
Oxidative Stress Plays an Important Role in Glutamatergic Excitotoxicity-Induced Cochlear Synaptopathy: Implication for Therapeutic Molecules Screening

 , ,
, ,  and
and 
Abstract
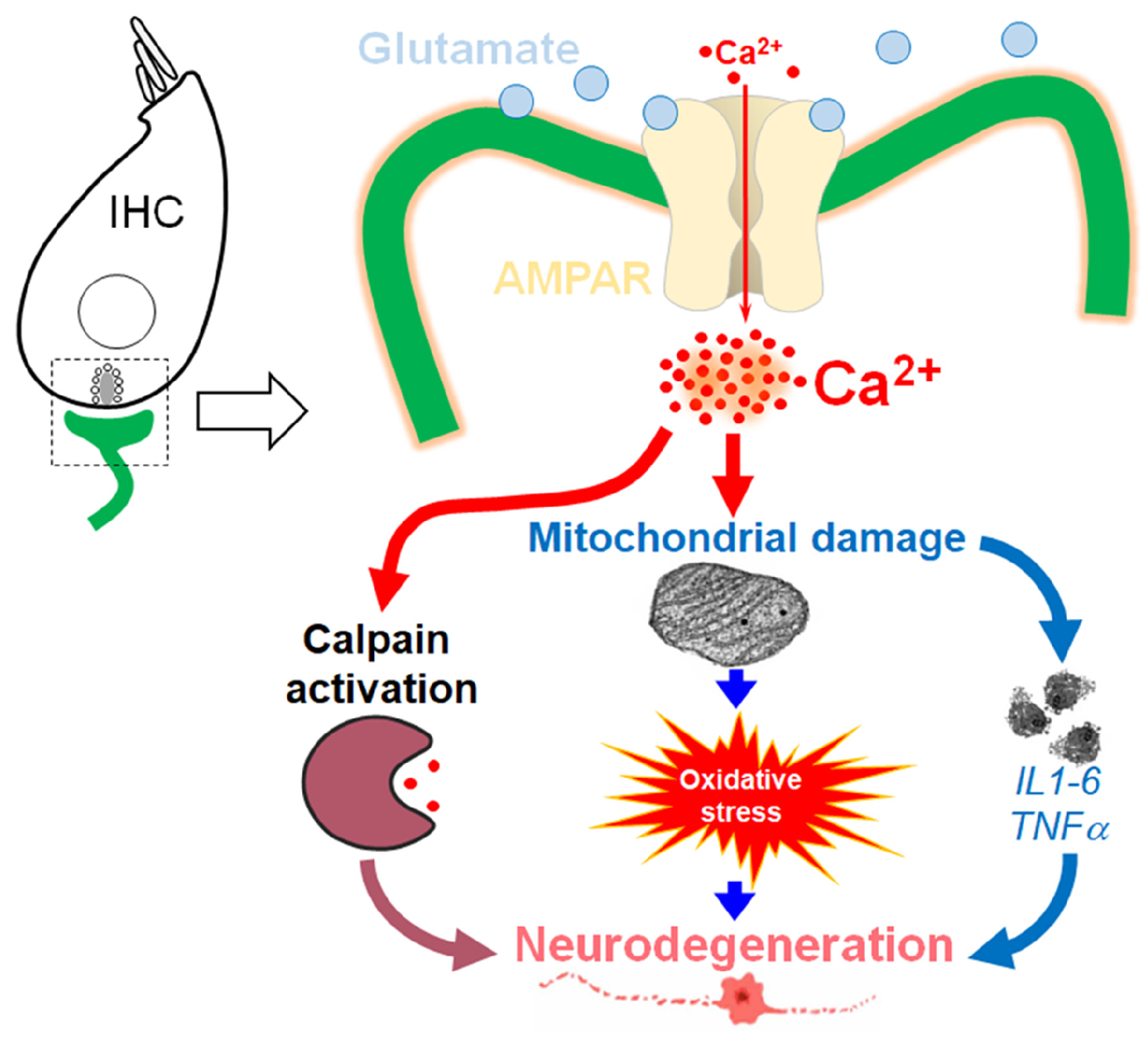
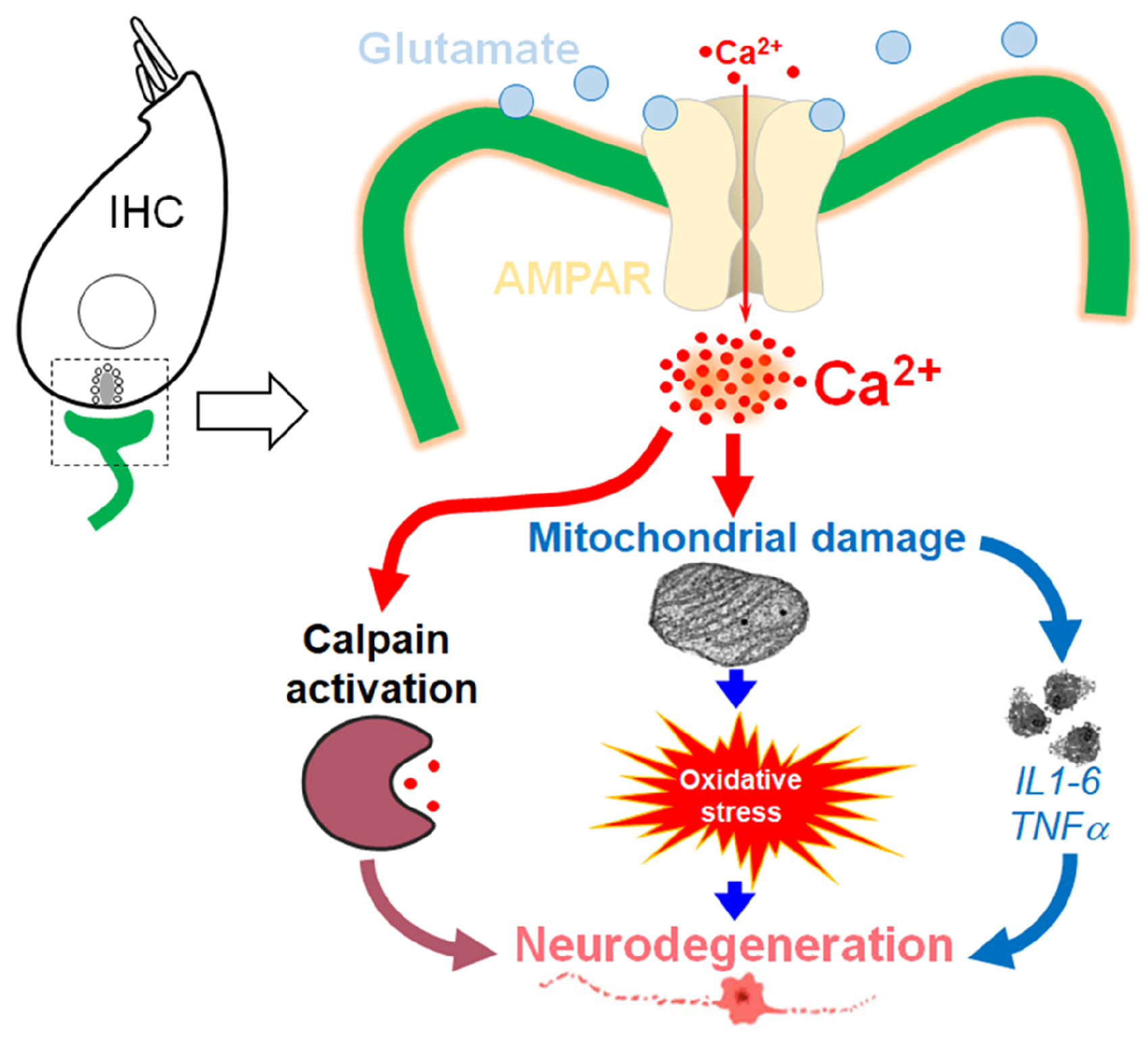
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Organ of Corti, and Whole Cochlea Cultures
2.3. KA-Induced Excitotoxic Damage
2.4. Pharmacological Drug Preparation and Interventions
2.5. Counting of Ribbon Synapses of IHCs
2.6. Counting of Terminals of Auditory Nerve Fibers
2.7. Measurement of Enzymatic Activities and Oxidative Stress
2.8. Immunocytochemistry
2.9. Immunoblotting
2.10. Quantitative PCR
3. Statistics
4. Results
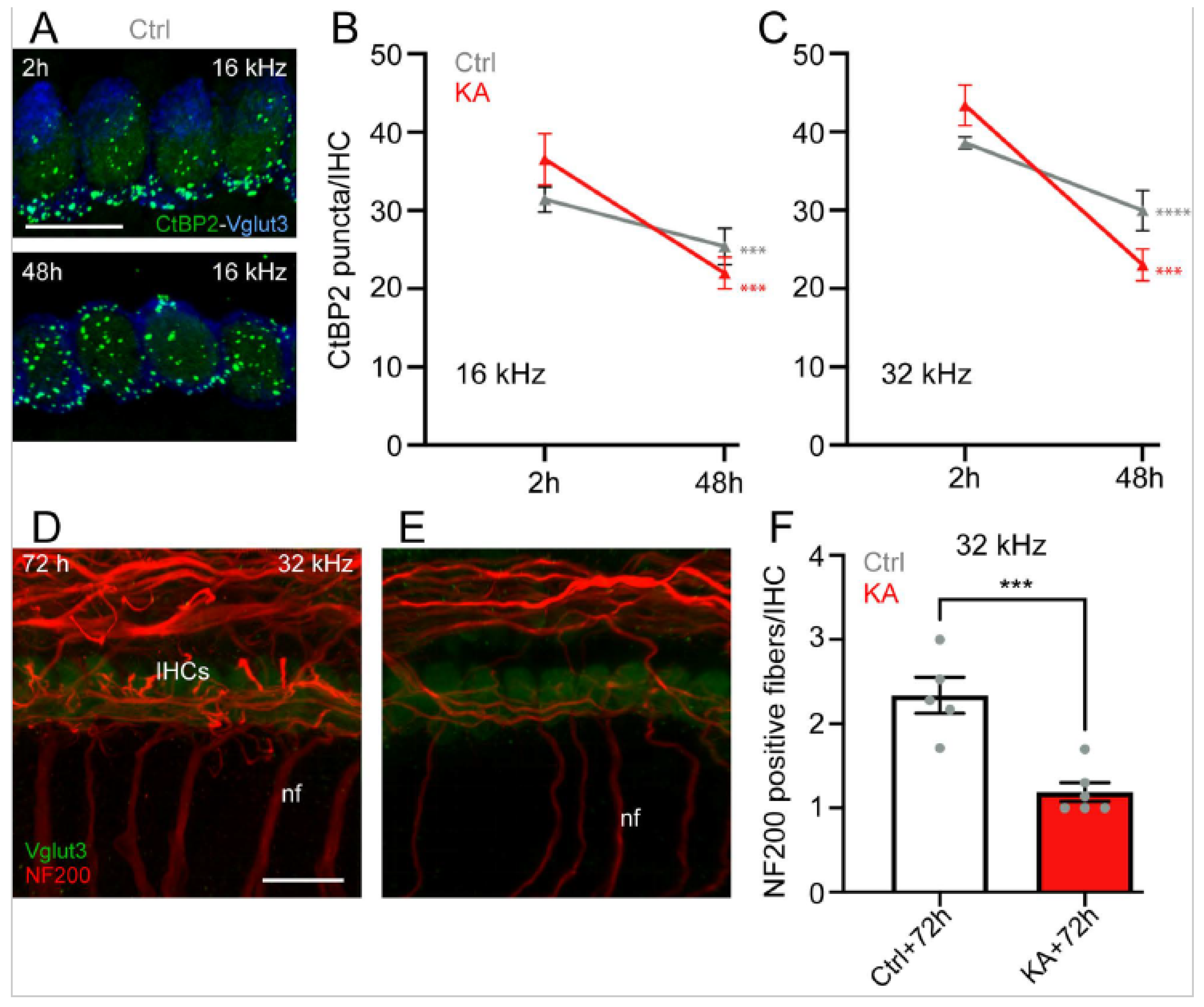
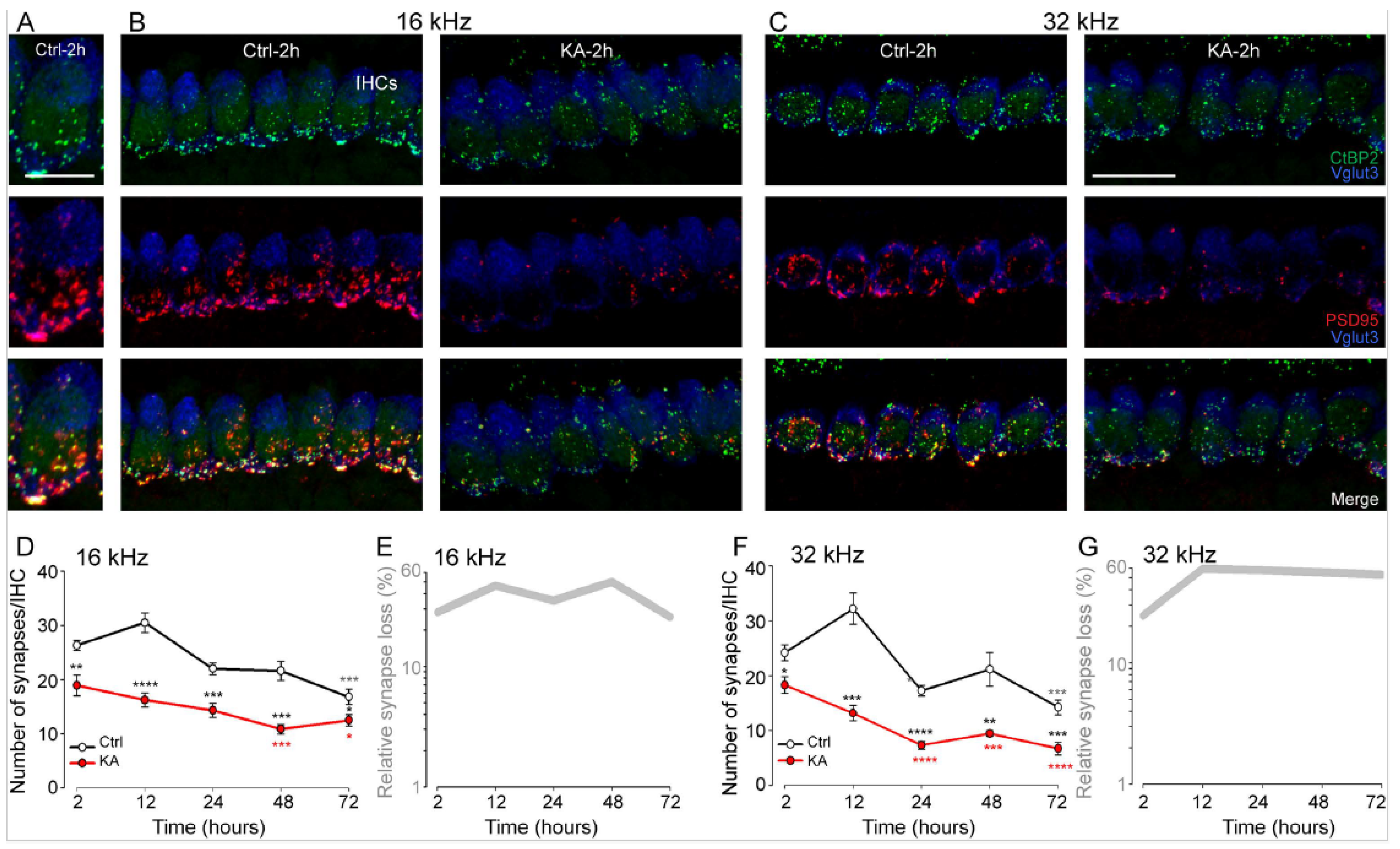
4.1. Short-Term KA Exposure Induces Rapid IHC Synaptopathy
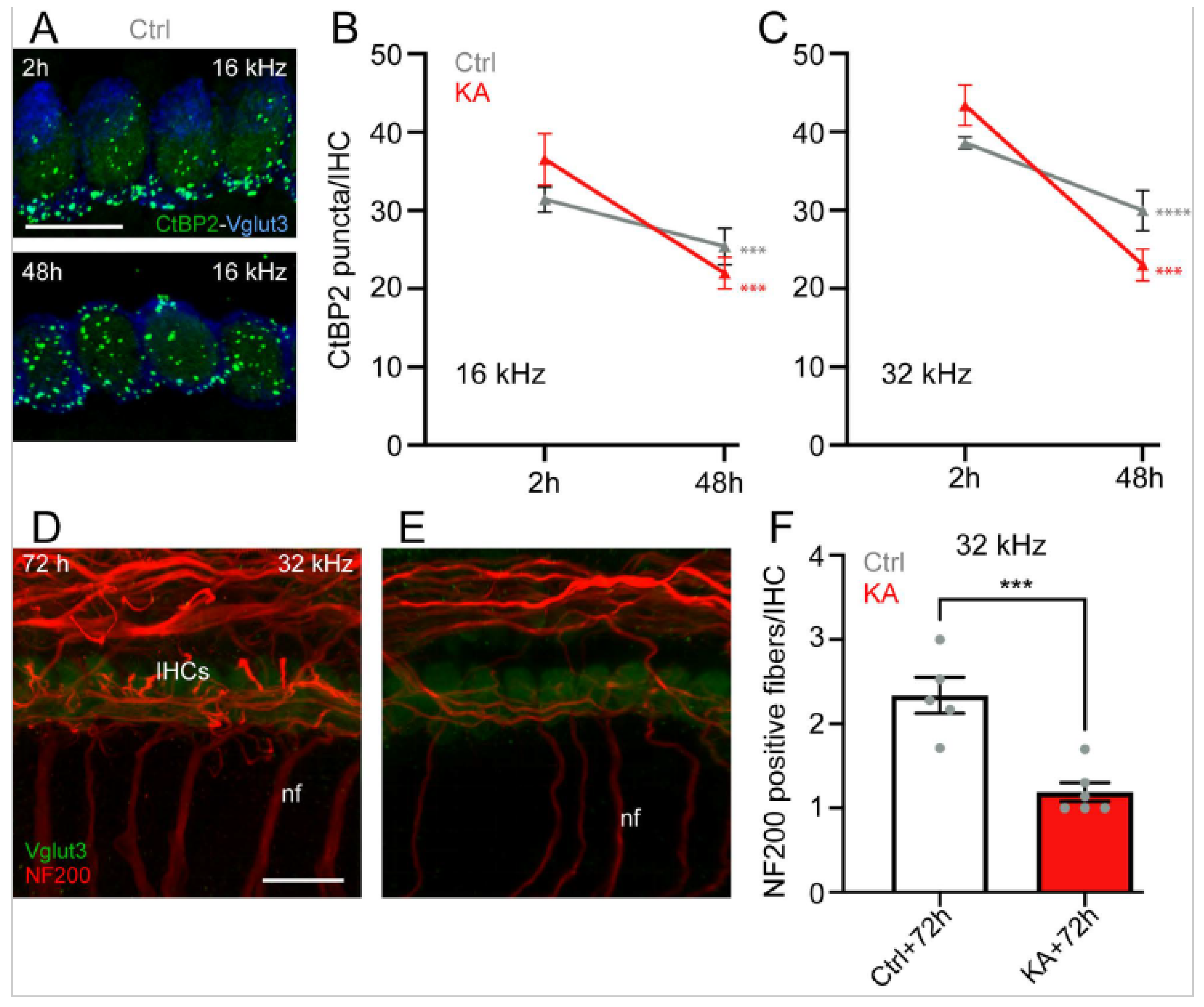
4.2. Time Course of KA-Induced IHC Synapse Loss
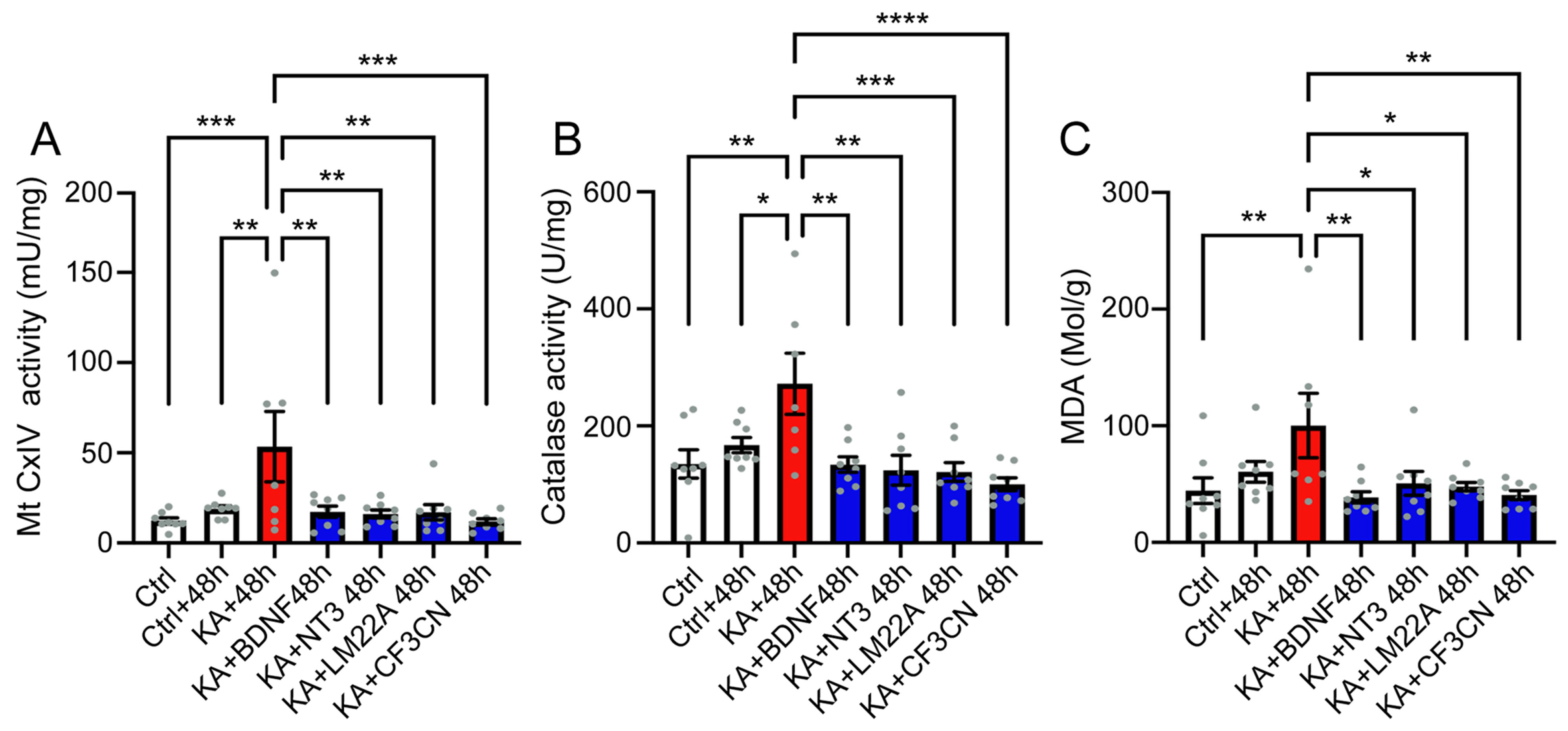
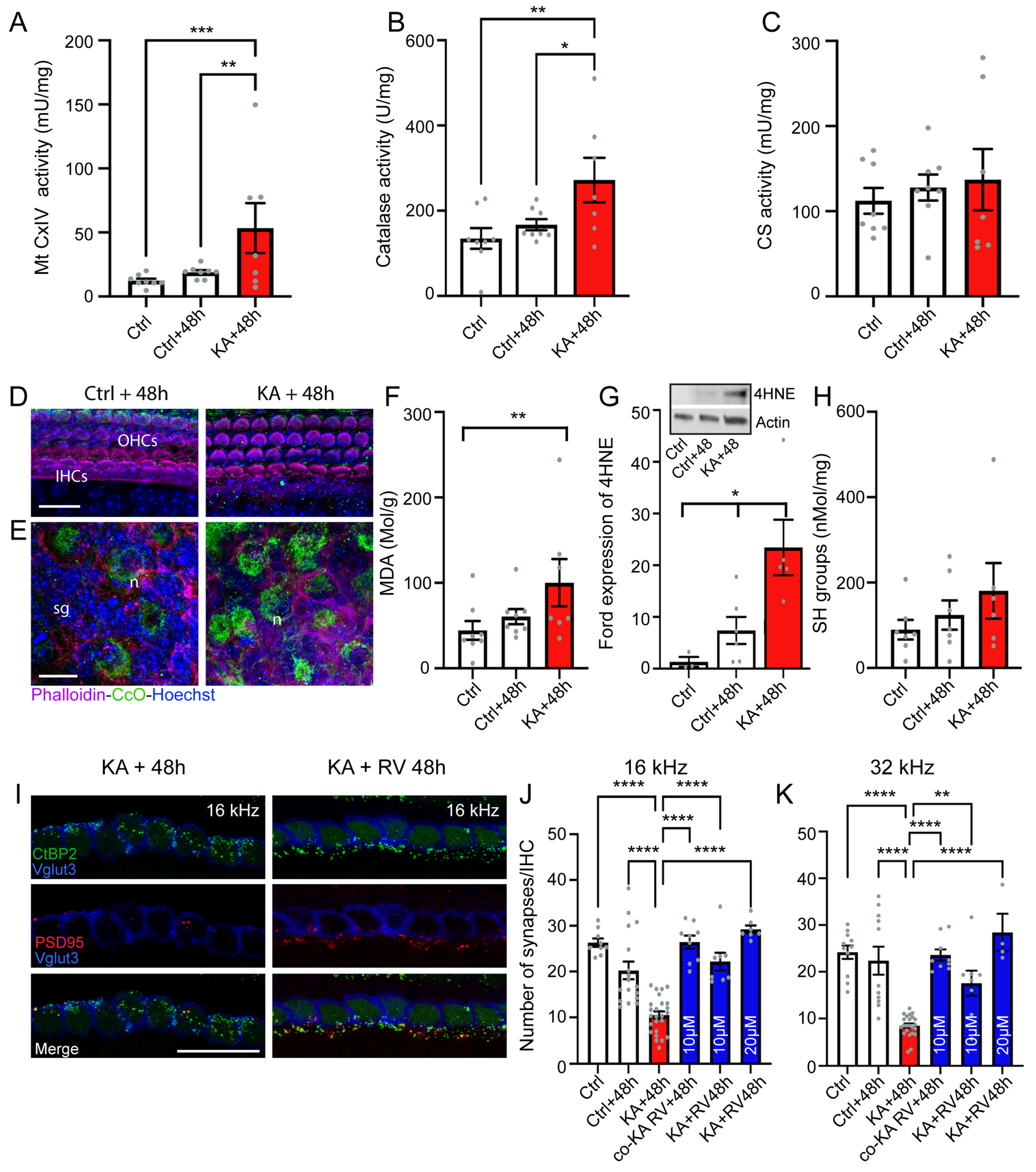
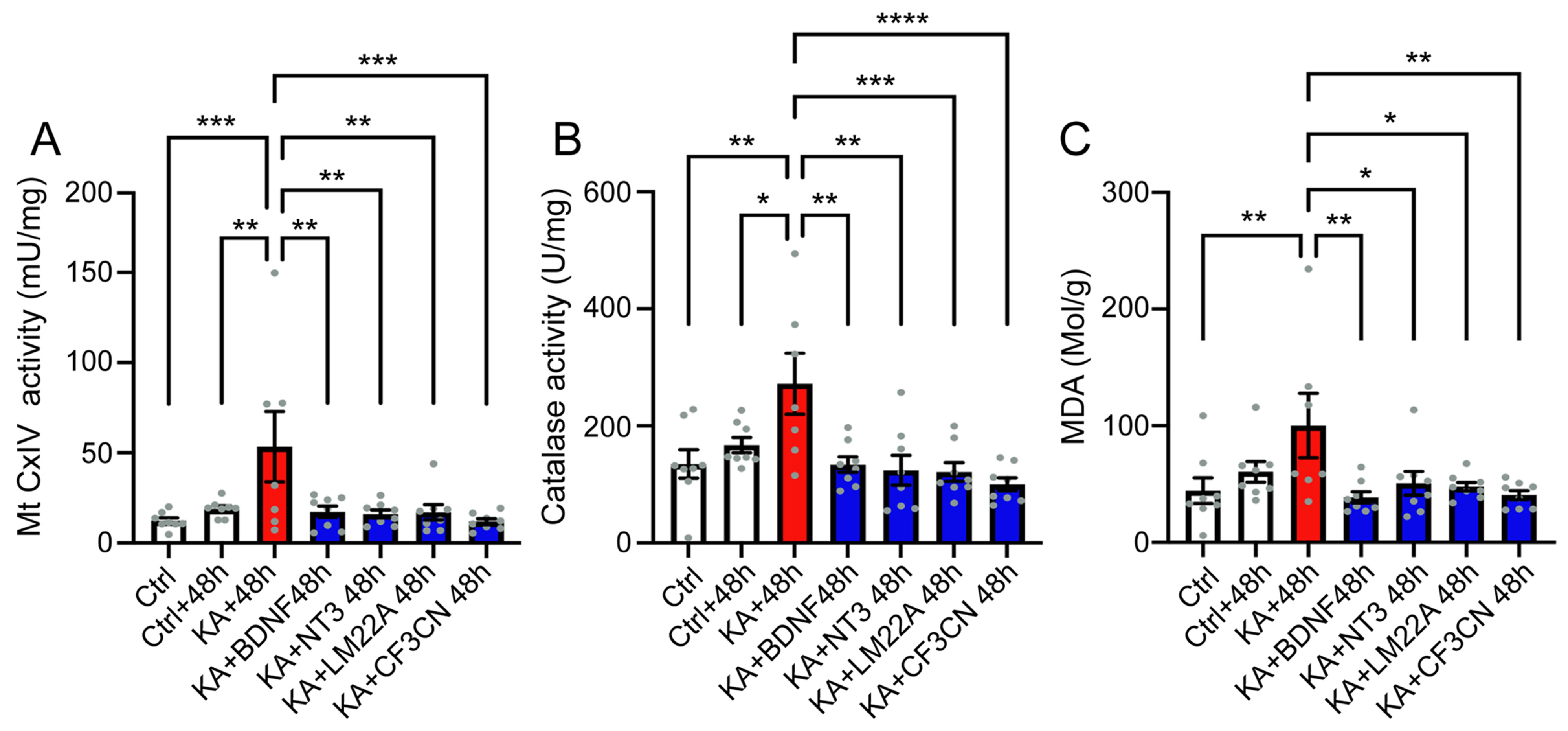
4.3. KA Causes Oxidative Stress
4.4. Antioxidant Treatment Prevent KA-Induced Acute Synaptopathy and Promote Synapse Regeneration
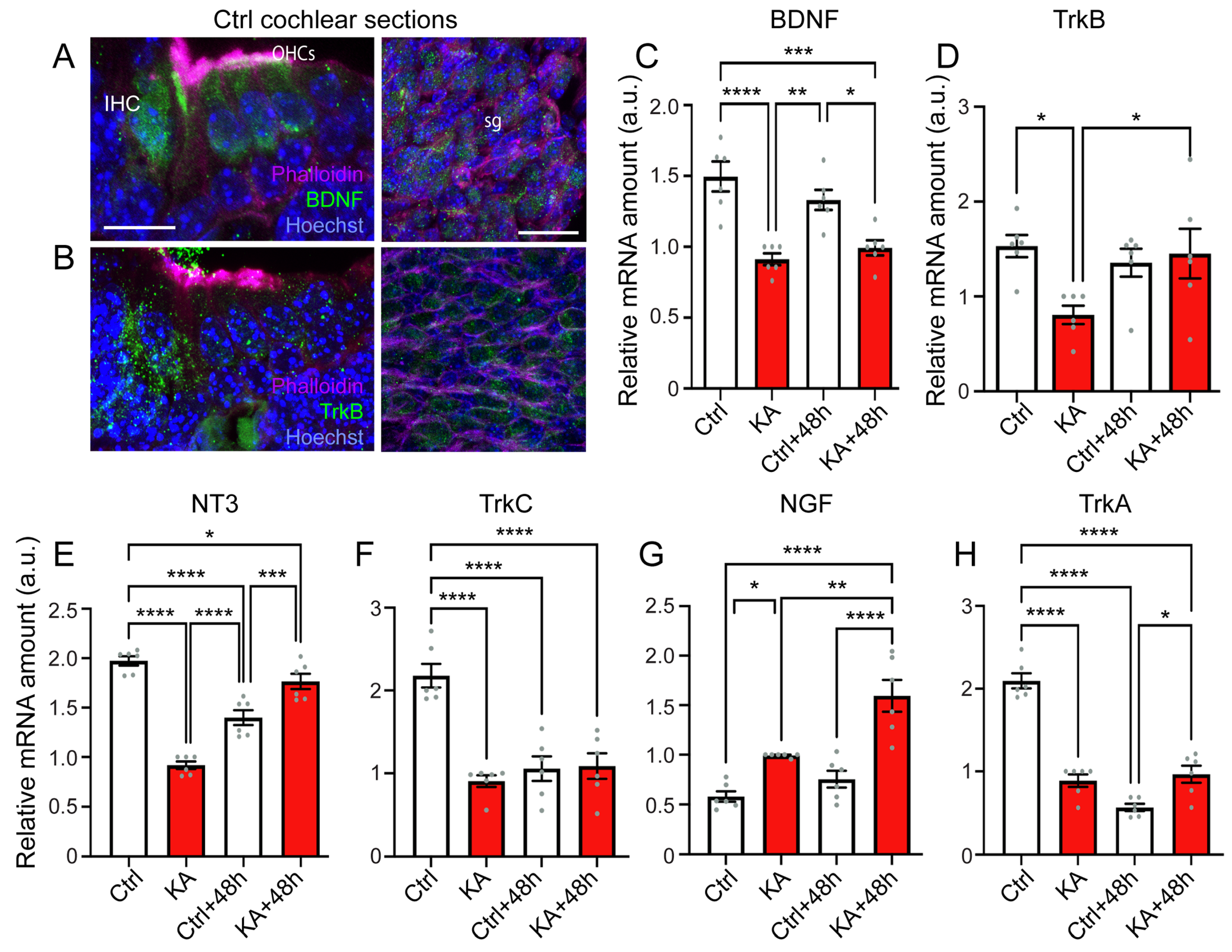
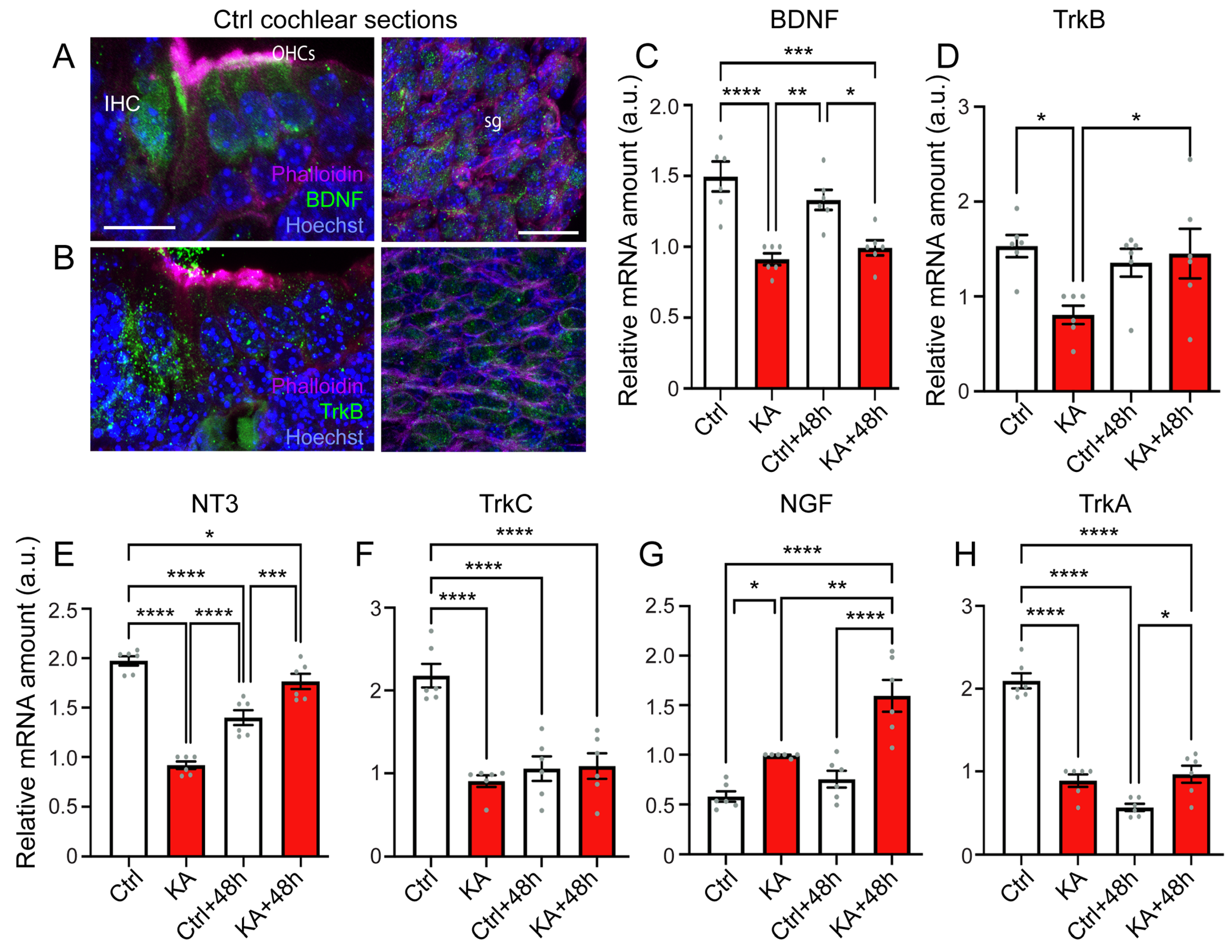
4.5. KA Causes Reduced Expression of BDNF, NT3, and Their Trk Receptors, While Increasing Expression of NGF
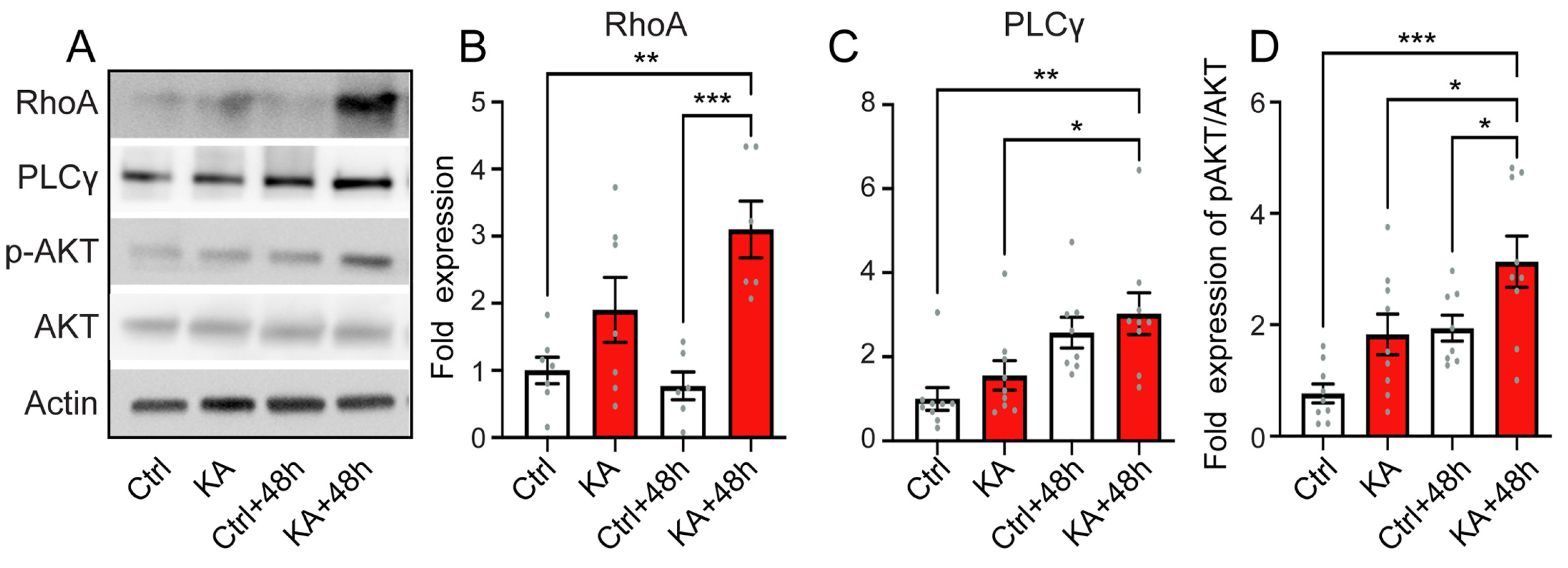
4.6. Activation of the Downstream Pathways of Trk
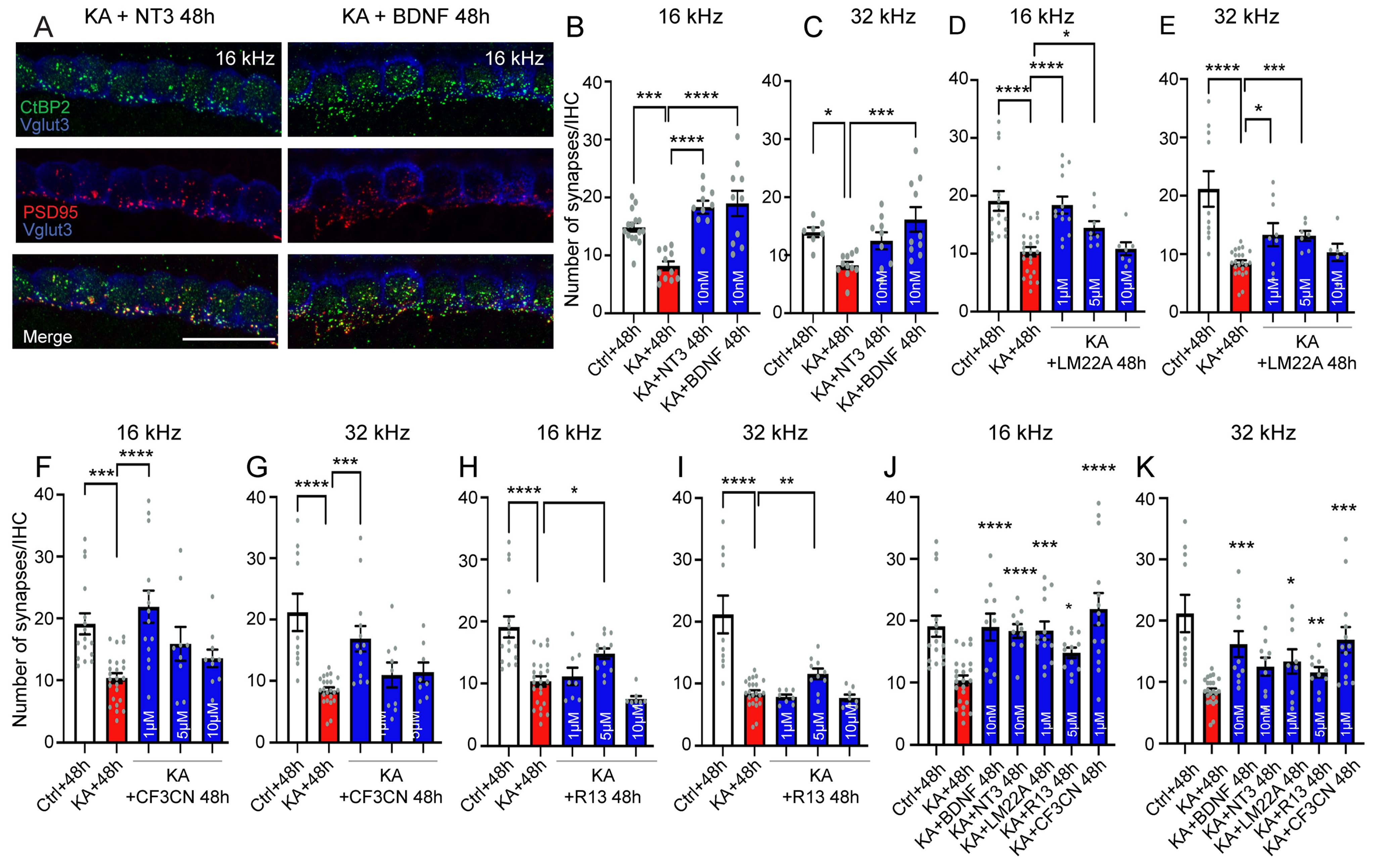
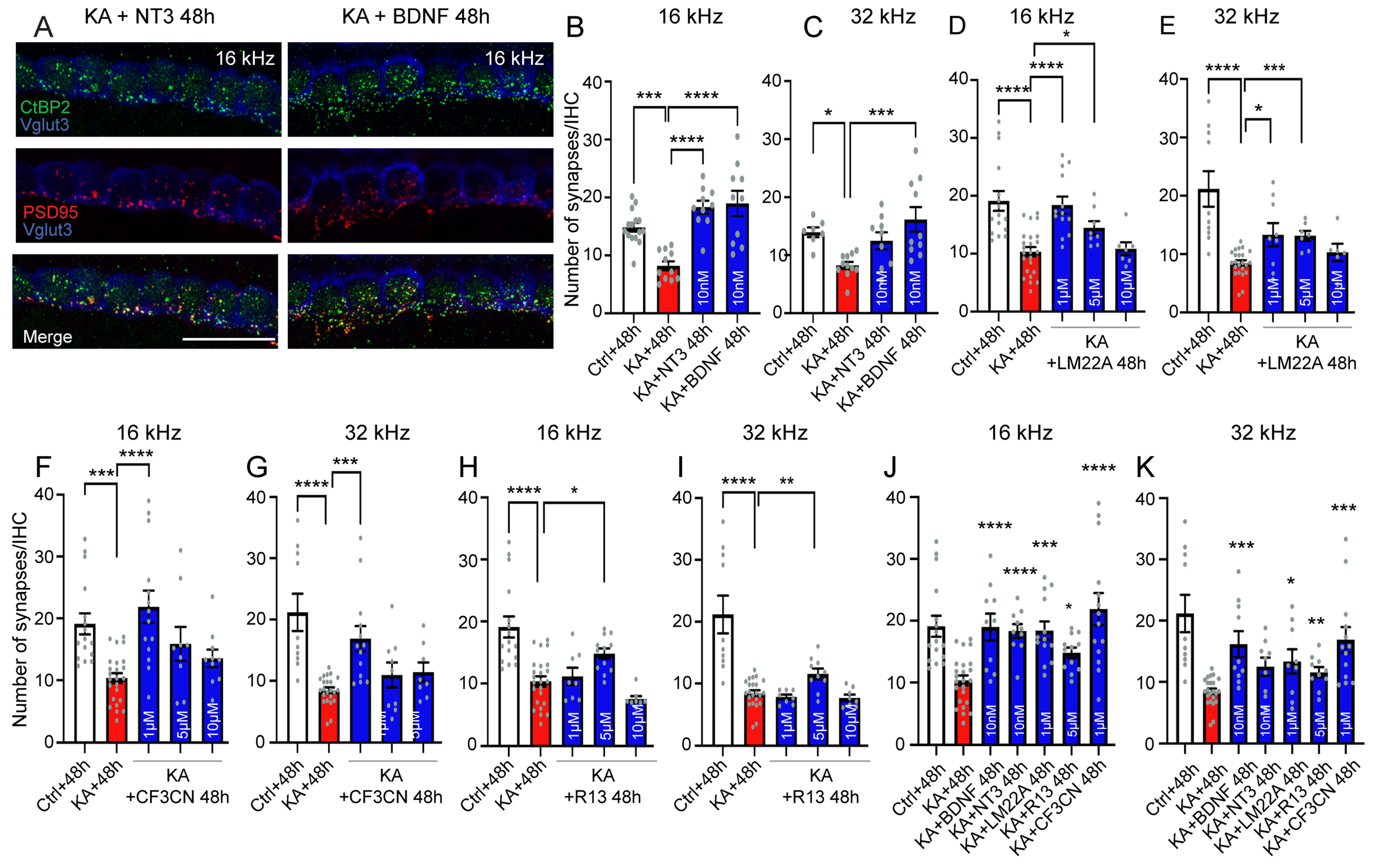
4.7. Neurotrophic Factor Treatment
5. Discussion
5.1. KA-Induced-Excitotoxicity Leads to Rapid and Irreversible Loss of IHC Synapses
5.2. Excitotoxicity-Induced Oxidative Stress and Antioxidant Treatment
5.3. Elicitation of Neurotrophic Effects for the Treatment of the Cochlear Synaptopathy
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eybalin, M.; Charachon, G.; Renard, N. Dopaminergic Lateral Efferent Innervation of the Guinea-Pig Cochlea: Immunoelectron Microscopy of Catecholamine-Synthesizing Enzymes and Effect of 6-Hydroxydopamine. Neuroscience 1993, 54, 133–142. [Google Scholar] [CrossRef]
- Spoendlin, H. Innervation Densities of the Cochlea. Acta Otolaryngol. 1972, 73, 235–248. [Google Scholar] [CrossRef]
- Ruggero, M.A.; Santi, P.A.; Rich, N.C. Type II Cochlear Ganglion Cells in the Chinchilla. Hear. Res. 1982, 8, 339–356. [Google Scholar] [CrossRef]
- Berglund, A.M.; Ryugo, D.K. A Monoclonal Antibody Labels Type II Neurons of the Spiral Ganglion. Brain Res. 1986, 383, 327–332. [Google Scholar] [CrossRef]
- Berglund, A.M.; Ryugo, D.K. Neurofilament Antibodies and Spiral Ganglion Neurons of the Mammalian Cochlea. J. Comp. Neurol. 1991, 306, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Carricondo, F.; Romero-Gómez, B. The Cochlear Spiral Ganglion Neurons: The Auditory Portion of the VIII Nerve. Anat. Rec. 2019, 302, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Safieddine, S.; El-Amraoui, A.; Petit, C. The Auditory Hair Cell Ribbon Synapse: From Assembly to Function. Annu. Rev. Neurosci. 2012, 35, 509–528. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, M.; Moser, T. The Ribbon Synapse between Type I Spiral Ganglion Neurons and Inner Hair Cells. In The Primary Auditory Neurons of the Mammalian Cochlea; Springer: New York, NY, USA, 2016; pp. 117–156. ISBN 978-1-4939-3030-2. [Google Scholar]
- Niedzielski, A.S.; Wenthold, R.J. Expression of AMPA, Kainate, and NMDA Receptor Subunits in Cochlear and Vestibular Ganglia. J. Neurosci. 1995, 15, 2338–2353. [Google Scholar] [CrossRef]
- Klotz, L.; Wendler, O.; Frischknecht, R.; Shigemoto, R.; Schulze, H.; Enz, R. Localization of Group II and III Metabotropic Glutamate Receptors at Pre- and Postsynaptic Sites of Inner Hair Cell Ribbon Synapses. FASEB J. 2019, 33, 13734–13746. [Google Scholar] [CrossRef]
- Ruel, J.; Emery, S.; Nouvian, R.; Bersot, T.; Amilhon, B.; Van Rybroek, J.M.; Rebillard, G.; Lenoir, M.; Eybalin, M.; Delprat, B.; et al. Impairment of SLC17A8 Encoding Vesicular Glutamate Transporter-3, VGLUT3, Underlies Nonsyndromic Deafness DFNA25 and Inner Hair Cell Dysfunction in Null Mice. Am. J. Hum. Genet. 2008, 83, 278–292. [Google Scholar] [CrossRef]
- Seal, R.P.; Akil, O.; Yi, E.; Weber, C.M.; Grant, L.; Yoo, J.; Clause, A.; Kandler, K.; Noebels, J.L.; Glowatzki, E.; et al. Sensorineural Deafness and Seizures in Mice Lacking Vesicular Glutamate Transporter 3. Neuron 2008, 57, 263–275. [Google Scholar] [CrossRef]
- Akil, O.; Seal, R.P.; Burke, K.; Wang, C.; Alemi, A.; During, M.; Edwards, R.H.; Lustig, L.R. Restoration of Hearing in the VGLUT3 Knockout Mouse Using Virally Mediated Gene Therapy. Neuron 2012, 75, 283–293. [Google Scholar] [CrossRef]
- Puel, J.L.; Ruel, J.; Gervais d’Aldin, C.; Pujol, R. Excitotoxicity and Repair of Cochlear Synapses after Noise-Trauma Induced Hearing Loss. Neuroreport 1998, 9, 2109–2114. [Google Scholar] [CrossRef]
- Pujol, R.; Puel, J.L.; Gervais d’Aldin, C.; Eybalin, M. Pathophysiology of the Glutamatergic Synapses in the Cochlea. Acta Otolaryngol. 1993, 113, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.J.T.; Thorne, P.R.; Vlajkovic, S.M. Characterisation of Cochlear Inflammation in Mice Following Acute and Chronic Noise Exposure. Histochem. Cell Biol. 2016, 146, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, K.; Nishimura, B.; Tanaka, S.; Hayashi, K.; Hirose, Y.; Hara, A. Ischemia-Reperfusion Injury of the Cochlea: Pharmacological Strategies for Cochlear Protection and Implications of Glutamate and Reactive Oxygen Species. Curr. Neuropharmacol. 2010, 8, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Ruel, J.; Bobbin, R.P.; Vidal, D.; Pujol, R.; Puel, J.L. The Selective AMPA Receptor Antagonist GYKI 53784 Blocks Action Potential Generation and Excitotoxicity in the Guinea Pig Cochlea. Neuropharmacology 2000, 39, 1959–1973. [Google Scholar] [CrossRef] [PubMed]
- Pujol, R.; Lenoir, M.; Robertson, D.; Eybalin, M.; Johnstone, B.M. Kainic Acid Selectively Alters Auditory Dendrites Connected with Cochlear Inner Hair Cells. Hear. Res. 1985, 18, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Juiz, J.M.; Rueda, J.; Merchán, J.A.; Sala, M.L. The Effects of Kainic Acid on the Cochlear Ganglion of the Rat. Hear. Res. 1989, 40, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Zhang, A.; She, X.; Yang, H.; Wang, K.; Zhu, Y.; Gao, X.; Cui, B. Disruption of Glutamate Release and Uptake-Related Protein Expression after Noise-Induced Synaptopathy in the Cochlea. Front. Cell Dev. Biol. 2021, 9, 720902. [Google Scholar] [CrossRef]
- Hakuba, N.; Koga, K.; Gyo, K.; Usami, S.I.; Tanaka, K. Exacerbation of Noise-Induced Hearing Loss in Mice Lacking the Glutamate Transporter GLAST. J. Neurosci. 2000, 20, 8750–8753. [Google Scholar] [CrossRef] [PubMed]
- Tserga, E.; Damberg, P.; Canlon, B.; Cederroth, C.R. Auditory Synaptopathy in Mice Lacking the Glutamate Transporter GLAST and Its Impact on Brain Activity. Prog. Brain Res. 2021, 262, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.X.; Payne, S.; Yang-Hood, A.; Li, S.-Z.; Davis, B.; Carlquist, J.; V-Ghaffari, B.; Gantz, J.A.; Kallogjeri, D.; Fitzpatrick, J.A.J.; et al. Vesicular Glutamatergic Transmission in Noise-Induced Loss and Repair of Cochlear Ribbon Synapses. J. Neurosci. 2019, 39, 4434–4447. [Google Scholar] [CrossRef] [PubMed]
- Kurabi, A.; Keithley, E.M.; Housley, G.D.; Ryan, A.F.; Wong, A.C.-Y. Cellular Mechanisms of Noise-Induced Hearing Loss. Hear. Res. 2017, 349, 129–137. [Google Scholar] [CrossRef]
- Sebe, J.Y.; Cho, S.; Sheets, L.; Rutherford, M.A.; von Gersdorff, H.; Raible, D.W. Ca2+-Permeable AMPARs Mediate Glutamatergic Transmission and Excitotoxic Damage at the Hair Cell Ribbon Synapse. J. Neurosci. 2017, 37, 6162–6175. [Google Scholar] [CrossRef]
- Hu, N.; Rutherford, M.A.; Green, S.H. Protection of Cochlear Synapses from Noise-Induced Excitotoxic Trauma by Blockade of Ca2+-Permeable AMPA Receptors. Proc. Natl. Acad. Sci. USA 2020, 117, 3828–3838. [Google Scholar] [CrossRef]
- Cassinotti, L.R.; Ji, L.; Borges, B.C.; Cass, N.D.; Desai, A.S.; Kohrman, D.C.; Liberman, M.C.; Corfas, G. Cochlear Neurotrophin-3 Overexpression at Mid-Life Prevents Age-Related Inner Hair Cell Synaptopathy and Slows Age-Related Hearing Loss. Aging Cell 2022, 21, e13708. [Google Scholar] [CrossRef] [PubMed]
- Kujawa, S.G.; Liberman, M.C. Adding Insult to Injury: Cochlear Nerve Degeneration after “Temporary” Noise-Induced Hearing Loss. J. Neurosci. 2009, 29, 14077–14085. [Google Scholar] [CrossRef]
- Furman, A.C.; Kujawa, S.G.; Liberman, M.C. Noise-Induced Cochlear Neuropathy Is Selective for Fibers with Low Spontaneous Rates. J. Neurophysiol. 2013, 110, 577–586. [Google Scholar] [CrossRef]
- Kujawa, S.G.; Liberman, M.C. Synaptopathy in the Noise-Exposed and Aging Cochlea: Primary Neural Degeneration in Acquired Sensorineural Hearing Loss. Hear. Res. 2015, 330, 191–199. [Google Scholar] [CrossRef]
- Hickox, A.E.; Larsen, E.; Heinz, M.G.; Shinobu, L.; Whitton, J.P. Translational Issues in Cochlear Synaptopathy. Hear. Res. 2017, 349, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Liu, L.; He, T.; Guo, X.; Yu, Z.; Yin, S.; Wang, J. Ribbon Synapse Plasticity in the Cochleae of Guinea Pigs after Noise-Induced Silent Damage. PLoS ONE 2013, 8, e81566. [Google Scholar] [CrossRef] [PubMed]
- Kobel, M.; Le Prell, C.G.; Liu, J.; Hawks, J.W.; Bao, J. Noise-Induced Cochlear Synaptopathy: Past Findings and Future Studies. Hear. Res. 2017, 349, 148–154. [Google Scholar] [CrossRef]
- Xiong, W.; Yu, S.; Liu, K.; Gong, S. Loss of Cochlear Ribbon Synapses in the Early Stage of Aging Causes Initial Hearing Impairment. Am. J. Transl. Res. 2020, 12, 7354–7366. [Google Scholar] [PubMed]
- Sergeyenko, Y.; Lall, K.; Liberman, M.C.; Kujawa, S.G. Age-Related Cochlear Synaptopathy: An Early-Onset Contributor to Auditory Functional Decline. J. Neurosci. 2013, 33, 13686–13694. [Google Scholar] [CrossRef] [PubMed]
- Altschuler, R.A.; Dolan, D.F.; Halsey, K.; Kanicki, A.; Deng, N.; Martin, C.; Eberle, J.; Kohrman, D.C.; Miller, R.A.; Schacht, J. Age-Related Changes in Auditory Nerve-Inner Hair Cell Connections, Hair Cell Numbers, Auditory Brain Stem Response and Gap Detection in UM-HET4 Mice. Neuroscience 2015, 292, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Makary, C.A.; Shin, J.; Kujawa, S.G.; Liberman, M.C.; Merchant, S.N. Age-Related Primary Cochlear Neuronal Degeneration in Human Temporal Bones. J. Assoc. Res. Otolaryngol. 2011, 12, 711–717. [Google Scholar] [CrossRef]
- Viana, L.M.; O’Malley, J.T.; Burgess, B.J.; Jones, D.D.; Oliveira, C.A.C.P.; Santos, F.; Merchant, S.N.; Liberman, L.D.; Liberman, M.C. Cochlear Neuropathy in Human Presbycusis: Confocal Analysis of Hidden Hearing Loss in Post-Mortem Tissue. Hear. Res. 2015, 327, 78–88. [Google Scholar] [CrossRef]
- Möhrle, D.; Ni, K.; Varakina, K.; Bing, D.; Lee, S.C.; Zimmermann, U.; Knipper, M.; Rüttiger, L. Loss of Auditory Sensitivity from Inner Hair Cell Synaptopathy Can Be Centrally Compensated in the Young but Not Old Brain. Neurobiol. Aging 2016, 44, 173–184. [Google Scholar] [CrossRef]
- Benkafadar, N.; Menardo, J.; Bourien, J.; Nouvian, R.; François, F.; Decaudin, D.; Maiorano, D.; Puel, J.-L.; Wang, J. Reversible P53 Inhibition Prevents Cisplatin Ototoxicity without Blocking Chemotherapeutic Efficacy. EMBO Mol. Med. 2017, 9, 7–26. [Google Scholar] [CrossRef]
- Benkafadar, N.; François, F.; Affortit, C.; Casas, F.; Ceccato, J.-C.; Menardo, J.; Venail, F.; Malfroy-Camine, B.; Puel, J.-L.; Wang, J. ROS-Induced Activation of DNA Damage Responses Drives Senescence-Like State in Postmitotic Cochlear Cells: Implication for Hearing Preservation. Mol. Neurobiol. 2019, 56, 5950–5969. [Google Scholar] [CrossRef]
- Wang, Q.; Green, S.H. Functional Role of Neurotrophin-3 in Synapse Regeneration by Spiral Ganglion Neurons on Inner Hair Cells after Excitotoxic Trauma In Vitro. J. Neurosci. 2011, 31, 7938–7949. [Google Scholar] [CrossRef]
- Szobota, S.; Mathur, P.D.; Siegel, S.; Black, K.; Saragovi, H.U.; Foster, A.C. BDNF, NT-3 and Trk Receptor Agonist Monoclonal Antibodies Promote Neuron Survival, Neurite Extension, and Synapse Restoration in Rat Cochlea Ex Vivo Models Relevant for Hidden Hearing Loss. PLoS ONE 2019, 14, e0224022. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Chen, C.; Ahn, E.H.; Liu, X.; Li, H.; Edgington-Mitchell, L.E.; Lu, Z.; Ming, S.; Ye, K. Targeting Both BDNF/TrkB Pathway and Delta-Secretase for Treating Alzheimer’s Disease. Neuropharmacology 2021, 197, 108737. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Wu, Z.; Liu, X.; Edgington-Mitchell, L.; Ye, K. Treating Parkinson’s Disease via Activation of BDNF/TrkB Signaling Pathways and Inhibition of Delta-Secretase. Neurotherapeutics 2022, 19, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; von Hünerbein, K.; Hoidis, S.; Smolders, J.W.T. A Physiological Place-Frequency Map of the Cochlea in the CBA/J Mouse. Hear. Res. 2005, 202, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Bourien, J.; Tang, Y.; Batrel, C.; Huet, A.; Lenoir, M.; Ladrech, S.; Desmadryl, G.; Nouvian, R.; Puel, J.-L.; Wang, J. Contribution of Auditory Nerve Fibers to Compound Action Potential of the Auditory Nerve. J. Neurophysiol. 2014, 112, 1025–1039. [Google Scholar] [CrossRef] [PubMed]
- Affortit, C.; Casas, F.; Ladrech, S.; Ceccato, J.-C.; Bourien, J.; Coyat, C.; Puel, J.-L.; Lenoir, M.; Wang, J. Exacerbated Age-Related Hearing Loss in Mice Lacking the P43 Mitochondrial T3 Receptor. BMC Biol. 2021, 19, 18. [Google Scholar] [CrossRef] [PubMed]
- Marklund, S. Spectrophotometric Study of Spontaneous Disproportionation of Superoxide Anion Radical and Sensitive Direct Assay for Superoxide Dismutase. J. Biol. Chem. 1976, 251, 7504–7507. [Google Scholar] [CrossRef]
- Wharton, D.C.; Tzagoloff, A. [45] Cytochrome Oxidase from Beef Heart Mitochondria. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1967; Volume 10, pp. 245–250. [Google Scholar]
- Wang, J.; Puel, J.-L. Presbycusis: An Update on Cochlear Mechanisms and Therapies. J. Clin. Med. 2020, 9, 218. [Google Scholar] [CrossRef]
- Sunderman, F.W.; Marzouk, A.; Hopfer, S.M.; Zaharia, O.; Reid, M.C. Increased Lipid Peroxidation in Tissues of Nickel Chloride-Treated Rats. Ann. Clin. Lab. Sci. 1985, 15, 229–236. [Google Scholar] [PubMed]
- Jocelyn, P.C. Spectrophotometric Assay of Thiols. Methods Enzymol. 1987, 143, 44–67. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate Normalization of Real-Time Quantitative RT-PCR Data by Geometric Averaging of Multiple Internal Control Genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [PubMed]
- Puel, J.L.; Pujol, R.; Tribillac, F.; Ladrech, S.; Eybalin, M. Excitatory Amino Acid Antagonists Protect Cochlear Auditory Neurons from Excitotoxicity. J. Comp. Neurol. 1994, 341, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Bononi, I.; Tedeschi, P.; Mantovani, V.; Maietti, A.; Mazzoni, E.; Pancaldi, C.; Brandolini, V.; Tognon, M. Antioxidant Activity of Resveratrol Diastereomeric Forms Assayed in Fluorescent-Engineered Human Keratinocytes. Antioxidants 2022, 11, 196. [Google Scholar] [CrossRef] [PubMed]
- Fritzsch, B.; Tessarollo, L.; Coppola, E.; Reichardt, L.F. Neurotrophins in the Ear: Their Roles in Sensory Neuron Survival and Fiber Guidance. Prog. Brain Res. 2004, 146, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Ramekers, D.; Versnel, H.; Grolman, W.; Klis, S.F.L. Neurotrophins and Their Role in the Cochlea. Hear. Res. 2012, 288, 19–33. [Google Scholar] [CrossRef]
- Wiechers, B.; Gestwa, G.; Mack, A.; Carroll, P.; Zenner, H.P.; Knipper, M. A Changing Pattern of Brain-Derived Neurotrophic Factor Expression Correlates with the Rearrangement of Fibers during Cochlear Development of Rats and Mice. J. Neurosci. 1999, 19, 3033–3042. [Google Scholar] [CrossRef]
- Shi, X.; Nuttall, A.L. Upregulated iNOS and Oxidative Damage to the Cochlear Stria Vascularis due to Noise Stress. Brain Res. 2003, 967, 1–10. [Google Scholar] [CrossRef]
- Gomes, J.R.; Costa, J.T.; Melo, C.V.; Felizzi, F.; Monteiro, P.; Pinto, M.J.; Inácio, A.R.; Wieloch, T.; Almeida, R.D.; Grãos, M.; et al. Excitotoxicity Downregulates TrkB.FL Signaling and Upregulates the Neuroprotective Truncated TrkB Receptors in Cultured Hippocampal and Striatal Neurons. J. Neurosci. 2012, 32, 4610–4622. [Google Scholar] [CrossRef]
- Simmons, D.A. Modulating Neurotrophin Receptor Signaling as a Therapeutic Strategy for Huntington’s Disease. J. Huntingtons Dis. 2017, 6, 303–325. [Google Scholar] [CrossRef]
- Nadol, J.B. Patterns of Neural Degeneration in the Human Cochlea and Auditory Nerve: Implications for Cochlear Implantation. Otolaryngol. Head Neck Surg. 1997, 117, 220–228. [Google Scholar] [CrossRef]
- Pujol, R.; Puel, J.L. Excitotoxicity, Synaptic Repair, and Functional Recovery in the Mammalian Cochlea: A Review of Recent Findings. Ann. N. Y. Acad. Sci. 1999, 884, 249–254. [Google Scholar] [CrossRef]
- Ruel, J.; Wang, J.; Rebillard, G.; Eybalin, M.; Lloyd, R.; Pujol, R.; Puel, J.-L. Physiology, Pharmacology and Plasticity at the Inner Hair Cell Synaptic Complex. Hear. Res. 2007, 227, 19–27. [Google Scholar] [CrossRef]
- Jia, H.; Wang, J.; François, F.; Uziel, A.; Puel, J.-L.; Venail, F. Molecular and Cellular Mechanisms of Loss of Residual Hearing after Cochlear Implantation. Ann. Otol. Rhinol. Laryngol. 2013, 122, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Someya, S. Mouse Models of Age-Related Mitochondrial Neurosensory Hearing Loss. Mol. Cell. Neurosci. 2013, 55, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Menardo, J.; Tang, Y.; Ladrech, S.; Lenoir, M.; Casas, F.; Michel, C.; Bourien, J.; Ruel, J.; Rebillard, G.; Maurice, T.; et al. Oxidative Stress, Inflammation, and Autophagic Stress as the Key Mechanisms of Premature Age-Related Hearing Loss in SAMP8 Mouse Cochlea. Antioxid. Redox Signal. 2012, 16, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Puel, J.-L. Toward Cochlear Therapies. Physiol. Rev. 2018, 98, 2477–2522. [Google Scholar] [CrossRef] [PubMed]
- Ohlemiller, K.K.; Wright, J.S.; Dugan, L.L. Early Elevation of Cochlear Reactive Oxygen Species Following Noise Exposure. Audiol. Neurootol. 1999, 4, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, D.; Jiang, H.-Y.; Schacht, J.; Miller, J.M. Delayed Production of Free Radicals Following Noise Exposure. Brain Res. 2004, 1019, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Ohinata, Y.; Miller, J.M.; Altschuler, R.A.; Schacht, J. Intense Noise Induces Formation of Vasoactive Lipid Peroxidation Products in the Cochlea. Brain Res. 2000, 878, 163–173. [Google Scholar] [CrossRef]
- Van Campen, L.E.; Murphy, W.J.; Franks, J.R.; Mathias, P.I.; Toraason, M.A. Oxidative DNA Damage Is Associated with Intense Noise Exposure in the Rat. Hear. Res. 2002, 164, 29–38. [Google Scholar] [CrossRef]
- Bondy, S.C.; LeBel, C.P. The Relationship between Excitotoxicity and Oxidative Stress in the Central Nervous System. Free Radic. Biol. Med. 1993, 14, 633–642. [Google Scholar] [CrossRef]
- Bondy, S.C.; Lee, D.K. Oxidative Stress Induced by Glutamate Receptor Agonists. Brain Res. 1993, 610, 229–233. [Google Scholar] [CrossRef]
- Naarala, J.T.; Loikkanen, J.J.; Ruotsalainen, M.H.; Savolainen, K.M. Lead Amplifies Glutamate-Induced Oxidative Stress. Free Radic. Biol. Med. 1995, 19, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Odaka, H. Brain-Derived Neurotrophic Factor Signaling in the Pathophysiology of Alzheimer’s Disease: Beneficial Effects of Flavonoids for Neuroprotection. Int. J. Mol. Sci. 2021, 22, 5719. [Google Scholar] [CrossRef] [PubMed]
- Urbina-Varela, R.; Soto-Espinoza, M.I.; Vargas, R.; Quiñones, L.; Del Campo, A. Influence of BDNF Genetic Polymorphisms in the Pathophysiology of Aging-Related Diseases. Aging Dis. 2020, 11, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Garzon, D.J.; Fahnestock, M. Oligomeric Amyloid Decreases Basal Levels of Brain-Derived Neurotrophic Factor (BDNF) mRNA via Specific Downregulation of BDNF Transcripts IV and V in Differentiated Human Neuroblastoma Cells. J. Neurosci. 2007, 27, 2628–2635. [Google Scholar] [CrossRef] [PubMed]
- Havenith, S.; Versnel, H.; Agterberg, M.J.H.; de Groot, J.C.M.J.; Sedee, R.-J.; Grolman, W.; Klis, S.F.L. Spiral Ganglion Cell Survival after Round Window Membrane Application of Brain-Derived Neurotrophic Factor Using Gelfoam as Carrier. Hear. Res. 2011, 272, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Leake, P.A.; Hradek, G.T.; Hetherington, A.M.; Stakhovskaya, O. Brain-Derived Neurotrophic Factor Promotes Cochlear Spiral Ganglion Cell Survival and Function in Deafened, Developing Cats. J. Comp. Neurol. 2011, 519, 1526–1545. [Google Scholar] [CrossRef]
- Wise, A.K.; Richardson, R.; Hardman, J.; Clark, G.; O’leary, S. Resprouting and Survival of Guinea Pig Cochlear Neurons in Response to the Administration of the Neurotrophins Brain-Derived Neurotrophic Factor and Neurotrophin-3. J. Comp. Neurol. 2005, 487, 147–165. [Google Scholar] [CrossRef]
- Sly, D.J.; Campbell, L.; Uschakov, A.; Saief, S.T.; Lam, M.; O’Leary, S.J. Applying Neurotrophins to the Round Window Rescues Auditory Function and Reduces Inner Hair Cell Synaptopathy After Noise-Induced Hearing Loss. Otol. Neurotol. 2016, 37, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Corfas, G.; Liberman, M.C. Round-Window Delivery of Neurotrophin 3 Regenerates Cochlear Synapses after Acoustic Overexposure. Sci. Rep. 2016, 6, 24907. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.; Gómez-Casati, M.E.; Gigliello, A.R.; Liberman, M.C.; Corfas, G. Neurotrophin-3 Regulates Ribbon Synapse Density in the Cochlea and Induces Synapse Regeneration after Acoustic Trauma. eLife 2014, 3, e03564. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | 5′ to 3′ Primer | Nucleotide Position | Product Size (bp) | GenBank Accession Number |
|---|---|---|---|---|
| Bdnf | F-AAAGTCCCGGTATCCAAAGG R-ATCGCCAGCCAATTCTCTTT | 879–1062 | 184 | NM_001048142 |
| Ngf | F-GCAGTGAGGTGCATAGCGTA R-CTGTGTCAAGGGAATGCTGA | 286–444 | 159 | NM_013609 |
| Nt3 | F-TCTGCCACGATCTTACAGGTG R-AGGGTGCTCTGGTAATTTTCCT | 304–521 | 218 | NM_001164034 |
| TrkA | F-GAACCCACTGCATTGTTCCT R-GCACTGCAGAAACACGTCAT | 459–669 | 211 | NM_001033124 |
| TrkB | F-ACTGTCCTGCTACCGCAGTT R-GTTCACAGTGGCTGGGACAT | 289–474 | 186 | NM_001025074 |
| TrkC | F-CCTGACACAGTGGTCATTGG R-CTTGTCTTTGGTGGGGCTTA | 1547–1753 | 207 | NM_008746 |
| Ddx48 | F-GGAGTTAGCGGTGCAGATTC R-AGCATCTTGATAGCCCGTGT | 395–598 | 204 | NM_138669 |
| Polr2j | F-ACCACACTCTGGGGAACATC R-CTCGCTGATGAGGTCTGTGA | 176–353 | 178 | NM_011293 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saidia, A.R.; François, F.; Casas, F.; Mechaly, I.; Venteo, S.; Veechi, J.T.; Ruel, J.; Puel, J.-L.; Wang, J. Oxidative Stress Plays an Important Role in Glutamatergic Excitotoxicity-Induced Cochlear Synaptopathy: Implication for Therapeutic Molecules Screening. Antioxidants 2024, 13, 149. https://doi.org/10.3390/antiox13020149
Saidia AR, François F, Casas F, Mechaly I, Venteo S, Veechi JT, Ruel J, Puel J-L, Wang J. Oxidative Stress Plays an Important Role in Glutamatergic Excitotoxicity-Induced Cochlear Synaptopathy: Implication for Therapeutic Molecules Screening. Antioxidants. 2024; 13(2):149. https://doi.org/10.3390/antiox13020149
Chicago/Turabian StyleSaidia, Anissa Rym, Florence François, François Casas, Ilana Mechaly, Stéphanie Venteo, Joseph T. Veechi, Jérôme Ruel, Jean-Luc Puel, and Jing Wang. 2024. "Oxidative Stress Plays an Important Role in Glutamatergic Excitotoxicity-Induced Cochlear Synaptopathy: Implication for Therapeutic Molecules Screening" Antioxidants 13, no. 2: 149. https://doi.org/10.3390/antiox13020149
APA StyleSaidia, A. R., François, F., Casas, F., Mechaly, I., Venteo, S., Veechi, J. T., Ruel, J., Puel, J.-L., & Wang, J. (2024). Oxidative Stress Plays an Important Role in Glutamatergic Excitotoxicity-Induced Cochlear Synaptopathy: Implication for Therapeutic Molecules Screening. Antioxidants, 13(2), 149. https://doi.org/10.3390/antiox13020149