Abstract
Diabetic retinopathy (DR) is the leading cause of visual impairment and blindness among the working-age population. Microglia, resident immune cells in the retina, are recognized as crucial drivers in the DR process. Microglia activation is a tightly regulated immunometabolic process. In the early stages of DR, the M1 phenotype commonly shifts from oxidative phosphorylation to aerobic glycolysis for energy production. Emerging evidence suggests that microglia in DR not only engage specific metabolic pathways but also rearrange their oxidation-reduction (redox) system. This redox adaptation supports metabolic reprogramming and offers potential therapeutic strategies using antioxidants. Here, we provide an overview of recent insights into the involvement of reactive oxygen species and the distinct roles played by key cellular antioxidant pathways, including the NADPH oxidase 2 system, which promotes glycolysis via enhanced glucose transporter 4 translocation to the cell membrane through the AKT/mTOR pathway, as well as the involvement of the thioredoxin and nuclear factor E2-related factor 2 antioxidant systems, which maintain microglia in an anti-inflammatory state. Therefore, we highlight the potential for targeting the modulation of microglial redox metabolism to offer new concepts for DR treatment.
1. Introduction
Diabetic retinopathy (DR) is a common secondary microvascular and neurodegenerative complication of diabetes mellitus, occurring in approximately 30% to 40% of diabetic patients [1,2,3]. As is estimated by 2030, the number of patients influenced by DR will reach 191 million [4]. DR is widely recognized as the leading cause of diabetes-related visual impairment among the working-age population all over the world [5,6]. If not prevented and controlled effectively, it will bring a great burden to society and the economy.
Recently, the administration of intravitreal injections of anti-vascular growth factor (VEGF) agents has demonstrated efficacy in mitigating retinal neovascularization and subsiding diabetic macular edema, representing significant advancements in therapeutic intervention [7]. However, anti-VEGF therapy is only effective in a fraction of patients with DR, requiring regular intravitreal injections, and its effects may wear off after a few years [8,9]. Therefore, as the incidence of diabetes continues to rise globally, we need to develop new treatments for this disease to prevent the early progression of DR. Nevertheless, the pathogenic mechanisms of DR have not been clarified yet, which hinders the development of new, effective treatments.
DR was originally described as a prototypical microvascular disease, manifesting vascular alterations such as microaneurysms in its advanced stage [10]. However, it is increasingly recognized that neuroinflammation and dysregulation of the immune system contribute significantly to the pathogenesis of DR at an early stage [11,12,13]. Microglia, the primary mediator of the inflammatory response, are present as the effector cells of innate immunity in the retina [14]. They are highly plastic and perform a diverse range of specialist functions in DR, including phagocytosis and removing toxic aggregates that contribute to inflammation and eventually neurodegeneration in DR [15]. Therefore, exploration of the immune regulatory mechanisms of microglia in the early stage of DR is expected to offer innovative insights for the development of preventive strategies against the onset of early-stage DR.
It is now recognized that microglia can be divided into two main phenotypes [16,17]. The classical, pro-inflammatory M1 phenotype is characterized by elevated production of reactive oxygen species (ROS) and cytokines such as interleukin (IL)-1β, IL-6, IL-33, and tumor necrosis factor (TNF)-α, while the anti-inflammatory M2 phenotype mitigates neuroinflammation by secreting IL-4, IL-10, and transforming growth factor (TGF)-β, promoting debris clearance, oxidative stress resolution, and tissue remodeling [18,19,20,21]. Recently, the field of immunometabolism has attracted considerable attention in the investigation of microglial polarization regulation. Immunometabolism is defined as the intracellular metabolic pathway that determines the fate and function of immune cells [22]. It describes changes in the interactions between immune and metabolic pathways during stress, which is a complex, dynamic process involving many regulators or signals [23,24]. It has been discovered that following activation, immune cells, especially microglia, reconfigure their redox systems, leading to the reprogramming of metabolism [25,26,27]. A great number of reports have shown that oxidative stress in microglia plays a crucial role in the pathogenesis of DR [28,29]. Neuroinflammation and excessive oxidative stress work together to cause damage to retinal blood vessels and neurons [30], eventually leading to neuronal degeneration, which is an early manifestation of DR [31]. However, the specific mechanism underlying the metabolism and redox regulation of microglia in DR remains to be further elucidated.
In this review, we initially introduce the concept of microglia polarization, followed by an analysis of the metabolic regulation of microglia polarization through immunometabolism in DR. Subsequently, we provide comprehensive roles played by redox systems (including the NADPH oxidase (NOX) system, the thioredoxin (TRX) system, and the nuclear factor E2-related factor 2 (NRF2) pathway) in the immunometabolism process associated with DR. Finally, we summarize the impact of redox signals on the metabolic status of microglia, along with potential therapeutic targets based on the redox regulation of immunometabolism, with the intention of offering innovative therapeutic insights and guidance for the clinical management of DR.
2. Immunometabolism in Microglia Polarization in DR
2.1. Phenotype of Microglia
Microglia can respond to endogenous stimuli after infection or injury and show pathogenic or protective effects. Conventionally, microglia can be divided into two distinct phenotypes: classically activated microglia, also known as the pro-inflammatory phenotype (M1 type), and alternately activated microglia, also known as the anti-inflammatory phenotype (M2 type). Different phenotypes of microglia exhibit completely distinct morphology, surface markers, secretory products, and functions.
The polarization of microglia towards the M1 phenotype is known as the classical activation pathway. Damage-induced cell debris, lipopolysaccharides (LPSs) derived from bacteria, and pro-inflammatory cytokines such as IL-1β and TNF-α can activate microglia and promote their polarization toward the M1 phenotype [32,33,34]. At this point, they begin to express unique surface markers such as cluster of differentiation (CD)14, CD16, CD32, CD80, and CD86. Upon activation, M1 microglia take on amoeboid morphology and acquire high phagocytosis and motility for killing foreign pathogens [17,35]. In the meantime, activated microglia, like peripheral macrophages, release various inflammatory factors (TNF-α, IL-1β, IL-6, IL-12, and IL-23), chemokines, redox molecules (ROS, phagocytosis oxidase, inducible nitric oxide synthase (iNOS)), cell adhesion molecules (ICAM-1), VEGF, and MHC-II [36]. Under physiological circumstances, these pro-inflammatory mediators help remove toxic aggregated proteins and cellular debris from the retina. However, in the case of pathological conditions such as DR, overactivated M1-type microglia release large amounts of neurotoxic inflammatory factors (TNF-α, IL-1α, IL-1β, IL-6, NO, hydrogen peroxide, superoxide anion, and glutamate), and these pro-inflammatory factors induce leukocyte infiltration into the retinal vasculature, leading to vascular inflammation and disruption of the blood retinal barrier (BRB) [37,38]. Early BRB breakdown is a typical event in the early stage of DR, leading to increased exposure of microglia to extracellular signals. Thereby initiating a pernicious cycle of inflammation.
The polarization of microglia towards the M2 phenotype is known as the alternative activation pathway. M2 microglia are activated by anti-inflammatory cytokines such as IL-4, IL-10, and IL-13 [39,40,41]. Their cytosol is thin, their protrusions are branched, and the cell surface expresses CD163, CD206, and arginase-1 (Arg-1) [42]. In contrast to pro-inflammatory microglia, M2-type microglia can release anti-inflammatory cytokines (IL-4, IL-13, IL-10, and TGF-β), growth factors (insulin-like growth factor 1), and neurotrophic mediators (brain-derived neurotrophic factor) to help reduce inflammation and promote neuronal survival [43,44].
2.2. The Activation and Polarization of Microglia in DR
The activation of microglia is stimulated by extracellular signals, among which glucose and its derivative metabolites figure prominently [45,46,47,48]. Chronic hyperglycemia activates complex pathophysiologic processes in DR, comprising ischemia, hypoxia, dyslipidemia, and advanced glycation end products (AGEs). It is recognized that sustained exposure to hyperglycemia significantly enhances the non-enzymatic glycosylation of large molecules such as proteins and lipids, ultimately leading to the accumulation of AGEs [49,50]. A comparative investigation of human brain cell populations has elucidated that the toxic AGE–albumin compound is mainly produced by microglia, with the synthesis rate of AGE–albumin being increased upon oxidative stress [51]. Wang et al.’s findings imply that AGEs are elevated in the serum of diabetic rats and may be associated with the activation of retinal microglia [52]. Hyperglycemia during early-stage diabetes induces BRB, leading to an augmentation of glucose and AGE levels due to vascular leakage. The efflux of AGEs may directly promote retinal microglial activation through the AGE receptor (RAGE) present on the microglial membrane, triggering the activation of NOX and thereby augmenting the intracellular synthesis of ROS [53]. In the feedback, ROS enhancement, in turn, may contribute to AGE formation [54] and RAGE expression [55].
In recent years, an abundance of research has shown that environmental perturbations in the retina activate microglia and contribute to retinal inflammation in DR [56,57]. Several animal models have been created to verify the polarization tendency of retinal microglia in DR. In a db/db mouse model, there was an increase in arginase-1/Iba-1 double immunopositive cells in the early stages of DR (5 weeks of age) [58]. This result suggests that the M2 anti-inflammatory phenotype is already present in microglia during the early stages of DR. With the prolongation of the disease course, at 8 weeks of age, the decrease of arginase-1 immunostaining and the high expression of iNOS suggested the possibility of microglia polarizing into the M1 state. More importantly, in addition to animal experiments, clinical studies have also shown that the appearance of retinal microvascular lesions in DR may be related to reduced M2 polarization and an increased M1/M2 ratio [59,60]. Since in vitro M1-polarized human adult microglia express the distinctive markers CD74, Iba-1, and iNOS [61], Ana et al. analyzed these markers in retinas from post-mortem diabetic and non-diabetic donors [58]. They found that the mRNA levels of Iba-1, CD74, and iNOS were markedly elevated in the retinas of diabetic donors, which revealed the transformation of microglia to the M1 type in patients with DR.
All in all, targeting microglia may be a strategy to treat DR, but the effectiveness of targeted therapy is rarely reported. Recent studies have shown that the synthetic progesterone analogue norgestrel reduces the CD68+ M1 phenotype in the rd10 model of retinitis pigmentosa through the presence of progesterone receptors on microglia cells and causes the expression of CD206/MRC1, arginase, etc. [62] Thus, it transforms into an M2 phenotype, ultimately preserving the activity and visual function of retinal photoreceptor cells. These observations may suggest that by converting microglia into an M2-polarized state, neuroinflammation in the early stage of DR could be alleviated, thereby reducing retinal blood vessel damage and neuronal degeneration, which may provide new insights for targeted treatment of DR.
2.3. The Immunometabolism of Microglia in DR
In recent years, the emerging literature on immunometabolism is defining new roles for cellular metabolic pathways in microglia polarization in DR [25,45,46,48,63]. Immunometabolism indicates the metabolic processes within immune cells, thereby influencing the destiny and functional outcomes of these cellular entities [64,65,66].
Under physiological conditions, the energy required by microglia mainly comes from the decomposition process of nutrients such as sugars, fats, and proteins, among which the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) of glucose are the most important processes [67]. However, this metabolic process is not constant. In previous studies, tumor cells still preferentially use glycolysis to produce lactic acid for energy under normal oxidation conditions rather than OXPHOS through aerobic respiration, a process known as the Warburg effect [68,69]. Similar to the “aerobic glycolysis” of tumor cells, the chemotaxis, phagocytosis, and proliferation of microglia after being activated into M1 type require dynamic recombination of the actin cytoskeleton and secretion of cytokines, so that M1 type microglia are in a state of energy deficiency. Although glycolysis produces much less ATP (2 ATP) than mitochondrial OXPHOS (36 ATP), glycolysis can be activated more quickly to match the energy demand of M1 microglia. In contrast, M2-type microglia, which are involved in anti-inflammatory repair, require a large amount of stable and sustained energy through mitochondrial OXPHOS to support normal life activities. During this process, the alterations in the expression levels and functionality of glucose transporters on the membranes of microglial cells are instrumental in the elevated glucose concentrations within the retina. Several reports have indicated that glucose uptake in microglia is predominately mediated by glucose transporter (GLUT) 1, especially under inflammatory conditions [70,71,72]. However, when treated with STF31, which impedes GLUT1-mediated glucose uptake in inflamed microglia, the metabolic pathway can be reprogrammed towards OXPHOS [73]. Therefore, targeting GLUT1 may emerge as an effective way to prevent DR.
As we mentioned earlier, integrated multi-omics reveals that activated microglia with an IL-1β increase in the early DR contribute to a metabolic bias favoring glycolysis, purine metabolism, and triacylglycerol synthesis, but less TCA. By establishing hypoxic-stimulated microglia cell line BV2, we observed that cytokine-related gene sets, including IL-1β, were significantly upregulated along with glycolysis gene sets [74]. These findings may indicate that activated microglia with intracellular metabolic reprogramming, especially glycolysis, in the retina, may contribute to pro-inflammation in the early DR. Similarly, in a mouse model of oxygen-induced retinopathy (OIR), high glycolysis of retinal microglia and endothelial cells is involved in the formation of retinal pathological blood vessels. Conversely, the knockout of glycolytic activators 6 phosphofructo 2 kinase/fructose 2,6 bisphosphatase (PFKFB3) in microglia inhibits the ability of microglia to release pro-inflammatory and pro-angiogenic factors and their transformation into pathological retinal angiogenesis-associated glycolytic microglia [75]. Moreover, the increased glycolytic activity of DR microglia appears to be more than a result of the proinflammatory state and may actually be a prerequisite for transformation into M1 proinflammatory microglia. As is demonstrated by two independent laboratories, blocking glycolysis with the glucose analogue 2-deoxyglucose can significantly inhibit pro-inflammatory microglial activation in vitro, which reduces the LPS-mediated increases in IL-6, IL-1β, and nitric oxide synthase 2 [76,77]. Taken together, these findings strongly support that changes in the metabolism of microglial cells in DR are closely related to the maintenance of their polarization state.
3. The Redox-Mediated Immunometabolic Regulation of Microglial Activation
In biological systems, redox reactions occur by transferring electrons from reduced donor molecules to acceptor molecules. During OXPHOS, the electron transport chain (ETC) transports electrons to molecular oxygen, and oxidative phosphorylation drives energy production in the mitochondria of aerobic organisms [78]. This inevitably produces ROS, such as superoxide anion (O2−) and hydrogen peroxide (H2O2) [79,80]. Microglia are major sources of free radicals such as superoxide in the retina and play crucial roles in various retinal diseases, especially DR [81,82]. In this process, some redox systems regulate the homeostasis of the redox microenvironment in microglia (Figure 1). Accumulating reports have shown that energy metabolism and redox state in microglia are tightly interwoven and interdependent to regulate the function of microglia ultimately [83], while the specific mechanism remains to be elucidated.

Figure 1.
Redox systems involved in microglia. The NADPH Oxidase (NOX), thioredoxin (TRX), and nuclear factor E2-related factor 2 (NRF2) systems constitute the redox homeostasis of the microglia’s internal environment. NOX2 is the main source of ROS generation in microglia. It reduces oxygen molecules to O2− or H2O2 by single-electron reduction dependent on NADPH. TRX-associated proteins are disulfide reductases that regulate the cellular redox state. Induced by various oxidative stresses, the TRX system scavenges ROS in association with TRX-dependent peroxiredoxins. As an endogenous inhibitor of the TRX system, thioredoxin-interacting protein (TXNIP) is transferred to the mitochondria after induction by ROS produced by NOX2. It activates the inflammatory body NOD-like receptor thermal protein domain associated protein 3 (NLRP3) and induces microglia to release the proinflammatory mediator IL-1β. In the meantime, the activation of the Wnt/β-catenin signaling pathway down-regulates the expression of antioxidant enzymes in microglia, further aggravating cellular oxidative stress. The transcription factor NRF2 binds to Kelch-like ECH-associated protein 1 (KEAP1) in the resting state and is fixed in the cytoplasm. Stimulated by oxidative stress, it separates from KEAP1 and translocates into the nucleus, binds to antioxidant response elements (ARE), and regulates the transcription-induced expression of multiple antioxidant genes.
3.1. Redox Systems in Microglia
3.1.1. NADPH Oxidase (NOX) System
NOX is the main functional enzyme for ROS production in the microglia [84]. It reduces oxygen molecules to O2− or H2O2 via NADPH-dependent single electron reduction, meanwhile generating ROS. The NOX family consists of seven isomers, including NOX1, NOX2, NOX3, NOX4, NOX5, Duox1, and Duox2 [85]. Most of them are located in different subcellular compartments within microglia [86,87,88]. Among them, NOX2, originally thought to be a phagocytic enzyme, is the first identified and most frequently explored in the field of DR research [89,90]. It is conventionally composed of membrane-bound catalytic subunit NOX2, regulatory small subunit p22phox, cytosolic subunits p47phox and p67phox, and a small G protein, Rac1 [91]. Numerous studies have shown that ROS generated by NOX2 can lead to microvascular dysfunction in the early stages of DR, possibly through specific mechanisms: (1) Positive feedback between high mobility group box-1 (HMGB1) and NOX-derived ROS mediates the diabetes-induced up-regulation of retinal apoptotic markers (e.g., PARP-1 and caspase-3), which leads to retinal apoptosis, causing vasculopathy and neuropathy [92]. (2) NOX2 leads to premature senescence of retinal endothelial cells through increased ROS formation, elevated arginase expression and activity, and decreased NO formation [93]. (3) NOX2 is highly activated in retinal cells, enhancing the expression of ICAM-1 and VEGF. This caused an increase in vascular permeability as well as leukocyte adhesion to the vessel wall [94]. In addition to NOX2, another isomer, NOX4, is also thought to be involved in diabetic retinopathy. On the one hand, it exacerbates the permeability and inflammatory response of the retinal vasculature [95], and on the other hand, it promotes retinal neovascularization through the H2O2/VEGFR2/ERK signaling pathway, which also plays a crucial role in proliferative DR [96]. Intriguingly, differing from NOX2 and NOX4, NOX1-derived ROS cause oxidative damage to the mitochondria of retinal endothelial cells in a high-glucose environment and trigger metabolic memory, giving rise to the progression of DR [97]. Thus, targeting the NOX system in microglia to reduce the toxic effects of derived ROS on retinal cells may be a potential way to reduce diabetic retinal damage.
3.1.2. Thioredoxin (TRX) System
The TRX system, consisting of TRX, thioredoxin reductase (TrxR), and peroxiredoxin (Prx), is an essential antioxidant system that regulates the protein dithiol/disulfide balance through the activity of disulfide reductase to resist oxidative stress [98]. TrxRs is a FAD-containing pyridine nucleotide disulfide oxidoreductase that uses NADPH to reduce the disulfide at the active site of TRXs [99]. Mammalian microglia have two TRX systems, with the TRX1 site in the cytoplasm and the TRX2 site in the mitochondria [83]. The function of TRX is to reduce oxidized cysteine (Cys) residues and break the disulfide bond by transferring electrons to Prxs and some redox-sensitive transcription factors. Prxs are widely distributed in different organs, including the retina, which receives electrons delivered by the TRX system to remove H2O2, ROOH, and ONOO− [100,101]. Meanwhile, as endogenous inhibitors of TRX, the cys63 and cys247 sites of thioredoxin-interacting protein (TXNIP) can form mixed disulfide bonds with thiols at the active site of TRX [102]. Various reports have pointed out that TXNIP plays a crucial role in glucose metabolism. Ao et al. confirmed that TXNIP is upregulated in both diabetic retinopathy and high glucose conditions [103]. In addition, TXNIP plays a number of physiological roles independent of TRX. TXNIP is likely transferred to the mitochondria after induction by ROS produced by NOX2 [104,105]. It activates the inflammatory body NOD-like receptor thermal protein domain associated protein 3 (NLRP3) and induces microglia to release the proinflammatory mediator IL-1β [106,107]. Recently, taxifolin has been reported to inhibit the inflammatory response of high-glucose-stimulated microglia in mice by attenuating the TXNIP–NLRP3 axis [108]. Therefore, we hypothesize that therapies targeting TXNIP may improve symptoms associated with DR by reducing intracellular ROS levels and neuroinflammation.
3.1.3. NRF2/KEAP Signaling
To cope with hyperglycemia, microglia are equipped with nuclear factor E2-related factor 2 (NRF2) in addition to antioxidant defenses in the organism [109]. NRF2 belongs to the basic leucine zip transcription factors and is a key player in the gene regulatory network of redox homeostasis [110,111]. In the resting state, NRF2 binds to Kelch-like ECH-associated protein 1 (KEAP1) and is fixed in the cytoplasm. When the body is oxidatively stressed, it separates from KEAP1 and translocates into the nucleus for transcriptional activity [112]. Modification of cysteine residue 151 (Cys151) of KEAP1, the primary sensor for sensing oxidative stress, alters the conformation of KEAP1 and inhibits ubiquitination of NRF2 [113]. Studies have shown that in diabetic retinal diseases, the redox sensing capacity of KEAP1 is altered due to increased oxidative stress, which prevents the separation of NRF2 from the KEAP1–NRF2 complex [109]. After entering the nucleus, stable NRF2 binds to antioxidant response elements (ARE) responsible for regulating the transcription and induced expression of a variety of genes, including antioxidant defense systems and inflammatory factors [114]. There are more than 250 NRF2-targeted genes, including NAD(P)H, heme oxygenase-1 (HO-1), peroxidase, superoxide dismutase (SOD), and glutamate cysteine ligase (GCL) [115]. Intriguingly, GCL is a heterodimer enzyme with catalytic and modified subunits that has been shown to be a key enzyme in catalyzing the synthesis of the antioxidant glutathione (GSH) from glutamate, cysteine, and glycine. The promoters of the GCL subunit contain ARE3 and ARE4, and NRF2 regulates GCL transcription by binding to ARE4. In diabetic retinas, NRF2 binding and transcription levels at the GCL promoter decrease, resulting in reduced GSH production. Therefore, targeting NRF2/KEAP1/GCL/GSH signals seems to be an effective means to correct the down-regulation of GSH levels in DR.
3.2. Redox Signaling Regulates Immunometabolism of Microglia in DR
3.2.1. Redox Regulation of Immunometabolism in M1 Microglia
During the period of DR, NOX2 is highly active in microglia [89,90]. Notably, polarization of M1 microglia during early inflammation of DR is dependent on metabolic alterations caused by NOX2 activation (Figure 2). It has been demonstrated in traumatic brain injury that NOX2 deficiency significantly reduces pro-inflammatory activation of microglia through an IL-10/STAT3-dependent mechanism, leading to an increase in the anti-inflammatory response [116]. Several inflammatory receptors in microglia are considered to couple with NOX2, including TLR4, which responds to bacterial LPS and pro-inflammatory cytokines [117,118]. After TLR4 is activated, NOX2 coupled with TLR4 is activated, and the superoxide anion radical (O2●−) is produced at the same time. O2●− can mutate into H2O2 under the mediation of SOD3 on the cell surface, which can freely penetrate the cell membrane to perform the second messenger function [119]. Intracellular H2O2 is a potent inhibitor of the protein tyrosine phosphatase (PTP), including phosphatase and tensin homolog (PTEN), a redox-sensitive protein [120,121]. Interestingly, PTEN is an inhibitor of AKT/PKB signaling activation [122,123], so AKT signaling increases when PTEN is inactivated, inhibiting microglia polarization from M1 to M2. Specifically, H2O2 produced by NOX2 inhibits PTEN activity under oxidative stress conditions. The activated AKT pathway promotes glucose absorption by upregulating the expression of GLUT4 on the membrane, thereby promoting microglial glycolysis [124]. In addition, the H2O2 signal activates the MAPK cascade simultaneously, triggering phosphorylation of IκK, degradation of IκB, and release of active NF-κB [125]. As a result, the secretion of iNOS and proinflammatory mediators increased. In the presence of arginine, iNOS catalyzes the formation of NO. NO produces high levels of peroxynitrite in mitochondria and can promote the nitrosylation of Fe–S clusters in the ETC, inhibit OXPHOS, and promote the occurrence of glycolysis.
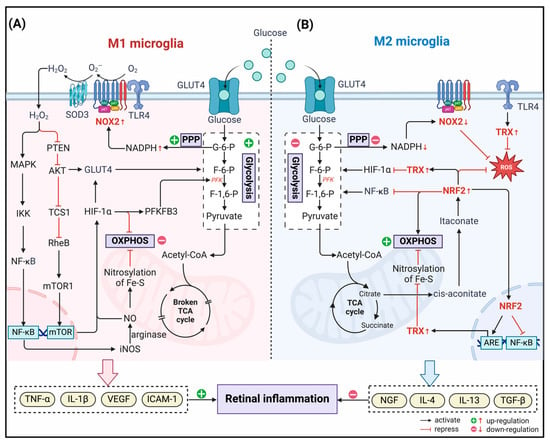
Figure 2.
Redox regulation of immunometabolism in the polarization of microglia in DR. (A) In M1 microglia, NADPH Oxidase (NOX) 2 couples to toll-like receptor 4 (TRL4) and generates H2O2 with the assistance of superoxide dismutase (SOD). H2O2, which enters the cytoplasm, acts as a second messenger. On the one hand, it activates NF-κB through the MAPK pathway, induces up-regulation of inducible nitric oxide synthase (iNOS) expression, and generates NO catalyzed by arginase, which nitrites Fe-S clusters in the mitochondrion and inhibits oxidative phosphorylation (OXPHOS) activity. On the other hand, by inhibiting phosphatase and tensin homolog (PTEN), H2O2 indirectly activates the AKT pathway, inducing glucose transporter (GLUT) 4 to transfer to the cell membrane and promoting glycolysis. Simultaneously, the mTOR signaling pathway is also activated, which contributes to the elevated expression of hypoxia-inducible factor (HIF)-1α. HIF-1α gained stability by nitroxylation and upregulated the expression of glycolysis-related genes, such as GLUT4 and 6 phosphofructo 2 kinase/fructose 2,6 bisphosphatase (PFKFB3), further promoting glycolysis. (B) In M2 microglia, NOX2 activity is reduced due to inhibition of the PPP pathway and decreased NADPH production. At the same time, the accumulated TCA cycle product citrate is isomerized in the cytoplasm to generate itaconate, which can covalently modify Kelch-like ECH-associated protein 1 (KEAP1) and activate nuclear factor E2-related factor 2 (NRF2). NRF2 enters the nucleus and, on the one hand, inhibits the transcriptional activity of NF-κB and reduces the production of pro-inflammatory mediators. On the other hand, it binds to ARE and induces the transcription of antioxidant molecule genes, especially thioredoxin (TRX). The activated TRX system can increase the efficiency of OXPHOS by removing the nitrosylation of Fe-S clusters, while down-regulating HIF-1α-induced glycolytic gene expression to inhibit the process of glycolysis.
In this process, hypoxia-inducible factor-1 α (HIF-1α) appears to coordinate redox signaling with microglial metabolic pathways and is thought to play a key role in DR. Following H2O2 produced by NOX2, HIF1-α is activated and stabilized under two oxidative regulatory pathways [126]. On the one hand, phosphorylated AKT signaling inactivates the tuberous sclerosis complex (TSC) ½ and mediates the accumulation of Ras homology (Rheb) in the form of GTP-binding, thereby activating the mammalian target of rapamycin complex 1 (mTORC1) and indirectly inducing HIF-1α upregulation [127,128]. On the other hand, due to the accumulation of nitrite in mitochondria caused by the increase of NO, Cys 533 was nitrated in the oxygen-dependent degradation domain of HIF-1α. HIF-1α therefore cannot be degraded by ubiquitination, thus achieving stability [129]. With the increase in HIF-1α transcriptional activity, the expression of glycolytic-related metabolic genes (hexokinase, PFKFB3, GLUT1, etc.) was up-regulated [130]. In addition, HIF-1α can also mediate the up-regulation of pyruvate dehydrogenase kinase 1 and the down-regulation of pyruvate dehydrogenase, thereby inhibiting the conversion of pyruvate to lactic acid and promoting the process of glycolysis [131,132]. In summary, NOX2-guided redox signaling induces microglia to transform in glycolytic and pro-inflammatory directions, which, to a large extent, further aggravates retinal vascular inflammation and nerve damage in DR.
3.2.2. Redox Regulation of Immunometabolism in M2 Microglia
As disease progresses into the middle to late stages, ROS production becomes less important. ATP and NO are depleted in microglia at this point, leading to a progressive accumulation of metabolites such as lactate, NAD+, and AMP. These metabolites send signals of nutrient deficiency and upregulate the expression of antioxidant genes to switch the metabolic phenotype of macrophages. Microglia then shift to the M2 phenotype and possess the ability to resist excessive neuroinflammation and promote tissue remodeling.
We have described that TCA cycling is restricted in M1 microglia, which gives rise to a large accumulation of metabolic substrates, especially citrate. Accumulated citrate is isomerized by aconitase to generate cis-aconitate, which is subsequently decarboxylated by immune-responsive gene 1 to generate itaconate [133,134]. Numerous studies have demonstrated the ability of itaconate to inhibit glycolysis through covalently modifying key glycolytic enzymes [135]. More importantly, itaconate can activate NRF2 by alkylating KEAP1 cysteine residues [136,137]. Activated NRF2 then fights oxidative stress by inducing the production of antioxidant molecules such as GSH, TRX, and SOD. At the same time, the activation of NRF2 is coupled with the inhibition of NF-κB, which inhibits the proinflammatory properties of microglia, including the production of NLRP3 and NO [138]. As another powerful antioxidant system, TRX is also activated and plays an essential role in the removal of nitro and mercaptan. It improves the efficiency of OXPHOS by decreasing the nitrite of Fe–S clusters within mitochondria and reducing the glycolysis flux by inhibiting the expression of the HIF-1α-induced glycolysis gene [139]. Overall, under the modulation of these antioxidant molecules, M2-type microglia exhibit low glycolysis and high OXPHOS and TCA properties for anti-inflammatory efficacy, which is undoubtedly beneficial for the cure of DR.
4. Promising Therapies for Diabetic Retinopathy Targeting Redox Regulation of Immunometabolism (Table 1)
4.1. NOX System Inhibitors
As previously described, in the DR, the NOX system coupled with TLR causes alterations in microglia immunometabolism through the AKT and MAPK signaling pathways, which, in turn, affect their polarization process. The down-regulation of the NOX system can effectively prevent the occurrence of retinal neovascularization and retinal oxidative stress. Therefore, inhibitors against the NOX system are naturally considered potential drug targets for the treatment of diabetic retinopathy.
GKT137831, also known as setanaxib, is currently recognized as a specific inhibitor of NOX1/4. Since they are structurally similar to NOX, GKT137831 may exert a competitive inhibitory effect on NOX1/4, although the exact mechanism has not been clarified [140]. Several studies have currently demonstrated the potential therapeutic effect of GKT137831 on diabetic retinopathy. Appukuttan et al. demonstrated that GKT137831 treatment significantly reduced dimethyl oxalylglycine (DMOG)-induced ROS generation and VEGFA expression in primary cultured human retinal endothelial cells [141]. Similarly, Jiao et al. also reported that treatment with GKT137831 significantly suppressed ROS levels and apoptosis, as well as caspase-3 activity in HREC, compared to the high-glucose (HG) group [142]. Among other retinal models, Wilkinson-Berka et al. described the beneficial effects of GKT137831 on OIR in rats in two reports [143]. Treatment with GKT137831 significantly reduced Iba1-positive microglia and decreased hypoxia-induced microglia production of ROS, inflammatory molecules (VEGF, IL-6, and TNFα), and leukocyte-recruiting molecules (MCP-1, RANTES, CINC2, CINC3, CXCL3, CXCL5, ICAM-1, and VCAM-1), which is suggestive of the drug’s ability to inhibit the strong pro-inflammatory signaling produced by activated retinal microglia. Currently, GKT137831 has already been administered to human subjects with diabetic nephropathy in a phase II clinical trial [144]. Although the progress of GKT137831 treatment in clinical trials in DR is relatively slow, its therapeutic potential cannot be ignored.
Another novel NOX inhibitor, GLX7013114, has been reported to have improved pharmacological properties in terms of efficacy and specificity in inhibiting NOX4. It has no affinity for the other NOX1, NOX2, and NOX5 isomers present in the retina. This highly selective NOX4 inhibitor has been shown to protect islet cells from cytokines and high glucose [145]. The latest study demonstrated that in STZ-induced rat DR models, topical administration of GLX7013114 prevented early events of DR associated with nitrative oxidative stress and apoptotic cell death [146]. More importantly, the administration of GLX7013114 reversed the activation of diabetic retinal microglia and reduced the expression of pro-inflammatory cytokines in the diabetic retina, thereby protecting the function of retinal neurons and retinal ganglion cells. This suggests the potential of GLX7013114 as a promising therapeutic candidate for the early treatment of DR, although a large number of clinical trials are required to confirm it.
4.2. TRX System Agonists
The TRX system is one of the important sulfur-dependent electron donors within the cell, fighting intracellular oxidative stress through disulfide reductase, which is essential for maintaining the oxidation/reduction balance of microglia. However, as an endogenous inhibitor of the TRX system, TXNIP induces oxidative stress by inhibiting the activity of TRX through direct binding to TRX1 and TRX2. It has been previously demonstrated that downregulation of TXNIP in vivo and in vitro reduced ROS production in microglia and alleviated retinal apoptosis [102,147]. These findings suggest that TXNIP may serve as a new potential therapeutic target for diabetic retinopathy.
Verapamil, an L-type calcium channel blocker, reduces the inward flow of extracellular calcium ions, which leads to vasodilation, lowers blood pressure, reduces myocardial contraction, and slows atrioventricular conduction [148,149]. Recent studies have found that Verapamil, the drug of choice for hypertension, has an equally positive impact on the treatment of diabetes and its serious complications, especially DR. Studies have demonstrated that verapamil can reduce intracellular calcium ion levels and reduce carb response element binding protein (ChREBP) into the nucleus, preventing its binding to TXNIP promoter E-box repeats, thereby inhibiting the transcription of TXNIP [150]. Eissa et al. also found that verapamil treatment enhanced TRX-R activity, significantly inhibited TLR4- and TXNIP-mediated NLRP3-inflammasome assembly, and subsequently reduced the release of inflammatory markers (TNF-α and IL-1β) into the vitreous humor and inhibited pathological angiogenesis [107]. It is worth noting that verapamil hydrochloride was approved by the FDA for the treatment of DR in 2016.
4.3. NRF2 Pathway Agonists
Accumulating evidence suggests that NRF2 has potential cytoprotective effects on both neurons and blood vessels in the injured retina [151]. In the DR, increased NRF2 activity in the microglial cytoplasm is expected to prevent the release of oxidative products and inflammatory factors. Many studies have shown that several natural compounds can inhibit oxidative stress and inflammation by targeting NRF2, thereby activating cytoprotective genes associated with antioxidant response elements. Thus, NRF2 may provide a new therapeutic target for the treatment of diabetic retinopathy.
Under normal conditions, NRF2 is kept at low levels because it binds to KEAP1 and is inactivated by KEAP1-dependent ubiquitination. Thus, interfering with KEAP1 activity is a rational strategy for agonizing NRF2 to translocate to the nucleus, where it binds to the ARE and mediates the expression of a range of antioxidant protein genes [152]. Interestingly, this is how some of the proven effective NRF2 activators work. Among them, CDDO-Me (RTA 402), one of the triterpenoids, is considered to be the most potent activator of NRF2 [153,154,155]. By interacting with and deubiquitinating specific cysteine residues on KEAP1, this, in turn, allows NRF2 to accumulate and translocate to the nucleus to bind to the ARE to drive transcription of antioxidant genes [156,157]. Notably, CDDO-Me has shown promise in initial clinical trials in patients with T2D and CKD. Ninety percent of T2D and CKD patients showed improvement in their estimated glomerular filtration rate (eGFR) after relatively short-term treatment with CDDO-Me. This suggests the promise of CDDO-Me as a clinical treatment for NRF2 agonists, although its effectiveness in other complications of diabetes, such as DR, remains to be further validated.
Curcumin is a natural polyphenol compound that has antioxidant and anti-inflammatory effects. Many animal experiments have confirmed that, in addition to acting as a direct antioxidant to remove ROS, curcumin can also act as an indirect antioxidant by mediating the activation of the NRF2 pathway [158,159,160]. According to mass spectrometry data, curcumin can modify Cys151 of KEAP1, resulting in conformational changes that inhibit protein interactions between NRF2 and KEAP1 [161]. Several in vivo and in vitro studies have shown its application in the treatment of diabetes and related complications, including DR [162,163]. In preclinical studies in rats, the use of curcumin, insulin, or combination therapy has been critical in regulating blood sugar, reducing oxidative stress, and improving histopathological damage in STZ-induced diabetic rats [163]. In vitro studies of RPE cells cultured under normal and HG conditions, Claudio et al. found that curcumin prevents HG-induced damage in RPE cells by activating Nrf2/HO-1 signaling involved in ERK pathway regulation [164]. However, to date, there are limited data on the pharmacological activity of curcumin in humans, which warrants further exploration.

Table 1.
The promising therapies targeting redox regulation of immunometabolism for DR.
Table 1.
The promising therapies targeting redox regulation of immunometabolism for DR.
| Classification | Drug | Current Use in Clinical Diseases | Potential Mechanisms for DR Treatment | References |
|---|---|---|---|---|
| NOX system inhibitor | Setanaxib (GKT137831) | Diabetic retinopathy, renal complication of diabetes, atherosclerosis, liver fibrosis, and idiopathic pulmonary fibrosis | Competitive inhibitor of NOX1/4. On the one hand, it inhibits retinal neovascularization and glial cell inflammation by modulating the VEGF/VEGFR2 pathway. On the other hand, it also reduces cell death and caspase-3 activity in the DR. | [140,141,142,143,144] |
| GLX7013114 | Diabetic retinopathy and type 2 diabetes | Highly selective inhibitor of NOX4. On the one hand, topical administration of GLX7013114 prevented early events of DR associated with nitrative oxidative stress and apoptotic cell death. On the other hand, GLX7013114 reversed the activation of diabetic retinal microglial and reduced the expression of pro-inflammatory cytokines in DR. | [145,146] | |
| TRX system agonist | Verapamil | Hypertension and diabetes | Verapamil improved oxidative stress damage in DR by inhibiting TXNIP protein expression, reducing its endogenous inhibition of the TRX system, while restoring beta cell health and improving metabolic abnormalities. | [107,148,149,150] |
| NRF2 pathway agonist | CDDO-Me | Type 2 diabetes and stage 4 chronic kidney disease | CDDO-Me interacts with specific cysteine residues on KEAP1, causing NRF2 to accumulate and translocate to the nucleus, leading to the up-regulation of ARE-responsive genes and promoting gene expression of antioxidant mole-cules. | [153,154,155,156,157] |
| Curcumin | Type 2 diabetes and non-alcoholic fatty liver disease | Curcumin can modify Cys151 of KEAP1, resulting in con-formational changes that inhibit protein interactions between NRF2 and KEAP1. | [158,159,160,161,162,163,164] |
5. Conclusions
Diabetic retinopathy, one of the most common complications of diabetes, has very limited treatment options currently. Well-recognized anti-VEGF therapies are effective in only one-third to one-half of patients with vision-threatening DR. Therefore, it is necessary to clarify the pathogenic mechanisms of DR and develop new, effective treatments. Accumulating studies have demonstrated that the direction of polarization of microglia in the retina largely influences the progress and prognosis of DR. There is no doubt that M1 microglia contribute to neuroinflammation and vascular injury in the early stages of DR. In contrast, disease remission and tissue healing are highly correlated with M2 microglia polarization. Thus, the goal of our current treatment is to convert microglia from M1 to M2 polarization, although whether this would increase VEGF output and exacerbate the lesions remains debatable.
The emerging literature on immunometabolism is defining a new role for cellular metabolic pathways in microglia reprogramming. In general, the M1-like and M2-like phenotypes of microglia have distinct metabolic profiles and may differentiate into distinct subtypes under the stimulus of chronic metabolic disorders. Therefore, regulation targeting microglial metabolic profiles may be a promising approach to moderate microglial cell polarization and function. In this process, redox signaling is gradually gaining attention. Redox response systems (including NOX, TRX, and NRF2) within microglia regulate cellular polarization direction and function by altering the metabolic balance of glycolysis and OXPHOS. Crosstalk between redox regulation and immunometabolism within microglia appears to open new doors for determining the fate of microglia in DR.
Although we have summarized the underlying mechanisms of redox regulation in microglial immunometabolism within the context of DR, numerous aspects remain to be further investigated. For instance, the AKT/mTOR pathway, which is instrumental in regulating the glycolytic activity of microglia via the NOX2 system [165], also triggers autophagy in various cell types, as documented in references [166,167]. Whether mTOR-induced autophagy is involved in the hyperglycolytic metabolism of microglia remains an open question. Furthermore, in the present review, our focus is on the oxidative stress generated within microglia cells themselves. It is noteworthy to consider the external sources of oxidative stress that contribute to damage beyond the microglia cell population during DR. Du et al. showed that the elimination of nerve photoreceptor cells in diabetes suppresses oxidative stress and inflammatory changes that lead to DR vasculopathy, demonstrating these cells also play an important role in the diabetes-induced increase in retinal superoxide and local inflammation in mice [168]. Similarly, in the rd1 mouse model, Daniela et al. emphasized that only photoreceptors undergo apoptosis, instead of the amacrine, bipolar, or horizontal retinal cells [169]. In fact, it has been known for a long time that oxygen is absorbed primarily at the photoreceptor level in cat retinas [170]. Further studies confirmed that the photoreceptors, especially the outer segments (OS), produce massive ROS because of the localization of ectopic enzymes in the respiratory chain [171]. The retinal rod OS disk membrane, despite the lack of mitochondria, performs OXPHOS and expresses ETC and TCA proteins [172,173]. The presence of this extra-mitochondrial oxidative metabolism lays the foundation for OS to be more susceptible to potential oxidative stress damage. However, whether antioxidant therapy targeting oxidative stress damage in the rod OS of the retina is a viable strategy for the early progression of DR remains to be clarified.
To conclude, in this review, we discuss the origin, phenotype, and function of microglia under physiological conditions. The effects of microglia polarization, immunometabolism, and intracellular redox systems on metabolic pathways have also been elaborated. Finally, we summarize potential therapeutic targets for microglial redox regulation, with the aim of providing new insights into emerging therapeutic approaches for DR.
Author Contributions
Conceptualization, F.Z. and L.C.; writing—original draft preparation, L.C. and M.X.; writing—review and editing, F.Z. and L.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (92357307 and 32171177) and the Key R&D project of the National Ministry of Science and Technology (2023YFA1801100).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
Figures were created with “BioRender.com”.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Wang, W.; Lo, A.C.Y. Diabetic Retinopathy: Pathophysiology and Treatments. Int. J. Mol. Sci. 2018, 19, 1816. [Google Scholar] [CrossRef] [PubMed]
- Simó, R.; Stitt, A.W.; Gardner, T.W. Neurodegeneration in diabetic retinopathy: Does it really matter? Diabetologia 2018, 61, 1902–1912. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.Y.; Hsih, W.H.; Lin, Y.B.; Wen, C.Y.; Chang, T.J. Update in the epidemiology, risk factors, screening, and treatment of diabetic retinopathy. J. Diabetes Investig. 2021, 12, 1322–1325. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.E.; Wong, T.Y. Diabetic retinopathy: Looking forward to 2030. Front. Endocrinol. 2022, 13, 1077669. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, J.D.; Bourne, R.R.; Briant, P.S.; Flaxman, S.R.; Taylor, H.R.; Jonas, J.B.; Abdoli, A.A.; Abrha, W.A.; Abualhasan, A.; Abu-Gharbieh, E.G.; et al. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: The Right to Sight: An analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e144–e160. [Google Scholar] [CrossRef] [PubMed]
- Al-Namaeh, M. Common causes of visual impairment in the elderly. Med. Hypothesis Discov. Innov. Ophthalmol. 2021, 10, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, A.; Aragona, E.; Bandello, F. VEGF-targeting drugs for the treatment of retinal neovascularization in diabetic retinopathy. Ann. Med. 2022, 54, 1089–1111. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Sun, Y.; Jiang, Y. Anti-VEGF therapy is not a magic bullet for diabetic retinopathy. Eye 2020, 34, 609–610. [Google Scholar] [CrossRef]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight 2017, 2, 93751. [Google Scholar] [CrossRef]
- Barot, M.; Gokulgandhi, M.R.; Patel, S.; Mitra, A.K. Microvascular complications and diabetic retinopathy: Recent advances and future implications. Future Med. Chem. 2013, 5, 301–314. [Google Scholar] [CrossRef]
- Zhang, T.; Ouyang, H.; Mei, X.; Lu, B.; Yu, Z.; Chen, K.; Wang, Z.; Ji, L. Erianin alleviates diabetic retinopathy by reducing retinal inflammation initiated by microglial cells via inhibiting hyperglycemia-mediated ERK1/2-NF-κB signaling pathway. FASEB J. 2019, 33, 11776–11790. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Xu, G.T.; Zhang, J.F. Inflammation in diabetic retinopathy: Possible roles in pathogenesis and potential implications for therapy. Neural Regen. Res. 2023, 18, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Kinuthia, U.M.; Wolf, A.; Langmann, T. Microglia and Inflammatory Responses in Diabetic Retinopathy. Front. Immunol. 2020, 11, 564077. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.X.; Ng, Y.K.; Ling, E.A. Neuronal and microglial response in the retina of streptozotocin-induced diabetic rats. Vis. Neurosci. 2000, 17, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Murenu, E.; Gerhardt, M.J.; Biel, M.; Michalakis, S. More than meets the eye: The role of microglia in healthy and diseased retina. Front. Immunol. 2022, 13, 1006897. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS−) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Funes, S.C.; Rios, M.; Escobar-Vera, J.; Kalergis, A.M. Implications of macrophage polarization in autoimmunity. Immunology 2018, 154, 186–195. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; El-Shishtawy, M.M.; Peña, A., Jr.; Liou, G.I. Genistein attenuates retinal inflammation associated with diabetes by targeting of microglial activation. Mol. Vis. 2010, 16, 2033–2042. [Google Scholar]
- Chen, T.; Zhu, W.; Wang, C.; Dong, X.; Yu, F.; Su, Y.; Huang, J.; Huo, L.; Wan, P. ALKBH5-Mediated m6A Modification of A20 Regulates Microglia Polarization in Diabetic Retinopathy. Front. Immunol. 2022, 13, 813979. [Google Scholar] [CrossRef] [PubMed]
- Lercher, A.; Baazim, H.; Bergthaler, A. Systemic Immunometabolism: Challenges and Opportunities. Immunity 2020, 53, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Saravia, J.; Raynor, J.L.; Chapman, N.M.; Lim, S.A.; Chi, H. Signaling networks in immunometabolism. Cell Res. 2020, 30, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Artyomov, M.N.; Van den Bossche, J. Immunometabolism in the Single-Cell Era. Cell Metab. 2020, 32, 710–725. [Google Scholar] [CrossRef] [PubMed]
- Bernier, L.P.; York, E.M.; MacVicar, B.A. Immunometabolism in the Brain: How Metabolism Shapes Microglial Function. Trends Neurosci. 2020, 43, 854–869. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Angiari, S. Microglia immunometabolism: From metabolic disorders to single cell metabolism. Semin. Cell Dev. Biol. 2019, 94, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.; Zhang, T.; Ouyang, H.; Lu, B.; Wang, Z.; Ji, L. Scutellarin alleviates blood-retina-barrier oxidative stress injury initiated by activated microglia cells during the development of diabetic retinopathy. Biochem. Pharmacol. 2019, 159, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Mendonca, H.R.; Carpi-Santos, R.; da Costa Calaza, K.; Blanco Martinez, A.M. Neuroinflammation and oxidative stress act in concert to promote neurodegeneration in the diabetic retina and optic nerve: Galectin-3 participation. Neural Regen. Res. 2020, 15, 625–635. [Google Scholar] [CrossRef]
- Lee, R.; Wong, T.Y.; Sabanayagam, C. Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis. 2015, 2, 17. [Google Scholar] [CrossRef]
- Gupta, S.K.; Kumar, B.; Nag, T.C.; Agrawal, S.S.; Agrawal, R.; Agrawal, P.; Saxena, R.; Srivastava, S. Curcumin prevents experimental diabetic retinopathy in rats through its hypoglycemic, antioxidant, and anti-inflammatory mechanisms. J. Ocul. Pharmacol. Ther. 2011, 27, 123–130. [Google Scholar] [CrossRef]
- Madore, C.; Yin, Z.; Leibowitz, J.; Butovsky, O. Microglia, Lifestyle Stress, and Neurodegeneration. Immunity 2020, 52, 222–240. [Google Scholar] [CrossRef]
- Atta, A.A.; Ibrahim, W.W.; Mohamed, A.F.; Abdelkader, N.F. Microglia polarization in nociplastic pain: Mechanisms and perspectives. Inflammopharmacology 2023, 31, 1053–1067. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, D.; Li, G. The role of microglia in viral encephalitis: A review. J. Neuroinflamm. 2019, 16, 76. [Google Scholar] [CrossRef]
- Ooi, K.; Hu, L.; Feng, Y.; Han, C.; Ren, X.; Qian, X.; Huang, H.; Chen, S.; Shi, Q.; Lin, H.; et al. Sigma-1 Receptor Activation Suppresses Microglia M1 Polarization via Regulating Endoplasmic Reticulum-Mitochondria Contact and Mitochondrial Functions in Stress-Induced Hypertension Rats. Mol. Neurobiol. 2021, 58, 6625–6646. [Google Scholar] [CrossRef]
- Boche, D.; Perry, V.H.; Nicoll, J.A. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef]
- Li, J.; Yu, S.; Lu, X.; Cui, K.; Tang, X.; Xu, Y.; Liang, X. The phase changes of M1/M2 phenotype of microglia/macrophage following oxygen-induced retinopathy in mice. Inflamm. Res. 2021, 70, 183–192. [Google Scholar] [CrossRef]
- Yao, Y.; Li, J.; Zhou, Y.; Wang, S.; Zhang, Z.; Jiang, Q.; Li, K. Macrophage/microglia polarization for the treatment of diabetic retinopathy. Front. Endocrinol. 2023, 14, 1276225. [Google Scholar] [CrossRef]
- Wang, J.; Xing, H.; Wan, L.; Jiang, X.; Wang, C.; Wu, Y. Treatment targets for M2 microglia polarization in ischemic stroke. Biomed. Pharmacother. 2018, 105, 518–525. [Google Scholar] [CrossRef]
- Fenn, A.M.; Henry, C.J.; Huang, Y.; Dugan, A.; Godbout, J.P. Lipopolysaccharide-induced interleukin (IL)-4 receptor-α expression and corresponding sensitivity to the M2 promoting effects of IL-4 are impaired in microglia of aged mice. Brain Behav. Immun. 2012, 26, 766–777. [Google Scholar] [CrossRef]
- Liu, X.; Liu, J.; Zhao, S.; Zhang, H.; Cai, W.; Cai, M.; Ji, X.; Leak, R.K.; Gao, Y.; Chen, J.; et al. Interleukin-4 Is Essential for Microglia/Macrophage M2 Polarization and Long-Term Recovery After Cerebral Ischemia. Stroke 2016, 47, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Ciotti, G.M.; Braun, D.; Kalinin, S.; Currò, D.; Dello Russo, C.; Coli, A.; Mangiola, A.; Anile, C.; Feinstein, D.L.; et al. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci. Lett. 2017, 645, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Li, R.; Han, C.; Lu, X.; Zhang, H. Minocycline promotes posthemorrhagic neurogenesis via M2 microglia polarization via upregulation of the TrkB/BDNF pathway in rats. J. Neurophysiol. 2018, 120, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Blaschke, S.J.; Demir, S.; König, A.; Abraham, J.A.; Vay, S.U.; Rabenstein, M.; Olschewski, D.N.; Hoffmann, C.; Hoffmann, M.; Hersch, N.; et al. Substrate Elasticity Exerts Functional Effects on Primary Microglia. Front. Cell. Neurosci. 2020, 14, 590500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xia, M.; Wu, Y.; Zhang, F. Branched-Chain Amino Acids Metabolism and Their Roles in Retinopathy: From Relevance to Mechanism. Nutrients 2023, 15, 2161. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Zhang, F. Amino Acids Metabolism in Retinopathy: From Clinical and Basic Research Perspective. Metabolites 2022, 12, 1244. [Google Scholar] [CrossRef] [PubMed]
- Jian, Q.; Wu, Y.; Zhang, F. Metabolomics in Diabetic Retinopathy: From Potential Biomarkers to Molecular Basis of Oxidative Stress. Cells 2022, 11, 3005. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Jian, Q.; Hu, G.; Du, R.; Xu, X.; Zhang, F. Integrated Metabolomics and Transcriptomics Reveal Metabolic Patterns in Retina of STZ-Induced Diabetic Retinopathy Mouse Model. Metabolites 2022, 12, 1245. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N. Advanced glycation endproducts—Role in pathology of diabetic complications. Diabetes Res. Clin. Pract. 2005, 67, 3–21. [Google Scholar] [CrossRef]
- Vargas-Soria, M.; García-Alloza, M.; Corraliza-Gómez, M. Effects of diabetes on microglial physiology: A systematic review of in vitro, preclinical and clinical studies. J. Neuroinflamm. 2023, 20, 57. [Google Scholar] [CrossRef]
- Byun, K.; Bayarsaikhan, E.; Kim, D.; Kim, C.Y.; Mook-Jung, I.; Paek, S.H.; Kim, S.U.; Yamamoto, T.; Won, M.H.; Song, B.J.; et al. Induction of neuronal death by microglial AGE-albumin: Implications for Alzheimer’s disease. PLoS ONE 2012, 7, e37917. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Yu, A.C.; He, Q.H.; Zhu, X.; Tso, M.O. AGEs mediated expression and secretion of TNF alpha in rat retinal microglia. Exp. Eye Res. 2007, 84, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Ma, Y.; Zeng, Z.; Yin, Q.; Hong, Y.; Hou, X.; Liu, X. RAGE-Specific Inhibitor FPS-ZM1 Attenuates AGEs-Induced Neuroinflammation and Oxidative Stress in Rat Primary Microglia. Neurochem. Res. 2017, 42, 2902–2911. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Mel’nikova, T.I.; Porozov, Y.B.; Terentiev, A.A. Oxidative Stress and Advanced Lipoxidation and Glycation End Products (ALEs and AGEs) in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 3085756. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Brownlee, M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef]
- Dong, N.; Chang, L.; Wang, B.; Chu, L. Retinal neuronal MCP-1 induced by AGEs stimulates TNF-α expression in rat microglia via p38, ERK, and NF-κB pathways. Mol. Vis. 2014, 20, 616–628. [Google Scholar] [PubMed]
- Khalid, M.; Petroianu, G.; Adem, A. Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives. Biomolecules 2022, 12, 542. [Google Scholar] [CrossRef] [PubMed]
- Arroba, A.I.; Alcalde-Estevez, E.; García-Ramírez, M.; Cazzoni, D.; de la Villa, P.; Sánchez-Fernández, E.M.; Mellet, C.O.; García Fernández, J.M.; Hernández, C.; Simó, R.; et al. Modulation of microglia polarization dynamics during diabetic retinopathy in db/db mice. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 1663–1674. [Google Scholar] [CrossRef]
- Yoshida, S.; Kobayashi, Y.; Nakama, T.; Zhou, Y.; Ishikawa, K.; Arita, R.; Nakao, S.; Miyazaki, M.; Sassa, Y.; Oshima, Y.; et al. Increased expression of M-CSF and IL-13 in vitreous of patients with proliferative diabetic retinopathy: Implications for M2 macrophage-involving fibrovascular membrane formation. Br. J. Ophthalmol. 2015, 99, 629–634. [Google Scholar] [CrossRef]
- Hernández, C.; Bogdanov, P.; Corraliza, L.; García-Ramírez, M.; Solà-Adell, C.; Arranz, J.A.; Arroba, A.I.; Valverde, A.M.; Simó, R. Topical Administration of GLP-1 Receptor Agonists Prevents Retinal Neurodegeneration in Experimental Diabetes. Diabetes 2016, 65, 172–187. [Google Scholar] [CrossRef]
- Walker, D.G.; Lue, L.F. Immune phenotypes of microglia in human neurodegenerative disease: Challenges to detecting microglial polarization in human brains. Alzheimer’s Res. Ther. 2015, 7, 56. [Google Scholar] [CrossRef]
- Roche, S.L.; Wyse-Jackson, A.C.; Gómez-Vicente, V.; Lax, P.; Ruiz-Lopez, A.M.; Byrne, A.M.; Cuenca, N.; Cotter, T.G. Progesterone Attenuates Microglial-Driven Retinal Degeneration and Stimulates Protective Fractalkine-CX3CR1 Signaling. PLoS ONE 2016, 11, e0165197. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Gu, L.; Wang, R.; Jian, Q.; Lv, K.; Xia, M.; Lai, M.; Shen, T.; Hu, J.; Yang, S.; et al. Ethanolamine as a biomarker and biomarker-based therapy for diabetic retinopathy in glucose-well-controlled diabetic patients. Sci. Bull. 2024, in press. [Google Scholar] [CrossRef]
- Fu, Y.; Wang, L.; Yu, B.; Xu, D.; Chu, Y. Immunometabolism shapes B cell fate and functions. Immunology 2022, 166, 444–457. [Google Scholar] [CrossRef]
- Chavakis, T. Immunometabolism: Where Immunology and Metabolism Meet. J. Innate Immun. 2022, 14, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Chi, H. Immunometabolism at the intersection of metabolic signaling, cell fate, and systems immunology. Cell. Mol. Immunol. 2022, 19, 299–302. [Google Scholar] [CrossRef]
- Borst, K.; Schwabenland, M.; Prinz, M. Microglia metabolism in health and disease. Neurochem. Int. 2019, 130, 104331. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Kang, H.; Kim, B.; Park, J.; Youn, H.; Youn, B. The Warburg effect on radioresistance: Survival beyond growth. Biochim. Biophys. Acta (BBA) Rev. Cancer 2023, 1878, 188988. [Google Scholar] [CrossRef]
- Klip, A.; Marette, A.; Dimitrakoudis, D.; Ramlal, T.; Giacca, A.; Shi, Z.Q.; Vranic, M. Effect of Diabetes on Glucoregulation: From glucose transporters to glucose metabolism in vivo. Diabetes Care 1992, 15, 1747–1766. [Google Scholar] [CrossRef]
- Lu, L.; Seidel, C.P.; Iwase, T.; Stevens, R.K.; Gong, Y.Y.; Wang, X.; Hackett, S.F.; Campochiaro, P.A. Suppression of GLUT1; a new strategy to prevent diabetic complications. J. Cell. Physiol. 2013, 228, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pavlou, S.; Du, X.; Bhuckory, M.; Xu, H.; Chen, M. Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol. Neurodegener. 2019, 14, 2. [Google Scholar] [CrossRef] [PubMed]
- Lv, K.; Ying, H.; Hu, G.; Hu, J.; Jian, Q.; Zhang, F. Integrated multi-omics reveals the activated retinal microglia with intracellular metabolic reprogramming contributes to inflammation in STZ-induced early diabetic retinopathy. Front. Immunol. 2022, 13, 942768. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, J.; Ma, Q.; Zhang, X.; Yang, Q.; Wang, L.; Cao, Y.; Xu, Z.; Tawfik, A.; Sun, Y.; et al. Glycolysis links reciprocal activation of myeloid cells and endothelial cells in the retinal angiogenic niche. Sci. Transl. Med. 2020, 12, eaay1371. [Google Scholar] [CrossRef] [PubMed]
- Fodelianaki, G.; Lansing, F.; Bhattarai, P.; Troullinaki, M.; Zeballos, M.A.; Charalampopoulos, I.; Gravanis, A.; Mirtschink, P.; Chavakis, T.; Alexaki, V.I. Nerve Growth Factor modulates LPS—Induced microglial glycolysis and inflammatory responses. Exp. Cell Res. 2019, 377, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhao, Y.; Sun, M.; Liu, S.; Li, B.; Zhang, L.; Yang, L. 2-Deoxy-d-glucose attenuates sevoflurane-induced neuroinflammation through nuclear factor-kappa B pathway in vitro. Toxicol. In Vitro 2014, 28, 1183–1189. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Sies, H. Oxidative eustress: On constant alert for redox homeostasis. Redox Biol. 2021, 41, 101867. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, M.; Gu, R.; Xu, G.; Wu, H. Activated microglia induce the production of reactive oxygen species and promote apoptosis of co-cultured retinal microvascular pericytes. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 777–788. [Google Scholar] [CrossRef]
- Murakami, Y.; Nakabeppu, Y.; Sonoda, K.H. Oxidative Stress and Microglial Response in Retinitis Pigmentosa. Int. J. Mol. Sci. 2020, 21, 7170. [Google Scholar] [CrossRef]
- Vilhardt, F.; Haslund-Vinding, J.; Jaquet, V.; McBean, G. Microglia antioxidant systems and redox signalling. Br. J. Pharmacol. 2017, 174, 1719–1732. [Google Scholar] [CrossRef] [PubMed]
- Haslund-Vinding, J.; McBean, G.; Jaquet, V.; Vilhardt, F. NADPH oxidases in oxidant production by microglia: Activating receptors, pharmacology and association with disease. Br. J. Pharmacol. 2017, 174, 1733–1749. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Ejlerskov, P.; Christensen, D.P.; Beyaie, D.; Burritt, J.B.; Paclet, M.H.; Gorlach, A.; van Deurs, B.; Vilhardt, F. NADPH oxidase is internalized by clathrin-coated pits and localizes to a Rab27A/B GTPase-regulated secretory compartment in activated macrophages. J. Biol. Chem. 2012, 287, 4835–4852. [Google Scholar] [CrossRef]
- Chéret, C.; Gervais, A.; Lelli, A.; Colin, C.; Amar, L.; Ravassard, P.; Mallet, J.; Cumano, A.; Krause, K.H.; Mallat, M. Neurotoxic activation of microglia is promoted by a nox1-dependent NADPH oxidase. J. Neurosci. 2008, 28, 12039–12051. [Google Scholar] [CrossRef] [PubMed]
- Mead, E.L.; Mosley, A.; Eaton, S.; Dobson, L.; Heales, S.J.; Pocock, J.M. Microglial neurotransmitter receptors trigger superoxide production in microglia; consequences for microglial-neuronal interactions. J. Neurochem. 2012, 121, 287–301. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kowluru, A.; Veluthakal, R.; Mohammad, G.; Syed, I.; Santos, J.M.; Mishra, M. TIAM1-RAC1 signalling axis-mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia 2014, 57, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A. Diabetic Retinopathy and NADPH Oxidase-2: A Sweet Slippery Road. Antioxidants 2021, 10, 783. [Google Scholar] [CrossRef]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Dürr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019, 26, 101272. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Alam, K.; Nawaz, M.I.; Siddiquei, M.M.; Mousa, A.; Abu El-Asrar, A.M. Mutual enhancement between high-mobility group box-1 and NADPH oxidase-derived reactive oxygen species mediates diabetes-induced upregulation of retinal apoptotic markers. J. Physiol. Biochem. 2015, 71, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Lemtalsi, T.; Toque, H.A.; Xu, Z.; Fulton, D.; Caldwell, R.W.; Caldwell, R.B. NOX2-Induced Activation of Arginase and Diabetes-Induced Retinal Endothelial Cell Senescence. Antioxidants 2017, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Zhang, W.; Xu, Z.; Lemtalsi, T.; Chandler, P.; Toque, H.A.; Caldwell, R.W.; Caldwell, R.B. Requirement of NOX2 expression in both retina and bone marrow for diabetes-induced retinal vascular injury. PLoS ONE 2013, 8, e84357. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, X.; Cheng, R.; Ma, J.X.; Yi, J.; Li, J. Salutary effect of fenofibrate on type 1 diabetic retinopathy via inhibiting oxidative stress-mediated Wnt/β-catenin pathway activation. Cell Tissue Res. 2019, 376, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.J.; Zhang, S.X. NADPH oxidase 4-derived H2O2 promotes aberrant retinal neovascularization via activation of VEGF receptor 2 pathway in oxygen-induced retinopathy. J. Diabetes Res. 2015, 2015, 963289. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, H.; Guan, W.; Kang, X.; Tai, X.; Shen, Y. Metabolic memory in mitochondrial oxidative damage triggers diabetic retinopathy. BMC Ophthalmol. 2018, 18, 258. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef]
- Álvarez-Barrios, A.; Álvarez, L.; García, M.; Artime, E.; Pereiro, R.; González-Iglesias, H. Antioxidant Defenses in the Human Eye: A Focus on Metallothioneins. Antioxidants 2021, 10, 89. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Li, N.; Wang, X.; He, S.; Li, W.; Fan, W.; Li, R.; Liu, J.; Hou, S. Icariin alleviates uveitis by targeting peroxiredoxin 3 to modulate retinal microglia M1/M2 phenotypic polarization. Redox Biol. 2022, 52, 102297. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Wang, N.N.; Qi, H.; Qiu, Y.Y.; Zhang, C.H.; Brown, E.; Kong, H.; Kong, L. Up-Regulation Thioredoxin Inhibits Advanced Glycation End Products-Induced Neurodegeneration. Cell. Physiol. Biochem. 2018, 50, 1673–1686. [Google Scholar] [CrossRef] [PubMed]
- Ao, H.; Li, H.; Zhao, X.; Liu, B.; Lu, L. TXNIP positively regulates the autophagy and apoptosis in the rat müller cell of diabetic retinopathy. Life Sci. 2021, 267, 118988. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Cai, W.; Jin, H.; Shen, T.; Yu, J. Downregulation of Inflammatory Response via Nrf2/Trx1/TXNIP Axis in Oxidative Stress-Induced ARPE-19 Cells and Mouse Model of AMD. Oxid. Med. Cell. Longev. 2022, 2022, 1497813. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lin, X.; Guan, M.; Zeng, Y.; Liu, Y. Verapamil Attenuated Prediabetic Neuropathy in High-Fat Diet-Fed Mice through Inhibiting TXNIP-Mediated Apoptosis and Inflammation. Oxid. Med. Cell. Longev. 2019, 2019, 1896041. [Google Scholar] [CrossRef] [PubMed]
- Saxena, G.; Chen, J.; Shalev, A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J. Biol. Chem. 2010, 285, 3997–4005. [Google Scholar] [CrossRef] [PubMed]
- Eissa, L.D.; Ghobashy, W.A.; El-Azab, M.F. Inhibition of thioredoxin-interacting protein and inflammasome assembly using verapamil mitigates diabetic retinopathy and pancreatic injury. Eur. J. Pharmacol. 2021, 901, 174061. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, M.; Kato, H.; Iwashita, K.; Yamakage, H.; Kato, S.; Saito, S.; Ihara, M.; Nishimura, H.; Kawamoto, A.; Suganami, T.; et al. Taxifolin Suppresses Inflammatory Responses of High-Glucose-Stimulated Mouse Microglia by Attenuating the TXNIP-NLRP3 Axis. Nutrients 2023, 15, 2738. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Mishra, M.; Kowluru, R.A. Transcription factor Nrf2-mediated antioxidant defense system in the development of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3941–3948. [Google Scholar] [CrossRef]
- Alfieri, A.; Srivastava, S.; Siow, R.C.; Modo, M.; Fraser, P.A.; Mann, G.E. Targeting the Nrf2-Keap1 antioxidant defence pathway for neurovascular protection in stroke. J. Physiol. 2011, 589, 4125–4136. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mishra, M. Epigenetic regulation of redox signaling in diabetic retinopathy: Role of Nrf2. Free Radic. Biol. Med. 2017, 103, 155–164. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Pi, J.; Zhang, Q. Signal amplification in the KEAP1-NRF2-ARE antioxidant response pathway. Redox Biol. 2022, 54, 102389. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.P.; Henry, R.J.; Villapol, S.; Stoica, B.A.; Kumar, A.; Burns, M.P.; Faden, A.I.; Loane, D.J. NOX2 deficiency alters macrophage phenotype through an IL-10/STAT3 dependent mechanism: Implications for traumatic brain injury. J. Neuroinflam. 2017, 14, 65. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xu, T.; Chen, T.; Liu, J.; Xu, S. Oxidative stress mediated by the TLR4/NOX2 signalling axis is involved in polystyrene microplastic-induced uterine fibrosis in mice. Sci. Total Environ. 2022, 838, 155825. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Jeong, J.M.; Kim, S.J.; Seo, W.; Kim, M.H.; Choi, W.M.; Yoo, W.; Lee, J.H.; Shim, Y.R.; Yi, H.S.; et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat. Commun. 2017, 8, 2247. [Google Scholar] [CrossRef]
- Petersen, S.V.; Poulsen, N.B.; Linneberg Matthiesen, C.; Vilhardt, F. Novel and Converging Ways of NOX2 and SOD3 in Trafficking and Redox Signaling in Macrophages. Antioxidants 2021, 10, 172. [Google Scholar] [CrossRef]
- Connor, K.M.; Subbaram, S.; Regan, K.J.; Nelson, K.K.; Mazurkiewicz, J.E.; Bartholomew, P.J.; Aplin, A.E.; Tai, Y.T.; Aguirre-Ghiso, J.; Flores, S.C.; et al. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J. Biol. Chem. 2005, 280, 16916–16924. [Google Scholar] [CrossRef]
- Lee, S.R.; Yang, K.S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef]
- Liu, W.; Xu, L.; Wang, X.; Zhang, D.; Sun, G.; Wang, M.; Wang, M.; Han, Y.; Chai, R.; Wang, H. PRDX1 activates autophagy via the PTEN-AKT signaling pathway to protect against cisplatin-induced spiral ganglion neuron damage. Autophagy 2021, 17, 4159–4181. [Google Scholar] [CrossRef] [PubMed]
- Haddadi, N.; Lin, Y.; Travis, G.; Simpson, A.M.; Nassif, N.T.; McGowan, E.M. PTEN/PTENP1: ‘Regulating the regulator of RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy. Mol. Cancer 2018, 17, 37. [Google Scholar] [CrossRef]
- Horie, T.; Ono, K.; Nagao, K.; Nishi, H.; Kinoshita, M.; Kawamura, T.; Wada, H.; Shimatsu, A.; Kita, T.; Hasegawa, K. Oxidative stress induces GLUT4 translocation by activation of PI3-K/Akt and dual AMPK kinase in cardiac myocytes. J. Cell. Physiol. 2008, 215, 733–742. [Google Scholar] [CrossRef]
- Guyton, K.Z.; Liu, Y.; Gorospe, M.; Xu, Q.; Holbrook, N.J. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J. Biol. Chem. 1996, 271, 4138–4142. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, H.R.; Gao, D.; Pararasa, C. Redox regulation in metabolic programming and inflammation. Redox Biol. 2017, 12, 50–57. [Google Scholar] [CrossRef]
- Howell, J.J.; Manning, B.D. mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol. Metab. 2011, 22, 94–102. [Google Scholar] [CrossRef]
- Casciano, F.; Zauli, E.; Rimondi, E.; Mura, M.; Previati, M.; Busin, M.; Zauli, G. The role of the mTOR pathway in diabetic retinopathy. Front. Med. 2022, 9, 973856. [Google Scholar] [CrossRef] [PubMed]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef]
- Cerychova, R.; Pavlinkova, G. HIF-1, Metabolism, and Diabetes in the Embryonic and Adult Heart. Front. Endocrinol. 2018, 9, 460. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Semba, H.; Takeda, N.; Isagawa, T.; Sugiura, Y.; Honda, K.; Wake, M.; Miyazawa, H.; Yamaguchi, Y.; Miura, M.; Jenkins, D.M.; et al. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat. Commun. 2016, 7, 11635. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Artyomov, M.N. Itaconate: The poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 2019, 19, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Peace, C.G.; O’Neill, L.A. The role of itaconate in host defense and inflammation. J. Clin. Investig. 2022, 132, 148548. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Qin, K.; Zhang, Y.; Jia, W.; Chen, Y.; Cheng, B.; Peng, L.; Chen, N.; Liu, Y.; Zhou, W.; et al. S-glycosylation-based cysteine profiling reveals regulation of glycolysis by itaconate. Nat. Chem. Biol. 2019, 15, 983–991. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Muri, J.; Wolleb, H.; Broz, P.; Carreira, E.M.; Kopf, M. Electrophilic Nrf2 activators and itaconate inhibit inflammation at low dose and promote IL-1β production and inflammatory apoptosis at high dose. Redox Biol. 2020, 36, 101647. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M.; Forrester, M.T.; Stamler, J.S. Protein denitrosylation: Enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol. 2009, 10, 721–732. [Google Scholar] [CrossRef]
- Zeng, S.-y.; Yang, L.; Yan, Q.-j.; Gao, L.; Lu, H.-q.; Yan, P.-k. Nox1/4 dual inhibitor GKT137831 attenuates hypertensive cardiac remodelling associating with the inhibition of ADAM17-dependent proinflammatory cytokines-induced signalling pathways in the rats with abdominal artery constriction. Biomed. Pharmacother. 2019, 109, 1907–1914. [Google Scholar] [CrossRef]
- Appukuttan, B.; Ma, Y.; Stempel, A.; Ashander, L.M.; Deliyanti, D.; Wilkinson-Berka, J.L.; Smith, J.R. Effect of NADPH oxidase 1 and 4 blockade in activated human retinal endothelial cells. Clin. Exp. Ophthalmol. 2018, 46, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Jiao, W.; Ji, J.; Li, F.; Guo, J.; Zheng, Y.; Li, S.; Xu, W. Activation of the Notch-Nox4-reactive oxygen species signaling pathway induces cell death in high glucose-treated human retinal endothelial cells. Mol. Med. Rep. 2019, 19, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson-Berka, J.L.; Deliyanti, D.; Rana, I.; Miller, A.G.; Agrotis, A.; Armani, R.; Szyndralewiez, C.; Wingler, K.; Touyz, R.M.; Cooper, M.E.; et al. NADPH oxidase, NOX1, mediates vascular injury in ischemic retinopathy. Antioxid. Redox Signal 2014, 20, 2726–2740. [Google Scholar] [CrossRef]
- Reutens, A.T.; Jandeleit-Dahm, K.; Thomas, M.; Salim, A.; De Livera, A.M.; Bach, L.A.; Colman, P.G.; Davis, T.M.E.; Ekinci, E.I.; Fulcher, G.; et al. A physician-initiated double-blind, randomised, placebo-controlled, phase 2 study evaluating the efficacy and safety of inhibition of NADPH oxidase with the first-in-class Nox-1/4 inhibitor, GKT137831, in adults with type 1 diabetes and persistently elevated urinary albumin excretion: Protocol and statistical considerations. Contemp. Clin. Trials 2020, 90, 105892. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Elksnis, A.; Wikström, P.; Walum, E.; Welsh, N.; Carlsson, P.O. The novel NADPH oxidase 4 selective inhibitor GLX7013114 counteracts human islet cell death in vitro. PLoS ONE 2018, 13, e0204271. [Google Scholar] [CrossRef] [PubMed]
- Dionysopoulou, S.; Wikstrom, P.; Bucolo, C.; Romano, G.L.; Micale, V.; Svensson, R.; Spyridakos, D.; Mastrodimou, N.; Georgakis, S.; Verginis, P.; et al. Topically Administered NOX4 Inhibitor, GLX7013114, Is Efficacious in Treating the Early Pathological Events of Diabetic Retinopathy. Diabetes 2023, 72, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.; Devi, T.S.; Hosoya, K.I.; Terasaki, T.; Singh, L.P. Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis. 2010, 1, e65. [Google Scholar] [CrossRef]
- Elliott, W.J.; Ram, C.V. Calcium channel blockers. J. Clin. Hypertens. 2011, 13, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, J.R.; Martin, S.S. Verapamil: Full spectrum calcium channel blocking agent: An overview. Med. Res. Rev. 1984, 4, 87–109. [Google Scholar] [CrossRef]
- Borowiec, A.M.; Właszczuk, A.; Olakowska, E.; Lewin-Kowalik, J. TXNIP inhibition in the treatment of diabetes. Verapamil as a novel therapeutic modality in diabetic patients. Med. Pharm. Rep. 2022, 95, 243–250. [Google Scholar] [CrossRef]
- Xu, Z.; Wei, Y.; Gong, J.; Cho, H.; Park, J.K.; Sung, E.R.; Huang, H.; Wu, L.; Eberhart, C.; Handa, J.T.; et al. NRF2 plays a protective role in diabetic retinopathy in mice. Diabetologia 2014, 57, 204–213. [Google Scholar] [CrossRef]
- Lu, M.C.; Ji, J.A.; Jiang, Z.Y.; You, Q.D. The Keap1-Nrf2-ARE Pathway As a Potential Preventive and Therapeutic Target: An Update. Med. Res. Rev. 2016, 36, 924–963. [Google Scholar] [CrossRef]
- Borella, R.; Forti, L.; Gibellini, L.; De Gaetano, A.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Synthesis and Anticancer Activity of CDDO and CDDO-Me, Two Derivatives of Natural Triterpenoids. Molecules 2019, 24, 4097. [Google Scholar] [CrossRef]
- Xue, P.; Hu, X.; Powers, J.; Nay, N.; Chang, E.; Kwon, J.; Wong, S.W.; Han, L.; Wu, T.H.; Lee, D.J.; et al. CDDO-Me, Sulforaphane and tBHQ attenuate the RANKL-induced osteoclast differentiation via activating the NRF2-mediated antioxidant response. Biochem. Biophys. Res. Commun. 2019, 511, 637–643. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Yang, Y.X.; Zhe, H.; He, Z.X.; Zhou, S.F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Dev. Ther. 2014, 8, 2075–2088. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar] [CrossRef]
- Sporn, M.B.; Liby, K.T.; Yore, M.M.; Fu, L.; Lopchuk, J.M.; Gribble, G.W. New synthetic triterpenoids: Potent agents for prevention and treatment of tissue injury caused by inflammatory and oxidative stress. J. Nat. Prod. 2011, 74, 537–545. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Shoorei, H.; Bahroudi, Z.; Hussen, B.M.; Talebi, S.F.; Taheri, M.; Ayatollahi, S.A. Nrf2-Related Therapeutic Effects of Curcumin in Different Disorders. Biomolecules 2022, 12, 82. [Google Scholar] [CrossRef]
- Shahcheraghi, S.H.; Salemi, F.; Peirovi, N.; Ayatollahi, J.; Alam, W.; Khan, H.; Saso, L. Nrf2 Regulation by Curcumin: Molecular Aspects for Therapeutic Prospects. Molecules 2021, 27, 167. [Google Scholar] [CrossRef]
- Jiménez-Osorio, A.S.; González-Reyes, S.; Pedraza-Chaverri, J. Natural Nrf2 activators in diabetes. Clin. Chim. Acta 2015, 448, 182–192. [Google Scholar] [CrossRef]
- Rahban, M.; Habibi-Rezaei, M.; Mazaheri, M.; Saso, L.; Moosavi-Movahedi, A.A. Anti-Viral Potential and Modulation of Nrf2 by Curcumin: Pharmacological Implications. Antioxidants 2020, 9, 1228. [Google Scholar] [CrossRef]
- Platania, C.B.M.; Fidilio, A.; Lazzara, F.; Piazza, C.; Geraci, F.; Giurdanella, G.; Leggio, G.M.; Salomone, S.; Drago, F.; Bucolo, C. Retinal Protection and Distribution of Curcumin In Vitro and In Vivo. Front. Pharmacol. 2018, 9, 670. [Google Scholar] [CrossRef]
- Xie, T.; Chen, X.; Chen, W.; Huang, S.; Peng, X.; Tian, L.; Wu, X.; Huang, Y. Curcumin is a Potential Adjuvant to Alleviates Diabetic Retinal Injury via Reducing Oxidative Stress and Maintaining Nrf2 Pathway Homeostasis. Front. Pharmacol. 2021, 12, 796565. [Google Scholar] [CrossRef]
- Bucolo, C.; Drago, F.; Maisto, R.; Romano, G.L.; D’Agata, V.; Maugeri, G.; Giunta, S. Curcumin prevents high glucose damage in retinal pigment epithelial cells through ERK1/2-mediated activation of the Nrf2/HO-1 pathway. J. Cell. Physiol. 2019, 234, 17295–17304. [Google Scholar] [CrossRef]
- Sun, X.G.; Chu, X.H.; Godje Godje, I.S.; Liu, S.Y.; Hu, H.Y.; Zhang, Y.B.; Zhu, L.J.; Wang, H.; Sui, C.; Huang, J.; et al. Aerobic Glycolysis Induced by mTOR/HIF-1α Promotes Early Brain Injury After Subarachnoid Hemorrhage via Activating M1 Microglia. Transl. Stroke Res. 2024, 15, 1–15. [Google Scholar] [CrossRef]
- Al-Bari, M.A.A.; Xu, P. Molecular regulation of autophagy machinery by mTOR-dependent and -independent pathways. Ann. N. Y. Acad. Sci. 2020, 1467, 3–20. [Google Scholar] [CrossRef]
- Ahumada-Castro, U.; Silva-Pavez, E.; Lovy, A.; Pardo, E.; Molgό, J.; Cárdenas, C. MTOR-independent autophagy induced by interrupted endoplasmic reticulum-mitochondrial Ca2+ communication: A dead end in cancer cells. Autophagy 2019, 15, 358–361. [Google Scholar] [CrossRef]
- Du, Y.; Veenstra, A.; Palczewski, K.; Kern, T.S. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc. Natl. Acad. Sci. USA 2013, 110, 16586–16591. [Google Scholar] [CrossRef]
- Sanges, D.; Comitato, A.; Tammaro, R.; Marigo, V. Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 17366–17371. [Google Scholar] [CrossRef]
- Alder, V.A.; Ben-Nun, J.; Cringle, S.J. PO2 profiles and oxygen consumption in cat retina with an occluded retinal circulation. Investig. Ophthalmol. Vis. Sci. 1990, 31, 1029–1034. [Google Scholar]
- Funk, R.H.; Schumann, U.; Engelmann, K.; Becker, K.A.; Roehlecke, C.; Anatomie, I.F. Blue light induced retinal oxidative stress:Implications for macular degeneration. World J. Ophthalmol. 2014, 4, 29–34. [Google Scholar] [CrossRef]
- Bruschi, M.; Bartolucci, M.; Petretto, A.; Calzia, D.; Caicci, F.; Manni, L.; Traverso, C.E.; Candiano, G.; Panfoli, I. Differential expression of the five redox complexes in the retinal mitochondria or rod outer segment disks is consistent with their different functionality. FASEB Bioadv. 2020, 2, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Panfoli, I.; Calzia, D.; Ravera, S.; Bruschi, M.; Tacchetti, C.; Candiani, S.; Morelli, A.; Candiano, G. Extramitochondrial tricarboxylic acid cycle in retinal rod outer segments. Biochimie 2011, 93, 1565–1575. [Google Scholar] [CrossRef]
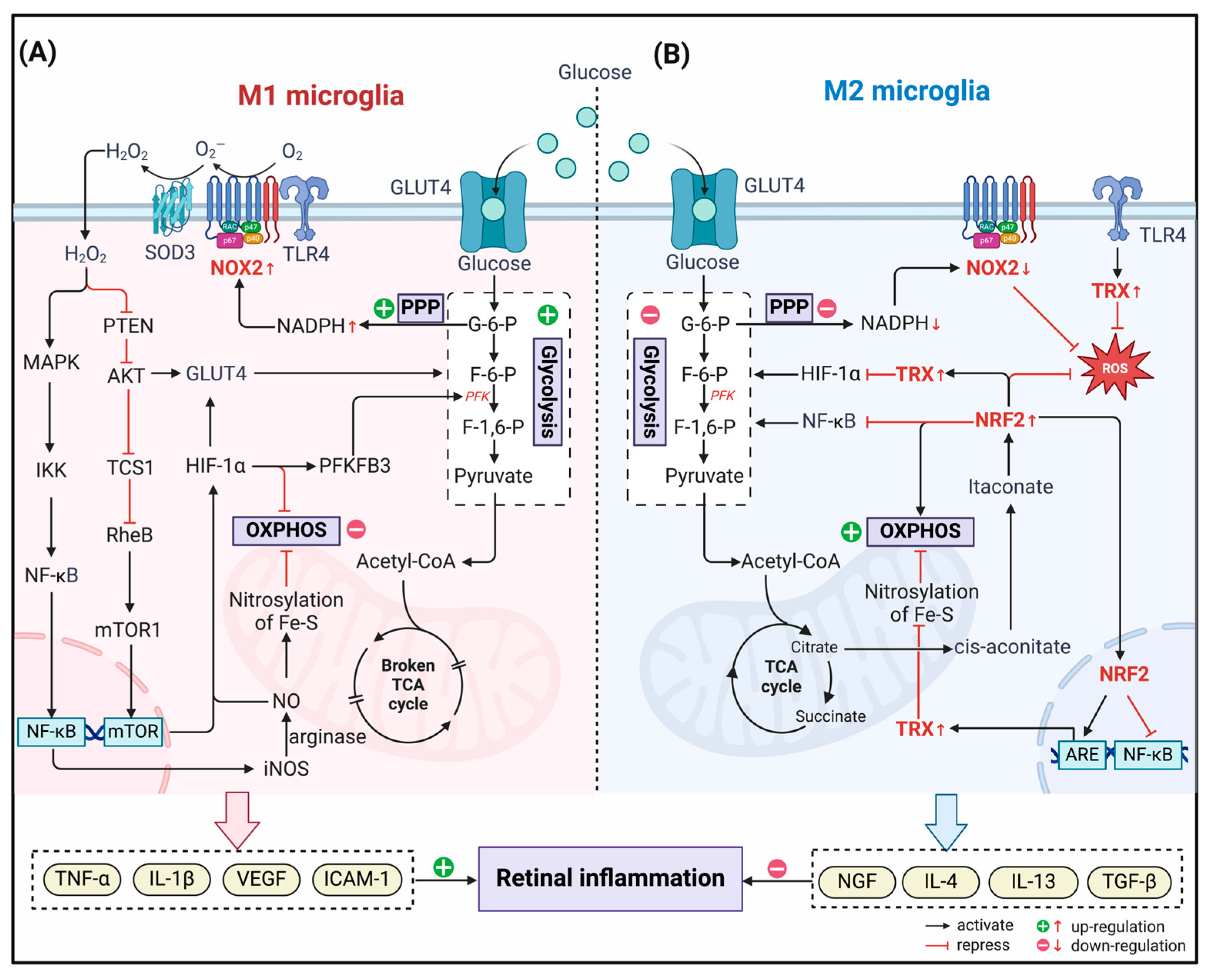
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).