

Liver Cell Mitophagy in Metabolic Dysfunction-Associated Steatotic Liver Disease and Liver Fibrosis

 ,
,
Abstract
1. Introduction
2. Mitophagy
2.1. Concept of Mitophagy
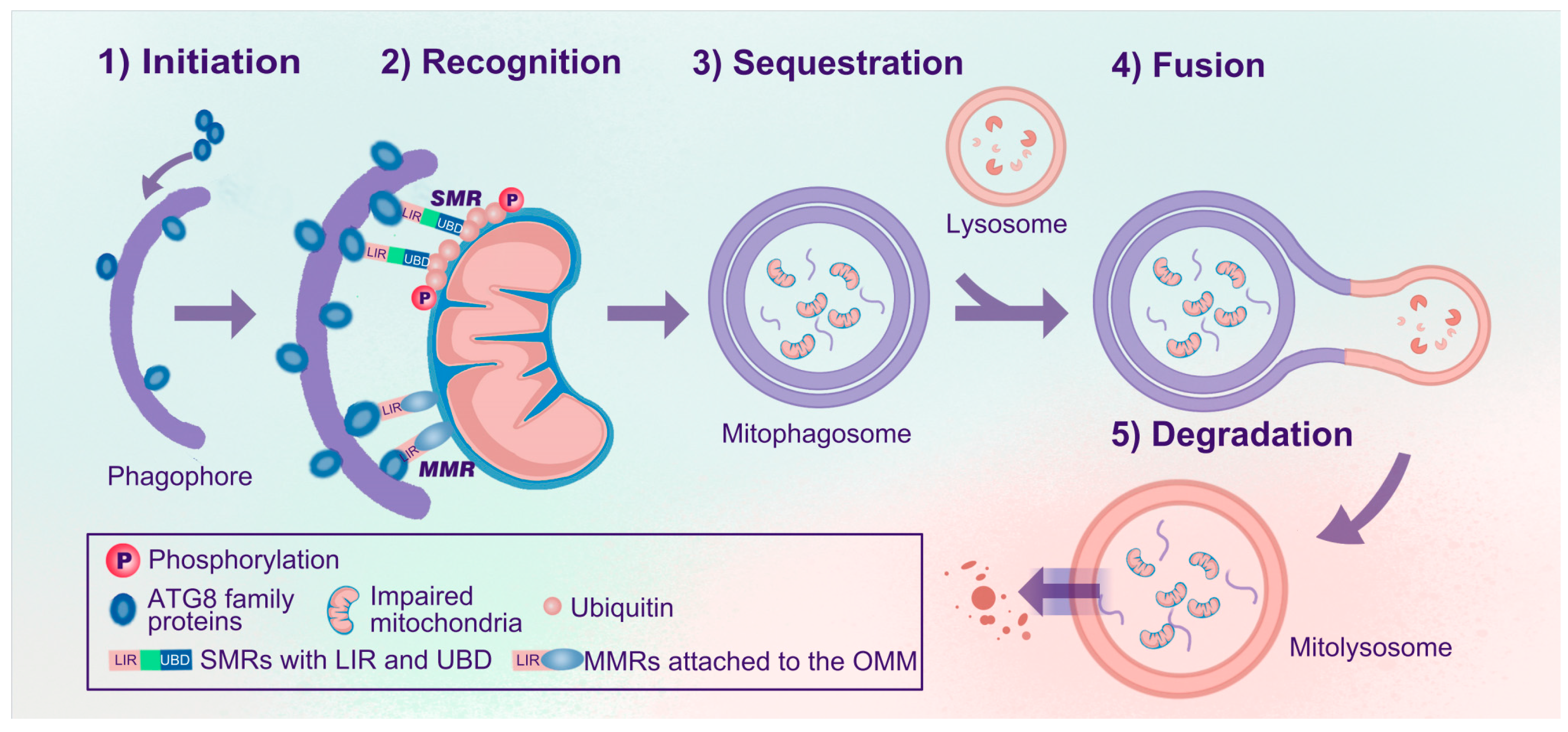
2.2. Processes of Mitophagy
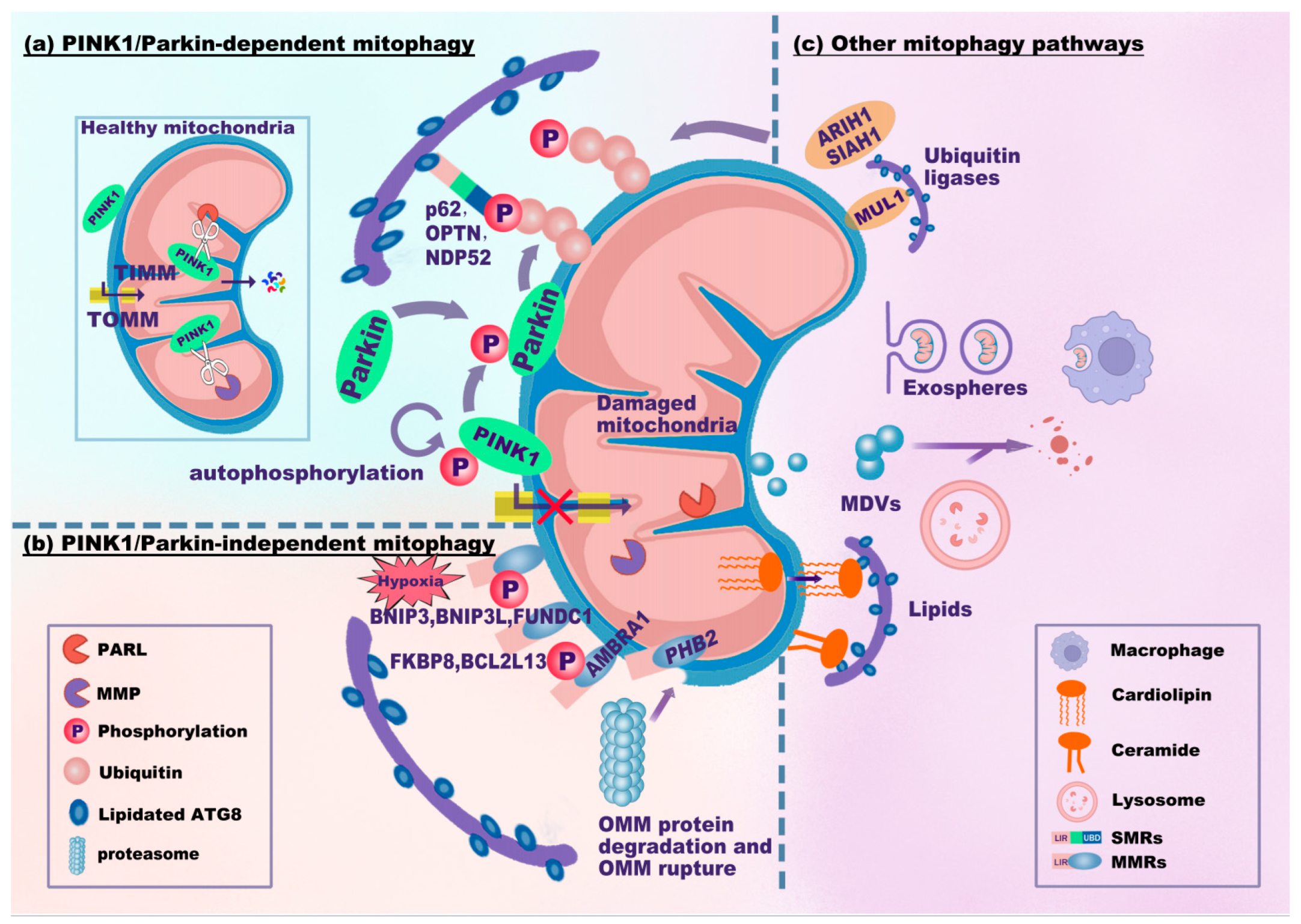
2.3. Signaling Pathway of Mitophagy
- (1)
- PINK1/Parkin-dependent mitophagy signaling pathway
- (2)
- PINK1/Parkin-independent mitophagy signaling pathway
- (3)
- Other mitophagy signaling pathways
3. Role of Liver Cell Mitophagy in MASLD and Liver Fibrosis
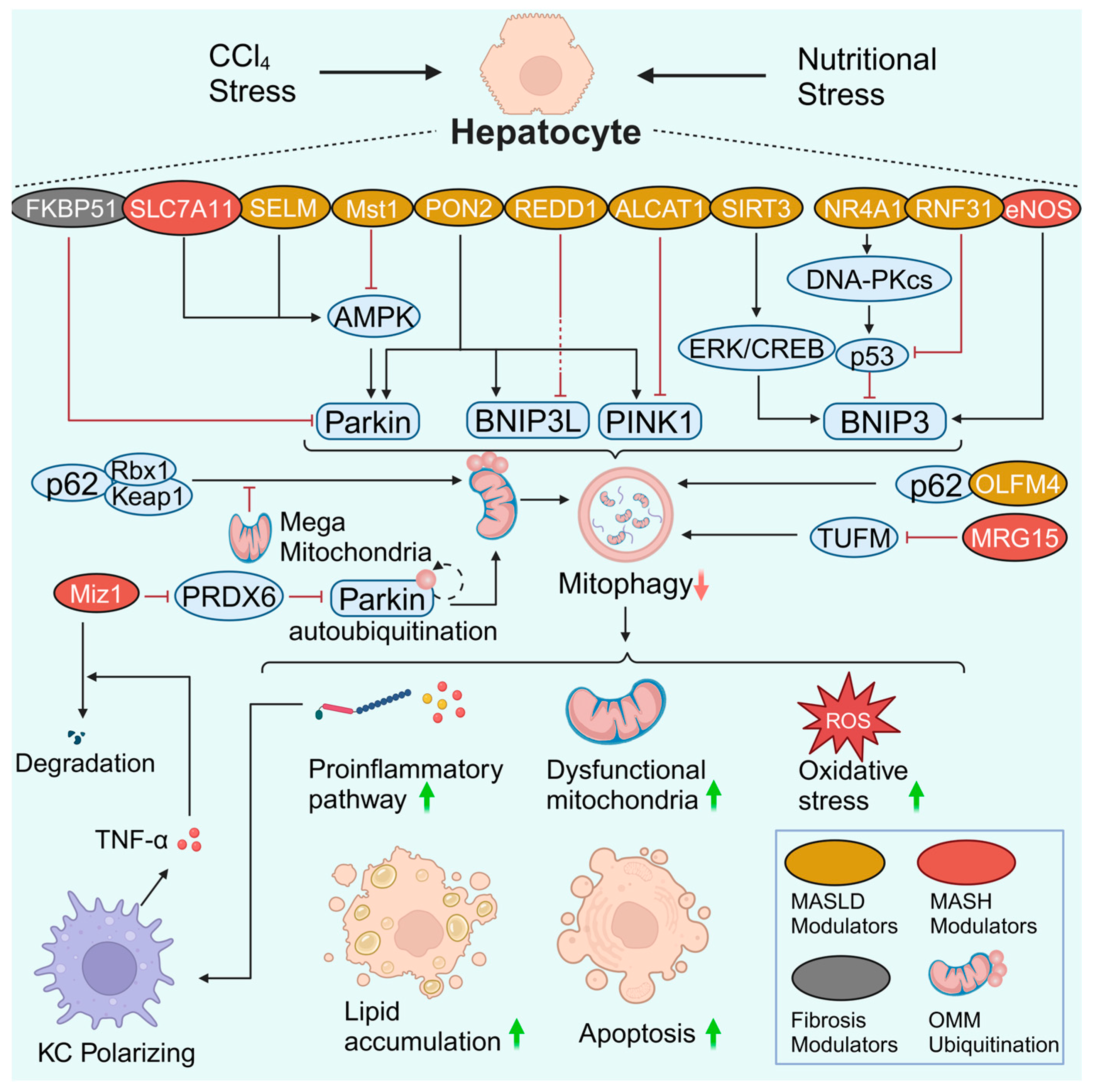
3.1. Hepatocyte Mitophagy in MASLD
3.2. Hepatocyte Mitophagy in Liver Fibrosis
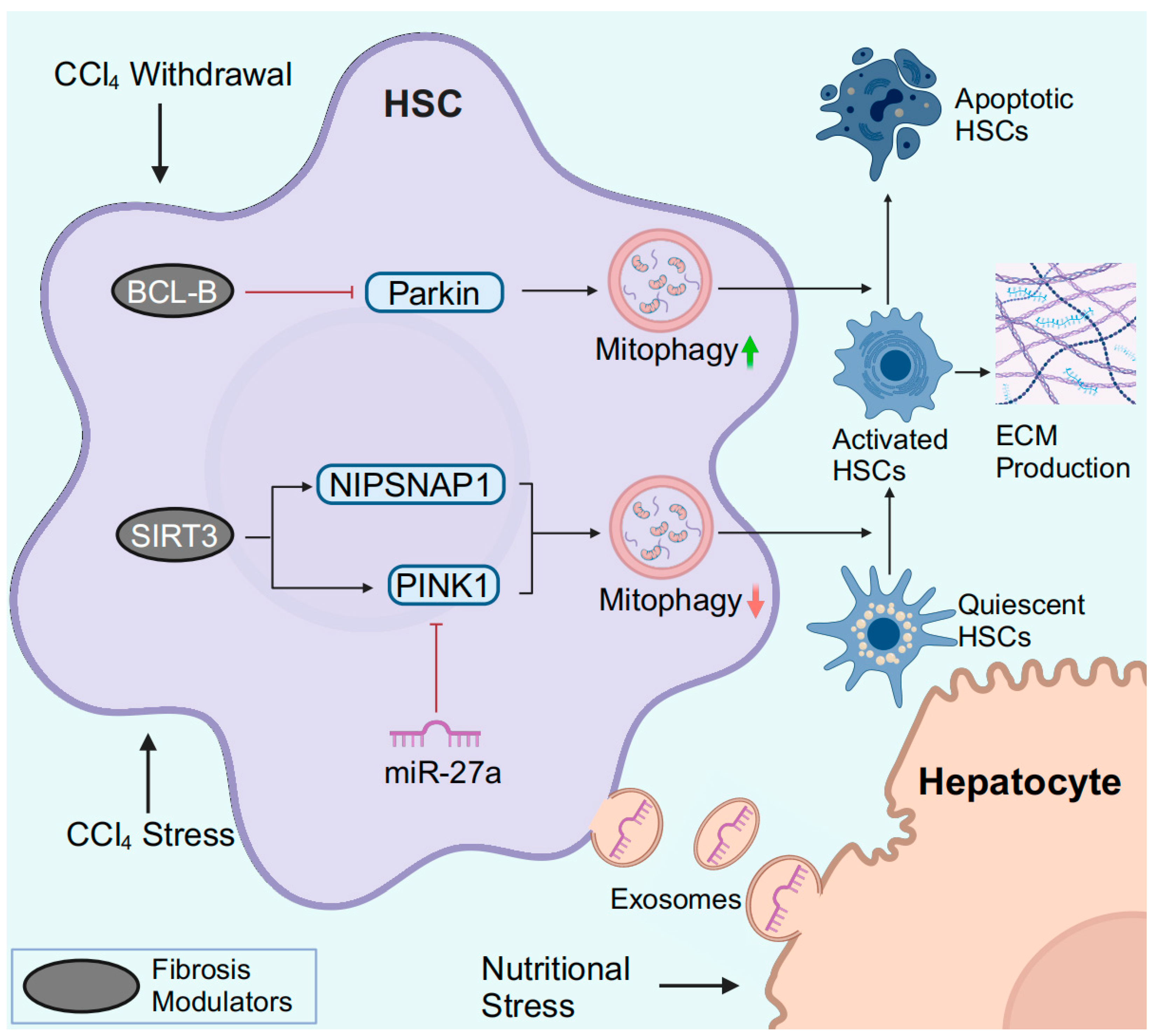
3.3. HSC Mitophagy in MASLD
3.4. HSC Mitophagy in Liver Fibrosis
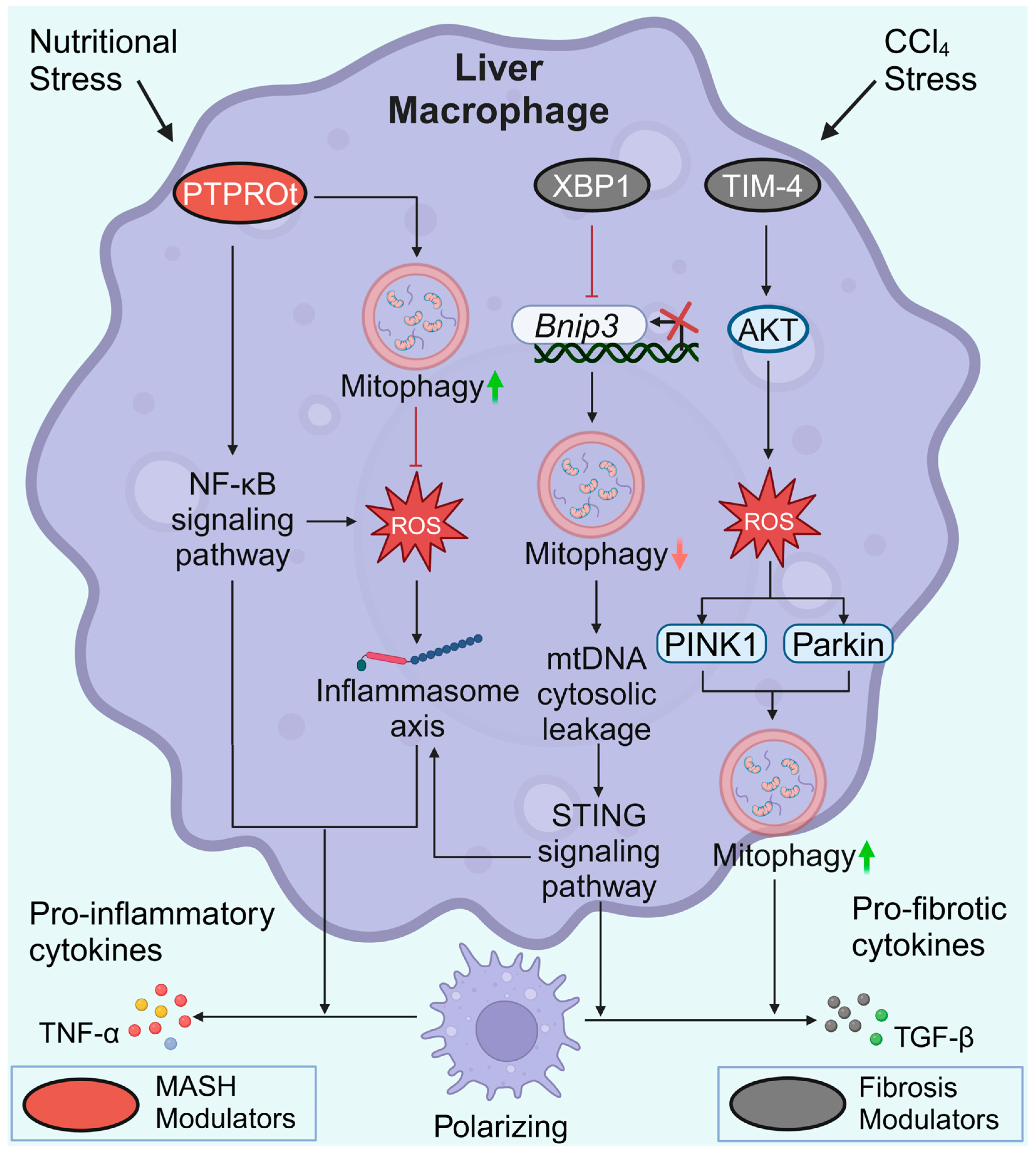
3.5. Macrophage Mitophagy in MASLD
3.6. Macrophage Mitophagy in Liver Fibrosis
3.7. LSEC Mitophagy in MASLD
3.8. LSEC Mitophagy in Liver Fibrosis
4. Mitophagy as a Target for the Treatment of MASLD and Liver Fibrosis
5. Prospects and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J. Hepatol. 2023, 79, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Ekstedt, M.; Wong, G.L.; Hagström, H. Changing epidemiology, global trends and implications for outcomes of NAFLD. J. Hepatol. 2023, 79, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.E.; Ong, E.Y.H.; Chung, C.H.; Ong, C.E.Y.; Koh, B.; Tan, D.J.H.; Lim, W.H.; Yong, J.N.; Xiao, J.; Wong, Z.Y.; et al. Longitudinal Outcomes Associated With Metabolic Dysfunction-Associated Steatotic Liver Disease: A Meta-analysis of 129 Studies. Clin. Gastroenterol. Hepatol. 2024, 22, 488–498.e414. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Makhlouf, H.R. Histology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in Adults and Children. Clin. Liver Dis. 2016, 20, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Bedossa, P.; Guy, C.D.; Schattenberg, J.M.; Loomba, R.; Taub, R.; Labriola, D.; Moussa, S.E.; Neff, G.W.; Rinella, M.E.; et al. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. N. Engl. J. Med. 2024, 390, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Roden, M. Mitochondrial alterations in fatty liver diseases. J. Hepatol. 2023, 78, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.; Ali, A.H.; Ibdah, J.A. Mitochondrial Dysfunction Plays Central Role in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7280. [Google Scholar] [CrossRef]
- Shin, S.; Kim, J.; Lee, J.Y.; Kim, J.; Oh, C.M. Mitochondrial Quality Control: Its Role in Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD). J. Obes. Metab. Syndr. 2023, 32, 289–302. [Google Scholar] [CrossRef]
- Picca, A.; Faitg, J.; Auwerx, J.; Ferrucci, L.; D’Amico, D. Mitophagy in human health, ageing and disease. Nat. Metab. 2023, 5, 2047–2061. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, Y.; Li, W.; Chen, H.; Du, L.; Liu, D.; Wang, X.; Xu, T.; Liu, L.; Chen, Q. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 2019, 15, 1882–1898. [Google Scholar] [CrossRef] [PubMed]
- Alim Al-Bari, A.; Ito, Y.; Thomes, P.G.; Menon, M.B.; García-Macia, M.; Fadel, R.; Stadlin, A.; Peake, N.; Faris, M.E.; Eid, N.; et al. Emerging mechanistic insights of selective autophagy in hepatic diseases. Front. Pharmacol. 2023, 14, 1149809. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.-Y. Mitophagy in the Pathogenesis of Liver Diseases. Cells 2020, 9, 831. [Google Scholar] [CrossRef]
- Ma, X.; McKeen, T.; Zhang, J.; Ding, W.-X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Y. The role and mechanism of action of mitophagy in various liver diseases. Antioxid. Redox Signal. 2022, 38, 529–549. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wu, X.; Liao, R. Mechanism and regulation of mitophagy in nonalcoholic fatty liver disease (NAFLD): A mini-review. Life Sci. 2023, 312, 121162. [Google Scholar] [CrossRef]
- Kitada, M.; Koya, D. Autophagy in metabolic disease and ageing. Nat. Rev. Endocrinol. 2021, 17, 647–661. [Google Scholar] [CrossRef]
- Kotani, T.; Sakai, Y.; Kirisako, H.; Kakuta, C.; Kakuta, S.; Ohsumi, Y.; Nakatogawa, H. A mechanism that ensures non-selective cytoplasm degradation by autophagy. Nat. Commun. 2023, 14, 5815. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2023, 24, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuv. Res. 2005, 8, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Terešak, P.; Lapao, A.; Subic, N.; Boya, P.; Elazar, Z.; Simonsen, A. Regulation of PRKN-independent mitophagy. Autophagy 2022, 18, 24–39. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Fedorowicz, M.A.; de Vries-Schneider, R.L.; Rüb, C.; Becker, D.; Huang, Y.; Zhou, C.; Alessi Wolken, D.M.; Voos, W.; Liu, Y.; Przedborski, S. Cytosolic cleaved PINK1 represses Parkin translocation to mitochondria and mitophagy. EMBO Rep. 2014, 15, 86–93. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Kishi, C.; Ishihara, N.; Mizushima, N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 2011, 286, 19630–19640. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.; Hess, S.; Chan, D.C. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Yamano, K.; Kikuchi, R.; Kojima, W.; Hayashida, R.; Koyano, F.; Kawawaki, J.; Shoda, T.; Demizu, Y.; Naito, M.; Tanaka, K.; et al. Critical role of mitochondrial ubiquitination and the OPTN-ATG9A axis in mitophagy. J. Cell Biol. 2020, 219, e201912144. [Google Scholar] [CrossRef]
- Padman, B.S.; Nguyen, T.N.; Uoselis, L.; Skulsuppaisarn, M.; Nguyen, L.K.; Lazarou, M. LC3/GABARAPs drive ubiquitin-independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat. Commun. 2019, 10, 408. [Google Scholar] [CrossRef]
- Yang, X.; Yin, H.; Zhang, Y.; Li, X.; Tong, H.; Zeng, Y.; Wang, Q.; He, W. Hypoxia-induced autophagy promotes gemcitabine resistance in human bladder cancer cells through hypoxia-inducible factor 1α activation. Int. J. Oncol. 2018, 53, 215–224. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xue, L.; Li, L.; Tang, C.; Wan, Z.; Wang, R.; Tan, J.; Tan, Y.; Han, H.; Tian, R.; et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J. Biol. Chem. 2016, 291, 21616–21629. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Massen, S.; Terenzio, M.; Lang, V.; Chen-Lindner, S.; Eils, R.; Novak, I.; Dikic, I.; Hamacher-Brady, A.; Brady, N.R. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 2013, 288, 1099–1113. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.V.; Suzuki, H.; Marinković, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubašić, M.; Šprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1131. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, H.Y.; Hanna, R.A.; Gustafsson, Å.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef]
- Gao, F.; Chen, D.; Si, J.; Hu, Q.; Qin, Z.; Fang, M.; Wang, G. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum. Mol. Genet. 2015, 24, 2528–2538. [Google Scholar] [CrossRef]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell 2014, 54, 362–377. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.; Ma, K.; Zhou, C.; Ding, P.; Zhu, Y.; Chen, Q.; Xia, B. Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy 2016, 12, 2363–2373. [Google Scholar] [CrossRef]
- Lv, M.; Wang, C.; Li, F.; Peng, J.; Wen, B.; Gong, Q.; Shi, Y.; Tang, Y. Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein Cell 2017, 8, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Otsu, K.; Murakawa, T.; Yamaguchi, O. BCL2L13 is a mammalian homolog of the yeast mitophagy receptor Atg32. Autophagy 2015, 11, 1932–1933. [Google Scholar] [CrossRef] [PubMed]
- Murakawa, T.; Yamaguchi, O.; Hashimoto, A.; Hikoso, S.; Takeda, T.; Oka, T.; Yasui, H.; Ueda, H.; Akazawa, Y.; Nakayama, H.; et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 2015, 6, 7527. [Google Scholar] [CrossRef] [PubMed]
- Bhujabal, Z.; Birgisdottir, Å.B.; Sjøttem, E.; Brenne, H.B.; Øvervatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238.e210. [Google Scholar] [CrossRef]
- Yan, C.; Gong, L.; Chen, L.; Xu, M.; Abou-Hamdan, H.; Tang, M.; Désaubry, L.; Song, Z. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 2020, 16, 419–434. [Google Scholar] [CrossRef]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; De Strooper, B.; Vandenberghe, W. Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef]
- Di Rita, A.; Peschiaroli, A.; D’Acunzo, P.; Strobbe, D.; Hu, Z.; Gruber, J.; Nygaard, M.; Lambrughi, M.; Melino, G.; Papaleo, E.; et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKα. Nat. Commun. 2018, 9, 3755. [Google Scholar] [CrossRef] [PubMed]
- Ambivero, C.T.; Cilenti, L.; Main, S.; Zervos, A.S. Mulan E3 ubiquitin ligase interacts with multiple E2 conjugating enzymes and participates in mitophagy by recruiting GABARAP. Cell Signal. 2014, 26, 2921–2929. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qi, W.; Chen, G.; Feng, D.; Liu, J.; Ma, B.; Zhou, C.; Mu, C.; Zhang, W.; Chen, Q.; et al. Mitochondrial outer-membrane E3 ligase MUL1 ubiquitinates ULK1 and regulates selenite-induced mitophagy. Autophagy 2015, 11, 1216–1229. [Google Scholar] [CrossRef] [PubMed]
- Szargel, R.; Shani, V.; Abd Elghani, F.; Mekies, L.N.; Liani, E.; Rott, R.; Engelender, S. The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum. Mol. Genet. 2016, 25, 3476–3490. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Proïcs, E.; Rubio-Patiño, C.; Obba, S.; Zunino, B.; Bossowski, J.P.; Rozier, R.M.; Chiche, J.; Mondragón, L.; Riley, J.S.; et al. Parkin-Independent Mitophagy Controls Chemotherapeutic Response in Cancer Cells. Cell Rep. 2017, 20, 2846–2859. [Google Scholar] [CrossRef] [PubMed]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Le Guerroué, F.; Eck, F.; Jung, J.; Starzetz, T.; Mittelbronn, M.; Kaulich, M.; Behrends, C. Autophagosomal Content Profiling Reveals an LC3C-Dependent Piecemeal Mitophagy Pathway. Mol. Cell 2017, 68, 786–796.e786. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Sagar, S.; Ravindran, R.; Najor, R.H.; Quiles, J.M.; Chi, L.; Diao, R.Y.; Woodall, B.P.; Leon, L.J.; Zumaya, E.; et al. Mitochondria are secreted in extracellular vesicles when lysosomal function is impaired. Nat. Commun. 2023, 14, 5031. [Google Scholar] [CrossRef]
- Nicolás-Ávila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martínez, L.; Sánchez-Díaz, M.; Díaz-García, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e123. [Google Scholar] [CrossRef]
- Soubannier, V.; McLelland, G.L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. CB 2012, 22, 135–141. [Google Scholar] [CrossRef]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; Rippstein, P.; Kaufman, B.A.; Shoubridge, E.A.; McBride, H.M. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE 2012, 7, e52830. [Google Scholar] [CrossRef]
- Sulkshane, P.; Ram, J.; Thakur, A.; Reis, N.; Kleifeld, O.; Glickman, M.H. Ubiquitination and receptor-mediated mitophagy converge to eliminate oxidation-damaged mitochondria during hypoxia. Redox Biol. 2021, 45, 102047. [Google Scholar] [CrossRef]
- Undamatla, R.; Fagunloye, O.G.; Chen, J.; Edmunds, L.R.; Murali, A.; Mills, A.; Xie, B.; Pangburn, M.M.; Sipula, I.; Gibson, G.; et al. Reduced mitophagy is an early feature of NAFLD and liver-specific PARKIN knockout hastens the onset of steatosis, inflammation and fibrosis. Sci. Rep. 2023, 13, 7575. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Huang, J.; Yang, J.; Chen, X.; Zhang, H.; Zhu, Y.; Liu, Q.; Zhang, Z. The protective effect of selenoprotein M on non-alcoholic fatty liver disease: The role of the AMPKα1-MFN2 pathway and Parkin mitophagy. Cell. Mol. Life Sci.CMLS 2022, 79, 354. [Google Scholar] [CrossRef]
- Zhou, T.; Chang, L.; Luo, Y.; Zhou, Y.; Zhang, J. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Biol. 2019, 21, 101120. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Lv, T.; Fan, X.; He, C.; Zhu, S.; Xiong, X.; Yan, W.; Liu, M.; Xu, H.; Shi, R.; He, Q. SLC7A11-ROS/αKG-AMPK axis regulates liver inflammation through mitophagy and impairs liver fibrosis and NASH progression. Redox Biol. 2024, 72, 103159. [Google Scholar] [CrossRef]
- Lee, D.H.; Jung, Y.Y.; Park, M.H.; Jo, M.R.; Han, S.B.; Yoon, D.Y.; Roh, Y.S.; Hong, J.T. Peroxiredoxin 6 Confers Protection Against Nonalcoholic Fatty Liver Disease Through Maintaining Mitochondrial Function. Antioxid. Redox Signal. 2019, 31, 387–402. [Google Scholar] [CrossRef]
- Jin, K.; Shi, Y.; Zhang, H.; Zhangyuan, G.; Wang, F.; Li, S.; Chen, C.; Zhang, J.; Wang, H.; Zhang, W.; et al. A TNFα/Miz1-positive feedback loop inhibits mitophagy in hepatocytes and propagates non-alcoholic steatohepatitis. J. Hepatol. 2023, 79, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.P.; Liu, X.J.; Xie, L.; Shen, X.Z.; Wu, J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Investig. J. Tech. Methods Pathol. 2019, 99, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Hao, M.; Liu, Y.; Ma, X.; Lin, W.; Xu, Q.; Zhou, H.; Shao, N.; Kuang, H. Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur. J. Pharmacol. 2019, 864, 172715. [Google Scholar] [CrossRef]
- Gao, X.; Ruan, Y.; Zhu, X.; Lin, X.; Xin, Y.; Li, X.; Mai, M.; Guo, H. Deoxycholic Acid Promotes Pyroptosis in Free Fatty Acid-Induced Steatotic Hepatocytes by Inhibiting PINK1-Mediated Mitophagy. Inflammation 2022, 45, 639–650. [Google Scholar] [CrossRef]
- Chen, S.; Wang, X.; Liu, Z.; Wang, J.; Guo, Y.; Wang, Q.; Huang, H.; Li, Y.; Yu, C.; Xu, C. Olfactomedin 4 deletion exacerbates nonalcoholic fatty liver disease through P62-dependent mitophagy in mice. Metab. Clin. Exp. 2023, 148, 155679. [Google Scholar] [CrossRef]
- Yamada, T.; Murata, D.; Adachi, Y.; Itoh, K.; Kameoka, S.; Igarashi, A.; Kato, T.; Araki, Y.; Huganir, R.L.; Dawson, T.M.; et al. Mitochondrial Stasis Reveals p62-Mediated Ubiquitination in Parkin-Independent Mitophagy and Mitigates Nonalcoholic Fatty Liver Disease. Cell Metab. 2018, 28, 588–604.e585. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Nie, J.; Zhang, J.; Kimball, S.R.; Zhang, H.; Zhang, W.J.; Jefferson, L.S.; Cheng, Z.; Ji, Q.; et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology 2015, 61, 486–496. [Google Scholar] [CrossRef]
- Tian, C.; Min, X.; Zhao, Y.; Wang, Y.; Wu, X.; Liu, S.; Dou, W.; Zhou, T.; Liu, Y.; Luo, R.; et al. MRG15 aggravates non-alcoholic steatohepatitis progression by regulating the mitochondrial proteolytic degradation of TUFM. J. Hepatol. 2022, 77, 1491–1503. [Google Scholar] [CrossRef]
- Glick, D.; Zhang, W.; Beaton, M.; Marsboom, G.; Gruber, M.; Simon, M.C.; Hart, J.; Dorn, G.W., 2nd; Brady, M.J.; Macleod, K.F. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol. Cell. Biol. 2012, 32, 2570–2584. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.D.; Meers, G.M.; Morris, E.M.; Linden, M.A.; Cunningham, R.P.; Ibdah, J.A.; Thyfault, J.P.; Laughlin, M.H.; Rector, R.S. eNOS deletion impairs mitochondrial quality control and exacerbates Western diet-induced NASH. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E605–E616. [Google Scholar] [CrossRef]
- Cunningham, R.P.; Moore, M.P.; Dashek, R.J.; Meers, G.M.; Takahashi, T.; Sheldon, R.D.; Wheeler, A.A.; Diaz-Arias, A.; Ibdah, J.A.; Parks, E.J.; et al. Critical Role for Hepatocyte-Specific eNOS in NAFLD and NASH. Diabetes 2021, 70, 2476–2491. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Du, W.; Li, Y.; Shi, C.; Hu, N.; Ma, S.; Wang, W.; Ren, J. Effects of melatonin on fatty liver disease: The role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J. Pineal Res. 2018, 64, e12450. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Xin, T.; Li, D.; Wang, C.; Zhu, H.; Zhou, H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: The role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018, 18, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, F.; Shi, Y.; Sheng, J.; Wang, Y.; Zhang, L.; Zhou, J.; Jin, Y.; Yan, Y. RNF31 alleviates liver steatosis by promoting p53/BNIP3-related mitophagy in hepatocytes. Free Radic. Biol. Med. 2024, 219, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Shin, G.C.; Lee, H.M.; Kim, N.; Yoo, S.K.; Park, H.S.; Choi, L.S.; Kim, K.P.; Lee, A.R.; Seo, S.U.; Kim, K.H. Paraoxonase-2 contributes to promoting lipid metabolism and mitochondrial function via autophagy activation. Sci. Rep. 2022, 12, 21483. [Google Scholar] [CrossRef] [PubMed]
- Dumas, K.; Ayachi, C.; Gilleron, J.; Lacas-Gervais, S.; Pastor, F.; Favier, F.B.; Peraldi, P.; Vaillant, N.; Yvan-Charvet, L.; Bonnafous, S.; et al. REDD1 deficiency protects against nonalcoholic hepatic steatosis induced by high-fat diet. FASEB J. 2020, 34, 5046–5060. [Google Scholar] [CrossRef] [PubMed]
- Grefhorst, A.; van de Peppel, I.P.; Larsen, L.E.; Jonker, J.W.; Holleboom, A.G. The Role of Lipophagy in the Development and Treatment of Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2020, 11, 601627. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Li, W.; He, P.; Huang, Y.; Li, Y.F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef]
- Dupont, N.; Chauhan, S.; Arko-Mensah, J.; Castillo, E.F.; Masedunskas, A.; Weigert, R.; Robenek, H.; Proikas-Cezanne, T.; Deretic, V. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr. Biol. CB 2014, 24, 609–620. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.W.; Hong, J.M.; Lee, S.M. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J. Pineal Res. 2016, 60, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Turkseven, S.; Bolognesi, M.; Brocca, A.; Pesce, P.; Angeli, P.; Di Pascoli, M. Mitochondria-targeted antioxidant mitoquinone attenuates liver inflammation and fibrosis in cirrhotic rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G298–G304. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Zhong, Z.; Dou, L.; Xu, Y.; Zou, Y.; Weldon, K.; Wang, J.; Zhang, L.; Liu, M.; Williams, K.E.; et al. Knocking out Fkbp51 decreases CCl(4)-induced liver injury through enhancement of mitochondrial function and Parkin activity. Cell Biosci. 2024, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, Z.; Wang, Y.; Sun, R.; Zou, B.; Tian, X.; Liu, D.; Zhao, X.; Zhou, J.; Zhao, Y.; et al. SIRT3 regulates mitophagy in liver fibrosis through deacetylation of PINK1/NIPSNAP1. J. Cell. Physiol. 2023, 238, 2090–2102. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Liu, S.; Qin, X.; Abudureyimu, M.; Wang, L.; Zou, R.; Ajoolabady, A.; Zhang, W.; Peng, H.; Ren, J.; et al. FUNDC1 interacts with GPx4 to govern hepatic ferroptosis and fibrotic injury through a mitophagy-dependent manner. J. Adv. Res. 2024, 55, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, K.M.; Jung, E.H.; Lee, H.R.; Yang, J.H.; Cho, S.S.; Ki, S.H. Parkin-Mediated Mitophagy by TGF-β Is Connected with Hepatic Stellate Cell Activation. Int. J. Mol. Sci. 2023, 24, 14826. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhang, Q.; Hua, C.; Ci, X. Melatonin alleviates particulate matter-induced liver fibrosis by inhibiting ROS-mediated mitophagy and inflammation via Nrf2 activation. Ecotoxicol. Environ. Saf. 2023, 268, 115717. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Gao, Y.L.; Jiang, S.; Qian, B.; Che, L.; Wu, J.S.; Du, Z.B.; Wang, M.Z.; Yang, Y.; Lin, Y.C.; et al. Aflatoxin B(1)-exposed hepatocyte-derived extracellular vesicles: Initiating hepatic stellate cell-mediated liver fibrosis through a p53-Parkin-dependent mitophagy pathway. Ecotoxicol. Environ. Saf. 2024, 277, 116363. [Google Scholar] [CrossRef]
- Luo, X.; Xu, Z.X.; Wu, J.C.; Luo, S.Z.; Xu, M.Y. Hepatocyte-derived exosomal miR-27a activateshepatic stellate cells through the inhibitionof PINK1-mediated mitophagy in MAFLD. Mol. Ther. Nucleic Acids 2021, 26, 1241–1254. [Google Scholar] [CrossRef]
- Qiu, Y.N.; Wang, G.H.; Zhou, F.; Hao, J.J.; Tian, L.; Guan, L.F.; Geng, X.K.; Ding, Y.C.; Wu, H.W.; Zhang, K.Z. PM2.5 induces liver fibrosis via triggering ROS-mediated mitophagy. Ecotoxicol. Environ. Saf. 2019, 167, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Yu, H.; Hao, J.J.; Peng, Y.; Yin, T.T.; Qiu, Y.N. PM(2.5) promotes Drp1-mediated mitophagy to induce hepatic stellate cell activation and hepatic fibrosis via regulating miR-411. Exp. Cell Res. 2021, 407, 112828. [Google Scholar] [CrossRef]
- Ding, Q.; Xie, X.L.; Wang, M.M.; Yin, J.; Tian, J.M.; Jiang, X.Y.; Zhang, D.; Han, J.; Bai, Y.; Cui, Z.J.; et al. The role of the apoptosis-related protein BCL-B in the regulation of mitophagy in hepatic stellate cells during the regression of liver fibrosis. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Lu, S.; Su, Y.; Ding, D.; Tao, L.; Wang, M.; Wang, Y.; Liu, X. C/EBP-α induces autophagy by binding to Beclin1 through its own acetylation modification in activated hepatic stellate cells. Exp. Cell Res. 2021, 405, 112721. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Liu, Y.; Shi, Y.; Zhang, H.; Sun, Y.; Zhangyuan, G.; Wang, F.; Yu, W.; Wang, J.; Tao, X.; et al. PTPROt aggravates inflammation by enhancing NF-κB activation in liver macrophages during nonalcoholic steatohepatitis. Theranostics 2020, 10, 5290–5304. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, G.; Wang, J.; Deng, M.; Yuan, F.; Gong, J. TIM-4 interference in Kupffer cells against CCL4-induced liver fibrosis by mediating Akt1/Mitophagy signalling pathway. Cell Prolif. 2020, 53, e12731. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bu, Q.; Liu, M.; Zhang, R.; Gu, J.; Li, L.; Zhou, J.; Liang, Y.; Su, W.; Liu, Z.; et al. XBP1-mediated activation of the STING signalling pathway in macrophages contributes to liver fibrosis progression. JHEP Rep. Innov. Hepatol. 2022, 4, 100555. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Caparrós, E.; Fernández-Iglesias, A.; Francés, R. Role of liver sinusoidal endothelial cells in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 411–431. [Google Scholar] [CrossRef]
- Gao, J.; Lan, T.; Kostallari, E.; Guo, Y.; Lai, E.; Guillot, A.; Ding, B.; Tacke, F.; Tang, C.; Shah, V.H. Angiocrine signaling in sinusoidal homeostasis and liver diseases. J. Hepatol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Gao, Y.X.; Wu, J.C.; Li, J.Z.; Fu, S.W.; Xu, M.Y. Single-cell transcriptome reveals a novel mechanism of C-Kit(+)-liver sinusoidal endothelial cells in NASH. Cell Biosci. 2024, 14, 31. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.Z.; Im, G.B.; Luo, A.C.; Zhu, Y.; Hong, X.; Neumeyer, J.; Tang, H.W.; Perrimon, N.; Melero-Martin, J.M. Mitochondrial transfer mediates endothelial cell engraftment through mitophagy. Nature 2024, 629, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Caldwell, M.E.; Poole, K.E.; Seija, A.; Harris, M.P.; Greene, N.P.; Wooten, J.S. Exercise during weight loss improves hepatic mitophagy. Sports Med. Health Sci. 2022, 4, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Caldwell, M.E.; Lee, D.E.; Brown, J.L.; Brown, L.A.; Perry, R.A., Jr.; Greene, E.S.; Carvallo Chaigneau, F.R.; Washington, T.A.; Greene, N.P. Moderate physical activity promotes basal hepatic autophagy in diet-induced obese mice. Appl. Physiol. Nutr. Metab. (Physiol. Appl. Nutr. Metab.) 2017, 42, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, I.O.; Passos, E.; Diogo, C.V.; Rocha-Rodrigues, S.; Santos-Alves, E.; Oliveira, P.J.; Ascensão, A.; Magalhães, J. Exercise mitigates mitochondrial permeability transition pore and quality control mechanisms alterations in nonalcoholic steatohepatitis. Appl. Physiol. Nutr. Metab. (Physiol. Appl. Nutr. Metab.) 2016, 41, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Amorim, R.; Simões, I.C.M.; Teixeira, J.; Cagide, F.; Potes, Y.; Soares, P.; Carvalho, A.; Tavares, L.C.; Benfeito, S.; Pereira, S.P.; et al. Mitochondria-targeted anti-oxidant AntiOxCIN(4) improved liver steatosis in Western diet-fed mice by preventing lipid accumulation due to upregulation of fatty acid oxidation, quality control mechanism and antioxidant defense systems. Redox Biol. 2022, 55, 102400. [Google Scholar] [CrossRef] [PubMed]
- Dou, S.D.; Zhang, J.N.; Xie, X.L.; Liu, T.; Hu, J.L.; Jiang, X.Y.; Wang, M.M.; Jiang, H.D. MitoQ inhibits hepatic stellate cell activation and liver fibrosis by enhancing PINK1/parkin-mediated mitophagy. Open Med. 2021, 16, 1718–1727. [Google Scholar] [CrossRef]
- Hang, W.; Shu, H.; Wen, Z.; Liu, J.; Jin, Z.; Shi, Z.; Chen, C.; Wang, D.W. N-Acetyl Cysteine Ameliorates High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease and Intracellular Triglyceride Accumulation by Preserving Mitochondrial Function. Front. Pharmacol. 2021, 12, 636204. [Google Scholar] [CrossRef]
- Chen, M.; Huang, F.; Chen, B.; Kang, J.; Yao, Y.; Liua, M.; Li, Y.; Li, Y.; Zhou, T.; Peng, D.; et al. A classical herbal formula alleviates high-fat diet induced nonalcoholic steatohepatitis (NASH) via targeting mitophagy to rehabilitate dysfunctional mitochondria, validated by UPLC-HRMS identification combined with in vivo experiment. Biomed. Pharmacother. 2023, 168, 115831. [Google Scholar] [CrossRef]
- Wang, C.H.; Liu, H.M.; Chang, Z.Y.; Lee, M.C.; Hsu, C.H.; Lee, T.Y. Antioxidants Rich Herbal Formula Ger-Gen-Chyn-Lian-Tang Protects Lipotoxicity and Ameliorates Inflammation Signaling through Regulation of Mitochondrial Biogenesis and Mitophagy in Nonalcoholic Fatty Liver Disease Mice. Front. Biosci. 2022, 27, 242. [Google Scholar] [CrossRef]
- Wu, Y.; Kuang, Y.; Wu, Y.; Dai, H.; Bi, R.; Hu, J.; Sun, L. Yang-Gan-Jiang-Mei formula alleviates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome through mitophagy. Biotechnol. Genet. Eng. Rev. 2023, 40, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Linden, M.A.; Lopez, K.T.; Fletcher, J.A.; Morris, E.M.; Meers, G.M.; Siddique, S.; Laughlin, M.H.; Sowers, J.R.; Thyfault, J.P.; Ibdah, J.A.; et al. Combining metformin therapy with caloric restriction for the management of type 2 diabetes and nonalcoholic fatty liver disease in obese rats. Appl. Physiol. Nutr. Metab. (Physiol. Appl. Nutr. Metab.) 2015, 40, 1038–1047. [Google Scholar] [CrossRef]
- Zhang, R.; Chu, K.; Zhao, N.; Wu, J.; Ma, L.; Zhu, C.; Chen, X.; Wei, G.; Liao, M. Corilagin Alleviates Nonalcoholic Fatty Liver Disease in High-Fat Diet-Induced C57BL/6 Mice by Ameliorating Oxidative Stress and Restoring Autophagic Flux. Front. Pharmacol. 2019, 10, 1693. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Wang, Y.; Zhang, C.; Sullivan, M.A.; Chen, W.; Jing, X.; Yu, H.; Li, F.; Wang, Q.; Zhou, Z.; et al. Quercetin ameliorates nonalcoholic fatty liver disease (NAFLD) via the promotion of AMPK-mediated hepatic mitophagy. J. Nutr. Biochem. 2023, 120, 109414. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Lin, H.; Xu, Y.; Zhou, F.; Wang, J.; Liu, J.; Zhu, X.; Guo, X.; Tang, Y.; Yao, P. Frataxin-Mediated PINK1-Parkin-Dependent Mitophagy in Hepatic Steatosis: The Protective Effects of Quercetin. Mol. Nutr. Food Res. 2018, 62, e1800164. [Google Scholar] [CrossRef]
- Yao, Z.; Li, X.; Wang, W.; Ren, P.; Song, S.; Wang, H.; Xie, Y.; Li, X.; Li, Z. Corn peptides attenuate non-alcoholic fatty liver disease via PINK1/Parkin-mediated mitochondrial autophagy. Food Nutr. Res. 2023, 67, 2023. [Google Scholar] [CrossRef]
- Li, X.; Shi, Z.; Zhu, Y.; Shen, T.; Wang, H.; Shui, G.; Loor, J.J.; Fang, Z.; Chen, M.; Wang, X.; et al. Cyanidin-3-O-glucoside improves non-alcoholic fatty liver disease by promoting PINK1-mediated mitophagy in mice. Br. J. Pharmacol. 2020, 177, 3591–3607. [Google Scholar] [CrossRef]
- Di Paola, R.; Modafferi, S.; Siracusa, R.; Cordaro, M.; D’Amico, R.; Ontario, M.L.; Interdonato, L.; Salinaro, A.T.; Fusco, R.; Impellizzeri, D.; et al. S-Acetyl-Glutathione Attenuates Carbon Tetrachloride-Induced Liver Injury by Modulating Oxidative Imbalance and Inflammation. Int. J. Mol. Sci. 2022, 23, 4429. [Google Scholar] [CrossRef]
- El-Derany, M.O.; El-Demerdash, E. Pyrvinium pamoate attenuates non-alcoholic steatohepatitis: Insight on hedgehog/Gli and Wnt/β-catenin signaling crosstalk. Biochem. Pharmacol. 2020, 177, 113942. [Google Scholar] [CrossRef]
- Song, N.; Xu, H.; Wu, S.; Luo, S.; Xu, J.; Zhao, Q.; Wang, R.; Jiang, X. Synergistic activation of AMPK by AdipoR1/2 agonist and inhibitor of EDPs-EBP interaction recover NAFLD through enhancing mitochondrial function in mice. Acta Pharm. Sin. B 2023, 13, 542–558. [Google Scholar] [CrossRef]
- Zou, Y.Y.; Tang, X.B.; Chen, Z.L.; Liu, B.; Zheng, L.; Song, M.Y.; Xiao, Q.; Zhou, Z.Q.; Peng, X.Y.; Tang, C.F. Exercise intervention improves mitochondrial quality in non-alcoholic fatty liver disease zebrafish. Front. Endocrinol. 2023, 14, 1162485. [Google Scholar] [CrossRef] [PubMed]
- Shao, N.; Yu, X.Y.; Ma, X.F.; Lin, W.J.; Hao, M.; Kuang, H.Y. Exenatide Delays the Progression of Nonalcoholic Fatty Liver Disease in C57BL/6 Mice, Which May Involve Inhibition of the NLRP3 Inflammasome through the Mitophagy Pathway. Gastroenterol. Res. Pract. 2018, 2018, 1864307. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Trauner, M. Gut-liver axis: Pathophysiological concepts and clinical implications. Cell Metab. 2022, 34, 1700–1718. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, X.; Elsabagh, M.; Zhang, Y.; Ma, Y.; Jin, Y.; Wang, M.; Wang, H.; Jiang, H. Effects of the Gut Microbiota and Barrier Function on Melatonin Efficacy in Alleviating Liver Injury. Antioxidants 2022, 11, 1727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yan, A.; Liu, X.; Ma, Y.; Zhao, F.; Wang, M.; Loor, J.J.; Wang, H. Melatonin ameliorates ochratoxin A induced liver inflammation, oxidative stress and mitophagy in mice involving in intestinal microbiota and restoring the intestinal barrier function. J. Hazard Mater. 2021, 407, 124489. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Liu, Y.; Yi, J.; Li, Y.; Li, Q.; Li, Y.; Shang, P.; Guo, J.; Hu, L.; Pan, J.; et al. Gut microbiota disturbance exaggerates battery wastewater-induced hepatotoxicity through a gut-liver axis. Sci. Total Environ. 2022, 809, 152188. [Google Scholar] [CrossRef]
- Yang, C.J.; Chang, H.C.; Sung, P.C.; Ge, M.C.; Tang, H.Y.; Cheng, M.L.; Cheng, H.T.; Chou, H.H.; Lin, C.Y.; Lin, W.R.; et al. Oral fecal transplantation enriches Lachnospiraceae and butyrate to mitigate acute liver injury. Cell Rep. 2024, 43, 113591. [Google Scholar] [CrossRef]
- Yang, C.W.; Liu, H.M.; Chang, Z.Y.; Liu, G.H.; Chang, H.H.; Huang, P.Y.; Lee, T.Y. Puerarin Modulates Hepatic Farnesoid X Receptor and Gut Microbiota in High-Fat Diet-Induced Obese Mice. Int. J. Mol. Sci. 2024, 25, 5274. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategies or Compounds | Disease | Model | Changes in Mitophagy | References | |
|---|---|---|---|---|---|
| Physical exercise and caloric restriction | WR plus CR | MASLD | Mice | Increased levels of BNIP3, PINK1, and Parkin | [124] |
| WR | MASLD | Mice | Induced upward trends in transcription and expression of Bnip3 | [125] | |
| Endurable WR | MASH | Rats | Increased expression of PINK1 and Parkin | [126] | |
| Antioxidant | AntiOxCIN4 | MASLD | Mice | Induced an upward trend in Parkin levels | [127] |
| MitoQ | Liver fibrosis | Rats, mice | Increased translocation of Parkin to mitochondria in rats; promoted PINK1/Parkin-mediated mitophagy in mouse HSCs | [103,128] | |
| NAC | MASLD | Mice | Inhibited highly activated expression of PINK1 | [129] | |
| Traditional Chinese medicine/classical herbal formula | SG formula | MASH | Mice | Increased BNIP3/BNIP3L-related mitophagy | [130] |
| GGCLT formula | MASLD | Mice, db/db mice | Increased Parkin-dependent/independent mitophagy | [131] | |
| YGJM formula | MASH | Rats | Promoted PINK1/Parkin-mediated mitophagy | [132] | |
| Diabetic medications | Metformin plus CR | MASLD | OLETF rats | Increased BNIP3 levels | [133] |
| Others | Corilagin | MASLD | Mice | Increased Parkin levels; enhanced colocalization of Parkin and mitochondria | [134] |
| Quercetin | MASLD | Mice | Increased PINK1/Parkin-mediated mitophagy | [135,136] | |
| Corn peptides | MASLD | Rats | Activated PINK1/Parkin-mediated mitophagy | [137] | |
| C3G | MASLD | Mice | Increased PINK1-mediated mitophagy | [138] | |
| Melatonin | MASLD | Mice | Restored BNIP3-related mitophagy in hepatocytes | [92] | |
| Liver fibrosis | Mice | Suppressed activated PINK1/Parkin-mediated mitophagy in hepatocytes treated with particulate matter | [108] | ||
| Liver fibrosis | Rats | Restored PINK1 and Parkin levels | [102] | ||
| SAG | Liver fibrosis | Mice | Restored PINK1 and Parkin levels | [139] | |
| PP | MASH | Rats | Restored PINK1/Parkin-mediated mitophagy | [140] | |
| JT003 plus V14 peptide | MASLD | Mice | Increased BNIP3 levels; expression of mitophagy-related genes (e.g., Park2, Bnip3l); and colocalization of lysosomes with mitochondria | [141] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Jian, L.; Guo, Y.; Tang, C.; Huang, Z.; Gao, J. Liver Cell Mitophagy in Metabolic Dysfunction-Associated Steatotic Liver Disease and Liver Fibrosis. Antioxidants 2024, 13, 729. https://doi.org/10.3390/antiox13060729
Chen J, Jian L, Guo Y, Tang C, Huang Z, Gao J. Liver Cell Mitophagy in Metabolic Dysfunction-Associated Steatotic Liver Disease and Liver Fibrosis. Antioxidants. 2024; 13(6):729. https://doi.org/10.3390/antiox13060729
Chicago/Turabian StyleChen, Jiaxin, Linge Jian, Yangkun Guo, Chengwei Tang, Zhiyin Huang, and Jinhang Gao. 2024. "Liver Cell Mitophagy in Metabolic Dysfunction-Associated Steatotic Liver Disease and Liver Fibrosis" Antioxidants 13, no. 6: 729. https://doi.org/10.3390/antiox13060729
APA StyleChen, J., Jian, L., Guo, Y., Tang, C., Huang, Z., & Gao, J. (2024). Liver Cell Mitophagy in Metabolic Dysfunction-Associated Steatotic Liver Disease and Liver Fibrosis. Antioxidants, 13(6), 729. https://doi.org/10.3390/antiox13060729