
ADP-Ribosylation Factor-Interacting Protein 2 Acts as a Novel Regulator of Mitophagy and Autophagy in Podocytes in Diabetic Nephropathy
 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Genotyping for Mice
2.3. Measurement of Urinary Albumin and Creatinine
2.4. Antibodies
2.5. Histologic Analyses
2.6. Electron Microscopy
2.7. Electron Microscopy Image Analyses
2.8. In Situ Hybridization and LacZ Staining
2.9. Immunofluorescence Stainings
2.10. Cell Culture
2.11. CRISPR/Cas9 and Lentiviral Transduction
2.12. Western Blot Analysis
2.13. Analysis of Cellular Reactive Oxygen Species (ROS) Levels
2.14. Analysis of Mitochondria Stress and Glycolytic Activity In Vitro
2.15. Image Analyses
2.16. Statistics
3. Results
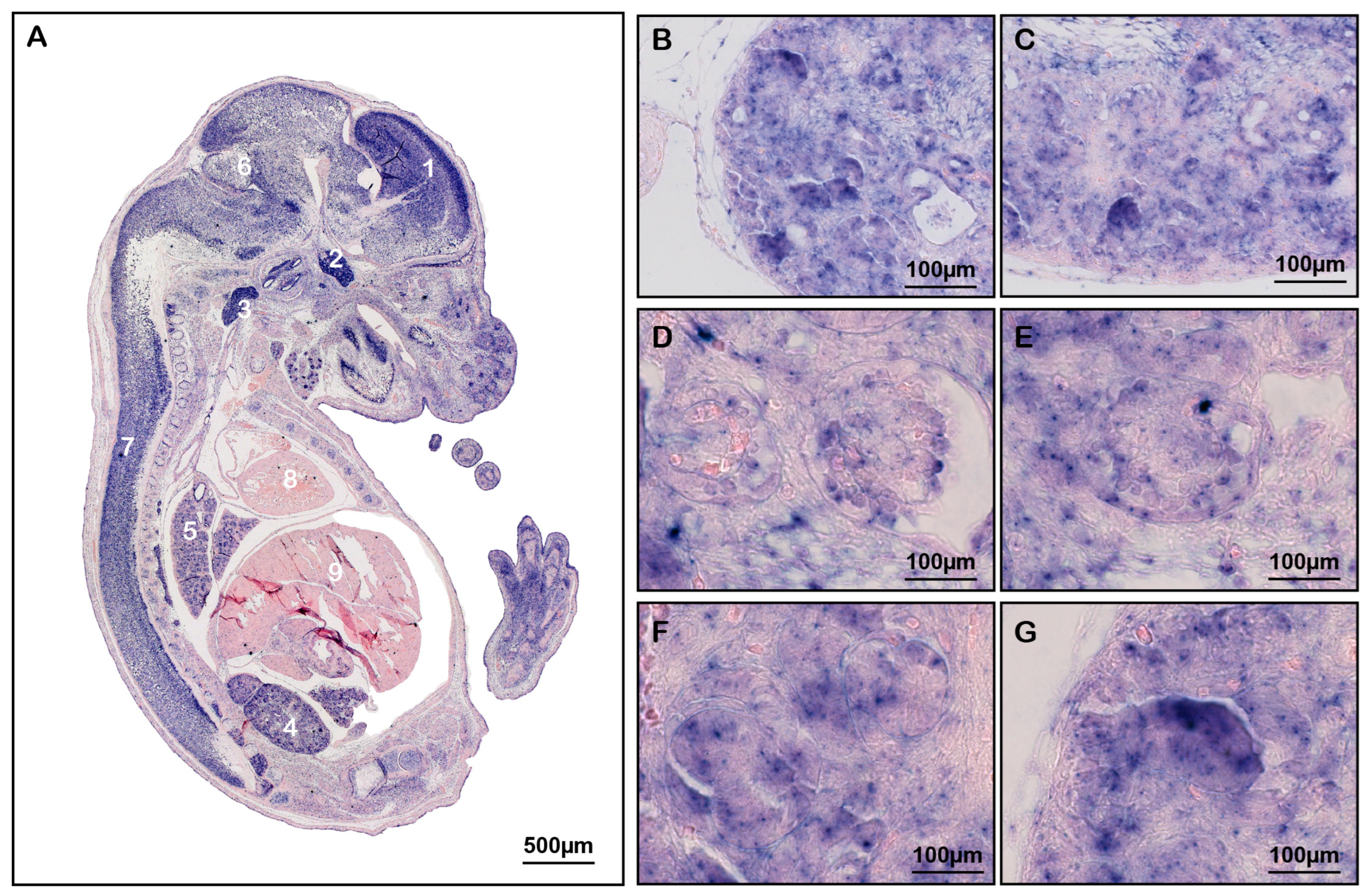
3.1. Arfip2 Is Expressed in Podocytes
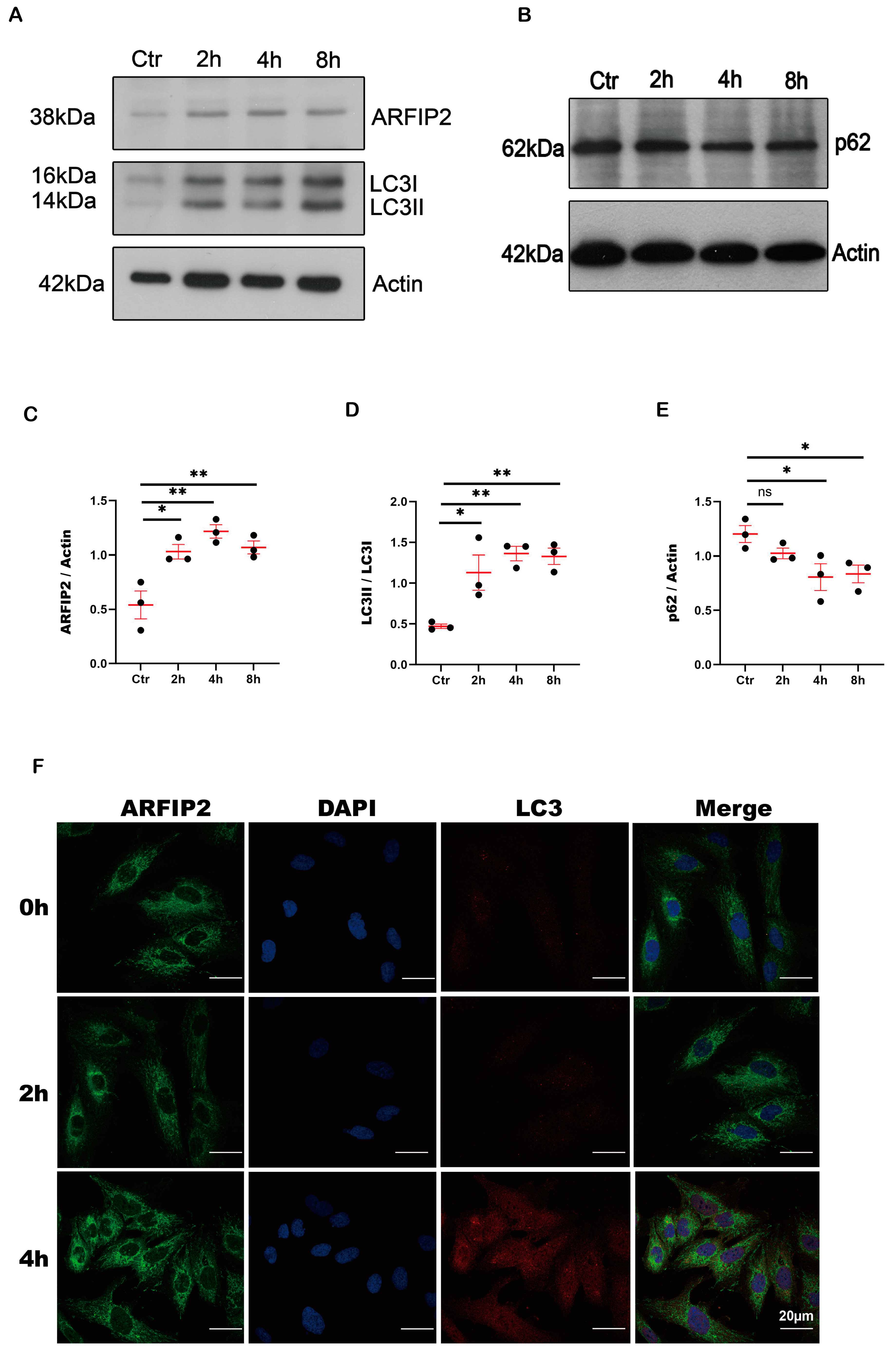
3.2. ARFIP2 Is a Regulator of Autophagy in Podocytes In Vitro
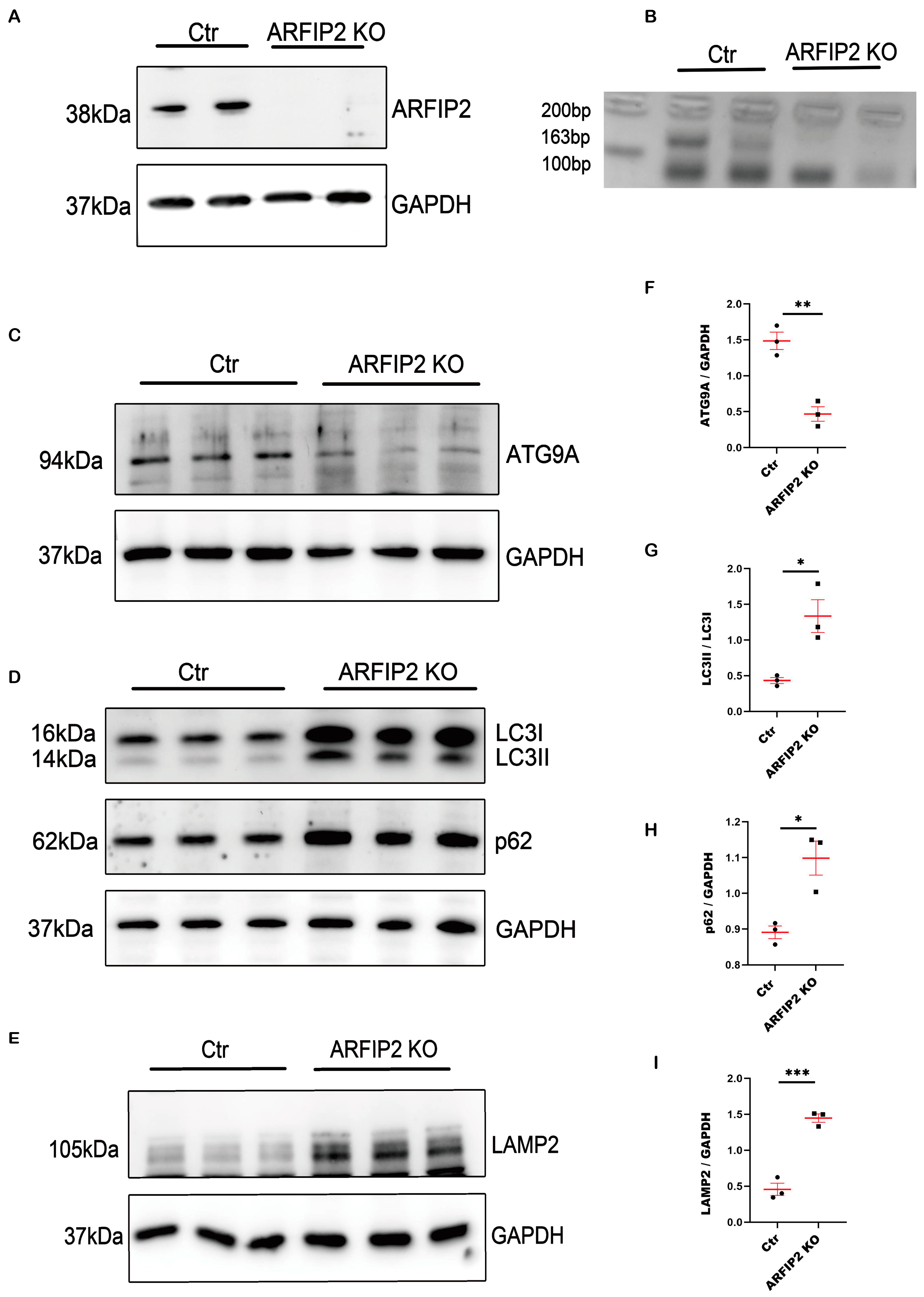
3.3. Autophagy Is Impaired in ARFIP2-Deficient Podocytes
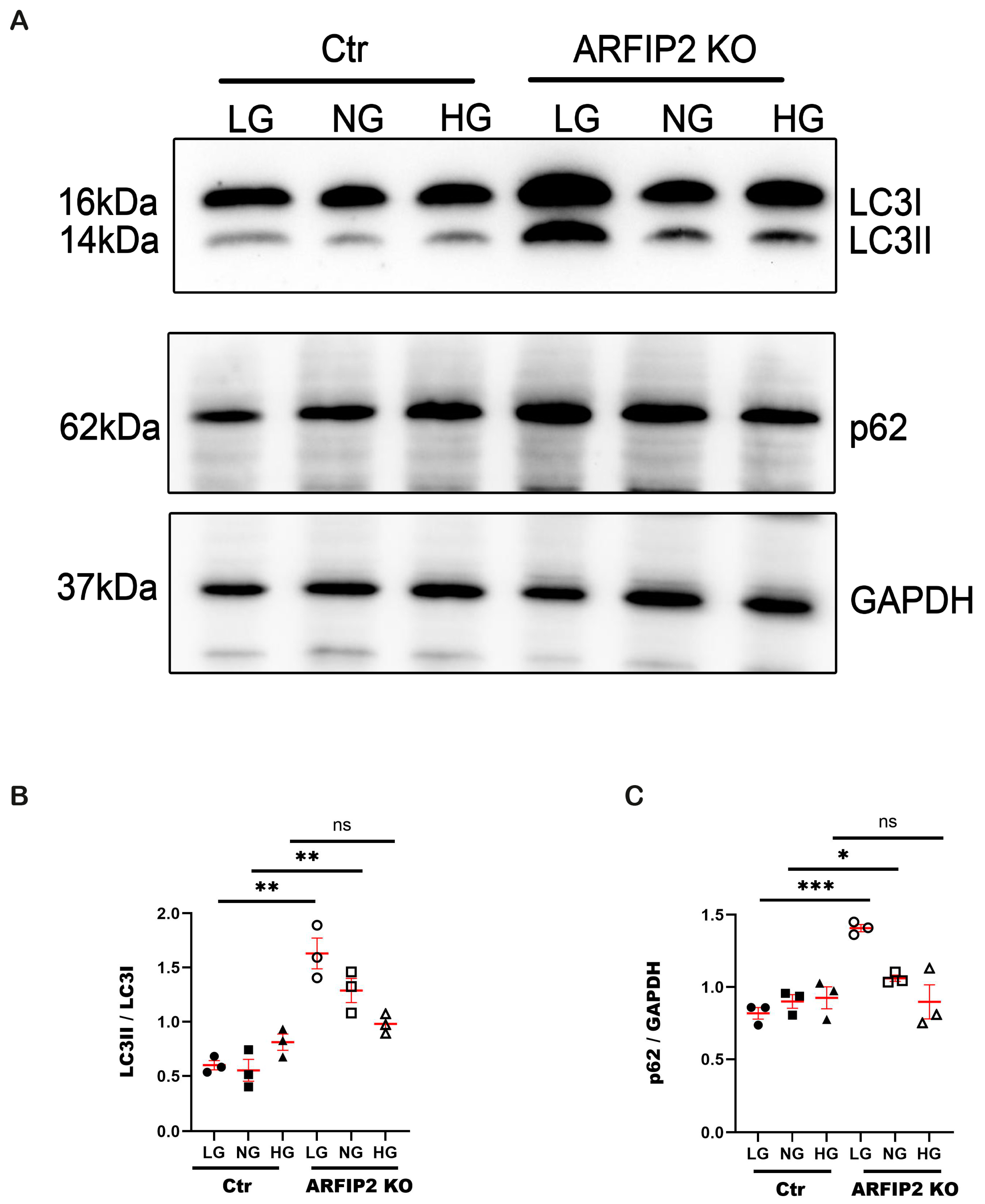
3.4. ARFIP2 Deficiency in Human Podocytes Impairs Autophagy under Low Glucose Conditions
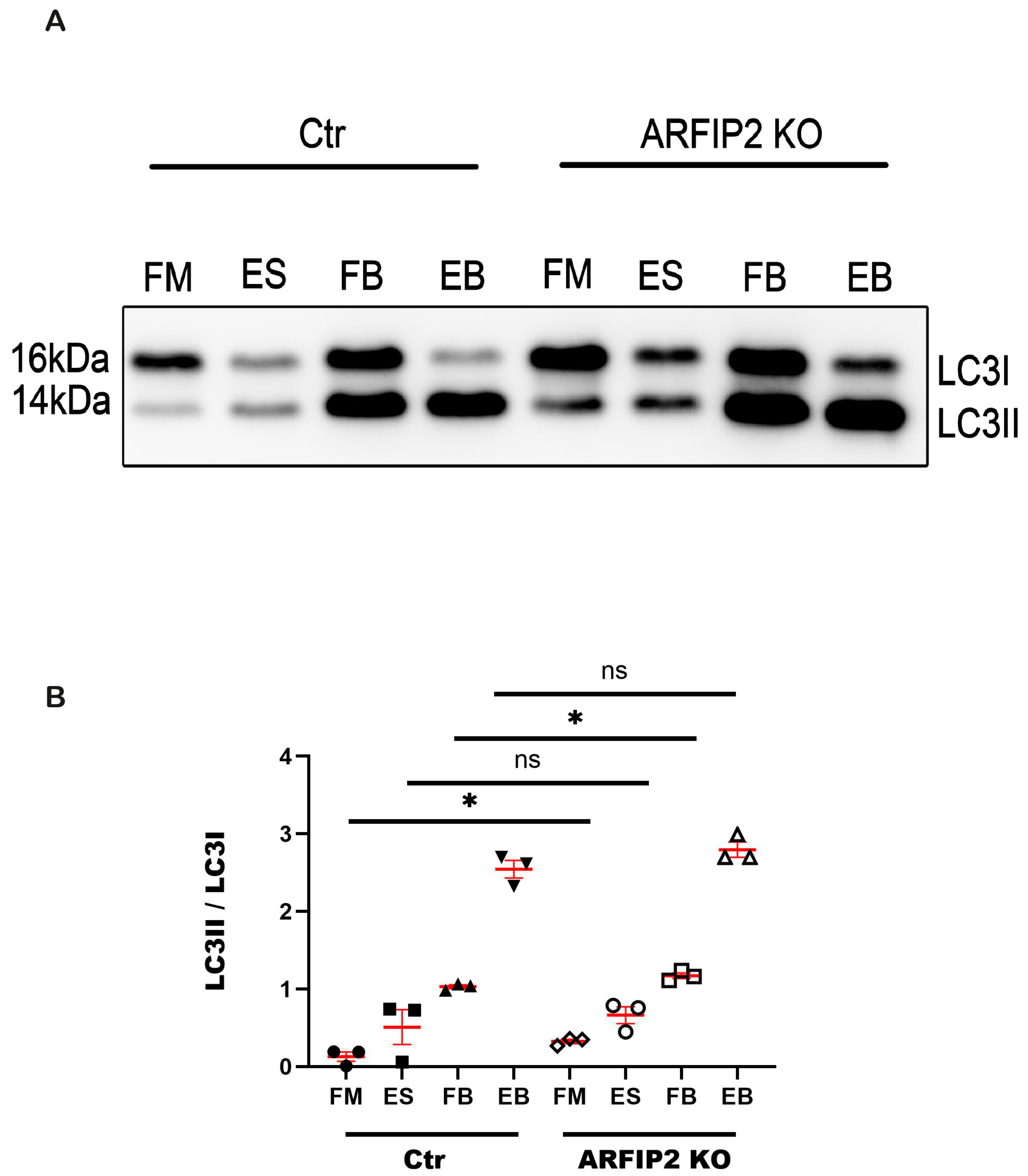
3.5. Autophagy Flux Is Disrupted in ARFIP2 Deficient Podocytes
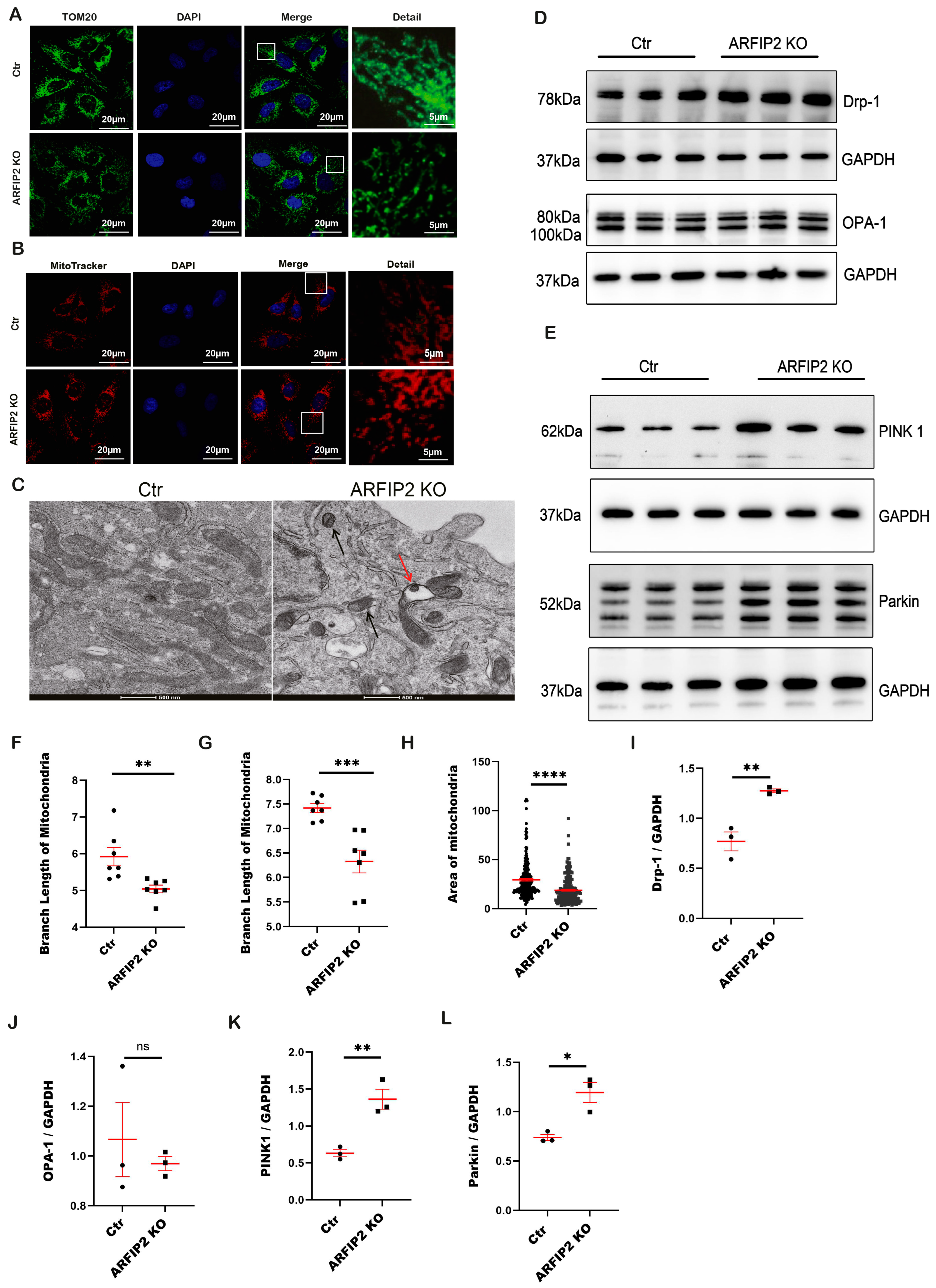
3.6. The PINK1-Parkin Pathway, Mitochondrial Fission and Mitophagy Are Affected in ARFIP2 Deficient Podocytes
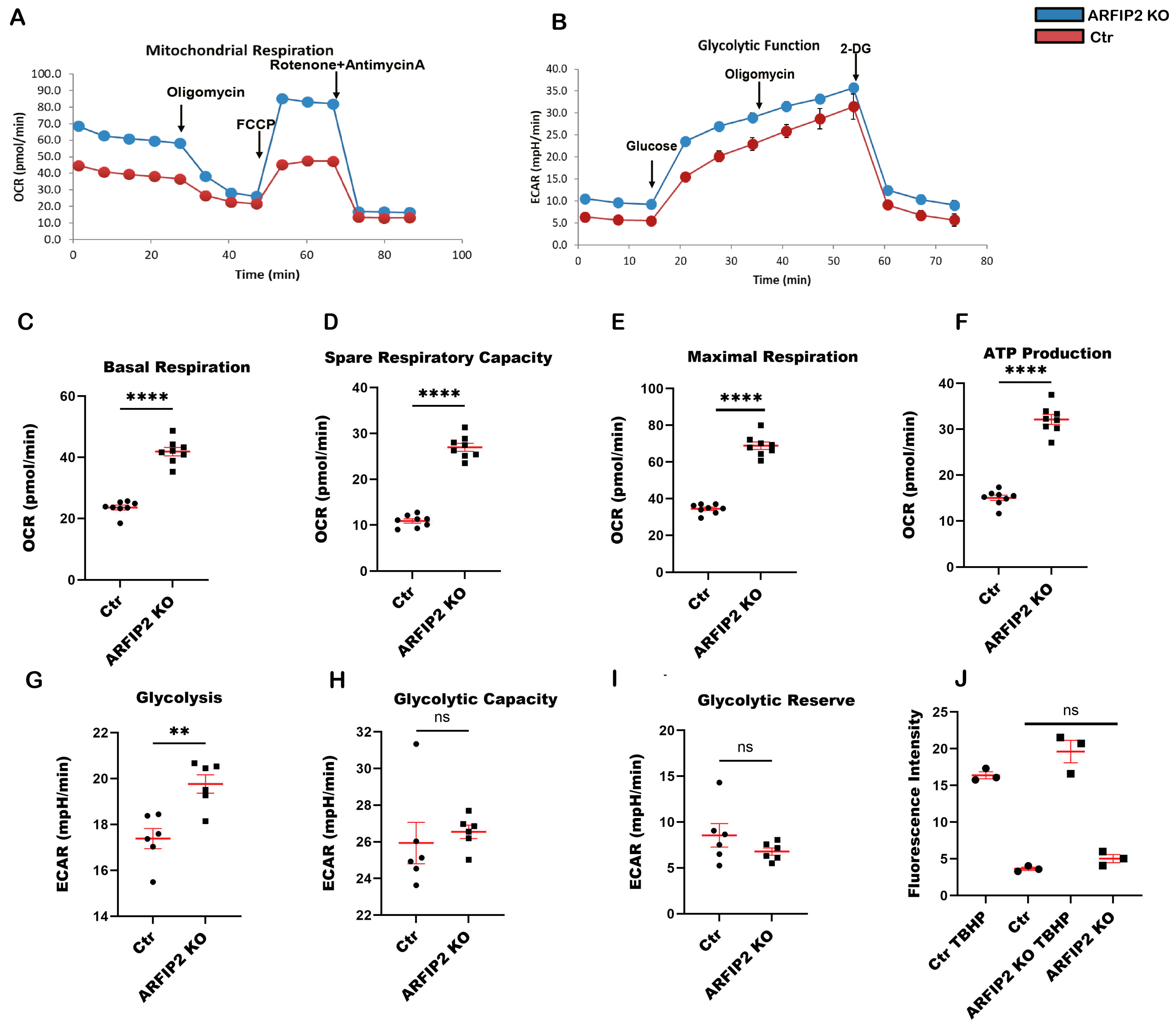
3.7. ARFIP2 Deficiency Leads to Increased Mitochondrial Respiration
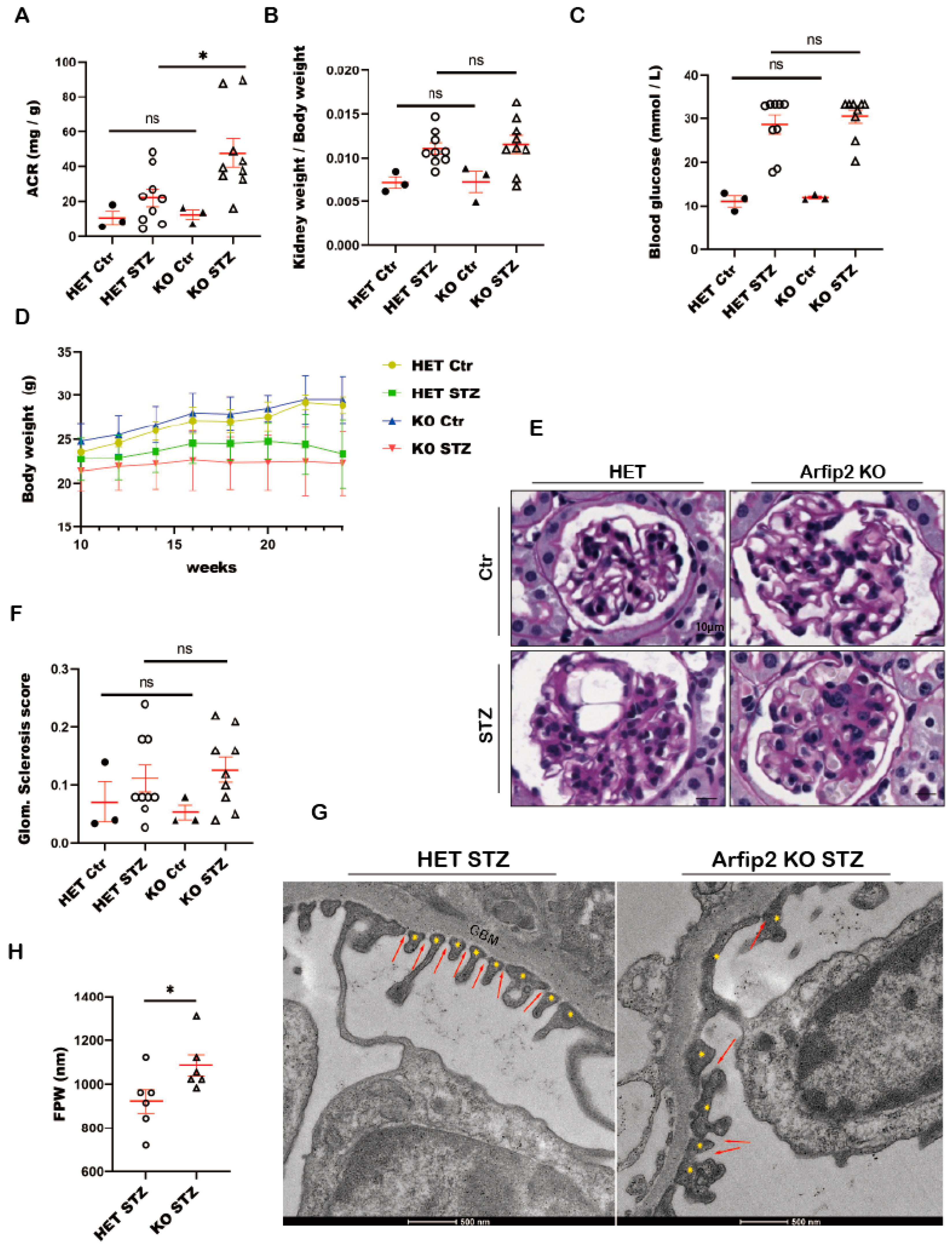
3.8. Arfip2 Deficiency Deteriorates Kidney Function in STZ Induced Type I Diabetic Nephropathy in Mice
3.9. Arfip2 Knockout Mice Show Subtle Glomerular Sclerosis in Type I Diabetic Nephropathy
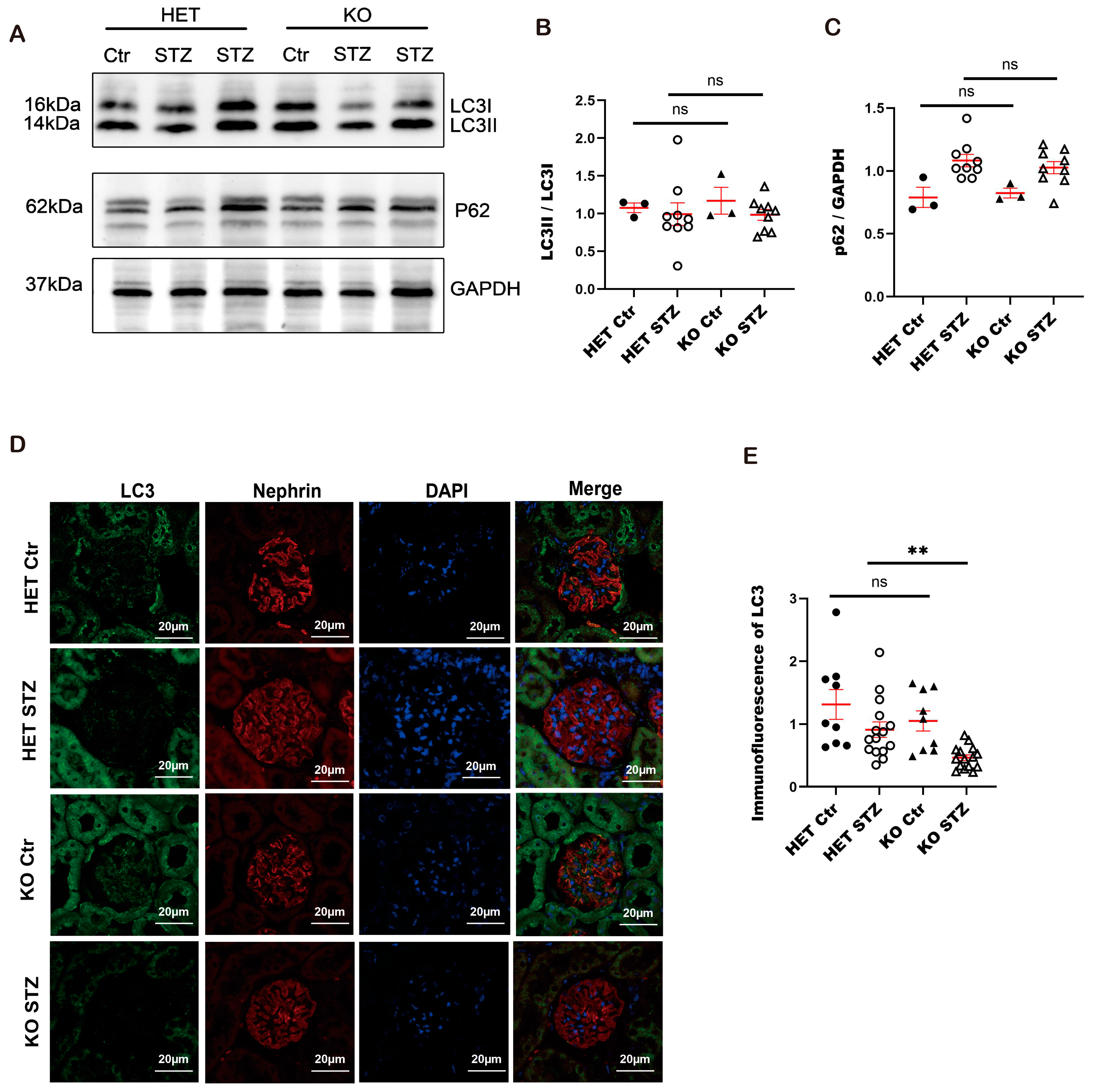
3.10. Autophagy Is Impaired in Constitutive Arfip2 Knockout Mice in Type I Diabetic Nephropathy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pavenstadt, H.; Kriz, W.; Kretzler, M. Cell biology of the glomerular podocyte. Physiol. Rev. 2003, 83, 253–307. [Google Scholar] [CrossRef]
- Wanner, N.; Hartleben, B.; Herbach, N.; Goedel, M.; Stickel, N.; Zeiser, R.; Walz, G.; Moeller, M.J.; Grahammer, F.; Huber, T.B. Unraveling the role of podocyte turnover in glomerular aging and injury. J. Am. Soc. Nephrol. 2014, 25, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.R.; Anderson, S. The aging kidney: Physiological changes. Adv. Chronic. Kidney Dis. 2010, 17, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; LeHir, M. Pathways to nephron loss starting from glomerular diseases-insights from animal models. Kidney Int. 2005, 67, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M. Podocyte injury and its consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Wang, G.H.; Lu, J.; Chen, P.P.; Zhang, Y.; Hu, Z.B.; Ma, K.L. Role of Podocyte Injury in Glomerulosclerosis. Adv. Exp. Med. Biol. 2019, 1165, 195–232. [Google Scholar] [PubMed]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005, 12 (Suppl. S2), 1542–1552. [Google Scholar] [CrossRef]
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr. 2007, 27, 19–40. [Google Scholar] [CrossRef]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef]
- Yan, L.; Vatner, D.E.; Kim, S.J.; Ge, H.; Masurekar, M.; Massover, W.H.; Yang, G.; Matsui, Y.; Sadoshima, J.; Vatner, S.F. Autophagy in chronically ischemic myocardium. Proc. Natl. Acad. Sci. USA 2005, 102, 13807–13812. [Google Scholar] [CrossRef]
- Huber, T.B.; Edelstein, C.L.; Hartleben, B.; Inoki, K.; Jiang, M.; Koya, D.; Kume, S.; Lieberthal, W.; Pallet, N.; Quiroga, A.; et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy 2012, 8, 1009–1031. [Google Scholar] [CrossRef] [PubMed]
- Kume, S.; Thomas, M.C.; Koya, D. Nutrient sensing, autophagy, and diabetic nephropathy. Diabetes 2012, 61, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, Z.; Zhang, S.; Zhang, T.; Liu, Y.; Zhang, L. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics 2023, 13, 736–766. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Garcia, D.; Ortega-Bellido, M.; Scarpa, M.; Villeneuve, J.; Jovic, M.; Porzner, M.; Balla, T.; Seufferlein, T.; Malhotra, V. Recruitment of arfaptins to the trans-Golgi network by PI(4)P and their involvement in cargo export. EMBO J. 2013, 32, 1717–1729. [Google Scholar] [CrossRef]
- Ambroggio, E.E.; Sillibourne, J.; Antonny, B.; Manneville, J.B.; Goud, B. Arf1 and membrane curvature cooperate to recruit Arfaptin2 to liposomes. PLoS ONE 2013, 8, e62963. [Google Scholar] [CrossRef]
- Rangone, H.; Pardo, R.; Colin, E.; Girault, J.A.; Saudou, F.; Humbert, S. Phosphorylation of arfaptin 2 at Ser260 by Akt Inhibits PolyQ-huntingtin-induced toxicity by rescuing proteasome impairment. J. Biol. Chem. 2005, 280, 22021–22028. [Google Scholar] [CrossRef]
- Judith, D.; Tooze, S.A. ATG9A supplies PtdIns4P to the autophagosome initiation site. Autophagy 2019, 15, 1660–1661. [Google Scholar] [CrossRef]
- Huang, K.; Lin, Y.; Wang, K.; Shen, J.; Wei, D. ARFIP2 Regulates EMT and Autophagy in Hepatocellular Carcinoma in Part Through the PI3K/Akt Signalling Pathway. J. Hepatocell. Carcinoma 2022, 9, 1323–1339. [Google Scholar] [CrossRef]
- Feng, R.; Wen, J. Overview of the roles of Sox2 in stem cell and development. Biol. Chem. 2015, 396, 883–891. [Google Scholar] [CrossRef]
- Rogg, M.; Maier, J.I.; Van Wymersch, C.; Helmstadter, M.; Sammarco, A.; Lindenmeyer, M.; Zareba, P.; Montanez, E.; Walz, G.; Werner, M.; et al. alpha-Parvin Defines a Specific Integrin Adhesome to Maintain the Glomerular Filtration Barrier. J. Am. Soc. Nephrol. JASN 2022, 33, 786–808. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, J.G.; van den Bergh Weerman, M.A.; Assmann, K.J.; Weening, J.J.; Florquin, S. Podocyte foot process effacement is not correlated with the level of proteinuria in human glomerulopathies. Kidney Int. 2004, 66, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Katti, P.; Biete, M.; Mungai, M.; AshShareef, S.; Neikirk, K.; Garza Lopez, E.; Vue, Z.; Christensen, T.A.; Beasley, H.K.; et al. A Universal Approach to Analyzing Transmission Electron Microscopy with ImageJ. Cells 2021, 10, 2177. [Google Scholar] [CrossRef] [PubMed]
- Rogg, M.; Yasuda-Yamahara, M.; Abed, A.; Dinse, P.; Helmstädter, M.; Conzelmann, A.C.; Frimmel, J.; Sellung, D.; Biniossek, M.L.; Kretz, O.; et al. The WD40-domain containing protein CORO2B is specifically enriched in glomerular podocytes and regulates the ventral actin cytoskeleton. Sci. Rep. 2017, 7, 15910. [Google Scholar] [CrossRef] [PubMed]
- Judith, D.; Jefferies, H.B.J.; Boeing, S.; Frith, D.; Snijders, A.P.; Tooze, S.A. ATG9A shapes the forming autophagosome through Arfaptin 2 and phosphatidylinositol 4-kinase IIIβ. J. Cell Biol. 2019, 218, 1634–1652. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Padman, B.S.; Zellner, S.; Khuu, G.; Uoselis, L.; Lam, W.K.; Skulsuppaisarn, M.; Lindblom, R.S.J.; Watts, E.M.; Behrends, C.; et al. ATG4 family proteins drive phagophore growth independently of the LC3/GABARAP lipidation system. Mol. Cell 2021, 81, 2013–2030.e2019. [Google Scholar] [CrossRef]
- Bork, T.; Liang, W.; Yamahara, K.; Lee, P.; Tian, Z.; Liu, S.; Schell, C.; Thedieck, K.; Hartleben, B.; Patel, K.; et al. Podocytes maintain high basal levels of autophagy independent of mtor signaling. Autophagy 2020, 16, 1932–1948. [Google Scholar] [CrossRef]
- Liu, M.; Ning, X.; Li, R.; Yang, Z.; Yang, X.; Sun, S.; Qian, Q. Signalling pathways involved in hypoxia-induced renal fibrosis. J. Cell. Mol. Med. 2017, 21, 1248–1259. [Google Scholar] [CrossRef]
- Bechtel, W.; Helmstädter, M.; Balica, J.; Hartleben, B.; Kiefer, B.; Hrnjic, F.; Schell, C.; Kretz, O.; Liu, S.; Geist, F.; et al. Vps34 deficiency reveals the importance of endocytosis for podocyte homeostasis. J. Am. Soc. Nephrol. JASN 2013, 24, 727–743. [Google Scholar] [CrossRef]
- Xin, W.; Li, Z.; Xu, Y.; Yu, Y.; Zhou, Q.; Chen, L.; Wan, Q. Autophagy protects human podocytes from high glucose-induced injury by preventing insulin resistance. Metab. Clin. Exp. 2016, 65, 1307–1315. [Google Scholar] [CrossRef]
- Al-Bari, M.A.A.; Xu, P. Molecular regulation of autophagy machinery by mTOR-dependent and -independent pathways. Ann. N. Y. Acad. Sci. 2020, 1467, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Van Buren, P.N.; Toto, R. Current update in the management of diabetic nephropathy. Curr. Diabetes Rev. 2013, 9, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Chimal, J.; Jaisser, F. Pathophysiologic mechanisms in diabetic kidney disease: A focus on current and future therapeutic targets. Diabetes Obes. Metab. 2020, 22 (Suppl. S1), 16–31. [Google Scholar] [CrossRef] [PubMed]
- Fioretto, P.; Mauer, M. Histopathology of diabetic nephropathy. Semin. Nephrol. 2007, 27, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Xu, X.; Tang, C.; Gao, P.; Chen, X.; Xiong, X.; Yang, M.; Yang, S.; Zhu, X.; Yuan, S.; et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol. 2018, 16, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Umanath, K.; Lewis, J.B. Update on Diabetic Nephropathy: Core Curriculum 2018. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2018, 71, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Samsu, N. Diabetic Nephropathy: Challenges in Pathogenesis, Diagnosis, and Treatment. BioMed Res. Int. 2021, 2021, 1497449. [Google Scholar] [CrossRef]
- Guo, H.; Wang, Y.; Zhang, X.; Zang, Y.; Zhang, Y.; Wang, L.; Wang, H.; Wang, Y.; Cao, A.; Peng, W. Astragaloside IV protects against podocyte injury via SERCA2-dependent ER stress reduction and AMPKalpha-regulated autophagy induction in streptozotocin-induced diabetic nephropathy. Sci. Rep. 2017, 7, 6852. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Kim, D.J.; Park, S.H.; Sung, M.; Kim, J.; Oh, J.; Yang, D.H.; et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol. Ren. Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef]
- Lenoir, O.; Jasiek, M.; Henique, C.; Guyonnet, L.; Hartleben, B.; Bork, T.; Chipont, A.; Flosseau, K.; Bensaada, I.; Schmitt, A.; et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 2015, 11, 1130–1145. [Google Scholar] [CrossRef]
- Fang, L.; Zhou, Y.; Cao, H.; Wen, P.; Jiang, L.; He, W.; Dai, C.; Yang, J. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS ONE 2013, 8, e60546. [Google Scholar] [CrossRef] [PubMed]
- Boerries, M.; Grahammer, F.; Eiselein, S.; Buck, M.; Meyer, C.; Goedel, M.; Bechtel, W.; Zschiedrich, S.; Pfeifer, D.; Laloe, D.; et al. Molecular fingerprinting of the podocyte reveals novel gene and protein regulatory networks. Kidney Int. 2013, 83, 1052–1064. [Google Scholar] [CrossRef] [PubMed]
- Barisoni, L.; Diomedi-Camassei, F.; Santorelli, F.M.; Caridi, G.; Thomas, D.B.; Emma, F.; Piemonte, F.; Ghiggeri, G.M. Collapsing glomerulopathy associated with inherited mitochondrial injury. Kidney Int. 2008, 74, 237–243. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Liu, M.; Fan, Y.; Li, D.; Han, B.; Meng, Y.; Chen, F.; Liu, T.; Song, Z.; Han, Y.; Huang, L.; et al. Ferroptosis inducer erastin sensitizes NSCLC cells to celastrol through activation of the ROS-mitochondrial fission-mitophagy axis. Mol. Oncol. 2021, 15, 2084–2105. [Google Scholar] [CrossRef]
- Pan, X.C.; Xiong, Y.L.; Hong, J.H.; Liu, Y.; Cen, Y.Y.; Liu, T.; Yang, Q.F.; Tao, H.; Li, Y.N.; Zhang, H.G. Cardiomyocytic FoxP3 is involved in Parkin-mediated mitophagy during cardiac remodeling and the regulatory role of triptolide. Theranostics 2022, 12, 2483–2501. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Names | Gene Accession Numbers | Sequence |
|---|---|---|
| Arfip2_sonde1_ml | NM_029802.4 | 5′ CGC GGG ACG CGT AGT CCC TGC TCA CAG TGC TT 3′ |
| Arfip2_sonde1_no | NM_029802.4 | 5′ CGC GGG GCG GCC GCC TGC AGT TGT CTT TGG ACC TG 3′ |
| Arfip2_sonde2_ml | NM_029802.4 | 5′ CGC GGG ACG CGT GTC AGG ACC CAA CCT CAA TG 3′ |
| Arfip2_sonde2_no | NM_029802.4 | 5′ CGC GGG GCG GCC GCC GTC TGT CCG GTA GGC ATC AT 3′ |
| in situ mArfip2 KO_f | NM_029802.4 | 5′ ATC CAT CTC ACA GCA CGT CA 3′ |
| in situ mArfip2 KO_r | NM_029802.4 | 5′ AAG CTC TGG GGA CTT CTG G 3′ |
| mArfip2 KO_f | JN948356.1 | 5′ TGC AGG CAC ATA GAC ACA TAC AGG AA 3′ |
| mArfip2 KO_r | JN948356.1 | 5′ GGA ACT TCA GCT TGA TGG CC 3′ |
| mArfip2 HET_f | JN948356.1 | 5′ TGC AGG CAC ATA GAC ACA TAC AGG AA 3′ |
| mArfip2 HET_r | JN948356.1 | 5′ TGT ACG AGA TAA GCG GGA ATA GAA GCA A 3′ |
| mArfip2 flox_f | JN948356.1 | 5′ AGC TTC TCA TAC TTG TCC CGA T 3′ |
| mArfip2 flox_r | JN948356.1 | 5′ AGC TTC TCA TAC TTG TCC CGA T 3′ |
| gRNA_Arfip2 | NM_012402.5 | 5′ GAG CGT GTC TTC CAT GGT CTT GG 3′ |
| Crispr/cas9 hArfip2_f | NM_012402.5 | 5′ AAG ACC ATG GAA GAC ACG CTC 3′ |
| Crispr/cas9 hArfip2_r | NM_012402.5 | 5′ CCC GAT GGG CCT GGA AAG 3′ |
| 5FRT_f | 5′ AGG CGC ATA AAC GAT ACC ACG AT 3′ | |
| 5FRT_r | 5′ CCA CAA CGG GTT CTT CTG TT 3′ | |
| Arfip Ef (forward) | JN948356.1 | 5′ TGC AGG CAC ATA GAC ACA TAC AGG AA 3′ |
| Arfip Er3 (reverse wildtype) | JN948356.1 | 5′ TGT ACG AGA TAA GCG GGA ATA GAA GCA A 3′ |
| Arfip Kr (reverse mutant) | JN948356.1 | 5′ GGG CAA GAA CAT AAA GTG ACC CTC C 3′ |
| Antibody/Fluorescent Compound | Dilution | Condition | Company |
|---|---|---|---|
| Rabbit anti-Arfapin 2 (40-2400) | 1:1000 | WB | Thermo Fisher Scientific, Germany |
| Rabbit anti-LC3B (#2775) | 1:1000 1:100 | WB IF | Cell Signaling Biotechnology Cambridge, UK |
| Guinea pig anti-nephrin (GP-N2) | 1:100 | IF | Progen Heidelberg, Germany |
| Guinea pig anti-p62 (GP62-C) | 1:1000 | WB | Progen Heidelberg, Germany |
| Rabbit anti-PINK 1 (BC100-494) | 1:1000 | WB | Novus Biologicals, USA |
| Rabbit anti-Parkin (14060-1-AP) | 1:1000 | WB | Proteintech, Manchester, UK |
| Mouse anti-TOM20 (F-10) | 1:100 | IF | Santa Cruz Biotech, Dallas, TX, USA |
| Rabbit anti-ATG9A (NB110-56893) | 1:1000 | WB | Novus Biologicals, USA |
| Rabbit anti-Lamp2 (ab37024) | 1:1000 | WB | Abcam, Berlin, Germany |
| Rabbit anti-GAPDH (14C10) | 1:1000 | WB | Cell Signaling Biotechnology, Cambridge, UK |
| Rabbit anti-Drp-1 (D6C7) | 1:1000 | WB | Cell Signaling Biotechnology, Cambridge, UK |
| Mouse anti-Opa-1(612607) | 1:1000 | WB | BD Transduction Laboratories™, Heidelberg, Germany |
| Alexa Fluor 488 donkey anti-mouse IgG | 1:500 | IF, pH9 | Invitrogen, Karlsruhe, Germany |
| Alexa Fluor 488 donkey anti-rabbit IgG | 1:500 | IF, pH9 | Invitrogen, Karlsruhe, Germany |
| Alexa Fluor 555 donkey anti-guinea pig IgG | 1:500 | IF, pH9 | Invitrogen, Karlsruhe, Germany |
| Hoechst | 1:5000 | IF, pH9 | Molecular Probes, Eugene, OR, USA |
| Goat anti-rabbit IgG-HRP (#7074S) | 1:3000 | WB | Cell Signaling Biotechnology Cambridge, UK |
| Goat anti-mouse IgG-HRP (#P0447) | 1:10,000 | WB | Dako, Glostrup, Denmark |
| Goat anti-guinea pig (A0564) | 1:8000 | WB | Dako, Glostrup, Denmark |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, H.; Rogg, M.; Keller, J.; Scherzinger, A.-K.; Jäckel, J.; Meyer, C.; Sammarco, A.; Helmstädter, M.; Gorka, O.; Groß, O.; et al. ADP-Ribosylation Factor-Interacting Protein 2 Acts as a Novel Regulator of Mitophagy and Autophagy in Podocytes in Diabetic Nephropathy. Antioxidants 2024, 13, 81. https://doi.org/10.3390/antiox13010081
Guo H, Rogg M, Keller J, Scherzinger A-K, Jäckel J, Meyer C, Sammarco A, Helmstädter M, Gorka O, Groß O, et al. ADP-Ribosylation Factor-Interacting Protein 2 Acts as a Novel Regulator of Mitophagy and Autophagy in Podocytes in Diabetic Nephropathy. Antioxidants. 2024; 13(1):81. https://doi.org/10.3390/antiox13010081
Chicago/Turabian StyleGuo, Haihua, Manuel Rogg, Julia Keller, Ann-Kathrin Scherzinger, Julia Jäckel, Charlotte Meyer, Alena Sammarco, Martin Helmstädter, Oliver Gorka, Olaf Groß, and et al. 2024. "ADP-Ribosylation Factor-Interacting Protein 2 Acts as a Novel Regulator of Mitophagy and Autophagy in Podocytes in Diabetic Nephropathy" Antioxidants 13, no. 1: 81. https://doi.org/10.3390/antiox13010081
APA StyleGuo, H., Rogg, M., Keller, J., Scherzinger, A.-K., Jäckel, J., Meyer, C., Sammarco, A., Helmstädter, M., Gorka, O., Groß, O., Schell, C., & Bechtel-Walz, W. (2024). ADP-Ribosylation Factor-Interacting Protein 2 Acts as a Novel Regulator of Mitophagy and Autophagy in Podocytes in Diabetic Nephropathy. Antioxidants, 13(1), 81. https://doi.org/10.3390/antiox13010081