E. Applications of Gene Therapy in Cardiomyopathies. JACC Heart Fail. 2024, 12, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, D.I.; Rider, D.A.; Yavari, A.; Wikström Lindholm, M.; Campion, G.V.; Nissen, S.E. Treatment and prevention of lipoprotein(a)-mediated cardiovascular disease: The emerging potential of RNA interference therapeutics. Cardiovasc. Res. 2022, 118, 1218–1231. [Google Scholar] [CrossRef] [PubMed]
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y.; et al. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 2020, 180, 862–877.e22. [Google Scholar] [CrossRef]
- Davis, J.R.; Banskota, S.; Levy, J.M.; Newby, G.A.; Wang, X.; Anzalone, A.V.; Nelson, A.T.; Chen, P.J.; Hennes, A.D.; An, M.; et al. Efficient prime editing in mouse brain, liver and heart with dual AAVs. Nat. Biotechnol. 2024, 42, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J.; Hodgkinson, C.P. RNA Therapeutics for the Cardiovascular System. Circulation 2024, 149, 707–716. [Google Scholar] [CrossRef]
- Menasché, P.; Vanneaux, V.; Fabreguettes, J.R.; Bel, A.; Tosca, L.; Garcia, S.; Bellamy, V.; Farouz, Y.; Pouly, J.; Damour, O.; et al. Towards a clinical use of human embryonic stem cell-derived cardiac progenitors: A translational experience. Eur. Heart J. 2015, 36, 743–750. [Google Scholar] [CrossRef]
- Bagno, L.; Hatzistergos, K.E.; Balkan, W.; Hare, J.M. Mesenchymal Stem Cell-Based Therapy for Cardiovascular Disease: Progress and Challenges. Mol. Ther. 2018, 26, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, F.; Ma, J.; Shi, J.; Chen, S.; Liu, Z.; Liu, J. Effect of stem cell transplantation on patients with ischemic heart failure: A systematic review and meta-analysis of randomized controlled trials. Stem Cell Res. Ther. 2019, 10, 125. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Xiong, W.; Sun, W.; Liu, W.; Zhang, Y.; Li, C.; Gu, N.; Shen, Y.; Qiu, Z.; Li, C. YTHDC1 Mitigates Apoptosis in Bone Marrow Mesenchymal Stem Cells by Inhibiting NfƙBiα and Augmenting Cardiac Function Following Myocardial Infarction. Cell Transplant. 2024, 33, 9636897241290910. [Google Scholar] [CrossRef]
- Mu, L.; Dong, R.; Li, C.; Chen, J.; Huang, Y.; Li, T.; Guo, B. ROS responsive conductive microspheres loaded with salvianolic acid B as adipose derived stem cell carriers for acute myocardial infarction treatment. Biomaterials 2025, 314, 122849. [Google Scholar] [CrossRef]
- Javaid, A.; Sharma, K.K.; Ka, A.; Verma, A.; Mudavath, S.L. Therapeutic Potential of Molsidomine-Loaded Liquid Crystal Nanoparticles for the Treatment and Management of Niacin-Induced Varicose Veins: In Vitro and In Vivo Studies. ACS Appl. Bio Mater. 2024, 7, 7306–7323. [Google Scholar] [CrossRef]
- Sahoo, S.; Adamiak, M.; Mathiyalagan, P.; Kenneweg, F.; Kafert-Kasting, S.; Thum, T. Therapeutic and Diagnostic Translation of Extracellular Vesicles in Cardiovascular Diseases: Roadmap to the Clinic. Circulation 2021, 143, 1426–1449. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Yellon, D.M.; Sivaraman, V.; Das, D.; Boi-Doku, C.; Arjun, S.; Zheng, Y.; Riquelme, J.A.; Kearney, J.; Sharma, V.; et al. Plasma exosomes protect the myocardium from ischemia-reperfusion injury. J. Am. Coll. Cardiol. 2015, 65, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Loyer, X.; Zlatanova, I.; Devue, C.; Yin, M.; Howangyin, K.Y.; Klaihmon, P.; Guerin, C.L.; Kheloufi, M.; Vilar, J.; Zannis, K.; et al. Intra-Cardiac Release of Extracellular Vesicles Shapes Inflammation Following Myocardial Infarction. Circ. Res. 2018, 123, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Skeik, N.; Patel, D.C. A review of troponins in ischemic heart disease and other conditions. Int. J. Angiol. 2007, 16, 53–58. [Google Scholar] [CrossRef]
- Li, B.; Cao, Y.; Sun, M.; Feng, H. Expression, regulation, and function of exosome-derived miRNAs in cancer progression and therapy. FASEB J. 2021, 35, e21916. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.; Moosavi, M.S.; Abed, H.M.; Dehghani, M.; Aalipour, M.; Heydari, E.A.; Behroozaghdam, M.; Entezari, M.; Salimimoghadam, S.; Gunduz, E.S.; et al. Long non-coding RNA (lncRNA) H19 in human cancer: From proliferation and metastasis to therapy. Pharmacol. Res. 2022, 184, 106418. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.L.; Shen, W.C.; Chen, Y.C.; Lai, T.C.; Lin, S.R.; Lin, S.W.; Yu, I.S.; Yeh, Y.H.; Li, T.K.; Lee, I.T.; et al. Mir221- and Mir222-enriched adsc-exosomes mitigate PM exposure-exacerbated cardiac ischemia-reperfusion injury through the modulation of the BNIP3-MAP1LC3B-BBC3/PUMA pathway. Autophagy 2024, 20, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Z.; Lin, A.; Zhang, N. Exosomes from Hypoxic Pretreatment ADSCs Ameliorate Cardiac Damage Post-MI via Activated circ-Stt3b/miR-15a-5p/GPX4 Signaling and Decreased Ferroptosis. Cardiovasc. Toxicol. 2024, 24, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Elhage, R.; Maret, A.; Pieraggi, M.T.; Thiers, J.C.; Arnal, J.F.; Bayard, F. Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation 1998, 97, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Rane, M. Interleukin-6 Signaling and Anti-Interleukin-6 Therapeutics in Cardiovascular Disease. Circ. Res. 2021, 128, 1728–1746. [Google Scholar] [CrossRef] [PubMed]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Ding, Z.; Pothineni, N.V.K.; Goel, A.; Lüscher, T.F.; Mehta, J.L. PCSK9 and inflammation: Role of shear stress, pro-inflammatory cytokines, and LOX-1. Cardiovasc. Res. 2020, 116, 908–915. [Google Scholar] [CrossRef]
- Newman, C.B.; Tobert, J.A. Targeting PCSK9 with Antibodies and Gene Silencing to Reduce LDL Cholesterol. J. Clin. Endocrinol. Metab. 2023, 108, 784–790. [Google Scholar] [CrossRef]
- Dutka, M.; Zimmer, K.; Ćwiertnia, M.; Ilczak, T.; Bobiński, R. The role of PCSK9 in heart failure and other cardiovascular diseases-mechanisms of action beyond its effect on LDL cholesterol. Heart Fail. Rev. 2024, 29, 917–937. [Google Scholar] [CrossRef] [PubMed]
- Neuen, B.L.; Heerspink, H.J.L.; Vart, P.; Claggett, B.L.; Fletcher, R.A.; Arnott, C.; de Oliveira Costa, J.; Falster, M.O.; Pearson, S.A.; Mahaffey, K.W.; et al. Estimated Lifetime Cardiovascular, Kidney, and Mortality Benefits of Combination Treatment with SGLT2 Inhibitors, GLP-1 Receptor Agonists, and Nonsteroidal MRA Compared with Conventional Care in Patients with Type 2 Diabetes and Albuminuria. Circulation 2024, 149, 450–462. [Google Scholar] [CrossRef]
- Nauck, M.A.; D’Alessio, D.A. Tirzepatide, a dual GIP/GLP-1 receptor co-agonist for the treatment of type 2 diabetes with unmatched effectiveness regrading glycaemic control and body weight reduction. Cardiovasc. Diabetol. 2022, 21, 169. [Google Scholar] [CrossRef]
- Marx, N.; Husain, M.; Lehrke, M.; Verma, S.; Sattar, N. GLP-1 Receptor Agonists for the Reduction of Atherosclerotic Cardiovascular Risk in Patients with Type 2 Diabetes. Circulation 2022, 146, 1882–1894. [Google Scholar] [CrossRef]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.; Heerspink, H.J.L.; Cuthbertson, D.J.; Wilding, J.P.H. SGLT2 inhibitors and GLP-1 receptor agonists: Established and emerging indications. Lancet 2021, 398, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Trepiccione, F.; Iervolino, A.; D’Acierno, M.; Siccardi, S.; Costanzo, V.; Sardella, D.; De La Motte, L.R.; D’Apolito, L.; Miele, A.; Perna, A.F.; et al. The SGLT2 inhibitor dapagliflozin improves kidney function in glycogen storage disease XI. Sci. Transl. Med. 2023, 15, eabn4214. [Google Scholar] [CrossRef]
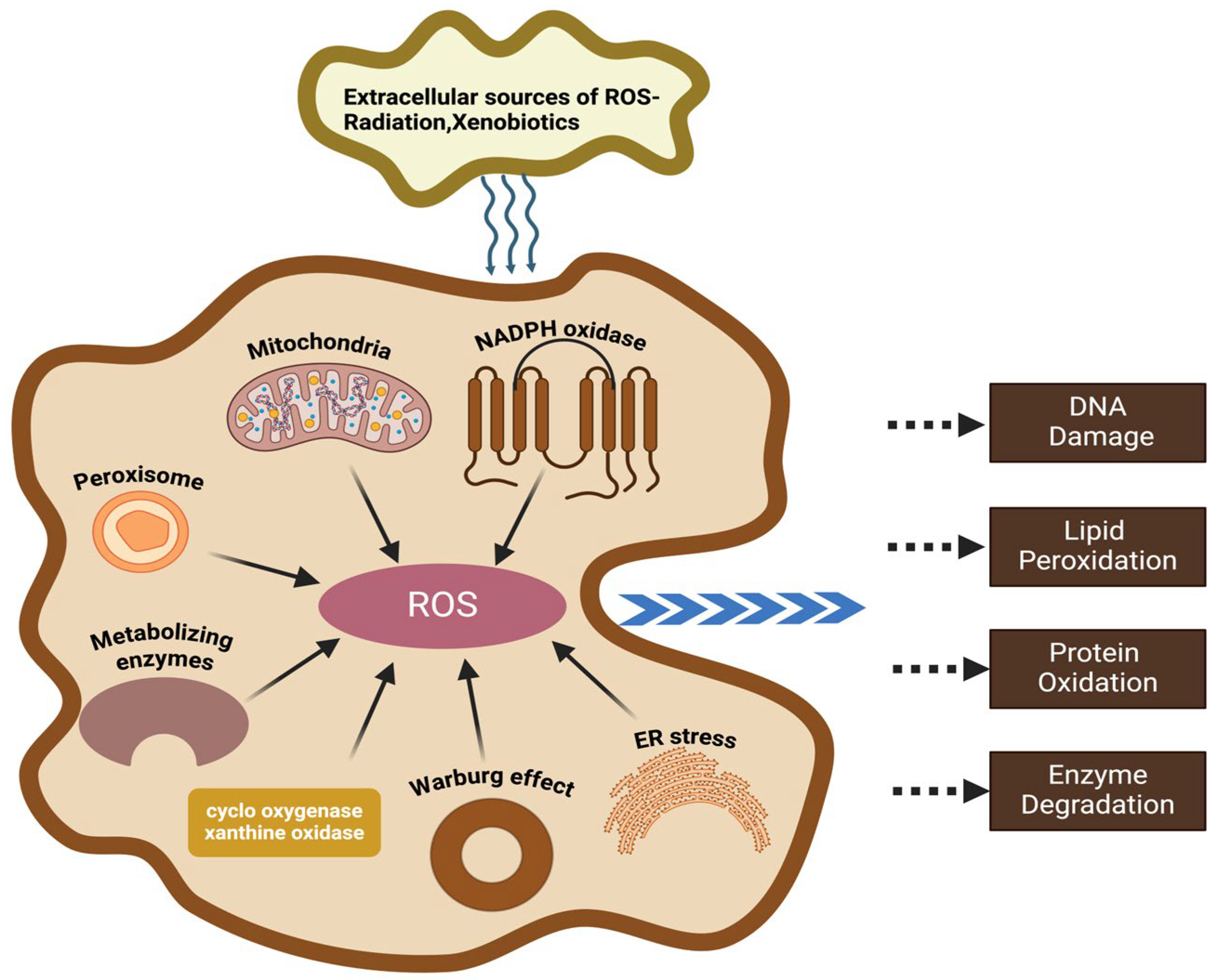

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Guo, J. Deciphering Oxidative Stress in Cardiovascular Disease Progression: A Blueprint for Mechanistic Understanding and Therapeutic Innovation. Antioxidants 2025, 14, 38. https://doi.org/10.3390/antiox14010038
Zhang Z, Guo J. Deciphering Oxidative Stress in Cardiovascular Disease Progression: A Blueprint for Mechanistic Understanding and Therapeutic Innovation. Antioxidants. 2025; 14(1):38. https://doi.org/10.3390/antiox14010038
Chicago/Turabian StyleZhang, Zhaoshan, and Jiawei Guo. 2025. "Deciphering Oxidative Stress in Cardiovascular Disease Progression: A Blueprint for Mechanistic Understanding and Therapeutic Innovation" Antioxidants 14, no. 1: 38. https://doi.org/10.3390/antiox14010038
APA StyleZhang, Z., & Guo, J. (2025). Deciphering Oxidative Stress in Cardiovascular Disease Progression: A Blueprint for Mechanistic Understanding and Therapeutic Innovation. Antioxidants, 14(1), 38. https://doi.org/10.3390/antiox14010038