
Flt3 Activation Mitigates Mitochondrial Fragmentation and Heart Dysfunction through Rebalanced L-OPA1 Processing by Hindering the Interaction between Acetylated p53 and PHB2 in Cardiac Remodeling

 , , , , ,
, , , , ,
Abstract
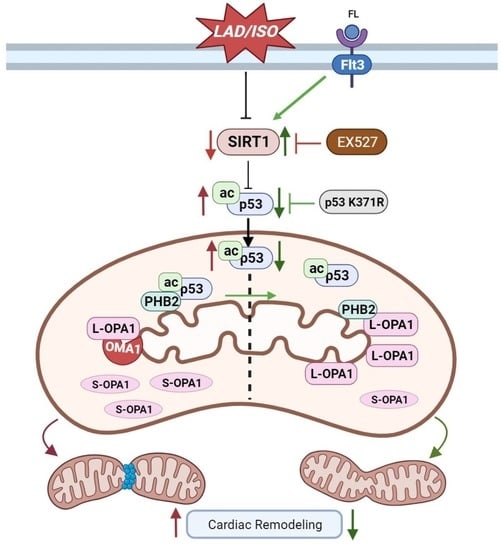
:
1. Introduction
2. Materials and Methods
2.1. Animal Experiment Protocol
2.2. Isolation, Culture, and Treatment of NRCMs
2.3. Echocardiography
2.4. Electrocardiographic Recordings
2.5. Histomorphology, Masson’s Trichrome Staining
2.6. Immunofluorescence Staining
2.7. Annexin V-FITC/PI Apoptosis Assay
2.8. Measurement of ROS
2.9. Mitochondrial Tracking
2.10. Quantitative Real-Time PCR (RT-qPCR)
2.11. Western Blotting Analysis
2.12. Transient cDNA Transfection
2.13. Immunoprecipitation (IP)
2.14. Statistical Analysis
3. Results
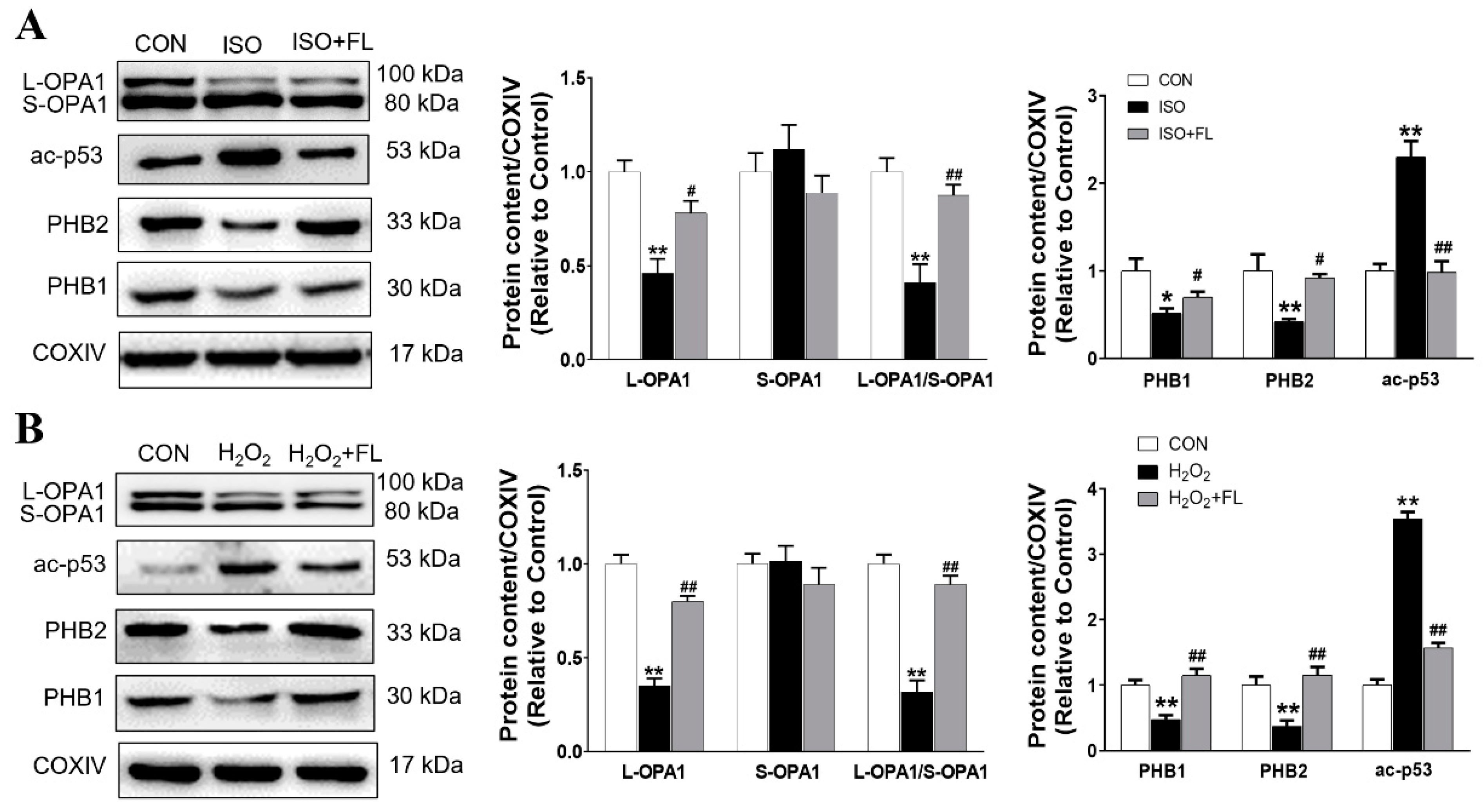
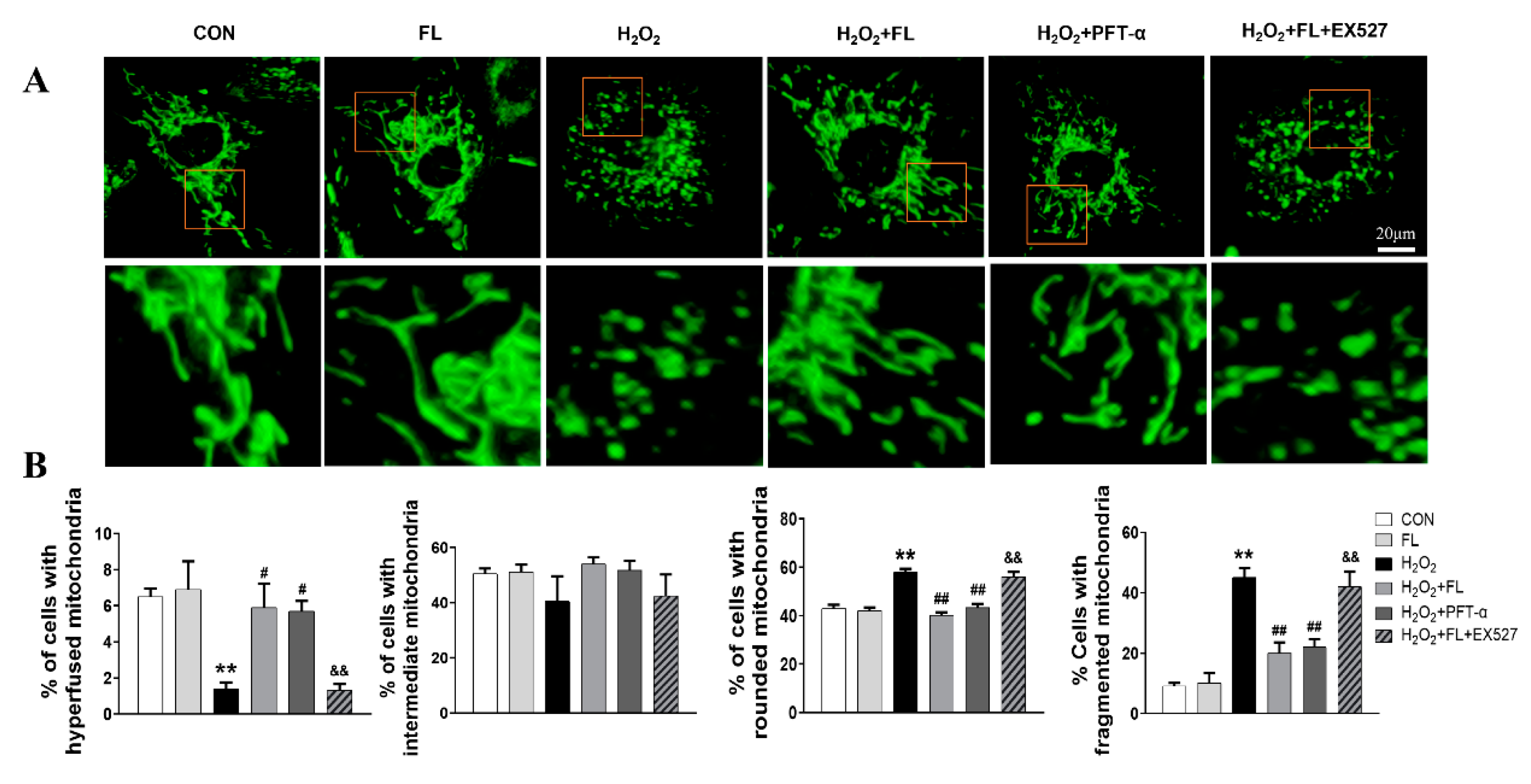
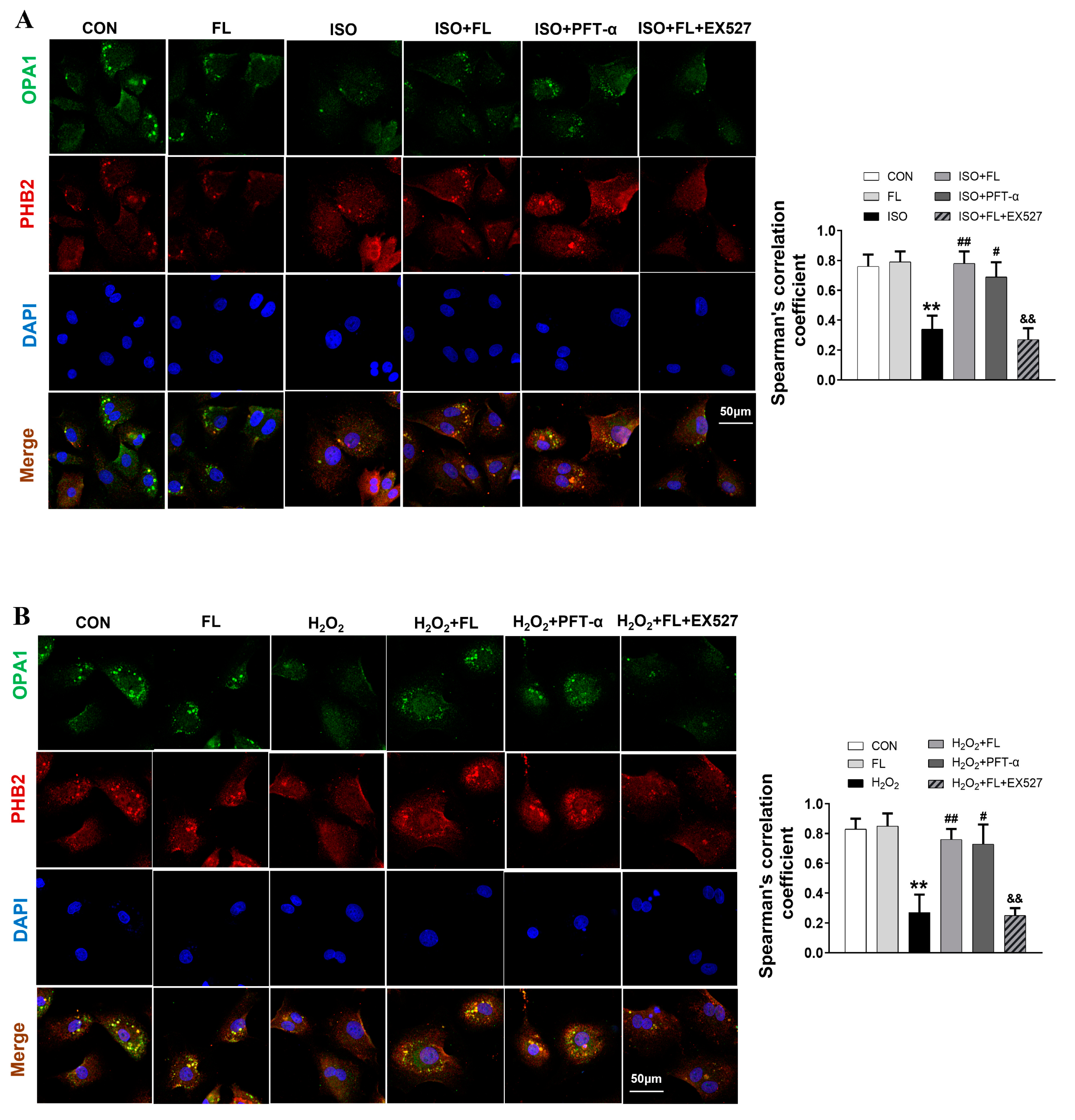
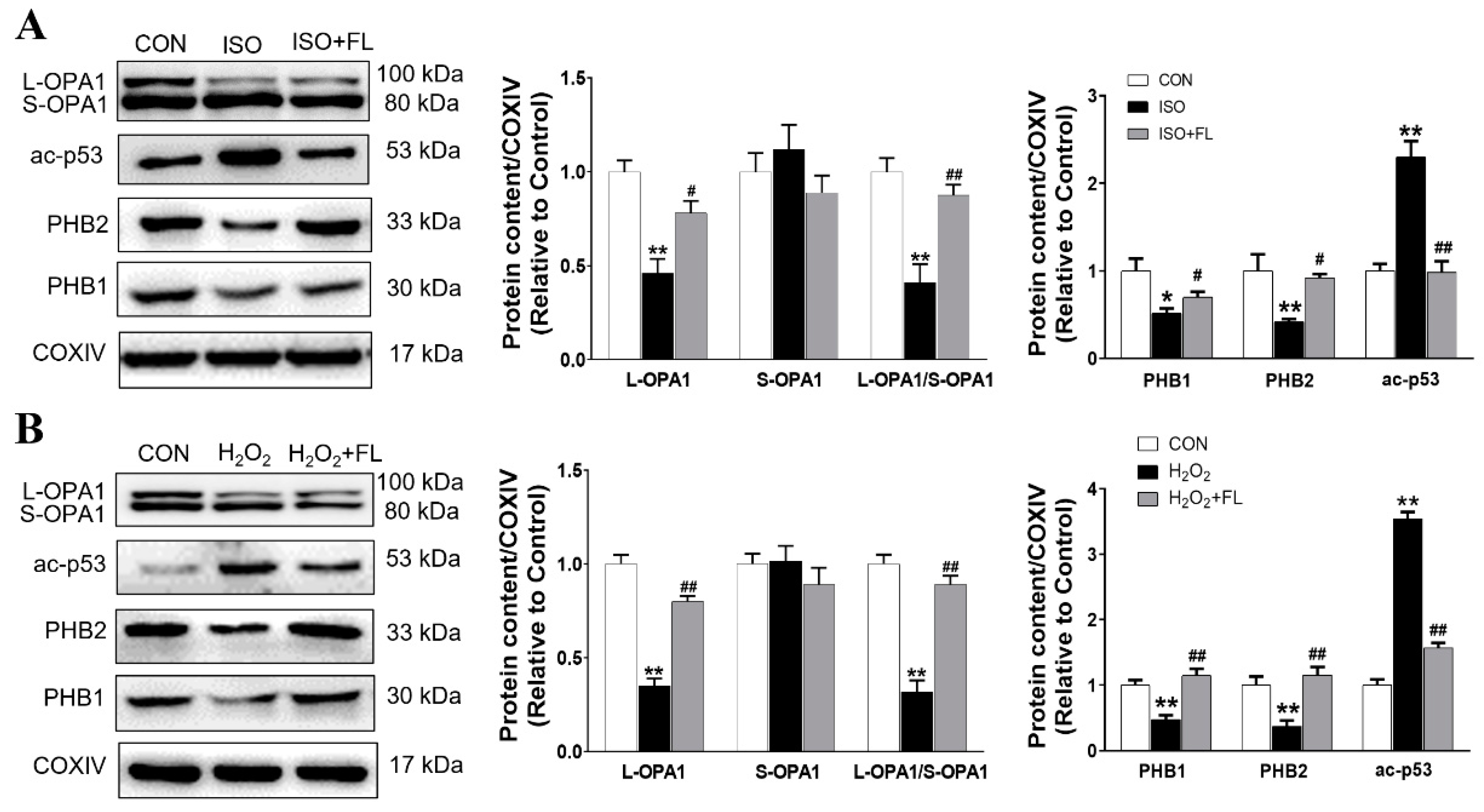
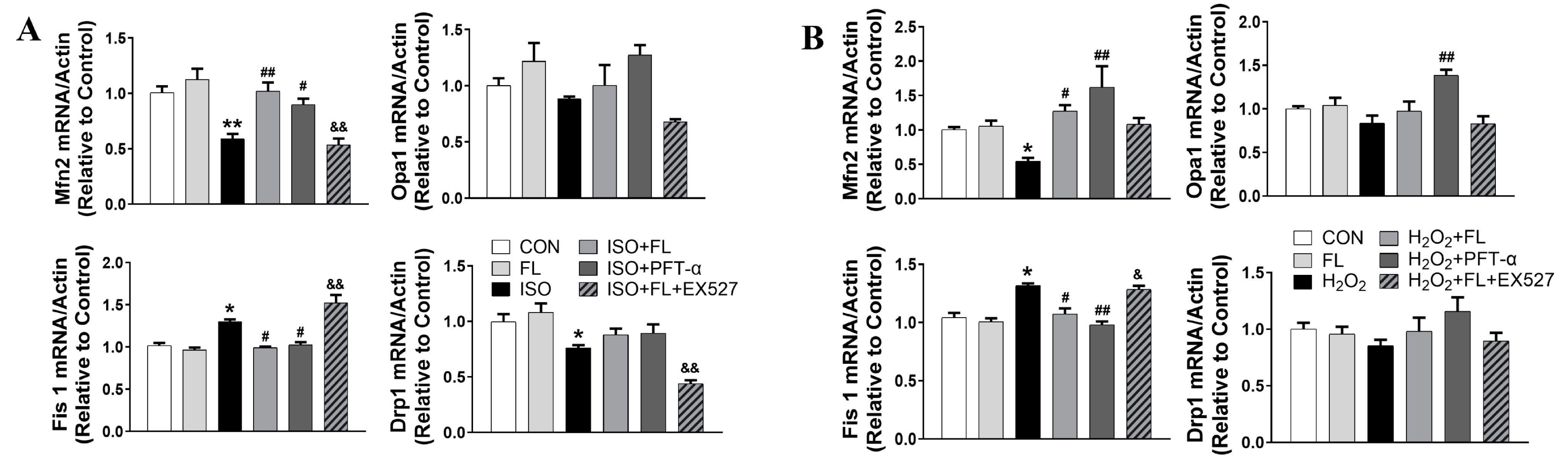
3.1. Flt3 Activation Restored ISO- or H2O2-Induced Mitochondrial Dynamics Imbalance by Reducing L-OPA1 Processing
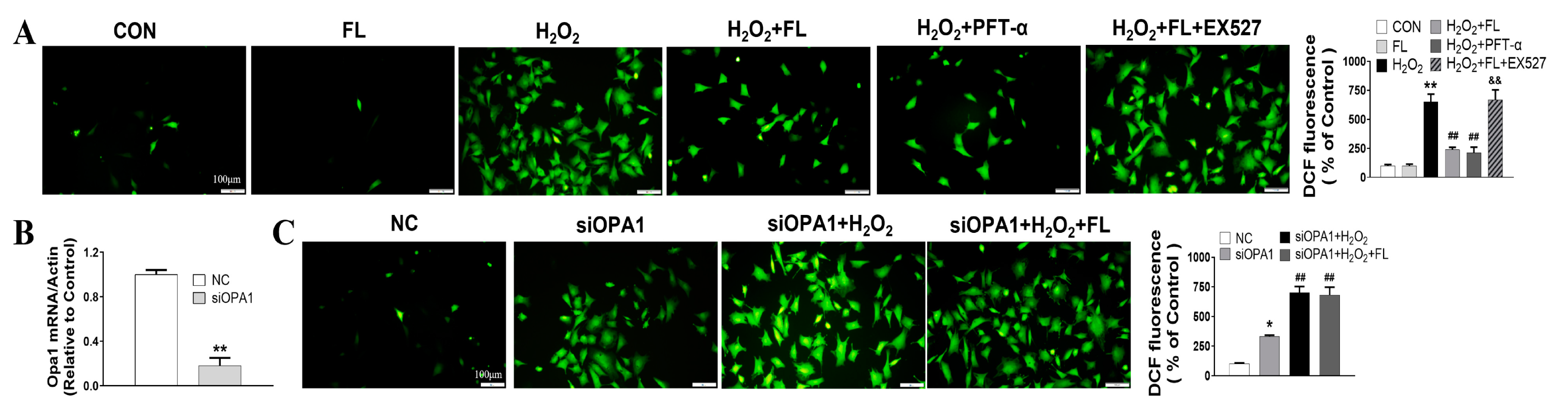
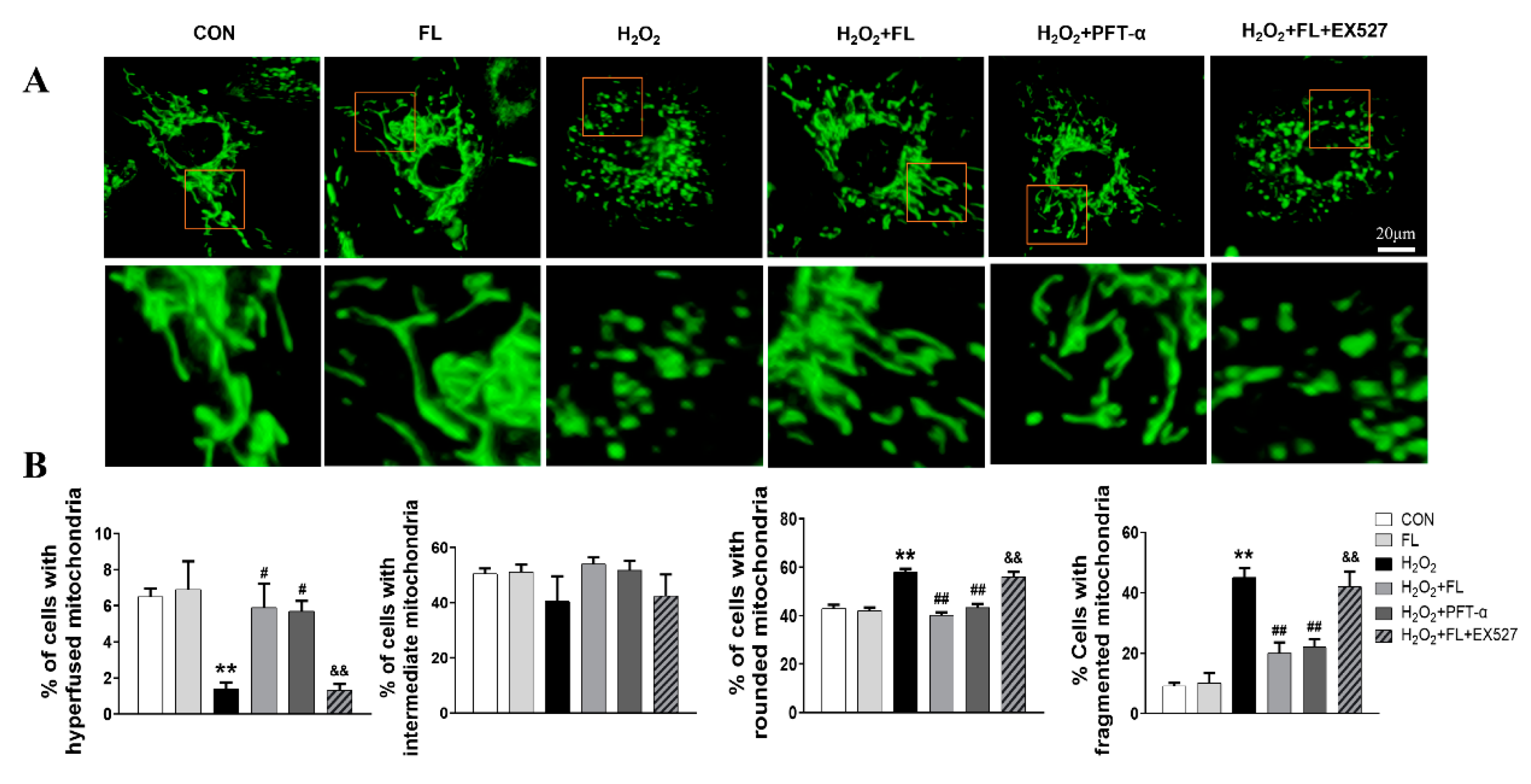
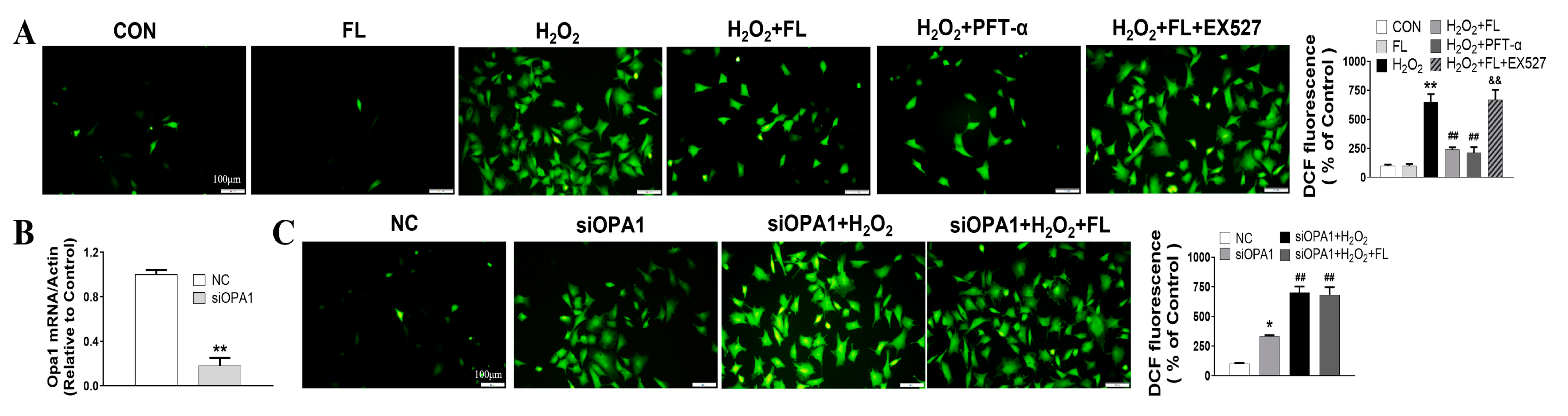
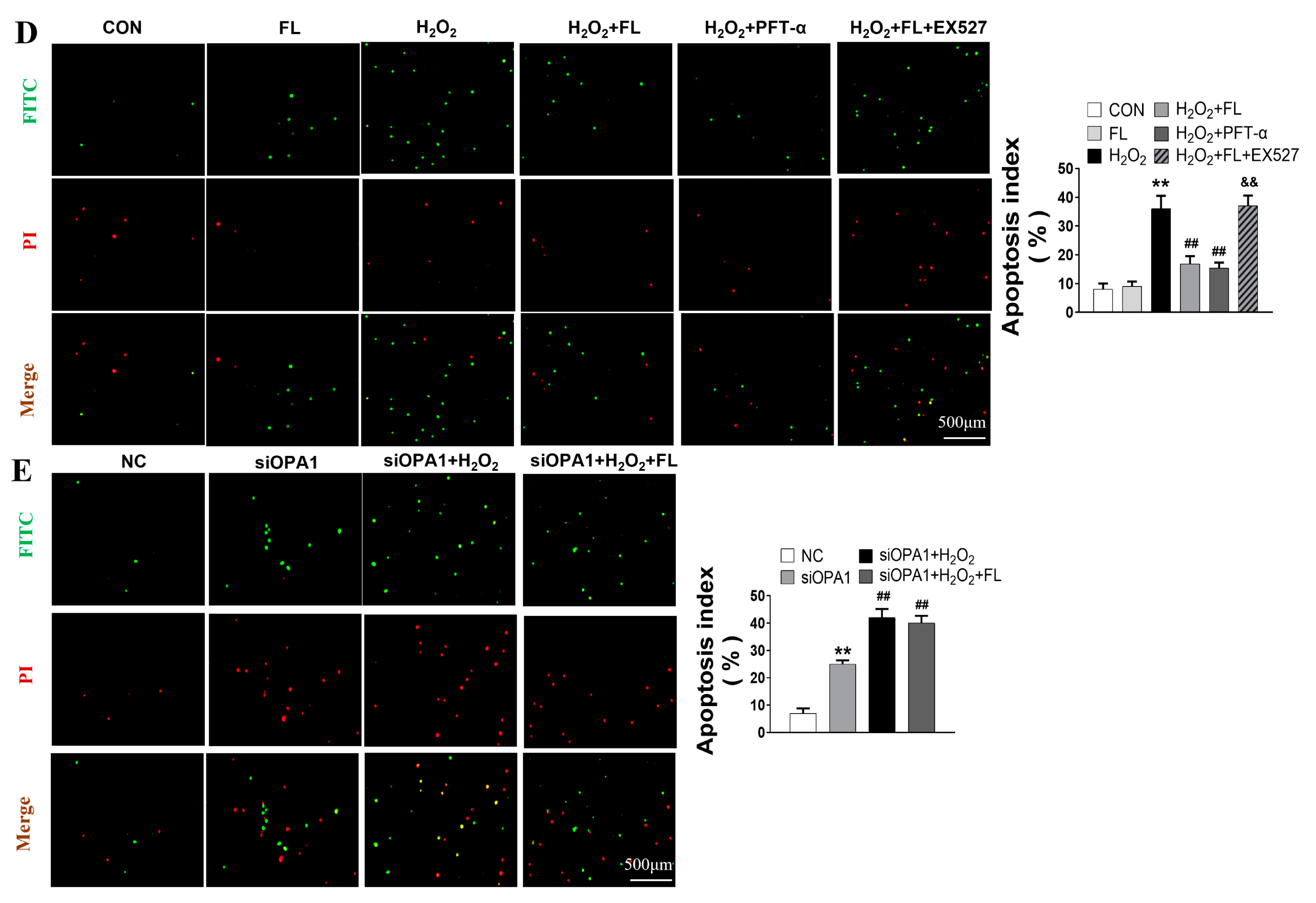
3.2. Flt3 Activation Inhibited H2O2-Induced ROS and Apoptosis by Improving Mitochondrial Dynamics Disturbance In Vitro
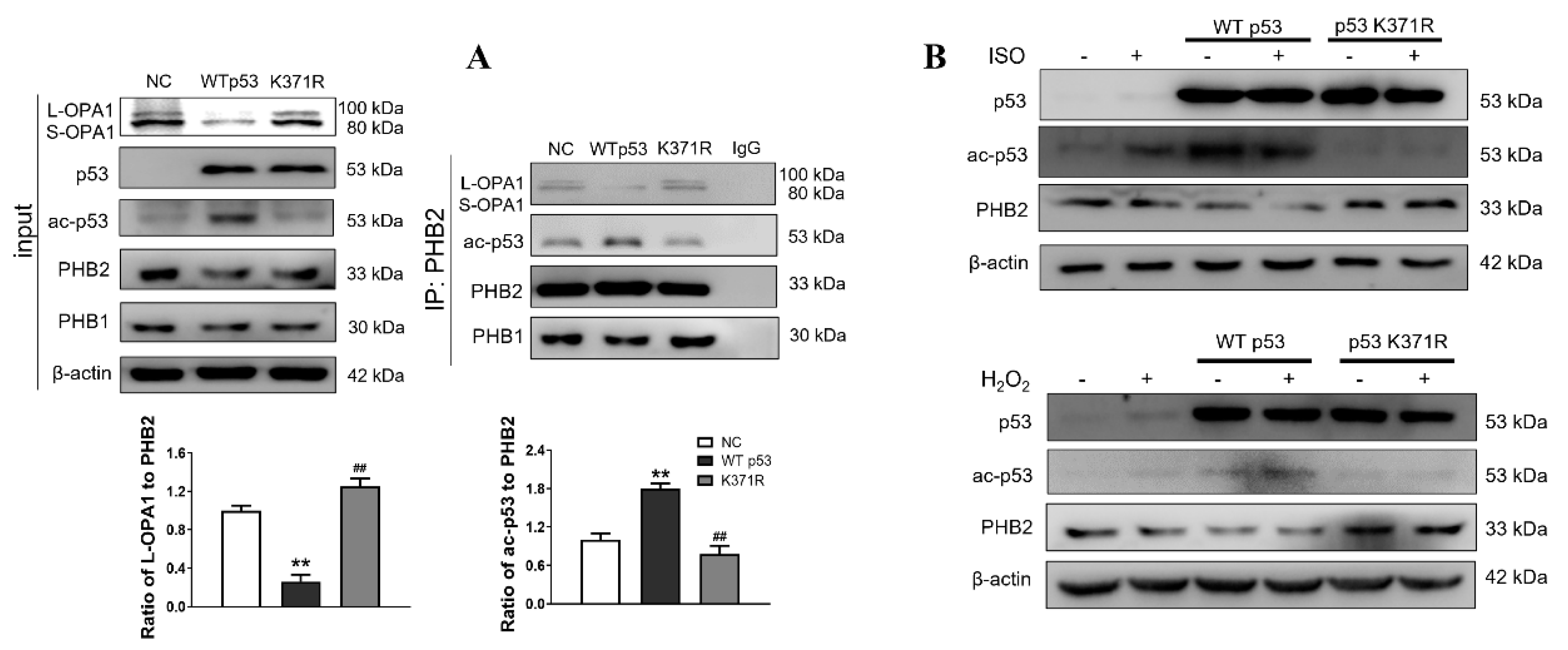
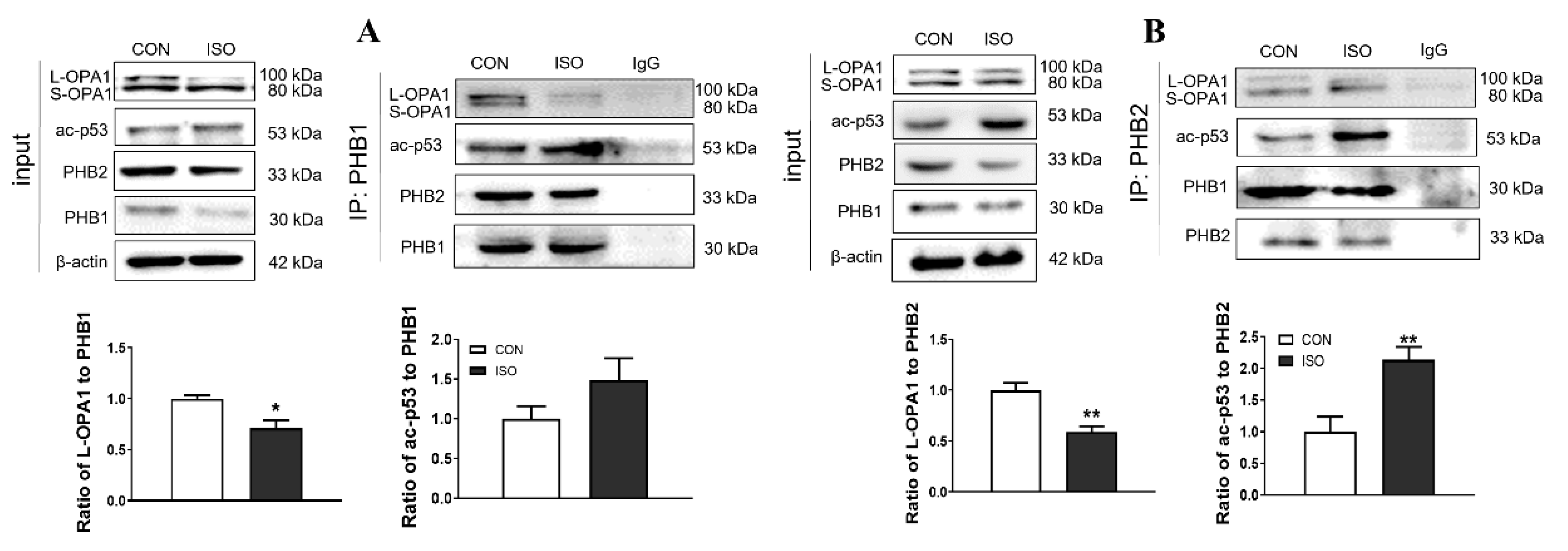
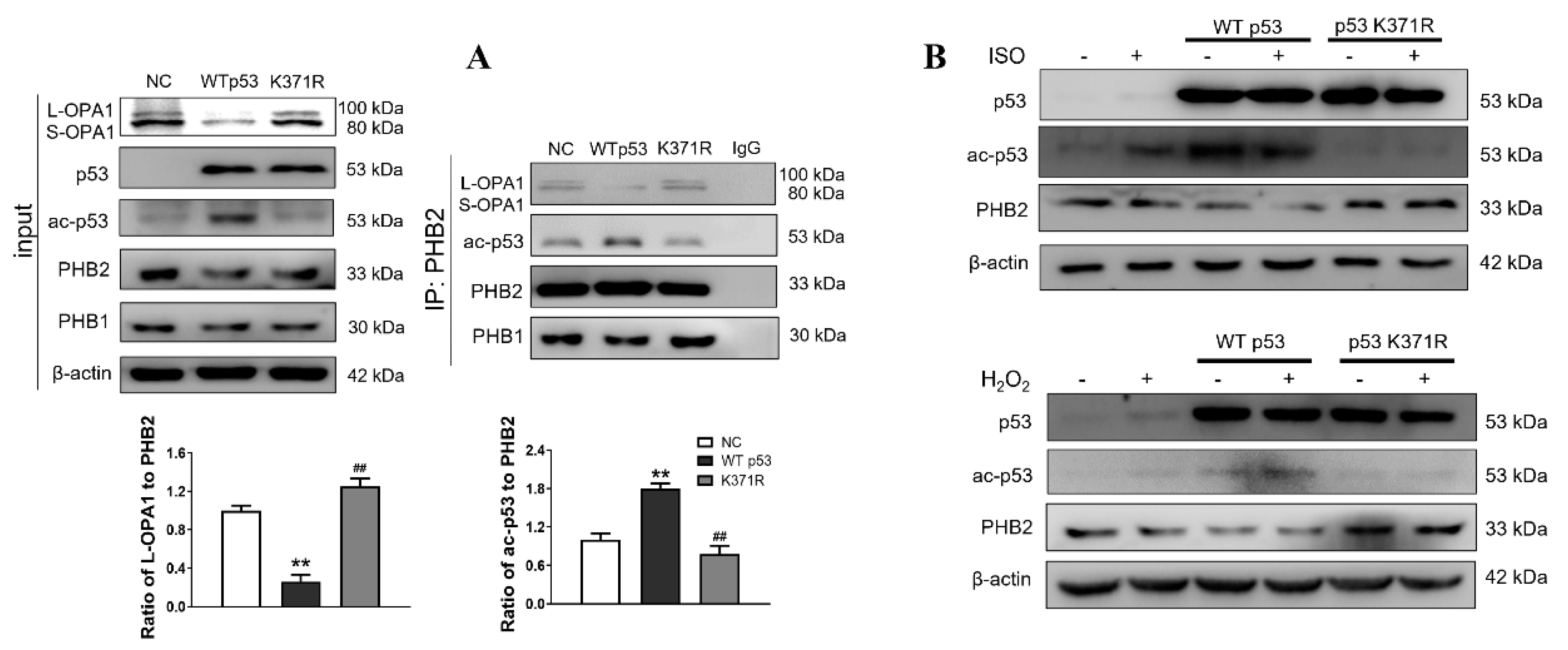
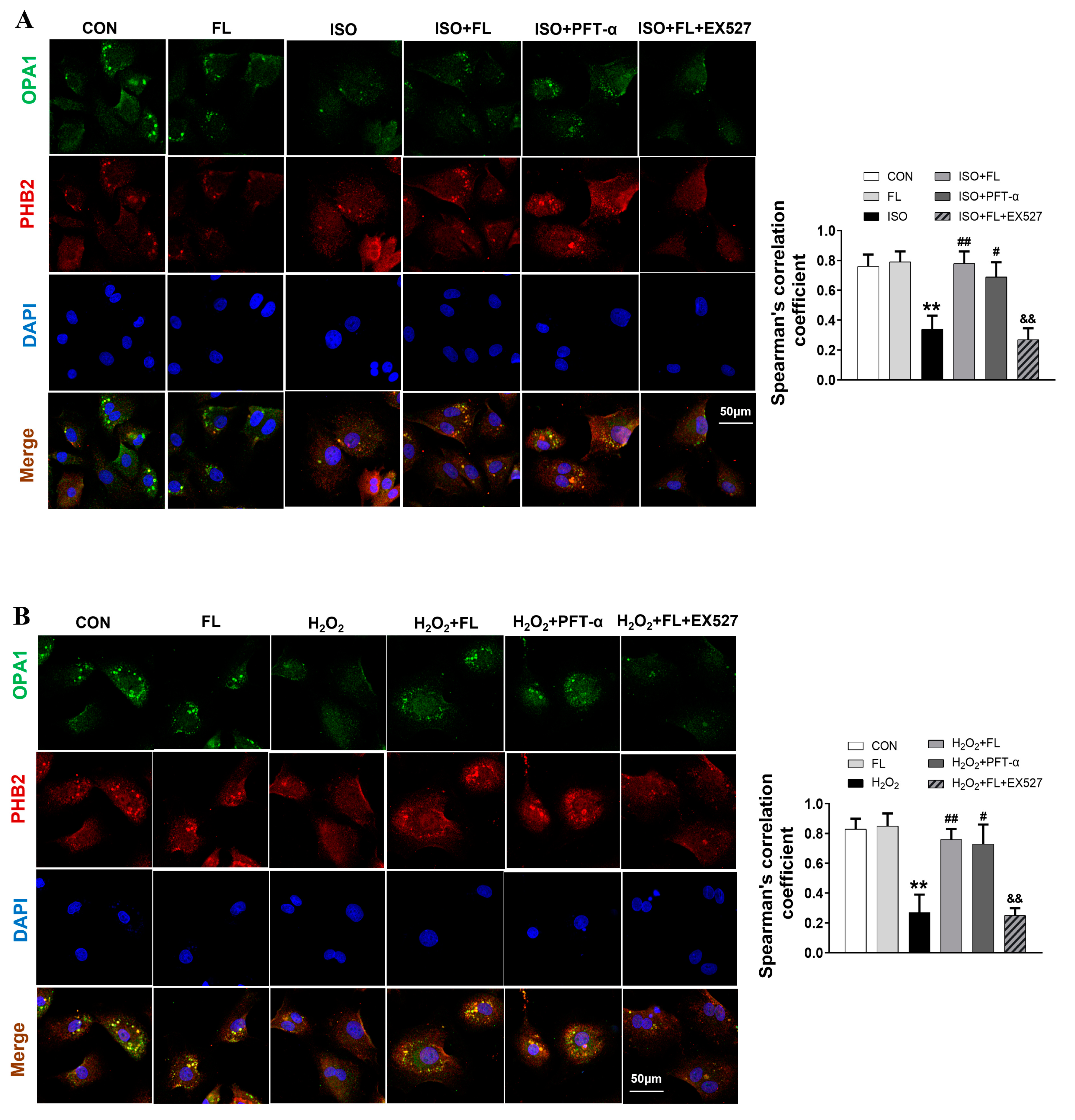
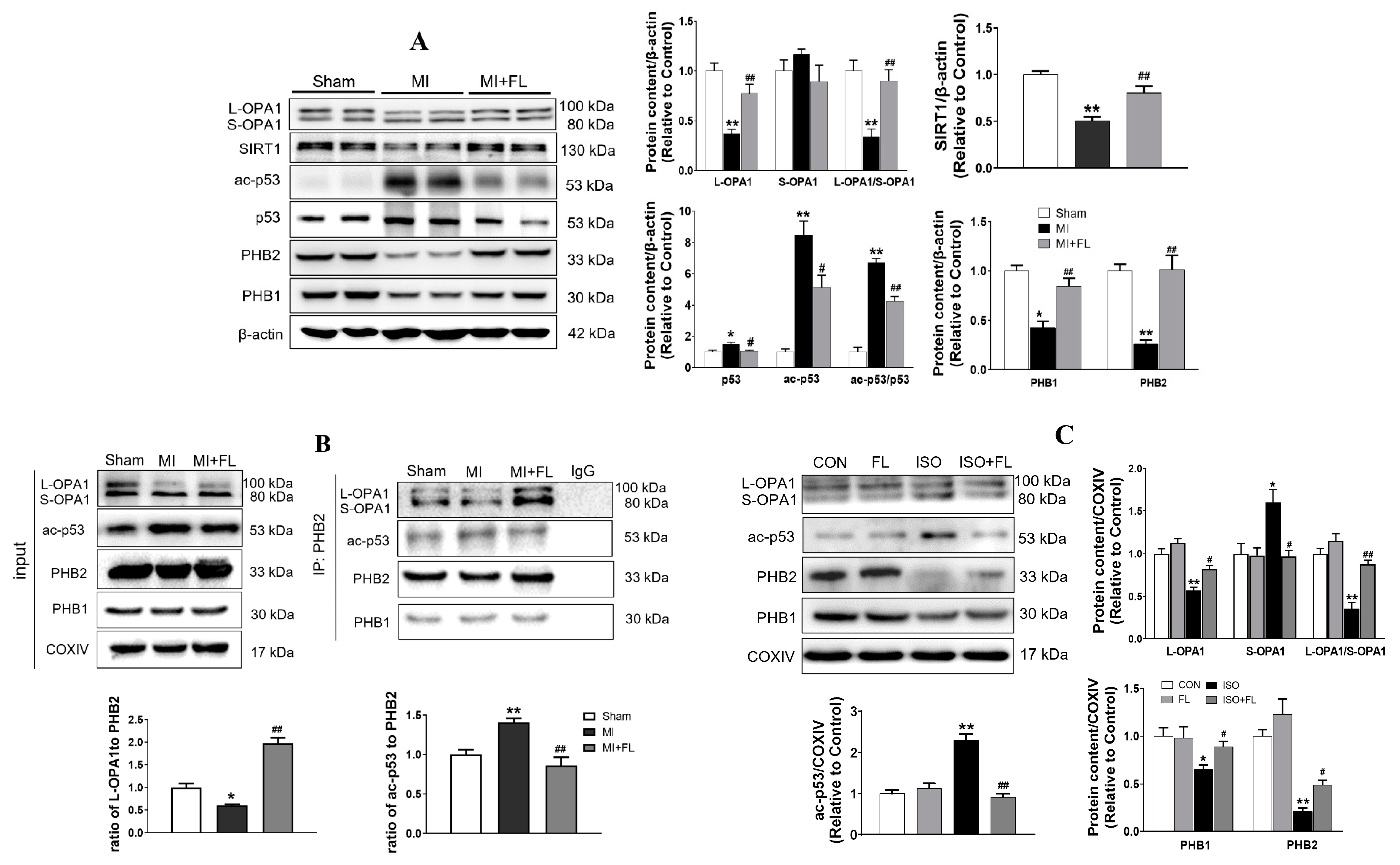
3.3. Interaction between ac-p53 and PHBs Contributed to Mitochondrial L-OPA1 Processing, Flt3 Activation Attenuated This Interaction, thus Increasing L-OPA1 and Improving ISO- or H2O2-Induced Mitochondrial Dynamics Disturbance
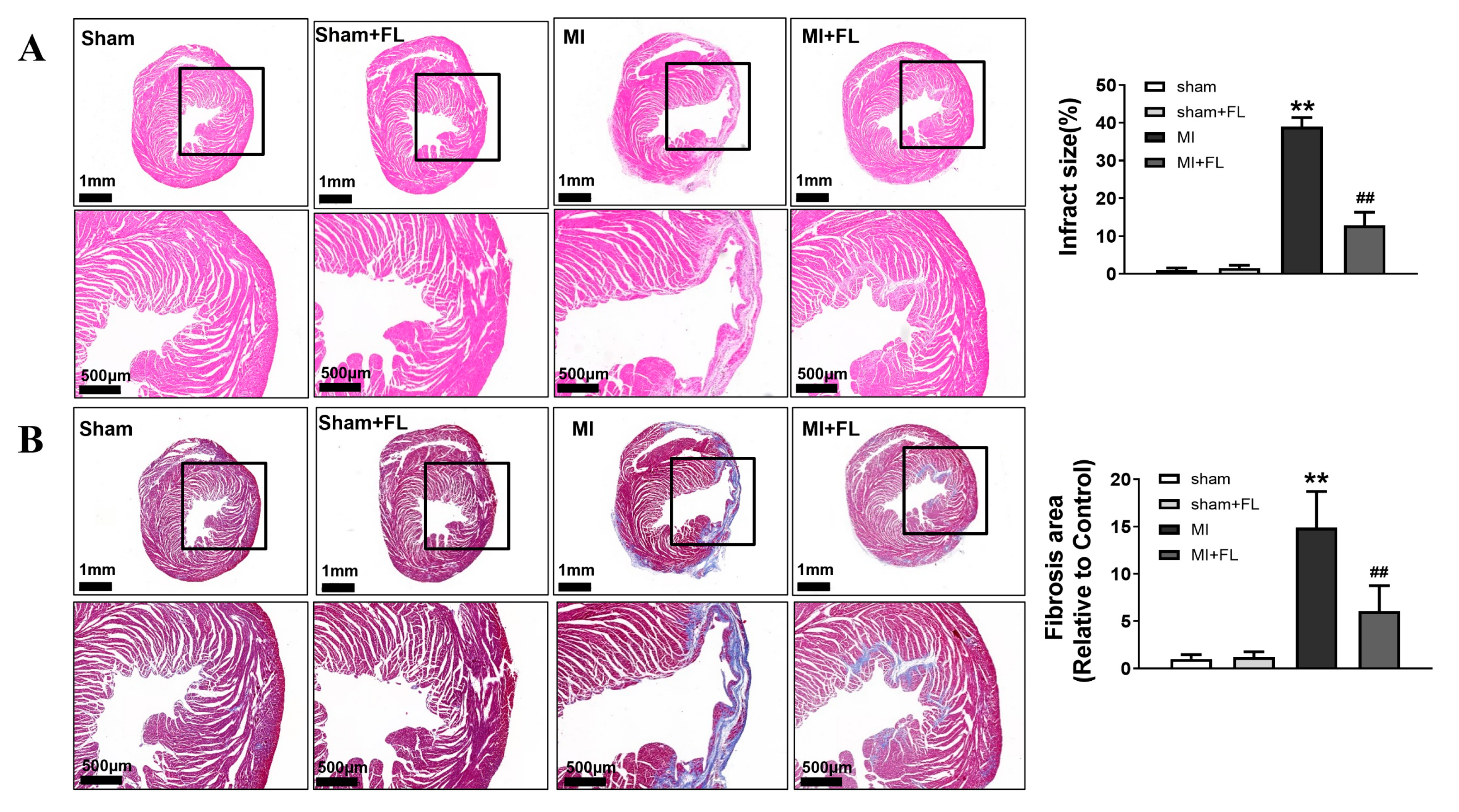
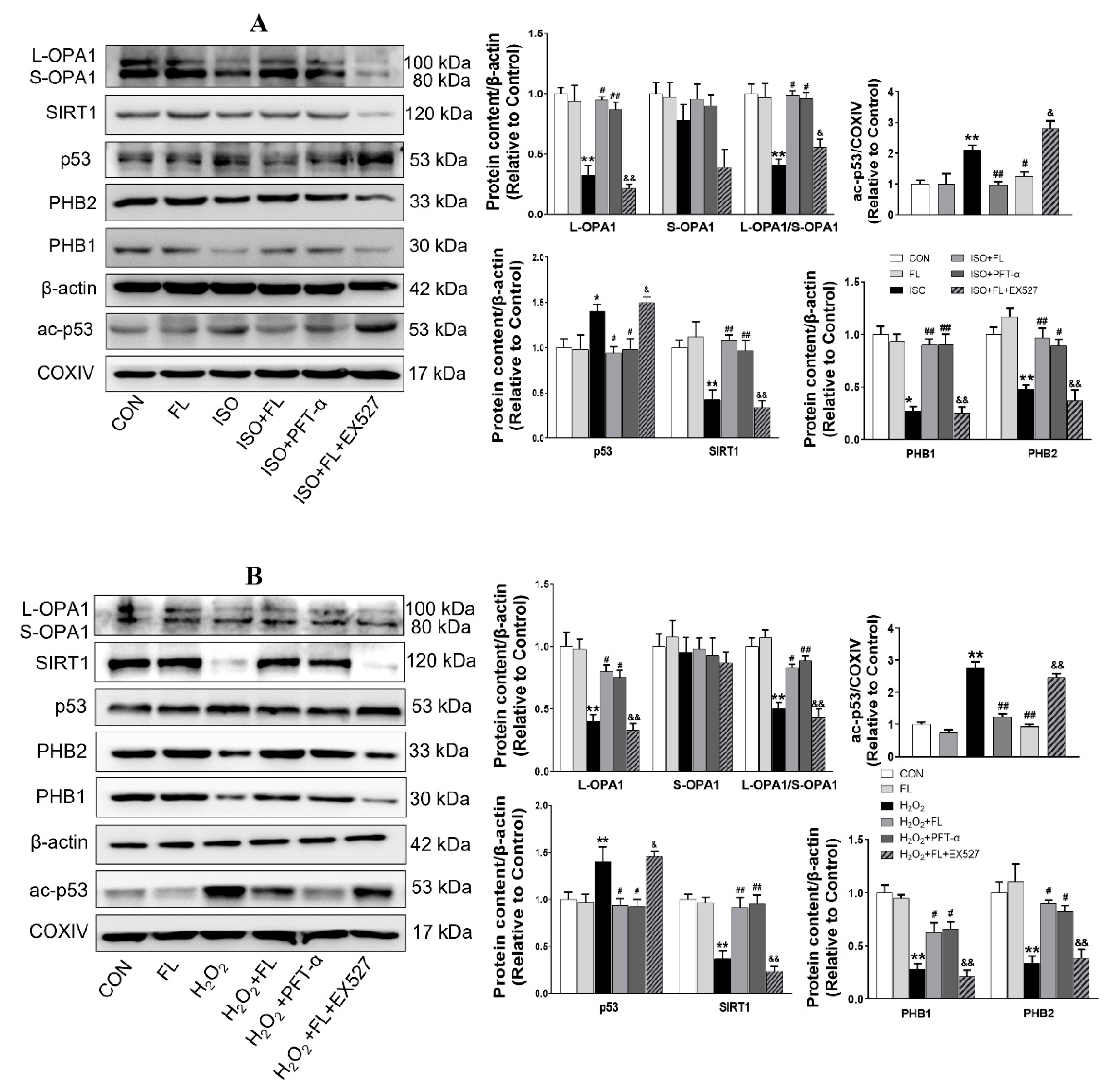
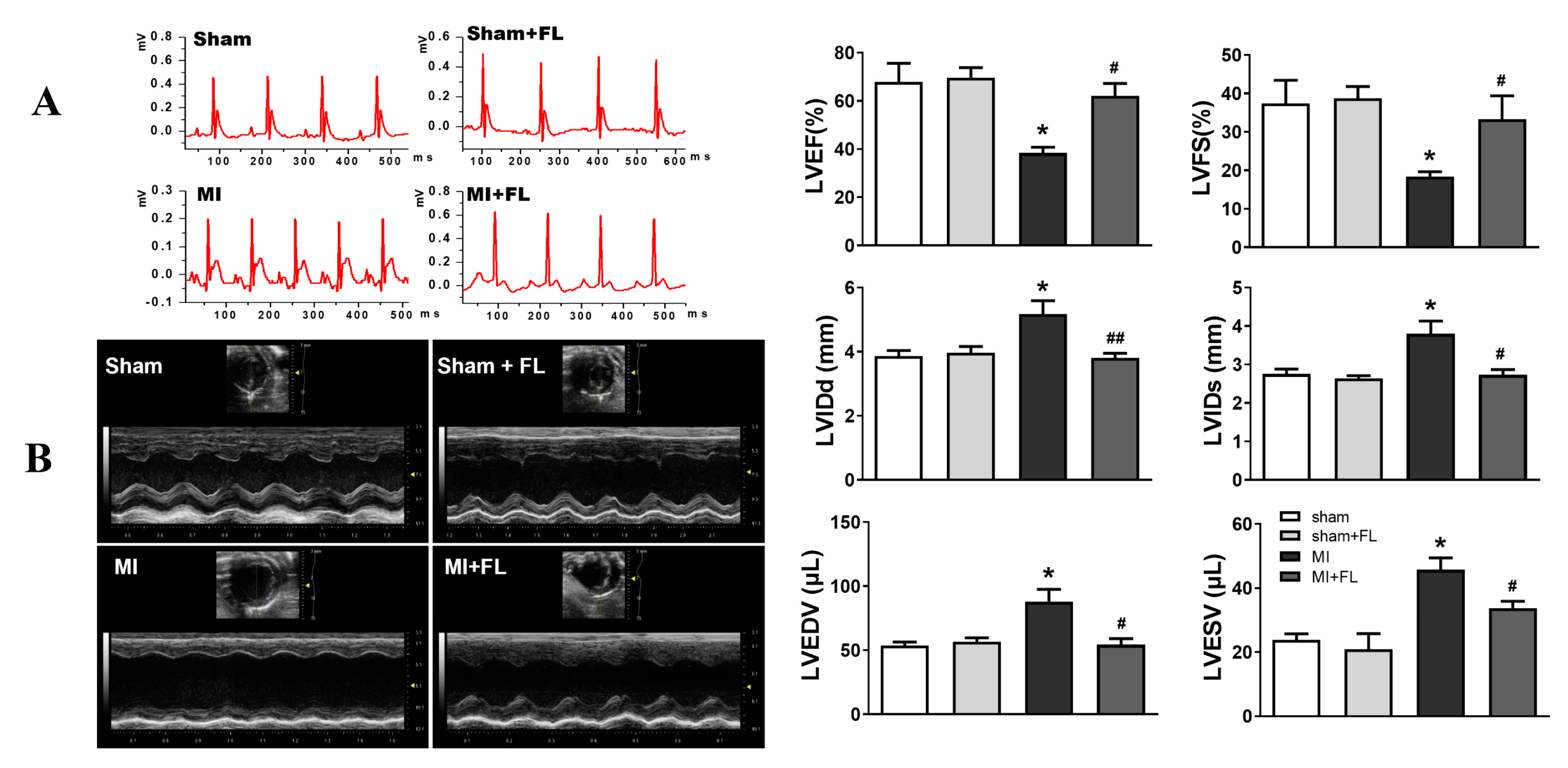
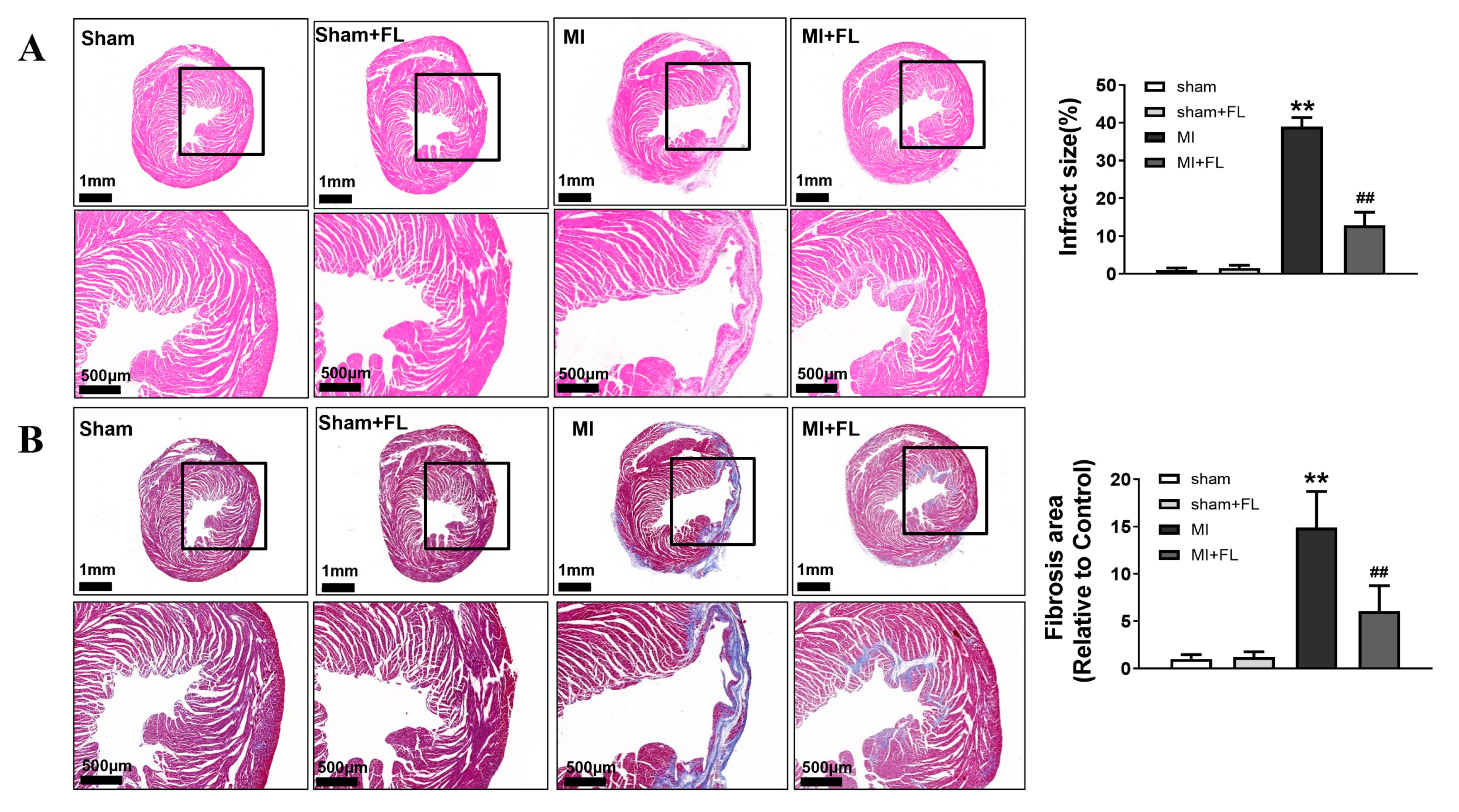
3.4. Flt3 Activation Ameliorates LAD-Induced Cardiac Remodeling
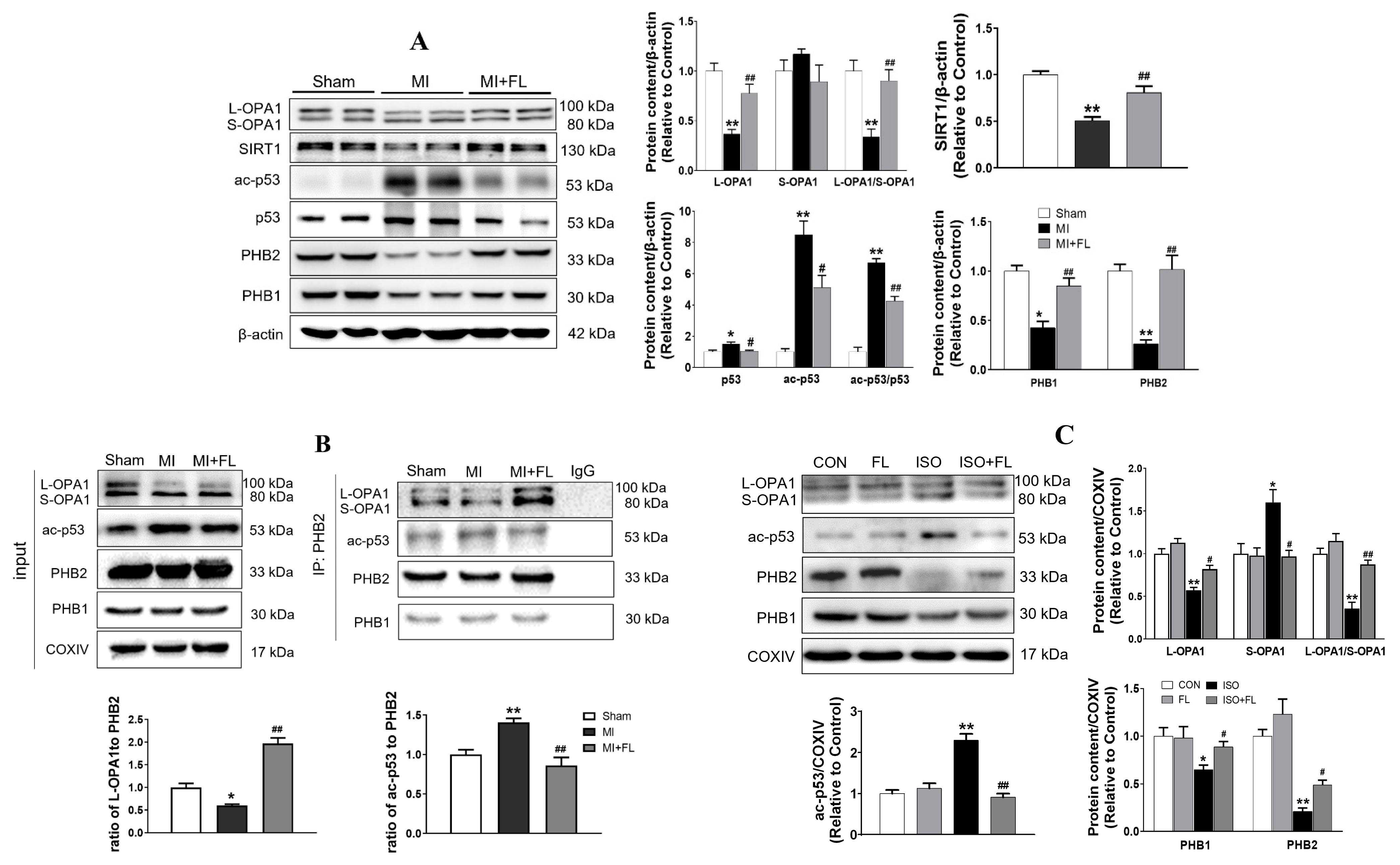
3.5. Flt3 Activation Reduced L-OPA1 Processing by Hindering the Interaction between p53 and PHBs in Mitochondria in LAD- or ISO-Induced Cardiac Remodeling
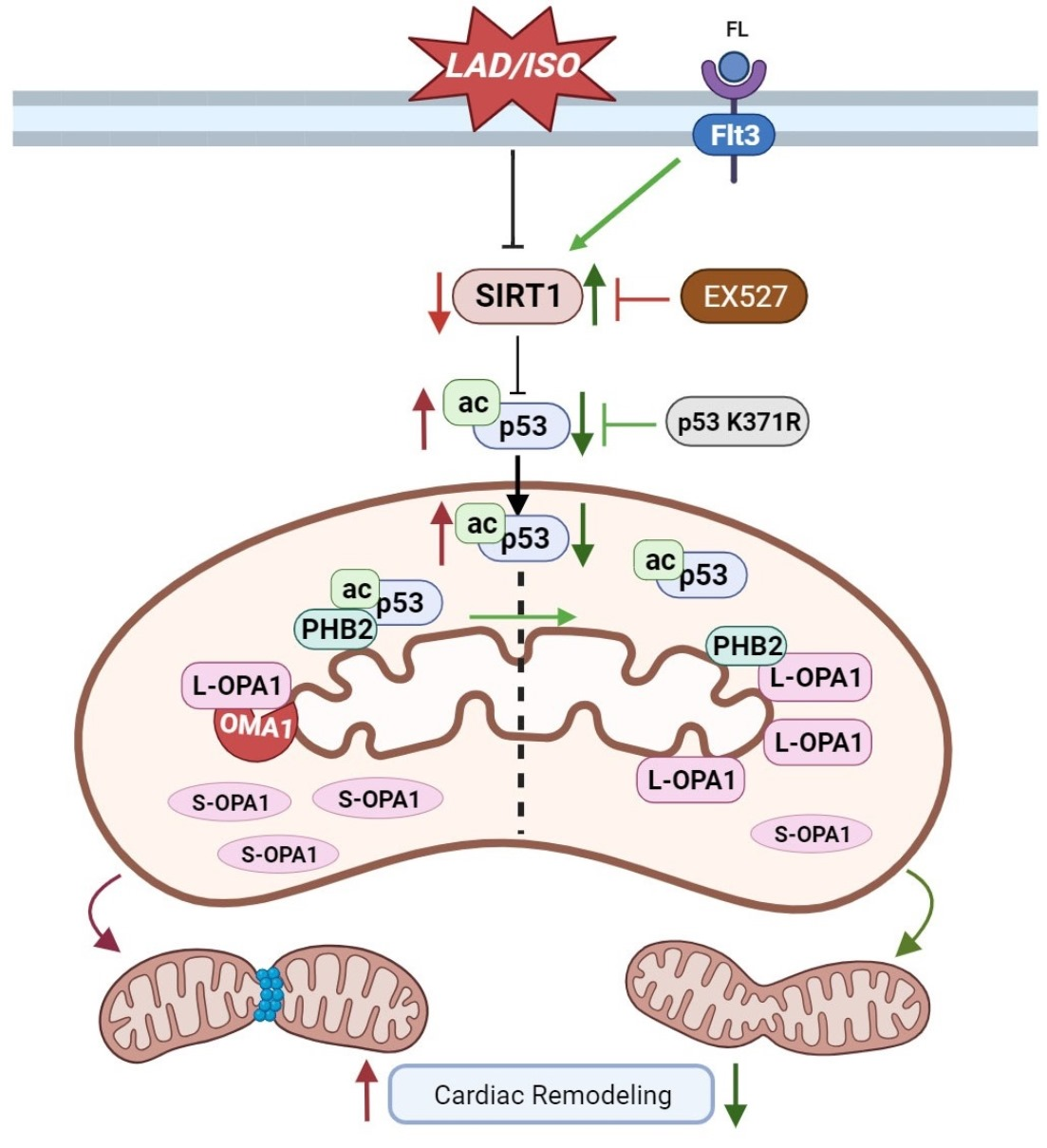
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torrealba, N.; Aranguiz, P.; Alonso, C.; Rothermel, B.A.; Lavandero, S. Mitochondria in Structural and Functional Cardiac Remodeling. Adv. Exp. Med. Biol. 2017, 982, 277–306. [Google Scholar]
- Weber, K.T.; Brilla, C.G. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 1991, 83, 1849–1865. [Google Scholar] [CrossRef] [PubMed]
- Swynghedauw, B. Molecular mechanisms of myocardial remodeling. Physiol. Rev. 1999, 79, 215–262. [Google Scholar] [CrossRef]
- Wai, T.; García-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.J.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, Z.; Qin, X.; Xu, J.; Hou, Z.; Yang, H.; Mao, X.; Xing, W.; Li, X.; Zhang, X.; et al. Enhancing fatty acid utilization ameliorates mitochondrial fragmentation and cardiac dysfunction via rebalancing optic atrophy 1 processing in the failing heart. Cardiovasc. Res. 2018, 114, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Alavi, M.V. OMA1-An integral membrane protease? Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140558. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, J.; Zhang, N.; Liu, Q.; Wang, Y.; Hu, X.; Chen, J.; Zhu, W.; Yu, H. GDF11 enhances therapeutic efficacy of mesenchymal stem cells for myocardial infarction via YME1L-mediated OPA1 processing. Stem Cells Transl. Med. 2020, 9, 1257–1271. [Google Scholar] [CrossRef]
- Yang, M.; Abudureyimu, M.; Wang, X.; Zhou, Y.; Zhang, Y.; Ren, J. PHB2 ameliorates Doxorubicin-induced cardiomyopathy through interaction with NDUFV2 and restoration of mitochondrial complex I function. Redox Biol. 2023, 65, 102812. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Kahl, A.; Fruitman, H.; Qian, L.; Zhou, P.; Manfredi, G.; Iadecola, C. Prohibitin levels regulate OMA1 activity and turnover in neurons. Cell Death Differ. 2020, 27, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Langer, T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochim. Biophys. Acta 2009, 1793, 27–32. [Google Scholar] [CrossRef]
- Li, L.; Martin-Levilain, J.; Jiménez-Sánchez, C.; Karaca, M.; Foti, M.; Martinou, J.C.; Maechler, P. In vivo stabilization of OPA1 in hepatocytes potentiates mitochondrial respiration and gluconeogenesis in a prohibitin-dependent way. J. Biol. Chem. 2019, 294, 12581–12598. [Google Scholar] [CrossRef] [PubMed]
- Kazi, J.U.; Rönnstrand, L. FMS-like Tyrosine Kinase 3/FLT3: From Basic Science to Clinical Implications. Physiol. Rev. 2019, 99, 1433–1466. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhang, K.; Gao, C.; Ma, W.; Liu, M.; Guo, X.; Bao, G.; Han, B.; Hu, H.; Zhao, Z. Activation of FMS-like tyrosine kinase 3 protects against isoprenaline-induced cardiac hypertrophy by improving autophagy and mitochondrial dynamics. FASEB J. 2022, 36, e22672. [Google Scholar] [CrossRef]
- Ma, W.; Liang, F.; Zhan, H.; Jiang, X.; Gao, C.; Zhang, X.; Zhang, K.; Sun, Q.; Hu, H.; Zhao, Z. Activated FMS-like tyrosine kinase 3 ameliorates angiotensin II-induced cardiac remodelling. Acta Physiol. 2020, 230, e13519. [Google Scholar] [CrossRef] [PubMed]
- Pfister, O.; Lorenz, V.; Oikonomopoulos, A.; Xu, L.; Häuselmann, S.P.; Mbah, C.; Kaufmann, B.A.; Liao, R.; Wodnar-Filipowicz, A.; Kuster, G.M. FLT3 activation improves post-myocardial infarction remodeling involving a cytoprotective effect on cardiomyocytes. J. Am. Coll. Cardiol. 2014, 63, 1011–1019. [Google Scholar] [CrossRef]
- Allawadhi, P.; Khurana, A.; Sayed, N.; Kumari, P.; Godugu, C. Isoproterenol-induced cardiac ischemia and fibrosis: Plant-based approaches for intervention. Phytother. Res. 2018, 32, 1908–1932. [Google Scholar] [CrossRef]
- Liu, Y.; Li, L.; Wang, Z.; Zhang, J.; Zhou, Z. Myocardial ischemia-reperfusion injury; Molecular mechanisms and prevention. Microvasc. Res. 2023, 149, 104565. [Google Scholar] [CrossRef]
- Kong, B.; Han, C.Y.; Kim, S.I.; Patten, D.A.; Han, Y.; Carmona, E.; Shieh, D.B.; Cheung, A.C.; Mes-Masson, A.M.; Harper, M.E.; et al. Prohibitin 1 interacts with p53 in the regulation of mitochondrial dynamics and chemoresistance in gynecologic cancers. J. Ovarian Res. 2022, 15, 70. [Google Scholar] [CrossRef]
- Chang, X.; Liu, R.; Li, R.; Peng, Y.; Zhu, P.; Zhou, H. Molecular Mechanisms of Mitochondrial Quality Control in Ischemic Cardiomyopathy. Int. J. Biol. Sci. 2023, 19, 426–448. [Google Scholar] [CrossRef]
- Sun, X.; Alford, J.; Qiu, H. Structural and Functional Remodeling of Mitochondria in Cardiac Diseases. Int. J. Mol. Sci. 2021, 22, 4167. [Google Scholar] [CrossRef]
- Cheng, Q.Q.; Wan, Y.W.; Yang, W.M.; Tian, M.H.; Wang, Y.C.; He, H.Y.; Zhang, W.D.; Liu, X. Gastrodin protects H9c2 cardiomyocytes against oxidative injury by ameliorating imbalanced mitochondrial dynamics and mitochondrial dysfunction. Acta Pharmacol. Sin. 2020, 41, 1314–1327. [Google Scholar] [CrossRef] [PubMed]
- Moulder, D.E.; Hatoum, D.; Tay, E.; Lin, Y.; McGowan, E.M. The Roles of p53 in Mitochondrial Dynamics and Cancer Metabolism: The Pendulum between Survival and Death in Breast Cancer? Cancers 2018, 10, 189. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Sesaki, H.; Qi, X. Drp1 stabilizes p53 on the mitochondria to trigger necrosis under oxidative stress conditions in vitro and in vivo. Biochem. J. 2014, 461, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Tetri, L.H.; Li, S.J.; Fajardo, G.; Ostberg, N.P.; Tsegay, K.B.; Gera, K.; Cornell, T.T.; Bernstein, D.; Mochly-Rosen, D.; et al. Drp1/p53 interaction mediates p53 mitochondrial localization and dysfunction in septic cardiomyopathy. J. Mol. Cell. Cardiol. 2023, 177, 28–37. [Google Scholar] [CrossRef]
- Kasashima, K.; Ohta, E.; Kagawa, Y.; Endo, H. Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2. J. Biol. Chem. 2006, 281, 36401–36410. [Google Scholar] [CrossRef]
- Cho, S.G.; Xiao, X.; Wang, S.; Gao, H.; Rafikov, R.; Black, S.; Huang, S.; Ding, H.F.; Yoon, Y.; Kirken, R.A.; et al. Bif-1 Interacts with Prohibitin-2 to Regulate Mitochondrial Inner Membrane during Cell Stress and Apoptosis. J. Am. Soc. Nephrol. 2019, 30, 1174–1191. [Google Scholar] [CrossRef]
- Kong, B.; Wang, Q.; Fung, E.; Xue, K.; Tsang, B.K. p53 is required for cisplatin-induced processing of the mitochondrial fusion protein L-Opa1 that is mediated by the mitochondrial metallopeptidase Oma1 in gynecologic cancers. J. Biol. Chem. 2014, 289, 27134–27145. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Forward | Reverse |
|---|---|---|
| Opa1 | CCGAAAGCCTCAGCTTGTTG | GCAGAAGTTCTTCCTGAAGTTGG |
| Mfn2 | GTGACGTGTTGGGTGTGAT | GGACATCTCGTTTCTAGCTGGT |
| Drp1 Fis 1 | CGAAAACTGTCTGCCCGAGA CCAGAACAACCAGGCCAAGGAG | GCATTACTGCCTTTGGGACG CACAGCCAGTCCAATGAGTCCAG |
| Actin | TGTCACCAACTGGGACGATA | GGGGTGTTGAAGGTCTCAAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, K.; Zheng, Y.; Bao, G.; Ma, W.; Han, B.; Shi, H.; Zhao, Z. Flt3 Activation Mitigates Mitochondrial Fragmentation and Heart Dysfunction through Rebalanced L-OPA1 Processing by Hindering the Interaction between Acetylated p53 and PHB2 in Cardiac Remodeling. Antioxidants 2023, 12, 1657. https://doi.org/10.3390/antiox12091657
Zhang K, Zheng Y, Bao G, Ma W, Han B, Shi H, Zhao Z. Flt3 Activation Mitigates Mitochondrial Fragmentation and Heart Dysfunction through Rebalanced L-OPA1 Processing by Hindering the Interaction between Acetylated p53 and PHB2 in Cardiac Remodeling. Antioxidants. 2023; 12(9):1657. https://doi.org/10.3390/antiox12091657
Chicago/Turabian StyleZhang, Kaina, Yeqing Zheng, Gaowa Bao, Wenzhuo Ma, Bing Han, Hongwen Shi, and Zhenghang Zhao. 2023. "Flt3 Activation Mitigates Mitochondrial Fragmentation and Heart Dysfunction through Rebalanced L-OPA1 Processing by Hindering the Interaction between Acetylated p53 and PHB2 in Cardiac Remodeling" Antioxidants 12, no. 9: 1657. https://doi.org/10.3390/antiox12091657
APA StyleZhang, K., Zheng, Y., Bao, G., Ma, W., Han, B., Shi, H., & Zhao, Z. (2023). Flt3 Activation Mitigates Mitochondrial Fragmentation and Heart Dysfunction through Rebalanced L-OPA1 Processing by Hindering the Interaction between Acetylated p53 and PHB2 in Cardiac Remodeling. Antioxidants, 12(9), 1657. https://doi.org/10.3390/antiox12091657