Efficacy and Safety of Coenzyme Q10 Supplementation in Neonates, Infants and Children: An Overview


Abstract
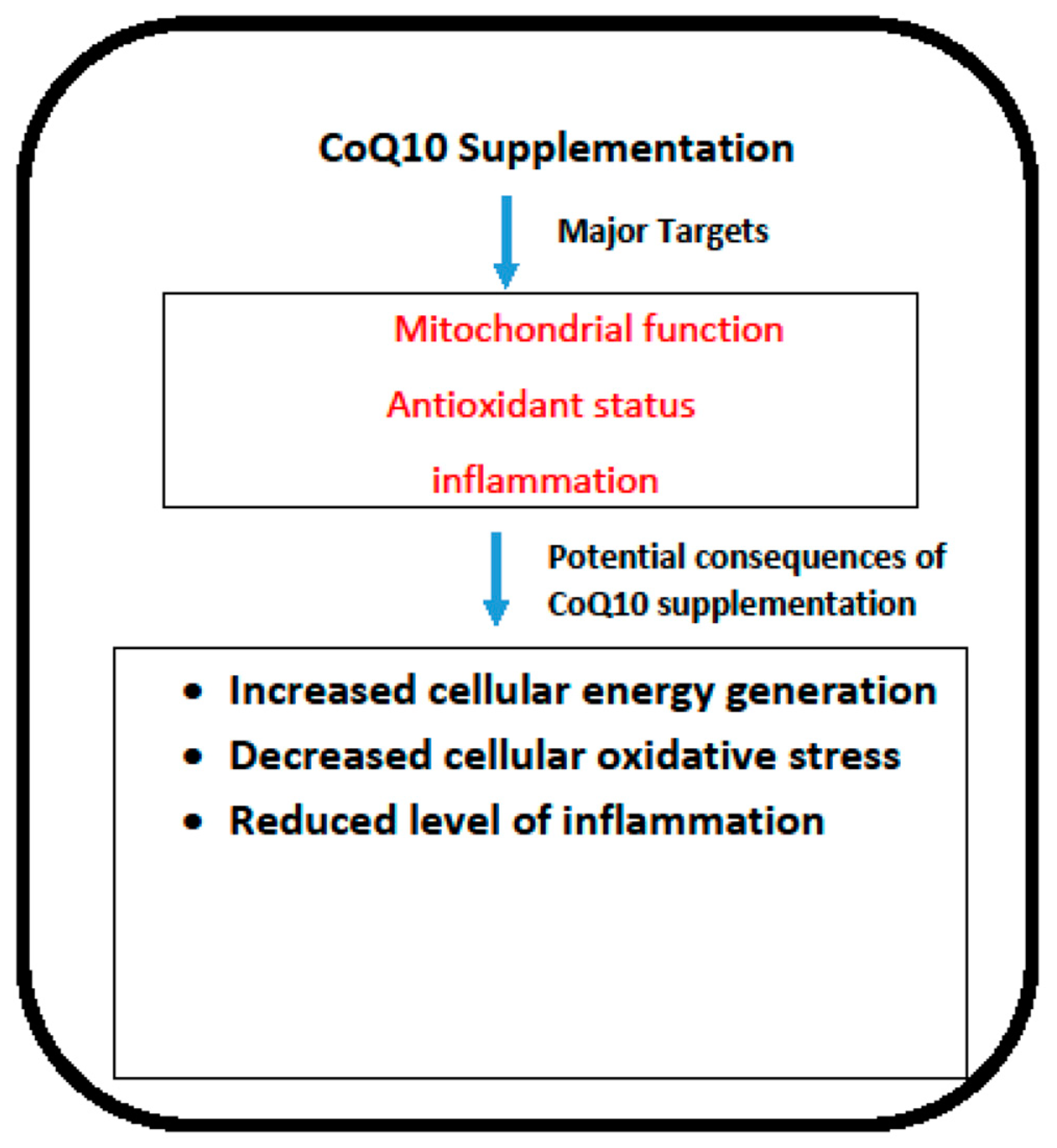
1. Introduction
2. Background to Disorders Covered by This Review

{kind=link}
| Study/Number of Patients | Mutation Type/Presentation | Age at Presentation/Supplementation | Outcome |
|---|---|---|---|
| Scalais et al. [25]/1 | COQ2/myoclonic seizures, cardiomyopathy | 3 weeks/2 months | Died at 5 months after 5–30 mg/kg/day |
| Eroglu et al. [26]/3 | COQ2/ | ||
| patient 1 | proteinuria | 4 days/3 months | Died at 4 months after 30 mg/kg/day |
| patient 2 | neonatal diabetes | 1 day/1 day | Died at 30 months after 30 mg/kg/day |
| patient 3 | proteinuria | 5 days/5 days | Died at 14 months after 30 mg/kg/day |
| Yu et al. [27]/5 | COQ4/ | ||
| patient 1 | metabolic acidosis | 7 days/5 months | Died at 8 months after 40 mg/kg/day |
| patient 2 | metabolic acidosis | 1 day/2 days | Died at 3 days after 100 mg/kg/day |
| patient 3 | cardiac failure | 22 days/22 days | Cardiac function normalised at 32 days; dose not specified |
| patient 4 | metabolic acidosis | 1 day/4 years 5 months | Less responsive to supplementation; dose not specified |
| patient 5 | metabolic acidosis | <1 month/1 year | Died at 13 months; dose not specified |
| Kwong et al. [28]/1 | COQ7/heart and respiratory failure | 4 days/2 months | Died at 1 year after 20–30 mg/kg/day |
| Duncan et al. [29]/1 | COQ9/seizures | 6 h/11.5 months | Died at 2 years after 300 mg/day |
| Study/Number of Patients | Mutation Type/Presentation | Age at Presentation/ Supplementation | Outcome of CoQ10 Supplementation |
|---|---|---|---|
| Bellusci et al. [30]/1 | COQ1 (PDSS2)/ Numerous morbidities | 5 months/5 months | 15–30 mg/kg/day. No effect on disease progression; died at 3 years. |
| Lopez et al. [31]/1 | COQ1 (PDSS2)/ Nephrotic syndrome and encephalopathy | 3 months/3 months | 50 mg/day. No effect on disease progression; died at 8 months |
| Xu et al. [32]/1 | COQ2/ nephrotic syndrome | 11 months/13 months | 30 mg/kg/day improved renal function |
| Starr et al. [33]/1 | COQ2/ nephrotic syndrome | 9 months/10 months | 50 mg/kg/day improved renal function |
| Li et al. [34]/1 | COQ2/proteinuria | 7 months/11 months | 30 mg/kg/day reduced proteinuria |
| Drovandi et al. [35]/32 | COQ2/nephrotic syndrome | 8 months/2.6 years | Up to 60 mg/kg/day improved proteinuria |
| Lu et al. [36]/1 | COQ4/Leigh syndrome | 12 months/12 months | Patient stabilised after 50 mg/kg/day |
| Yu et al. [27]/3 | COQ4/ | ||
| patient 1 | metabolic acidosis, dystonia | 5 months/2 years | no improvement after supplementation (dose not specified), died at 3 years 6 months |
| patient 2 | metabolic acidosis, spasms | 6 months/9 months | improved responsiveness (dose not specified) |
| patient 3 | seizures | 2 months/11 months | Seizure control improved after 30 mg/kg/day |
| Cao et al. [37]/1 | COQ6/nephrotic syndrome | 10 months/10 months | Normal renal function restored after 30 mg/kg/day for 3 months |
| Heeringa et al. [38]/1 | COQ6/nephrotic syndrome | 2 months/2 months | Proteinuria improved after 15 mg/kg/day |
| Ashraf et al. [39]/1 | COQ8B/nephrotic syndrome | <12 months/12 months | Partial remission of nephropathy after 100 mg/day |
| Feng et al. [40]/1 | COQ8B/seizures | 9 months/9 months | Proteinuria showed good response after 15–30 mg/kg/day |
| Olgac et al. [41]/1 | COQ9/seizures | 9 months/10 months | No neurological improvement after 5–50 mg/kg/day |
| Study/Number of Patients | Mutation Type/Presentation | Age at Presentation/Supplementation | Outcome of CoQ10 Supplementation |
|---|---|---|---|
| Diomedi-Camassei et al. [42]/1 | COQ2/nephrotic syndrome | 18 months/18 months | Clinical stabilisation after 30 mg/kg/day |
| Starr et al. [33]/2 | COQ2/ nephrotic syndrome | (1) 10 years/10 years 8 months (2) 2 years/2 years 9 months | (1) No improvement after 50 mg/kg/day (2) Improved renal function after 50 mg/kg/day |
| Salviati et al. [43]/1 | COQ4/neuromuscular symptoms | 3 years/3 years | Significant symptomatic improvement after 30 mg/kg/day |
| Bosch et al. [44]/2 | COQ4/spinocerebellar ataxia and stroke-like episodes | (1) 4 years/13 years (2) 9 years/11 years | No improvement in either patient after 1000 mg/day |
| Caglayan et al. [45]/1 | COQ4/ataxia | 8 years/26 years | Ataxia improved after 2000 mg/day |
| Malicdan et al. [46]/3 | COQ5/ataxia, developmental delay | Early childhood/twenty years | Improved ataxia/cognitive function after 15 mg/kg/day ubiquinol |
| Heeringa et al. [38]/1 | COQ6/nephrotic syndrome | 2.5 years/5.5 years | Proteinuria improved after 15 mg/kg/day |
| Stanczyk et al. [47]/1 | COQ6/nephropathy | 2 years/4 years | Complete remission after 30 mg/kg/day |
| Nam et al. [48]/12 | COQ6/nephropathy, hearing loss | 2.7 years (mean)/5.6 years (mean) | Hearing loss in 50% of patients responded well after 30 mg/kg CoQ10 |
| Mollet et al. [49]/3 | COQ8A/ (1) cerebellar ataxia (2) developmental delay, seizures, ataxia (3) as for patient 2 | (1) 18 months/3 years 8 months (2) 2 years/14 years (3) as for patient 2 | No improvement in any of 3 patients following 10–20 mg/kg/day |
| Blumkin et al. [50]/1 | COQ8A/cerebellar ataxia | 2 years/5 years | Improved motor and cognitive abilities after 20 mg/kg/day |
| Jacobsen et al. [51]/2 | COQ8A/ataxia, cerebellar atrophy | 26–30 months/not specified | Ataxia improved after 20 mg/kg/day |
| Ucella et al. [52]/1 | COQ8A/ataxia, seizures | 8 years 6 months/14 years | Disease progression slowed after 15 mg/kg/day |
| Paprocka et al. [53]/1 | COQ8A/developmental regression | 22 months/22 months | Improved communication after 300 mg/day |
| Atmaca et al. [54]/8 | COQ8B/proteinuria | 16.8 years/18.9 years | Improved proteinuria after 30 mg/kg/day |
| Feng et al. [40]/1 | COQ8B/nephrotic syndrome | 9 years/11 years | No improvement after 30 mg/kg/day |
| Song et al. [55]/20 | COQ8B/nephrotic syndrome | 5–9 years/not specified | Reduced proteinuria in trial group of 5 subjects after 15–30 mg/kg/day |
3. CoQ10 Normal Ranges in Neonates, Infants and Children
4. CoQ10 Supplementation in Neonates
5. CoQ10 Supplementation in Infants
6. CoQ10 Supplementation in Children
7. Safety of CoQ10 Supplementation
8. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hargreaves, I.; Heaton, R.A.; Mantle, D. Disorders of Human Coenzyme Q10 Metabolism: An Overview. Int. J. Mol. Sci. 2020, 21, 6695. [Google Scholar] [CrossRef] [PubMed]
- Mantle, D.; Millichap, L.; Castro-Marrero, J.; Hargreaves, I.P. Primary Coenzyme Q10 Deficiency: An Update. Antioxidants 2023, 12, 1652. [Google Scholar] [CrossRef] [PubMed]
- Mantle, D.; Turton, N.; Hargreaves, I.P. Depletion and Supplementation of Coenzyme Q10 in Secondary Deficiency Disorders. Front. Biosci. (Landmark Ed.) 2022, 27, 322. [Google Scholar] [CrossRef] [PubMed]
- Kalén, A.; Appelkvist, E.L.; Dallner, G. Age-related changes in the lipid compositions of rat and human tissues. Lipids 1989, 24, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Osawa, Y.; Kobayashi, H.; Bo, R.; Mushimoto, Y.; Hasegawa, Y.; Yamaguchi, S.; Taketani, T. Clinical and molecular investigation of 37 Japanese patients with multiple acyl-CoA dehydrogenase deficiency: P.Y507D in ETFDH, a common Japanese variant, causes a mortal phenotype. Mol. Genet. Metab. Rep. 2022, 33, 100940. [Google Scholar] [CrossRef]
- Cornelius, N.; Byron, C.; Hargreaves, I.; Guerra, P.F.; Furdek, A.K.; Land, J.; Radford, W.W.; Frerman, F.; Corydon, T.J.; Gregersen, N.; et al. Secondary coenzyme Q10 deficiency and oxidative stress in cultured fibroblasts from patients with riboflavin responsive multiple Acyl-CoA dehydrogenation deficiency. Hum. Mol. Genet. 2013, 22, 3819–3827. [Google Scholar] [CrossRef]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.G.; Hans, V.H.; Pálmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Béhin, A.; Acquaviva-Bourdain, C.; Souvannanorath, S.; Streichenberger, N.; Attarian, S.; Bassez, G.; Brivet, M.; Fouilhoux, A.; Labarre-Villa, A.; Laquerrière, A.; et al. Multiple acyl-CoA dehydrogenase deficiency (MADD) as a cause of late-onset treatable metabolic disease. Rev. Neurol. 2016, 172, 231–241. [Google Scholar] [CrossRef]
- Liang, W.C.; Ohkuma, A.; Hayashi, Y.K.; López, L.C.; Hirano, M.; Nonaka, I.; Noguchi, S.; Chen, L.H.; Jong, Y.J.; Nishino, I. ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul. Disord. 2009, 19, 212–216. [Google Scholar] [CrossRef]
- Yiu, E.M.; Kornberg, A.J. Duchenne muscular dystrophy. J. Paediatr. Child. Health 2015, 51, 759–764. [Google Scholar] [CrossRef]
- Yorns, W.R., Jr.; Hardison, H.H. Mitochondrial dysfunction in migraine. Semin. Pediatr. Neurol. 2013, 20, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Li, Y.; Lu, S.; Huang, W.; Di, W. Efficacy of CoQ10 as supplementation for migraine: A meta-analysis. Acta Neurol. Scand. 2019, 139, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Parohan, M.; Sarraf, P.; Javanbakht, M.H.; Ranji-Burachaloo, S.; Djalali, M. Effect of coenzyme Q10 supplementation on clinical features of migraine: A systematic review and dose-response meta-analysis of randomized controlled trials. Nutr. Neurosci. 2020, 23, 868–875. [Google Scholar] [CrossRef]
- Smith, A.; Bussmann, N.; Breatnach, C.; Levy, P.; Molloy, E.; Miletin, J.; Curley, A.; McCallion, N.; Franklin, O.; El-Khuffash, A. Serial Assessment of Cardiac Function and Pulmonary Hemodynamics in Infants with Down Syndrome. J. Am. Soc. Echocardiogr. 2022, 35, 1176–1183.e5. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Castello, G. Oxidative stress and mitochondrial dysfunction in Down syndrome. Adv. Exp. Med. Biol. 2012, 724, 291–299. [Google Scholar] [CrossRef]
- Floet, A.M.; Scheiner, C.; Grossman, L. Attention deficit hyperactivity disorder. Pediatr. Rev. 2010, 31, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Zwaigenbaum, L.; Penner, M. Autism spectrum disorder: Advances in diagnosis and evaluation. BMJ 2018, 361, k1674. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef]
- Thorsen, M. Oxidative stress, metabolic and mitochondrial abnormalities associated with autism spectrum disorder. Prog. Mol. Biol. Transl. Sci. 2020, 173, 331–354. [Google Scholar] [CrossRef]
- Öğütlü, H.; Kaşak, M.; Tutku Tabur, S. Mitochondrial Dysfunction in Attention Deficit Hyperactivity Disorder. Eurasian J. Med. 2022, 54 (Suppl. S1), 187–195. [Google Scholar] [CrossRef]
- Anderson, H.N.; Cetta, F.; Driscoll, D.J.; Olson, T.M.; Ackerman, M.J.; Johnson, J.N. Idiopathic Restrictive Cardiomyopathy in Children and Young Adults. Am. J. Cardiol. 2018, 121, 1266–1270. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.; Giunti, P. Friedreich’s ataxia: Clinical features, pathogenesis and management. Br. Med. Bull. 2017, 124, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Lodi, R.; Tonon, C.; D’Agata, V.; Sapienza, M.; Scapagnini, G.; Mangiameli, A.; Pennisi, G.; Stella, A.M.; Butterfield, D.A. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J. Neurol. Sci. 2005, 233, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.G.; Napierala, M.; Pébay, A.; Dottori, M.; Lim, S.Y. Cellular pathophysiology of Friedreich’s ataxia cardiomyopathy. Int. J. Cardiol. 2022, 346, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Scalais, E.; Chafai, R.; Van Coster, R.; Bindl, L.; Nuttin, C.; Panagiotaraki, C.; Seneca, S.; Lissens, W.; Ribes, A.; Geers, C.; et al. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2). Eur. J. Paediatr. Neurol. 2013, 17, 625–630. [Google Scholar] [CrossRef]
- Eroglu, F.K.; Ozaltin, F.; Gönç, N.; Nalçacıoğlu, H.; Özçakar, Z.B.; Yalnızoğlu, D.; Güçer, Ş.; Orhan, D.; Eminoğlu, F.T.; Göçmen, R.; et al. Response to Early Coenzyme Q10 Supplementation Is not Sustained in CoQ10 Deficiency Caused by CoQ2 Mutation. Pediatr. Neurol. 2018, 88, 71–74. [Google Scholar] [CrossRef]
- Yu, M.H.; Tsang, M.H.; Lai, S.; Ho, M.S.; Tse, D.M.L.; Willis, B.; Kwong, A.K.; Chou, Y.Y.; Lin, S.P.; Quinzii, C.M.; et al. Primary coenzyme Q10 deficiency-7: Expanded phenotypic spectrum and a founder mutation in southern Chinese. NPJ Genom. Med. 2019, 4, 18. [Google Scholar] [CrossRef]
- Kwong, A.K.; Chiu, A.T.; Tsang, M.H.; Lun, K.S.; Rodenburg, R.J.T.; Smeitink, J.; Chung, B.H.; Fung, C.W. A fatal case of COQ7-associated primary coenzyme Q10 deficiency. JIMD Rep. 2019, 47, 23–29. [Google Scholar] [CrossRef]
- Duncan, A.J.; Bitner-Glindzicz, M.; Meunier, B.; Costello, H.; Hargreaves, I.P.; López, L.C.; Hirano, M.; Quinzii, C.M.; Sadowski, M.I.; Hardy, J.; et al. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: A potentially treatable form of mitochondrial disease. Am. J. Hum. Genet. 2009, 84, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Bellusci, M.; García-Silva, M.T.; Martínez de Aragón, A.; Martín, M.A. Distal phalangeal erythema in an infant with biallelic PDSS1 mutations: Expanding the phenotype of primary Coenzyme Q10 deficiency. JIMD Rep. 2021, 62, 3–5. [Google Scholar] [CrossRef]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; Dimauro, S.; Hirano, M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef]
- Xu, K.; Mao, X.Y.; Yao, Y.; Cheng, H.; Zhang, X.J. Clinical analysis of one infantile nephrotic syndrome caused by COQ2 gene mutation and literature review. Zhonghua Er Ke Za Zhi 2018, 56, 662–666. [Google Scholar] [CrossRef]
- Starr, M.C.; Chang, I.J.; Finn, L.S.; Sun, A.; Larson, A.A.; Goebel, J.; Hanevold, C.; Thies, J.; Van Hove, J.L.K.; Hingorani, S.R.; et al. COQ2 nephropathy: A treatable cause of nephrotic syndrome in children. Pediatr. Nephrol. 2018, 33, 1257–1261. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yue, Z.; Lin, H.; Wang, H.; Chen, H.; Sun, L. COQ2 mutation associated isolated nephropathy in two siblings from a Chinese pedigree. Ren. Fail. 2021, 43, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Drovandi, S.; Lipska-Ziętkiewicz, B.S.; Ozaltin, F.; Emma, F.; Gulhan, B.; Boyer, O.; Trautmann, A.; Xu, H.; Shen, Q.; Rao, J.; et al. Oral Coenzyme Q10 supplementation leads to better preservation of kidney function in steroid-resistant nephrotic syndrome due to primary Coenzyme Q10 deficiency. Kidney Int. 2022, 102, 604–612. [Google Scholar] [CrossRef]
- Lu, M.; Zhou, Y.; Wang, Z.; Xia, Z.; Ren, J.; Guo, Q. Clinical phenotype, in silico and biomedical analyses, and intervention for an East Asian population-specific c.370G>A (p.G124S) COQ4 mutation in a Chinese family with CoQ10 deficiency-associated Leigh syndrome. J. Hum. Genet. 2019, 64, 297–304. [Google Scholar] [CrossRef]
- Cao, Q.; Li, G.M.; Xu, H.; Shen, Q.; Sun, L.; Fang, X.Y.; Liu, H.M.; Guo, W.; Zhai, Y.H.; Wu, B.B. Coenzyme Q(10) treatment for one child with COQ6 gene mutation induced nephrotic syndrome and literature review. Zhonghua Er Ke Za Zhi 2017, 55, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef]
- Ashraf, S.; Gee, H.Y.; Woerner, S.; Xie, L.X.; Vega-Warner, V.; Lovric, S.; Fang, H.; Song, X.; Cattran, D.C.; Avila-Casado, C.; et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Investig. 2013, 123, 5179–5189. [Google Scholar] [CrossRef]
- Feng, C.; Wang, Q.; Wang, J.; Liu, F.; Shen, H.; Fu, H.; Mao, J. Coenzyme Q10 supplementation therapy for 2 children with proteinuria renal disease and ADCK4 mutation: Case reports and literature review. Medicine 2017, 96, e8880. [Google Scholar] [CrossRef]
- Olgac, A.; Öztoprak, Ü.; Kasapkara, Ç.S.; Kılıç, M.; Yüksel, D.; Derinkuyu, E.B.; Taşçı Yıldız, Y.; Ceylaner, S.; Ezgu, F.S. A rare case of primary coenzyme Q10 deficiency due to COQ9 mutation. J. Pediatr. Endocrinol. Metab. 2020, 33, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Diomedi-Camassei, F.; Di Giandomenico, S.; Santorelli, F.M.; Caridi, G.; Piemonte, F.; Montini, G.; Ghiggeri, G.M.; Murer, L.; Barisoni, L.; Pastore, A.; et al. COQ2 nephropathy: A newly described inherited mitochondriopathy with primary renal involvement. J. Am. Soc. Nephrol. 2007, 18, 2773–2780. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L.; Trevisson, E.; Rodriguez Hernandez, M.A.; Casarin, A.; Pertegato, V.; Doimo, M.; Cassina, M.; Agosto, C.; Desbats, M.A.; Sartori, G.; et al. Haploinsufficiency of COQ4 causes coenzyme Q10 deficiency. J. Med. Genet. 2012, 49, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.M.; Kamsteeg, E.J.; Rodenburg, R.J.; van Deutekom, A.W.; Buis, D.R.; Engelen, M.; Cobben, J.M. Coenzyme Q10 deficiency due to a COQ4 gene defect causes childhood-onset spinocerebellar ataxia and stroke-like episodes. Mol. Genet. Metab. Rep. 2018, 17, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Caglayan, A.O.; Gumus, H.; Sandford, E.; Kubisiak, T.L.; Ma, Q.; Ozel, A.B.; Per, H.; Li, J.Z.; Shakkottai, V.G.; Burmeister, M. COQ4 Mutation Leads to Childhood-Onset Ataxia Improved by CoQ10 Administration. Cerebellum 2019, 18, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Malicdan, M.C.V.; Vilboux, T.; Ben-Zeev, B.; Guo, J.; Eliyahu, A.; Pode-Shakked, B.; Dori, A.; Kakani, S.; Chandrasekharappa, S.C.; Ferreira, C.R.; et al. A novel inborn error of the coenzyme Q10 biosynthesis pathway: Cerebellar ataxia and static encephalomyopathy due to COQ5 C-methyltransferase deficiency. Hum. Mutat. 2018, 39, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Stańczyk, M.; Bałasz-Chmielewska, I.; Lipska-Ziętkiewicz, B.; Tkaczyk, M. CoQ10-related sustained remission of proteinuria in a child with COQ6 glomerulopathy-a case report. Pediatr. Nephrol. 2018, 33, 2383–2387. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.W.; Park, S.S.; Lee, S.M.; Suh, M.W.; Park, M.K.; Song, J.J.; Choi, B.Y.; Lee, J.H.; Oh, S.H.; Moon, K.C.; et al. Effects of CoQ10 Replacement Therapy on the Audiological Characteristics of Pediatric Patients with COQ6 Variants. Biomed. Res. Int. 2022, 2022, 5250254. [Google Scholar] [CrossRef] [PubMed]
- Mollet, J.; Delahodde, A.; Serre, V.; Chretien, D.; Schlemmer, D.; Lombes, A.; Boddaert, N.; Desguerre, I.; de Lonlay, P.; de Baulny, H.O.; et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am. J. Hum. Genet. 2008, 82, 623–630. [Google Scholar] [CrossRef]
- Blumkin, L.; Leshinsky-Silver, E.; Zerem, A.; Yosovich, K.; Lerman-Sagie, T.; Lev, D. Heterozygous Mutations in the ADCK3 Gene in Siblings with Cerebellar Atrophy and Extreme Phenotypic Variability. JIMD Rep. 2014, 12, 103–107. [Google Scholar] [CrossRef]
- Jacobsen, J.C.; Whitford, W.; Swan, B.; Taylor, J.; Love, D.R.; Hill, R.; Molyneux, S.; George, P.M.; Mackay, R.; Robertson, S.P.; et al. Compound Heterozygous Inheritance of Mutations in Coenzyme Q8A Results in Autosomal Recessive Cerebellar Ataxia and Coenzyme Q10 Deficiency in a Female Sib-Pair. JIMD Rep. 2018, 42, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Uccella, S.; Pisciotta, L.; Severino, M.; Bertini, E.; Giacomini, T.; Zanni, G.; Prato, G.; De Grandis, E.; Nobili, L.; Mancardi, M.M. Photoparoxysmal response in ADCK3 autosomal recessive ataxia: A case report and literature review. Epileptic. Disord. 2021, 23, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Paprocka, J.; Nowak, M.; Chuchra, P.; Śmigiel, R. COQ8A-Ataxia as a Manifestation of Primary Coenzyme Q Deficiency. Metabolites 2022, 12, 955. [Google Scholar] [CrossRef] [PubMed]
- Atmaca, M.; Gülhan, B.; Atayar, E.; Bayazıt, A.K.; Candan, C.; Arıcı, M.; Topaloğlu, R.; Özaltın, F. Long-term follow-up results of patients with ADCK4 mutations who have been diagnosed in the asymptomatic period: Effects of early initiation of CoQ10 supplementation. Turk. J. Pediatr. 2019, 61, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Fang, X.; Tang, X.; Cao, Q.; Zhai, Y.; Chen, J.; Liu, J.; Zhang, Z.; Xiang, T.; Qian, Y.; et al. COQ8B nephropathy: Early detection and optimal treatment. Mol. Genet. Genom. Med. 2020, 8, e1360. [Google Scholar] [CrossRef] [PubMed]
- Finckh, B.; Kontush, A.; Commentz, J.; Hübner, C.; Burdelski, M.; Kohlschütter, A. Monitoring of ubiquinol-10, ubiquinone-10, carotenoids, and tocopherols in neonatal plasma microsamples using high-performance liquid chromatography with coulometric electrochemical detection. Anal. Biochem. 1995, 232, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Niklowitz, P.; Menke, T.; Andler, W.; Okun, J.G. Simultaneous analysis of coenzyme Q10 in plasma, erythrocytes and platelets: Comparison of the antioxidant level in blood cells and their environment in healthy children and after oral supplementation in adults. Clin. Chim. Acta 2004, 342, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Artuch, R.; Moreno, J.; Quintana, M.; Puig, R.M.; Vilaseca, M.A. Serum ubiquinone-10 in a pediatric population. Clin. Chem. 1998, 44, 2378–2379. [Google Scholar] [CrossRef] [PubMed]
- Miles, M.V.; Horn, P.S.; Tang, P.H.; Morrison, J.A.; Miles, L.; DeGrauw, T.; Pesce, A.J. Age-related changes in plasma coenzyme Q10 concentrations and redox state in apparently healthy children and adults. Clin. Chim. Acta 2004, 347, 139–144. [Google Scholar] [CrossRef]
- Wittenstein, B.; Klein, M.; Finckh, B.; Ullrich, K.; Kohlschütter, A. Plasma antioxidants in pediatric patients with glycogen storage disease, diabetes mellitus, and hypercholesterolemia. Free Radic. Biol. Med. 2002, 33, 103–110. [Google Scholar] [CrossRef]
- Menke, T.; Niklowitz, P.; de Sousa, G.; Reinehr, T.; Andler, W. Comparison of coenzyme Q10 plasma levels in obese and normal weight children. Clin. Chim. Acta 2004, 349, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Boetticher, D.; Leichsenring, M. Ubiquinone-10 plasma concentrations in healthy European children. Redox Rep. 1995, 1, 97–98. [Google Scholar] [CrossRef] [PubMed]
- Miles, M.V.; Miles, L.; Tang, P.H.; Horn, P.S.; Steele, P.E.; DeGrauw, A.J.; Wong, B.L.; Bove, K.E. Systematic evaluation of muscle coenzyme Q10 content in children with mitochondrial respiratory chain enzyme deficiencies. Mitochondrion 2008, 8, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Niklowitz, P.; Scherer, J.; Döring, F.; Paulussen, M.; Menke, T. Oxidized proportion of muscle coenzyme Q10 increases with age in healthy children. Pediatr. Res. 2015, 78, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Di Giovamberardino, G.; Petrillo, S.; Boenzi, S.; Bertini, E.; Dionisi-Vici, C.; Piemonte, F. Pediatric reference intervals for muscle coenzyme Q(10). Biomarkers 2012, 17, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Stanescu, S.; Belanger-Quintana, A.; Alcalde Martin, C.; Pérez-Cerdá Silvestre, C.; Merinero Cortés, B.; Gonzalez Pérez, B.; Fernández García-Abril, C.; Arrieta Blanco, F.; Palacios Valverde, E.; Martínez-Pardo Casanova, M. Beneficial Effect of N-Carbamylglutamate in a Neonatal Form of Multiple Acyl-CoA Dehydrogenase Deficiency. Case Rep. Pediatr. 2020, 2020, 1370293. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Zhang, X.; Chen, T.; Lin, J. Case report: A novel c.1842_1845dup mutation of ETFDH in two Chinese siblings with multiple acyl-CoA dehydrogenase deficiency. Front. Pediatr. 2023, 10, 1038440. [Google Scholar] [CrossRef] [PubMed]
- Kadoya, T.; Sakakibara, A.; Kitayama, K.; Yamada, Y.; Higuchi, S.; Kawakita, R.; Kawasaki, Y.; Fujino, M.; Murakami, Y.; Shimura, M.; et al. Successful treatment of infantile-onset ACAD9-related cardiomyopathy with a combination of sodium pyruvate, beta-blocker, and coenzyme Q10. J. Pediatr. Endocrinol. Metab. 2019, 32, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Saral, N.Y.; Aksungar, F.B.; Aktuglu-Zeybek, C.; Coskun, J.; Demirelce, O.; Serteser, M. Glutaric acidemia type II patient with thalassemia minor and novel electron transfer flavoprotein-A gene mutations: A case report and review of literature. World J. Clin. Cases 2018, 6, 786–790. [Google Scholar] [CrossRef]
- Tummolo, A.; Leone, P.; Tolomeo, M.; Solito, R.; Mattiuzzo, M.; Lepri, F.R.; Lorè, T.; Cardinali, R.; De Giovanni, D.; Simonetti, S.; et al. Combined isobutyryl-CoA and multiple acyl-CoA dehydrogenase deficiency in a boy with altered riboflavin homeostasis. JIMD Rep. 2022, 63, 276–291. [Google Scholar] [CrossRef]
- Spurney, C.F.; Rocha, C.T.; Henricson, E.; Florence, J.; Mayhew, J.; Gorni, K.; Pasquali, L.; Pestronk, A.; Martin, G.R.; Hu, F.; et al. CINRG pilot trial of coenzyme Q10 in steroid-treated Duchenne muscular dystrophy. Muscle Nerve 2011, 44, 174–178. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Meier, T.; Voit, T.; Schara, U.; Straathof, C.S.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.M.; Finkel, R.S.; Goemans, N.; et al. Idebenone reduces respiratory complications in patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 2016, 26, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Buyse, G.M.; Voit, T.; Schara, U.; Straathof, C.S.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.M.; Finkel, R.S.; Goemans, N.; Rummey, C.; et al. Treatment effect of idebenone on inspiratory function in patients with Duchenne muscular dystrophy. Pediatr. Pulmonol. 2017, 52, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Servais, L.; Straathof, C.S.M.; Schara, U.; Klein, A.; Leinonen, M.; Hasham, S.; Meier, T.; De Waele, L.; Gordish-Dressman, H.; McDonald, C.M.; et al. Long-term data with idebenone on respiratory function outcomes in patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 2020, 30, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Buyse, G.M.; Van der Mieren, G.; Erb, M.; D’hooge, J.; Herijgers, P.; Verbeken, E.; Jara, A.; Van Den Bergh, A.; Mertens, L.; Courdier-Fruh, I.; et al. Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: Cardiac protection and improved exercise performance. Eur. Heart J. 2009, 30, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Salehi, F.; Zeinaloo, A.; Riasi, H.R.; Shamloo, A.S. Effectiveness of Coenzyme Q10 on echocardiographic parameters of patients with Duchenne muscular dystrophy. Electron. Physician 2017, 9, 3896–3904. [Google Scholar] [CrossRef] [PubMed]
- Folkers, K.; Wolaniuk, J.; Simonsen, R.; Morishita, M.; Vadhanavikit, S. Biochemical rationale and the cardiac response of patients with muscle disease to therapy with coenzyme Q10. Proc. Natl. Acad. Sci. USA 1985, 82, 4513–4516. [Google Scholar] [CrossRef] [PubMed]
- Hershey, A.D.; Powers, S.W.; Vockell, A.L.; Lecates, S.L.; Ellinor, P.L.; Segers, A.; Burdine, D.; Manning, P.; Kabbouche, M.A. Coenzyme Q10 deficiency and response to supplementation in pediatric and adolescent migraine. Headache 2007, 47, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Yaghini, O.; Hoseini, N.; Ghazavi, M.R.; Mansouri, V.; Nasiri, J.; Moosavian, T.; Salehi, M.M. A Comparative Study on the Efficacy of Coenzyme Q10 and Amitriptyline in the Prophylactic Treatment of Migraine Headaches in Children: A Randomized Controlled Trial. Adv. Biomed. Res. 2022, 11, 43. [Google Scholar] [CrossRef]
- Slater, S.K.; Nelson, T.D.; Kabbouche, M.A.; LeCates, S.L.; Horn, P.; Segers, A.; Manning, P.; Powers, S.W.; Hershey, A.D. A randomized, double-blinded, placebo-controlled, crossover, add-on study of CoEnzyme Q10 in the prevention of pediatric and adolescent migraine. Cephalalgia 2011, 31, 897–905. [Google Scholar] [CrossRef]
- Zaki, M.E.; El-Bassyouni, H.T.; Tosson, A.M.; Youness, E.; Hussein, J. Coenzyme Q10 and pro-inflammatory markers in children with Down syndrome: Clinical and biochemical aspects. J. Pediatr. 2017, 93, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Tiano, L.; Carnevali, P.; Padella, L.; Santoro, L.; Principi, F.; Brugè, F.; Carle, F.; Gesuita, R.; Gabrielli, O.; Littarru, G.P. Effect of Coenzyme Q10 in mitigating oxidative DNA damage in Down syndrome patients, a double blind randomized controlled trial. Neurobiol. Aging 2011, 32, 2103–2105. [Google Scholar] [CrossRef]
- Larsen, E.L.; Padella, L.; Bergholdt, H.K.M.; Henriksen, T.; Santoro, L.; Gabrielli, O.; Poulsen, H.E.; Littarru, G.P.; Orlando, P.; Tiano, L. The effect of long-term treatment with coenzyme Q10 on nucleic acid modifications by oxidation in children with Down syndrome. Neurobiol. Aging 2018, 67, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Karagöz, Y.S.; Doğan, Ö.; Elgün, S.; Öztop, D.B.; Kılıç, B.G. Ubiquinone Levels as a Marker of Antioxidant System in Children with Attention Deficit Hyperactivity Disorder. J. Mol. Neurosci. 2021, 71, 2173–2178. [Google Scholar] [CrossRef] [PubMed]
- Gamal, F.; El Agami, O.; Salamah, A. Coenzyme Q10 in the Treatment of Attention Deficit Hyperactivity Disorder in Children: A Randomized Controlled Trial. CNS Neurol. Disord. Drug Targets 2022, 21, 717–723. [Google Scholar] [CrossRef]
- Mousavinejad, E.; Ghaffari, M.A.; Riahi, F.; Hajmohammadi, M.; Tiznobeyk, Z.; Mousavinejad, M. Coenzyme Q10 supplementation reduces oxidative stress and decreases antioxidant enzyme activity in children with autism spectrum disorders. Psychiatry Res. 2018, 265, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Soongswang, J.; Sangtawesin, C.; Durongpisitkul, K.; Laohaprasitiporn, D.; Nana, A.; Punlee, K.; Kangkagate, C. The effect of coenzyme Q10 on idiopathic chronic dilated cardiomyopathy in children. Pediatr. Cardiol. 2005, 26, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.L.; Chang, P.S.; Lin, Y.C.; Lin, P.T. A Pilot Clinical Study of Liquid Ubiquinol Supplementation on Cardiac Function in Pediatric Dilated Cardiomyopathy. Nutrients 2018, 10, 1697. [Google Scholar] [CrossRef] [PubMed]
- Hausse, A.O.; Aggoun, Y.; Bonnet, D.; Sidi, D.; Munnich, A.; Rötig, A.; Rustin, P. Idebenone and reduced cardiac hypertrophy in Friedreich’s ataxia. Heart 2002, 87, 346–349. [Google Scholar] [CrossRef]
- Paredes-Fuentes, A.J.; Cesar, S.; Montero, R.; Latre, C.; Genovès, J.; Martorell, L.; Cuadras, D.; Colom, H.; Pineda, M.; Del Mar O’Callaghan, M.; et al. Plasma idebenone monitoring in Friedreich’s ataxia patients during a long-term follow-up. Biomed. Pharmacother. 2021, 143, 112143. [Google Scholar] [CrossRef]
- Lagedrost, S.J.; Sutton, M.S.; Cohen, M.S.; Satou, G.M.; Kaufman, B.D.; Perlman, S.L.; Rummey, C.; Meier, T.; Lynch, D.R. Idebenone in Friedreich ataxia cardiomyopathy-results from a 6-month phase III study (IONIA). Am. Heart J. 2011, 161, 639–645.e1. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Arpa, J.; Montero, R.; Aracil, A.; Domínguez, F.; Galván, M.; Mas, A.; Martorell, L.; Sierra, C.; Brandi, N.; et al. Idebenone treatment in paediatric and adult patients with Friedreich ataxia: Long-term follow-up. Eur. J. Paediatr. Neurol. 2008, 12, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R.; Perlman, S.L.; Meier, T. A phase 3, double-blind, placebo-controlled trial of idebenone in friedreich ataxia. Arch. Neurol. 2010, 67, 941–947. [Google Scholar] [CrossRef]
- Drinkard, B.E.; Keyser, R.E.; Paul, S.M.; Arena, R.; Plehn, J.F.; Yanovski, J.A.; Di Prospero, N.A. Exercise capacity and idebenone intervention in children and adolescents with Friedreich ataxia. Arch. Phys. Med. Rehabil. 2010, 91, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Serag, H.; El Wakeel, L.; Adly, A. Coenzyme Q10 administration has no effect on sICAM-1 and metabolic parameters of pediatrics with type 1 diabetes mellitus. Int. J. Vitam. Nutr. Res. 2021, 91, 315–324. [Google Scholar] [CrossRef] [PubMed]
- López, L.C.; Quinzii, C.M.; Area, E.; Naini, A.; Rahman, S.; Schuelke, M.; Salviati, L.; Dimauro, S.; Hirano, M. Treatment of CoQ(10) deficient fibroblasts with ubiquinone, CoQ analogs, and vitamin C: Time- and compound-dependent effects. PLoS ONE 2010, 5, e11897. [Google Scholar] [CrossRef] [PubMed]
- Duberley, K.E.; Heales, S.J.; Abramov, A.Y.; Chalasani, A.; Land, J.M.; Rahman, S.; Hargreaves, I.P. Effect of Coenzyme Q10 supplementation on mitochondrial electron transport chain activity and mitochondrial oxidative stress in Coenzyme Q10 deficient human neuronal cells. Int. J. Biochem. Cell Biol. 2014, 50, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Mantle, D.; Dybring, A. Bioavailability of Coenzyme Q10: An Overview of the Absorption Process and Subsequent Metabolism. Antioxidants 2020, 9, 386. [Google Scholar] [CrossRef]
- Mantle, D.; Lopez-Lluch, G.; Hargreaves, I.P. Coenzyme Q10 Metabolism: A Review of Unresolved Issues. Int. J. Mol. Sci. 2023, 24, 2585. [Google Scholar] [CrossRef]
- Hidaka, T.; Fujii, K.; Funahashi, I.; Fukutomi, N.; Hosoe, K. Safety assessment of coenzyme Q10 (CoQ10). Biofactors 2008, 32, 199–208. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mantle, D.; Hargreaves, I.P. Efficacy and Safety of Coenzyme Q10 Supplementation in Neonates, Infants and Children: An Overview. Antioxidants 2024, 13, 530. https://doi.org/10.3390/antiox13050530
Mantle D, Hargreaves IP. Efficacy and Safety of Coenzyme Q10 Supplementation in Neonates, Infants and Children: An Overview. Antioxidants. 2024; 13(5):530. https://doi.org/10.3390/antiox13050530
Chicago/Turabian StyleMantle, David, and Iain Parry Hargreaves. 2024. "Efficacy and Safety of Coenzyme Q10 Supplementation in Neonates, Infants and Children: An Overview" Antioxidants 13, no. 5: 530. https://doi.org/10.3390/antiox13050530
APA StyleMantle, D., & Hargreaves, I. P. (2024). Efficacy and Safety of Coenzyme Q10 Supplementation in Neonates, Infants and Children: An Overview. Antioxidants, 13(5), 530. https://doi.org/10.3390/antiox13050530