
TNF-α Levels Are Increased in Patients with Subjective Cognitive Impairment and Are Negatively Correlated with β Amyloid-42

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Blood Sampling
2.3. Enzyme-Linked Immunosorbent Assay (ELISA)
2.4. Microglia Cell Culture
2.5. Western Blot and Immunoprecipitation (IP)
2.6. Immunofluorescence
2.7. Image Acquisition and Processing
2.8. Statistical Methodology
3. Results
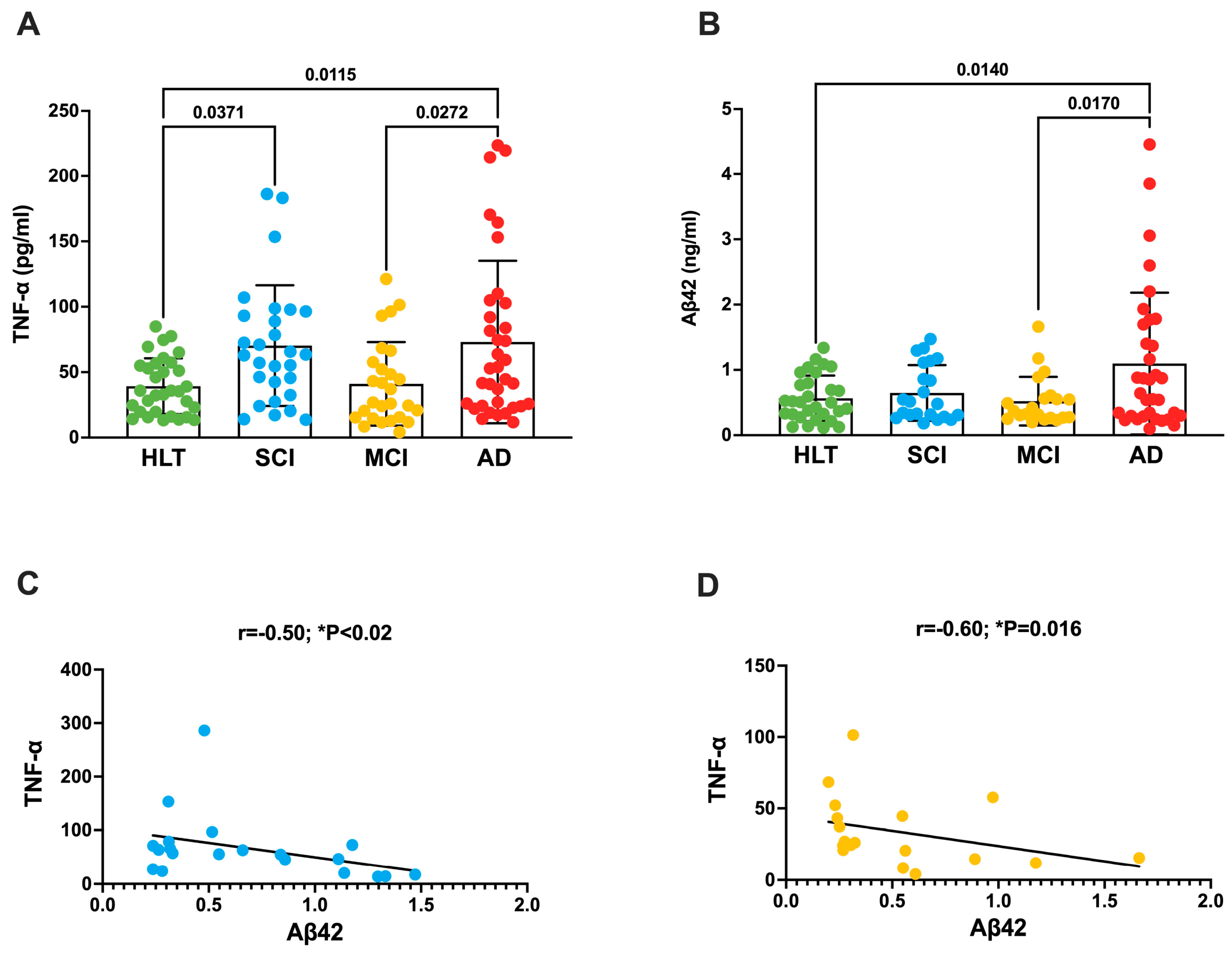
3.1. TNF-a Increases in the Plasma of Patients with SCI and AD
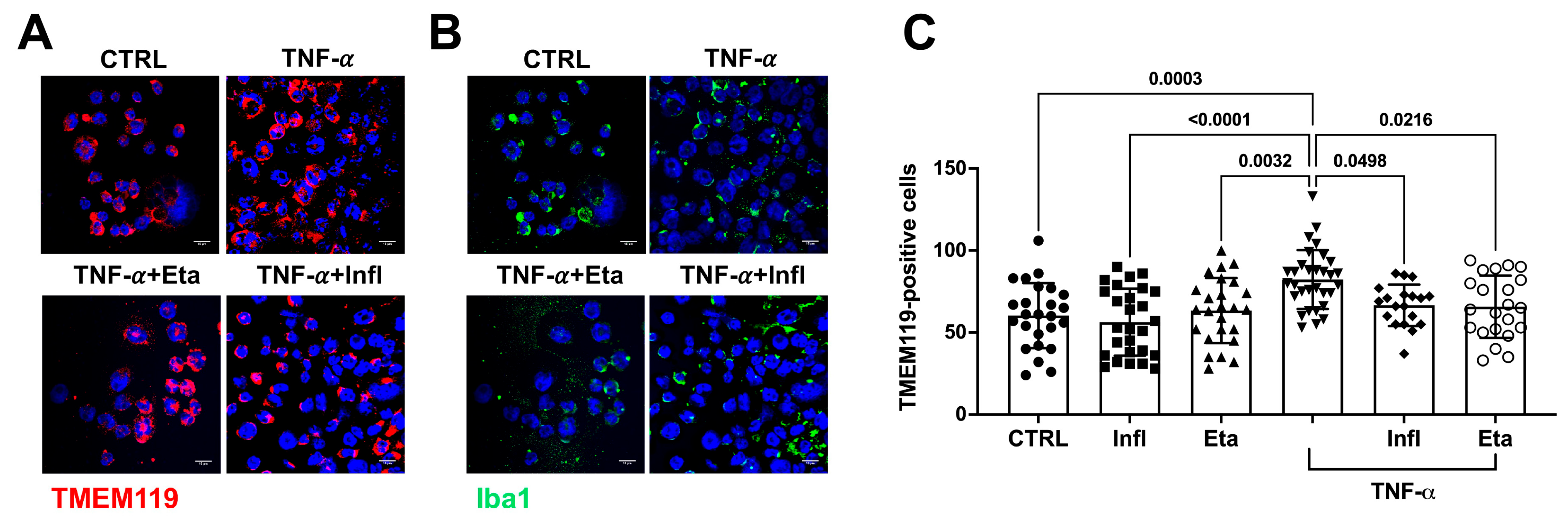
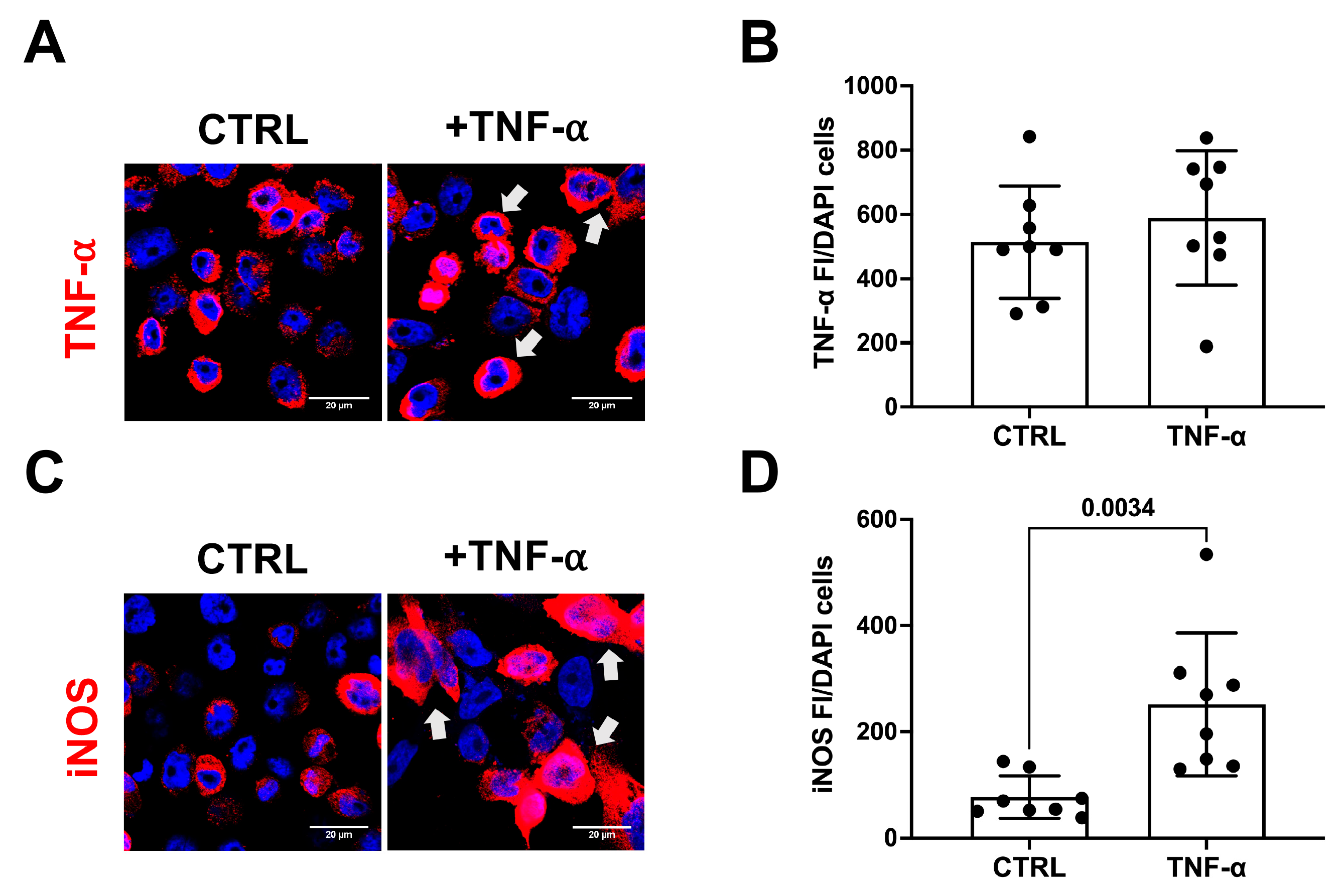
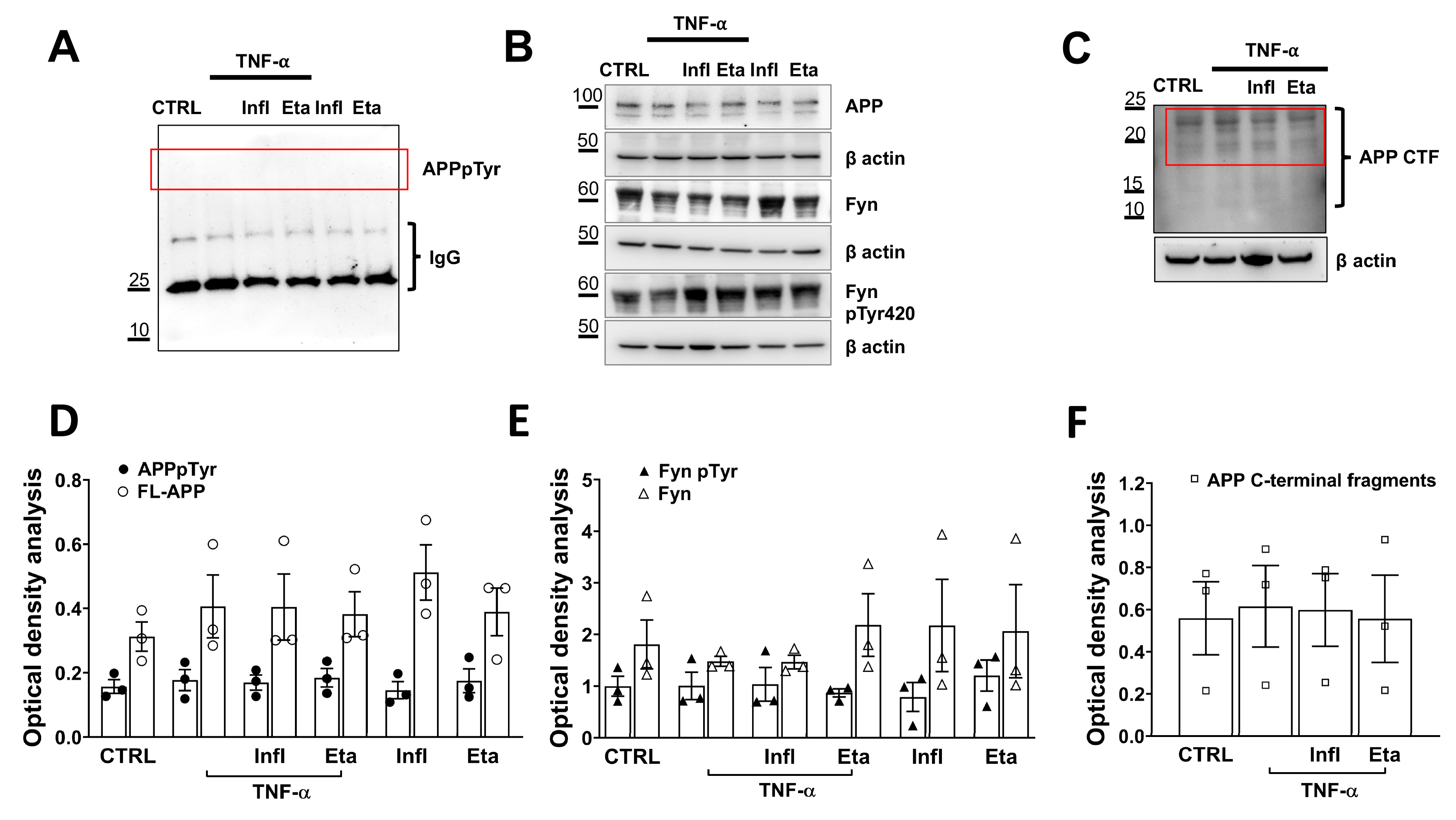
3.2. TNF-α Does Not Initiate Amyloidogenic Processes in Human Microglial Cell Cultures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rogers, C.J.; Ayuso, J.; Hackney, M.E.; Penza, C. Alzheimer Disease and Related Cognitive Impairment in Older Adults: A Narrative Review of Screening, Prevention, and Management for Manual Therapy Providers. J. Chiropr. Med. 2023, 22, 148–156. [Google Scholar] [CrossRef]
- Cohen, S.; Cummings, J.; Knox, S.; Potashman, M.; Harrison, J. Clinical Trial Endpoints and Their Clinical Meaningfulness in Early Stages of Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2022, 9, 507–522. [Google Scholar] [CrossRef]
- Eisenmenger, L.B.; Peret, A.; Famakin, B.M.; Spahic, A.; Roberts, G.S.; Bockholt, J.H.; Johnson, K.M.; Paulsen, J.S. Vascular contributions to Alzheimer’s disease. Transl. Res. 2022, 254, 41–53. [Google Scholar] [CrossRef]
- Licastro, F. Special Issue Editorial: “Infections, Inflammation and Neurodegeneration in Alzheimer Disease” Infections, Neuronal Senescence, and Dementia. Int. J. Mol. Sci. 2022, 23, 5865. [Google Scholar] [CrossRef]
- van Loo, G.; Bertrand, M.J.M. Death by TNF: A road to inflammation. Nat. Rev. Immunol. 2022, 23, 289–303. [Google Scholar] [CrossRef]
- Novoa, C.; Salazar, P.; Cisternas, P.; Gherardelli, C.; Vera-Salazar, R.; Zolezzi, J.M.; Inestrosa, N.C. Inflammation context in Alzheimer’s disease, a relationship intricate to define. Biol. Res. 2022, 55, 39. [Google Scholar] [CrossRef]
- Ekert, J.O.; Gould, R.L.; Reynolds, G.; Howard, R.J. TNF alpha inhibitors in Alzheimer’s disease: A systematic review. Int. J. Geriatr. Psychiatry 2018, 33, 688–694. [Google Scholar] [CrossRef]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xu, R.; Kaelber, D.C.; Gurney, M.E. Tumor Necrosis Factor (TNF) blocking agents are associated with lower risk for Alzheimer’s disease in patients with rheumatoid arthritis and psoriasis. PLoS ONE 2020, 15, e0229819. [Google Scholar] [CrossRef] [PubMed]
- Wang, T. TNF-alpha G308A polymorphism and the susceptibility to Alzheimer’s disease: An updated meta-analysis. Arch. Med. Res. 2015, 46, 24–30.e21. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.M.; Lin, M.T.; Larson, E.B.; Maezawa, I.; Tseng, L.H.; Edwards, K.L.; Schellenberg, G.D.; Hansen, J.A.; Kukull, W.A.; Jin, L.W. Tumor necrosis factor alpha and interleukin 10 promoter region polymorphisms and risk of late-onset Alzheimer disease. Arch. Neurol. 2006, 63, 1165–1169. [Google Scholar] [CrossRef][Green Version]
- Alvarez, V.; Mata, I.F.; González, P.; Lahoz, C.H.; MartÃnez, C.; Peña, J.; Guisasola, L.M.; Coto, E. Association between the TNFalpha-308 A/G polymorphism and the onset-age of Alzheimer disease. Am. J. Med. Genet. 2002, 114, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fan, H.; Ni, M.; Zhang, W.; Fang, F.; Sun, J.; Lyu, P.; Ma, P. Etanercept Reduces Neuron Injury and Neuroinflammation via Inactivating c-Jun N-terminal Kinase and Nuclear Factor-κB Pathways in Alzheimer’s Disease: An In Vitro and In Vivo Investigation. Neuroscience 2022, 484, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Paouri, E.; Tzara, O.; Kartalou, G.I.; Zenelak, S.; Georgopoulos, S. Peripheral Tumor Necrosis Factor-Alpha (TNF-α) Modulates Amyloid Pathology by Regulating Blood-Derived Immune Cells and Glial Response in the Brain of AD/TNF Transgenic Mice. J. Neurosci. 2017, 37, 5155–5171. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Herring, A.; Ceballos-Diaz, C.; Das, P.; Golde, T.E. Hippocampal expression of murine TNFα results in attenuation of amyloid deposition in vivo. Mol. Neurodegener. 2011, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Janelsins, M.C.; Mastrangelo, M.A.; Park, K.M.; Sudol, K.L.; Narrow, W.C.; Oddo, S.; LaFerla, F.M.; Callahan, L.M.; Federoff, H.J.; Bowers, W.J. Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. Am. J. Pathol. 2008, 173, 1768–1782. [Google Scholar] [CrossRef]
- Jayaraman, A.; Htike, T.T.; James, R.; Picon, C.; Reynolds, R. TNF-mediated neuroinflammation is linked to neuronal necroptosis in Alzheimer’s disease hippocampus. Acta Neuropathol. Commun. 2021, 9, 159. [Google Scholar] [CrossRef]
- Lee, K.H.; Cha, M.; Lee, B.H. Crosstalk between Neuron and Glial Cells in Oxidative Injury and Neuroprotection. Int. J. Mol. Sci. 2021, 22, 13315. [Google Scholar] [CrossRef]
- Fischer, R.; Maier, O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of TNF. Oxidative Med. Cell. Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef]
- Ionescu-Tucker, A.; Cotman, C.W. Emerging roles of oxidative stress in brain aging and Alzheimer’s disease. Neurobiol. Aging 2021, 107, 86–95. [Google Scholar] [CrossRef]
- Ferretti, G.; Serafini, S.; Angiolillo, A.; Monterosso, P.; Di Costanzo, A.; Matrone, C. Advances in peripheral blood biomarkers of patients with Alzheimer’s disease: Moving closer to personalized therapies. Biomed. Pharmacother. 2023, 165, 115094. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.J.; Lee, J.H. Subjective Cognitive Decline and Alzheimer’s Disease Spectrum Disorder. Dement. Neurocogn. Disord. 2017, 16, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Flores-Cordero, J.A.; Pérez-Pérez, A.; Jiménez-Cortegana, C.; Alba, G.; Flores-Barragán, A.; Sánchez-Margalet, V. Obesity as a Risk Factor for Dementia and Alzheimer’s Disease: The Role of Leptin. Int. J. Mol. Sci. 2022, 23, 5202. [Google Scholar] [CrossRef] [PubMed]
- Abdi, S.; Alghamdi, A.A.; AlGhunaim, N.N.A.; Almutairi, R.M.; Ataya, F.S.; Ansari, M.G.A.; Hussain, S.D.; Masoud, M.; Alamro, A.; Almutairi, O.T.; et al. Association of Alzheimer’s Disease with Genetic Variants of Apolipoprotein E, Clusterin, TNF-α, and IL-6 Among Elderly Saudis. Curr. Pharm. Biotechnol. 2022, 23, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Reveglia, P.; Nasso, R.; Angiolillo, A.; Lecce, L.; Paolillo, C.; De Tullio, S.; Gelzo, M.; Di Costanzo, A.; Matrone, C.; Corso, G. Tandem Mass Spectrometry as Strategy for the Selective Identification and Quantification of the Amyloid Precursor Protein Tyr682 Residue Phosphorylation Status in Human Blood Mononuclear Cells. Biomolecules 2021, 11, 1297. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzi, F.; Frisardi, V.; Annunziato, L.; Matrone, C. Might Fibroblasts from Patients with Alzheimer’s Disease Reflect the Brain Pathology? A Focus on the Increased Phosphorylation of Amyloid Precursor Protein Tyr. Brain Sci. 2021, 11, 103. [Google Scholar] [CrossRef]
- Iannuzzi, F.; Sirabella, R.; Canu, N.; Maier, T.J.; Annunziato, L.; Matrone, C. Fyn Tyrosine Kinase Elicits Amyloid Precursor Protein Tyr682 Phosphorylation in Neurons from Alzheimer’s Disease Patients. Cells 2020, 9, 1807. [Google Scholar] [CrossRef]
- Haass, C.; Mandelkow, E. Fyn-tau-amyloid: A toxic triad. Cell 2010, 142, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef]
- Ferretti, G.; Romano, A.; Sirabella, R.; Serafini, S.; Maier, T.J.; Matrone, C. An increase in Semaphorin 3A biases the axonal direction and induces an aberrant dendritic arborization in an in vitro model of human neural progenitor differentiation. Cell Biosci. 2022, 12, 182. [Google Scholar] [CrossRef]
- Dello Russo, C.; Cappoli, N.; Coletta, I.; Mezzogori, D.; Paciello, F.; Pozzoli, G.; Navarra, P.; Battaglia, A. The human microglial HMC3 cell line: Where do we stand? A systematic literature review. J. Neuroinflamm. 2018, 15, 259. [Google Scholar] [CrossRef]
- Matrone, C.; Barbagallo, A.P.; La Rosa, L.R.; Florenzano, F.; Ciotti, M.T.; Mercanti, D.; Chao, M.V.; Calissano, P.; D’Adamio, L. APP is phosphorylated by TrkA and regulates NGF/TrkA signaling. J. Neurosci. 2011, 31, 11756–11761. [Google Scholar] [CrossRef]
- Satoh, J.; Kino, Y.; Asahina, N.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2016, 36, 39–49. [Google Scholar] [CrossRef]
- Matrone, C.; Iannuzzi, F.; Annunziato, L. The Y682ENPTY687 motif of APP: Progress and insights toward a targeted therapy for Alzheimer’s disease patients. Ageing Res. Rev. 2019, 52, 120–128. [Google Scholar] [CrossRef]
- Lee, G.; Thangavel, R.; Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Fang, S.M.; Do, L.H.; Andreadis, A.; Van Hoesen, G.; Ksiezak-Reding, H. Phosphorylation of tau by fyn: Implications for Alzheimer’s disease. J. Neurosci. 2004, 24, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Haytural, H.; Lundgren, J.L.; Köse, T.B.; Jordà-Siquier, T.; Kalcheva, M.; Seed Ahmed, M.; Winblad, B.; Sundström, E.; Barthet, G.; Tjernberg, L.O.; et al. Non-specific Detection of a Major Western Blotting Band in Human Brain Homogenates by a Multitude of Amyloid Precursor Protein Antibodies. Front. Aging Neurosci. 2019, 11, 273. [Google Scholar] [CrossRef] [PubMed]
- Padala, S.P.; Newhouse, P.A. Blood-based biomarkers in Alzheimer’s disease: A mini-review. Metab. Brain Dis. 2023, 38, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.T.; Howell, J.C.; Ozturk, T.; Gangishetti, U.; Kollhoff, A.L.; Hatcher-Martin, J.M.; Anderson, A.M.; Tyor, W.R. CSF Cytokines in Aging, Multiple Sclerosis, and Dementia. Front. Immunol. 2019, 10, 480. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Cribbs, D.H.; Anderson, A.J.; Cummings, B.J.; Su, J.H.; Wasserman, A.J.; Cotman, C.W. The induction of the TNFalpha death domain signaling pathway in Alzheimer’s disease brain. Neurochem. Res. 2003, 28, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Versele, R.; Sevin, E.; Gosselet, F.; Fenart, L.; Candela, P. TNF-α and IL-1β Modulate Blood-Brain Barrier Permeability and Decrease Amyloid-β Peptide Efflux in a Human Blood-Brain Barrier Model. Int. J. Mol. Sci. 2022, 23, 10235. [Google Scholar] [CrossRef]
- Parada, H.; Bergstrom, J.; Bangen, K.J.; Reas, E.T. Pulse pressure trajectories predict brain microstructure in community-dwelling older adults: Associations with executive function and modification by APOE. Alzheimers Dement. 2023, 19, 1963–1973. [Google Scholar] [CrossRef]
- Elman, J.A.; Puckett, O.K.; Hagler, D.J.; Pearce, R.C.; Fennema-Notestine, C.; Hatton, S.N.; Lyons, M.J.; McEvoy, L.K.; Panizzon, M.S.; Reas, E.T.; et al. Associations between MRI-assessed locus coeruleus integrity and cortical gray matter microstructure. Cereb. Cortex 2022, 32, 4191–4203. [Google Scholar] [CrossRef]
- Zheng, C.; Fillmore, N.R.; Ramos-Cejudo, J.; Brophy, M.; Osorio, R.; Gurney, M.E.; Qiu, W.Q.; Au, R.; Perry, G.; Dubreuil, M.; et al. Potential long-term effect of tumor necrosis factor inhibitors on dementia risk: A propensity score matched retrospective cohort study in US veterans. Alzheimers Dement. 2022, 18, 1248–1259. [Google Scholar] [CrossRef]
- Torres-Acosta, N.; O’Keefe, J.H.; O’Keefe, E.L.; Isaacson, R.; Small, G. Therapeutic Potential of TNF-α Inhibition for Alzheimer’s Disease Prevention. J. Alzheimers Dis. 2020, 78, 619–626. [Google Scholar] [CrossRef]
- Desai, R.J.; Varma, V.R.; Gerhard, T.; Segal, J.; Mahesri, M.; Chin, K.; Horton, D.B.; Kim, S.C.; Schneeweiss, S.; Thambisetty, M. Comparative Risk of Alzheimer Disease and Related Dementia Among Medicare Beneficiaries with Rheumatoid Arthritis Treated with Targeted Disease-Modifying Antirheumatic Agents. JAMA Netw. Open 2022, 5, e226567. [Google Scholar] [CrossRef]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef]
- Tarkowski, E.; Andreasen, N.; Tarkowski, A.; Blennow, K. Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Cunningham, C.; Zotova, E.; Culliford, D.; Perry, V.H. Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology 2011, 77, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Meng, L.; Shen, L. Multiple roles of short-chain fatty acids in Alzheimer disease. Nutrition 2021, 93, 111499. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xun, K.; Chen, L.; Wang, Y. TNF-alpha, a potent lipid metabolism regulator. Cell Biochem. Funct. 2009, 27, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Matrone, C.; Petrillo, F.; Nasso, R.; Ferretti, G. Fyn Tyrosine Kinase as Harmonizing Factor in Neuronal Functions and Dysfunctions. Int. J. Mol. Sci. 2020, 21, 4444. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lei, S.; Cai, T.; Cheng, Y.; Bai, J.; Fu, W.; Huang, M. Inducible nitric oxide synthase activity mediates TNF-α-induced endothelial cell dysfunction. Am. J. Physiol. Cell Physiol. 2023, 325, C780–C795. [Google Scholar] [CrossRef] [PubMed]
- Bernath, A.K.; Murray, T.E.; Shirley Yang, S.; Gibon, J.; Klegeris, A. Microglia secrete distinct sets of neurotoxins in a stimulus-dependent manner. Brain Res. 2023, 1807, 148315. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HLT (n.34) | SCI (n.30) | MCI (n.30) | AD (n.39) | Chi-Square Test or Unpaired T Test (p Value) | |
|---|---|---|---|---|---|
| Gender | 15F/19M | 17F/13M | 18F/12M | 20F/19M | NS |
| Age (years, Mean ± SEM) | 67.9 ± 1.5 | 71.2 ± 1.2 | 68.2 ± 1.9 | 78.4 ± 1.3 | NS |
| Education (years, Mean ± SEM) | 10.7 ± 0.7 | 11.7 ± 0.9 | 10.7 ± 0.9 | 7.97 ± 1.0 | NS |
| BMI (Mean ± SEM) | 28.5 ± 0.73 | 27.1 ± 0.8 | 27.6 ± 0.7 | 25.2 ± 0.8 | NS |
| MMSE (Mean ± SEM) | 29.55 ± 0.31 * | 29.8 ± 0.3 * | 27.9 ± 0.3 * | 10.7 ± 1.3 | * (p < 0.05 vs. AD) |
| Risk factors | |||||
| Smokers | 19/34 | 11/30 | 9/30 | 11/39 | NS |
| Alcohol (more than 3 glasses) | 0/34 | 1/30 | 2/30 | 4/39 | NS |
| Comorbidities | |||||
| Hypertension | 18/34 | 18/30 | 16/30 | 17/39 | NS |
| Myocardial infarction | 1/34 | 1/30 | 0/30 | 3/39 | NS |
| Tia/stroke | 0/34 | 0/30 | 2/30 | 1/39 | NS |
| Dyslipidemia | 8/34 | 10/30 | 11/30 | 9/39 | NS |
| Diabetes | * 0/34 | 2/30 | 3/30 | 8/39 | * (p = 0.04 vs. AD) |
| TNF-α | 39.30 ± 3.8 | 73.9 ± 10.9 * | 41.0 ± 6.15 § | 73.0 ± 10.5 * | * p < 0.05 vs. HLT § p < 0.05 vs. AD |
| Aβ42 | 0.57 ± 0.063 # | 0.65 ± 0.09 | 0.52 ± 0.08 # | 1.09 ± 0.18 | # p < 0.05 vs. AD |
| TNF-α vs. | HLT | SCI | MCI | AD | |
| Age | |||||
| r | 0.04 | −0.21 | −0.02 | 0.05 | |
| p | 0.82 | 0.28 | 0.9 | 0.77 | |
| 95% CI | −0.3 to 0.4 | −0.5 to 0.2 | −0.4 to 0.4 | −0.3 to 0.3 | |
| n | 32 | 28 | 27 | 35 | |
| BMI | |||||
| r | 0.17 | 0.08 | −0.17 | −0.16 | |
| p | 0.35 | 0.65 | 0.39 | 0.39 | |
| 95% CI | −0.2 to 0.5 | −0.3 to 0.5 | −0.5 to 0.2 | −0.5 to 0.2 | |
| n | 32 | 28 | 27 | 29 | |
| MMSE | |||||
| r | −0.12 | −0.11 | 0.16 | 0.13 | |
| p | 0.52 | 0.57 | 0.41 | 0.45 | |
| 95% CI | −0.4 to 0.2 | −0.47 to 0.28 | −0.2 to 0.5 | −0.2 to 0.5 | |
| n | 32 | 28 | 27 | 35 | |
| Aβ42 | |||||
| r | −0.30 | −0.50 | −0.60 | −0.20 | |
| p | 0.17 | 0.03 | 0.016 | 0.32 | |
| 95% CI | −0.6 to 0.1 | −0.8 to 0.05 | −0.8 to −0.1 | −0.5 to 0.2 | |
| n | 29 | 20 | 18 | 32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serafini, S.; Ferretti, G.; Monterosso, P.; Angiolillo, A.; Di Costanzo, A.; Matrone, C. TNF-α Levels Are Increased in Patients with Subjective Cognitive Impairment and Are Negatively Correlated with β Amyloid-42. Antioxidants 2024, 13, 216. https://doi.org/10.3390/antiox13020216
Serafini S, Ferretti G, Monterosso P, Angiolillo A, Di Costanzo A, Matrone C. TNF-α Levels Are Increased in Patients with Subjective Cognitive Impairment and Are Negatively Correlated with β Amyloid-42. Antioxidants. 2024; 13(2):216. https://doi.org/10.3390/antiox13020216
Chicago/Turabian StyleSerafini, Sara, Gabriella Ferretti, Paola Monterosso, Antonella Angiolillo, Alfonso Di Costanzo, and Carmela Matrone. 2024. "TNF-α Levels Are Increased in Patients with Subjective Cognitive Impairment and Are Negatively Correlated with β Amyloid-42" Antioxidants 13, no. 2: 216. https://doi.org/10.3390/antiox13020216
APA StyleSerafini, S., Ferretti, G., Monterosso, P., Angiolillo, A., Di Costanzo, A., & Matrone, C. (2024). TNF-α Levels Are Increased in Patients with Subjective Cognitive Impairment and Are Negatively Correlated with β Amyloid-42. Antioxidants, 13(2), 216. https://doi.org/10.3390/antiox13020216