
Mitochondrial ROS Triggers KIN Pathogenesis in FAN1-Deficient Kidneys

 , , and
, , and 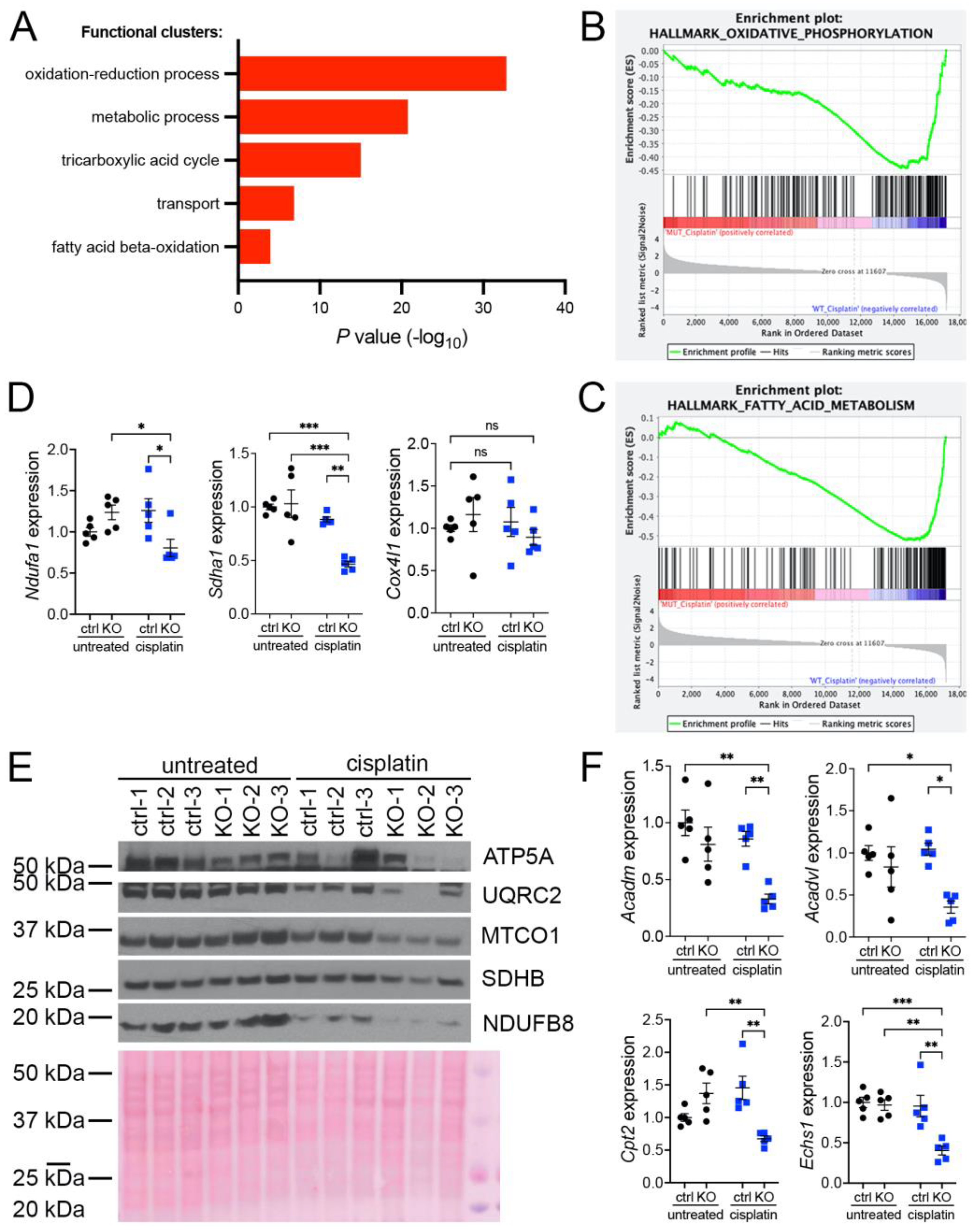{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mouse Lines Used and Study Approval
2.2. Measurement of Renal Function
2.3. Histological and Immunofluorescence Analysis of Kidney
2.4. Immunohistochemistry
2.5. Antibodies
2.6. RNA Extraction and qRT–PCR
2.7. Cell Culture
2.8. YSI Metabolite Measurements
2.9. Oroboros Respirometry
2.10. Immunofluorescence
2.11. Subcellular Fractionation
2.12. Western Blotting
2.13. Measurement of Mitochondrial ROS
2.14. Measurement of Cellular ROS
2.15. ATP Measurement
2.16. Oil Red O Staining
2.17. Quantification
2.18. Statistical Methods
2.19. Data sharing Statement
3. Results
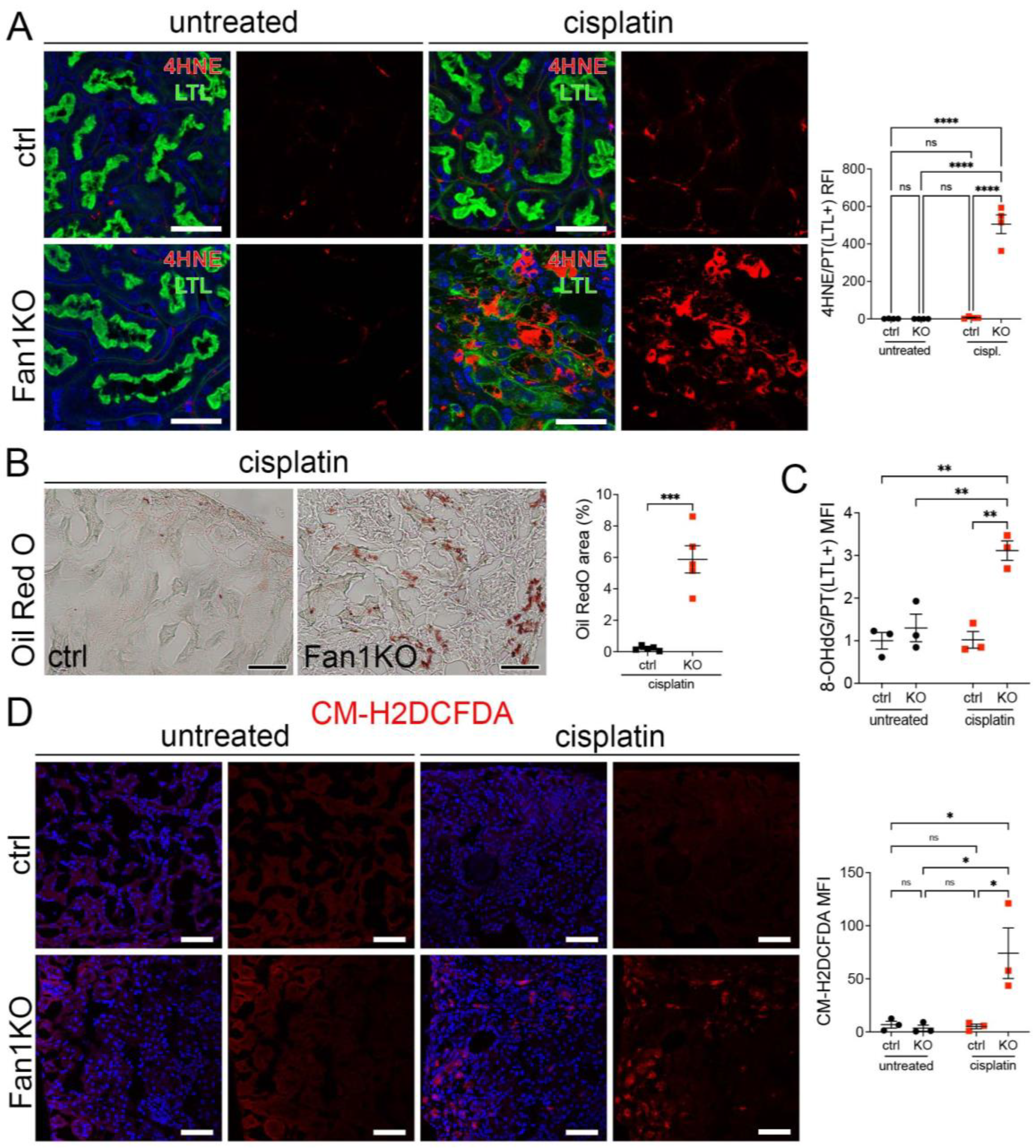
3.1. KIN Is Associated with Impaired Mitochondrial Metabolism and Increased Oxidative Stress in FAN1-Deficient Kidneys
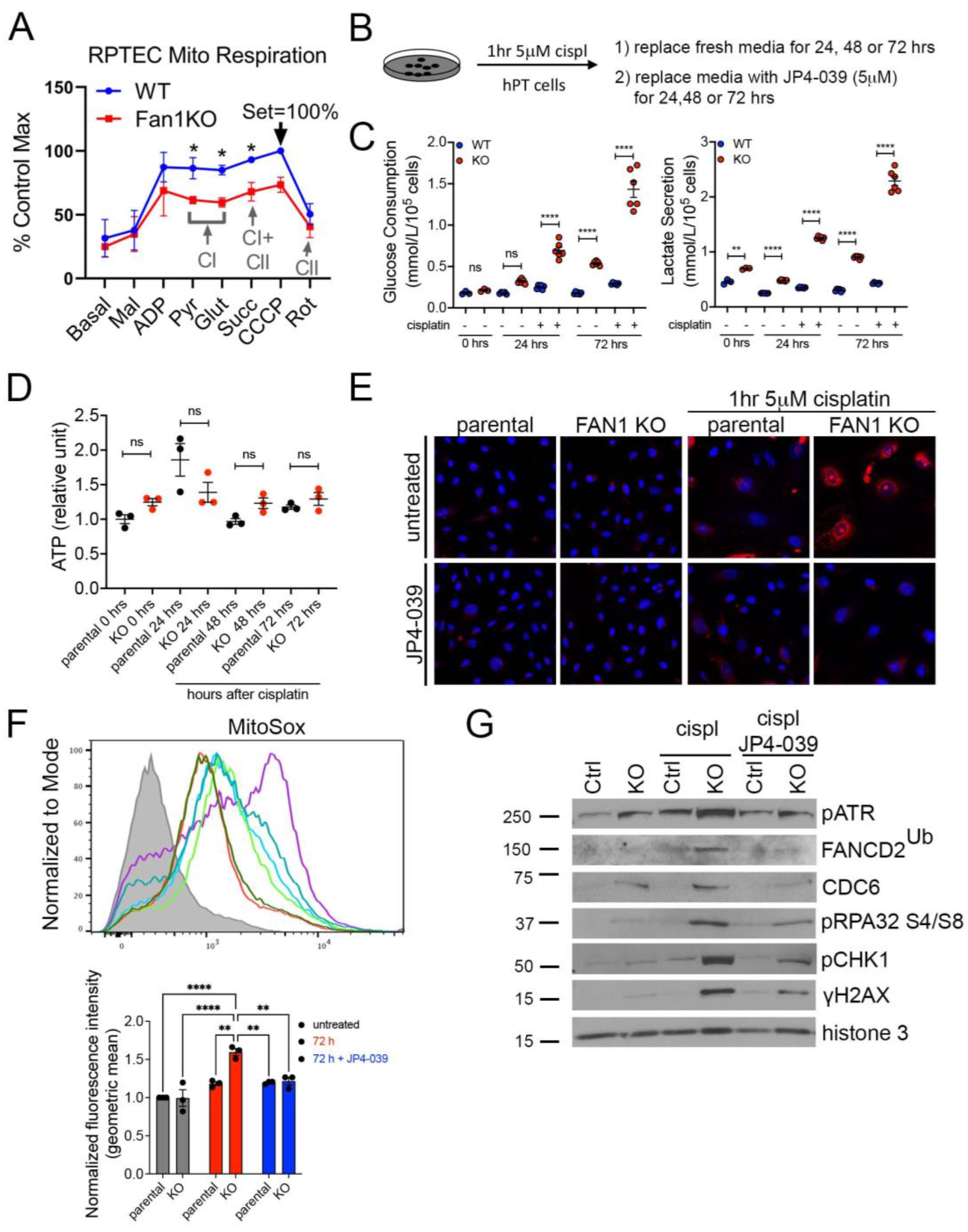
3.2. FAN1KO Proximal Tubule Cells Are Hypersensitive to Cisplatin-Induced Oxidative Stress
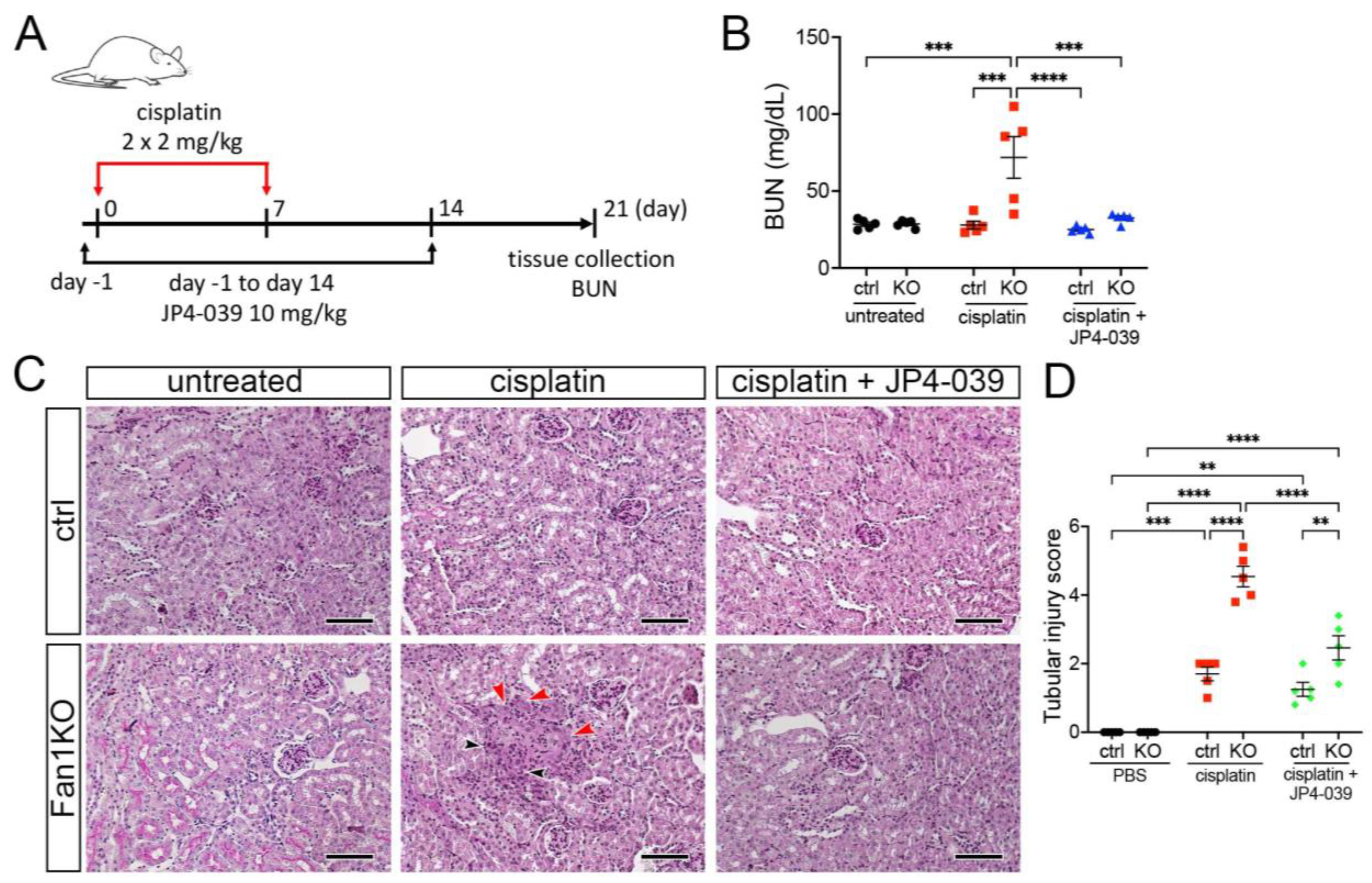
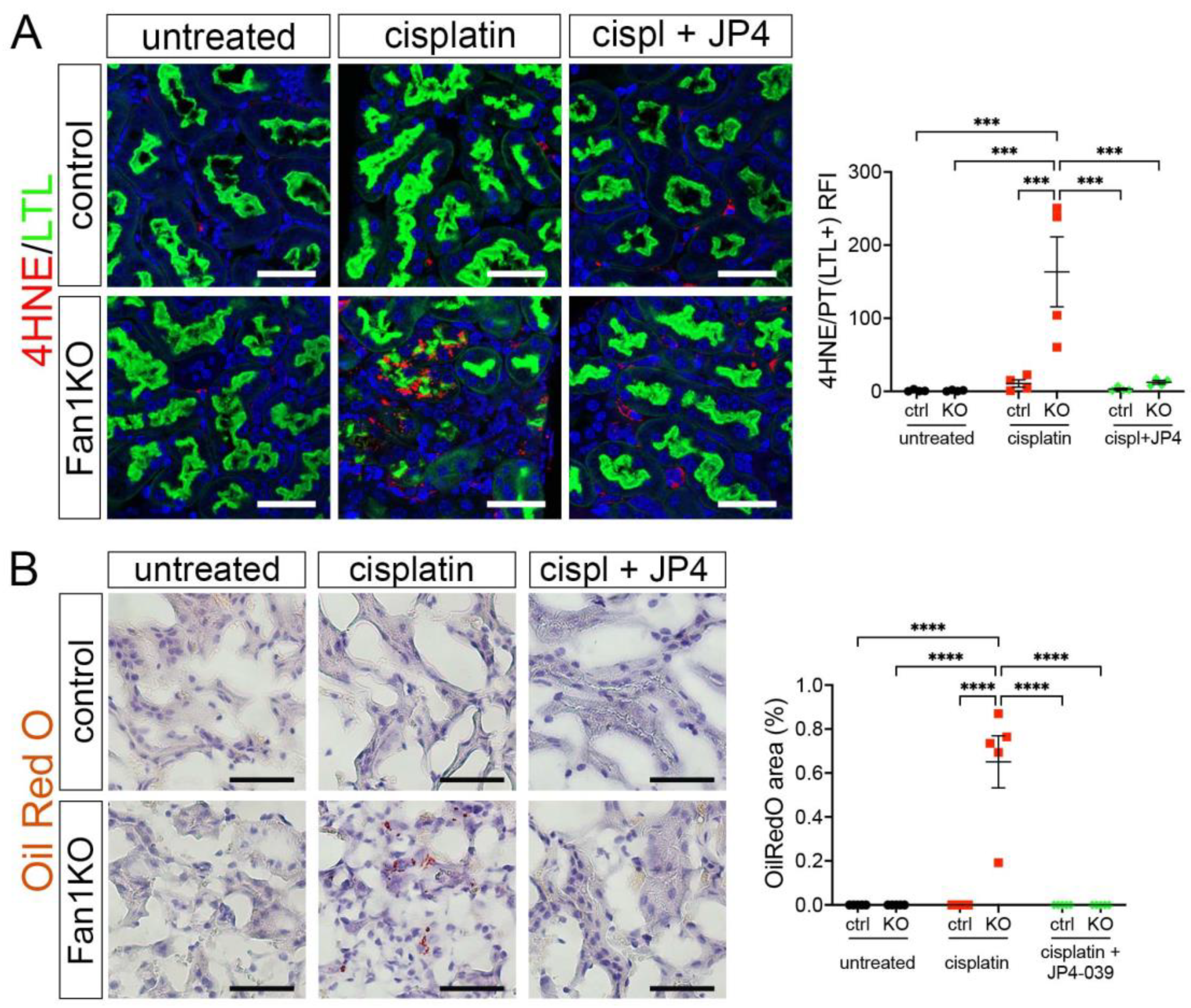
3.3. Treatment with Mitochondrial ROS Scavenger JP4-039 Reduces Tubular Damage and Prevents KIN Pathogenesis in FAN1-Null Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, W.; Otto, E.A.; Cluckey, A.; Airik, R.; Hurd, T.W.; Chaki, M.; Diaz, K.; Lach, F.P.; Bennett, G.R.; Gee, H.Y.; et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet. 2012, 44, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Airik, R.; Schueler, M.; Airik, M.; Cho, J.; Porath, J.D.; Mukherjee, E.; Sims-Lucas, S.; Hildebrandt, F. A FANCD2/FANCI-Associated Nuclease 1-Knockout Model Develops Karyomegalic Interstitial Nephritis. J. Am. Soc. Nephrol. 2016, 27, 3552–3559. [Google Scholar] [CrossRef]
- Spoendlin, M.; Moch, H.; Brunner, F.; Brunner, W.; Burger, H.R.; Kiss, D.; Wegmann, W.; Dalquen, P.; Oberholzer, M.; Thiel, G.; et al. Karyomegalic interstitial nephritis: Further support for a distinct entity and evidence for a genetic defect. Am. J. Kidney Dis. 1995, 25, 242–252. [Google Scholar] [CrossRef]
- Dash, J.; Saudan, P.; Paoloni-Giacobino, A.; Moll, S.; de Seigneux, S. Case report: A 58-year-old man with small kidneys and elevated liver enzymes. BMC Nephrol. 2020, 21, 107. [Google Scholar] [CrossRef]
- Gueguen, L.; Delaval, R.; Blanluet, M.; Sartelet, H.; Leou, S.; Dubois d’Enghien, C.; Golmard, L.; Stoppa-Lyonnet, D.; Testevuide, P.; Faguer, S. Recurrent FAN1 p.W707X Pathogenic Variant Originating Before ad 1800 Underlies High Frequency of Karyomegalic Interstitial Nephritis in South Pacific Islands. Kidney Int. Rep. 2021, 6, 2207–2211. [Google Scholar] [CrossRef] [PubMed]
- Segui, N.; Mina, L.B.; Lazaro, C.; Sanz-Pamplona, R.; Pons, T.; Navarro, M.; Bellido, F.; Lopez-Doriga, A.; Valdes-Mas, R.; Pineda, M.; et al. Germline Mutations in FAN1 Cause Hereditary Colorectal Cancer by Impairing DNA Repair. Gastroenterology 2015, 149, 563–566. [Google Scholar] [CrossRef]
- Lachaud, C.; Moreno, A.; Marchesi, F.; Toth, R.; Blow, J.J.; Rouse, J. Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science 2016, 351, 846–849. [Google Scholar] [CrossRef]
- Murray, S.L.; Connaughton, D.M.; Fennelly, N.K.; Kay, E.W.; Dorman, A.; Doyle, B.; Conlon, P.J. Karyomegalic Interstitial Nephritis: Cancer Risk Following Transplantation. Nephron 2020, 144, 49–54. [Google Scholar] [CrossRef]
- Burry, A.F. Extreme dysplasia in renal epithelium of a young woman dying from hepatocarcinoma. J. Pathol. 1974, 113, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ghosal, G.; Yuan, J.; Chen, J.; Huang, J. FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 2010, 329, 693–696. [Google Scholar] [CrossRef]
- Kratz, K.; Schopf, B.; Kaden, S.; Sendoel, A.; Eberhard, R.; Lademann, C.; Cannavo, E.; Sartori, A.A.; Hengartner, M.O.; Jiricny, J. Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell 2010, 142, 77–88. [Google Scholar] [CrossRef]
- MacKay, C.; Declais, A.C.; Lundin, C.; Agostinho, A.; Deans, A.J.; MacArtney, T.J.; Hofmann, K.; Gartner, A.; West, S.C.; Helleday, T.; et al. Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 2010, 142, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Smogorzewska, A.; Desetty, R.; Saito, T.T.; Schlabach, M.; Lach, F.P.; Sowa, M.E.; Clark, A.B.; Kunkel, T.A.; Harper, J.W.; Colaiacovo, M.P.; et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell 2010, 39, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, I.; Stroik, D.R.; Sobeck, A. FANCD2-controlled chromatin access of the Fanconi-associated nuclease FAN1 is crucial for the recovery of stalled replication forks. Mol. Cell. Biol. 2014, 34, 3939–3954. [Google Scholar] [CrossRef]
- Porro, A.; Berti, M.; Pizzolato, J.; Bologna, S.; Kaden, S.; Saxer, A.; Ma, Y.; Nagasawa, K.; Sartori, A.A.; Jiricny, J. FAN1 interaction with ubiquitylated PCNA alleviates replication stress and preserves genomic integrity independently of BRCA2. Nat. Commun. 2017, 8, 1073. [Google Scholar] [CrossRef]
- Longobardi, C.; Ferrara, G.; Andretta, E.; Montagnaro, S.; Damiano, S.; Ciarcia, R. Ochratoxin A and Kidney Oxidative Stress: The Role of Nutraceuticals in Veterinary Medicine—A Review. Toxins 2022, 14, 398. [Google Scholar] [CrossRef]
- Damiano, S.; Jarriyawattanachaikul, W.; Girolami, F.; Longobardi, C.; Nebbia, C.; Andretta, E.; Lauritano, C.; Dabbou, S.; Avantaggiato, G.; Schiavone, A.; et al. Curcumin Supplementation Protects Broiler Chickens Against the Renal Oxidative Stress Induced by the Dietary Exposure to Low Levels of Aflatoxin B1. Front. Vet. Sci. 2021, 8, 822227. [Google Scholar] [CrossRef] [PubMed]
- Schupp, N.; Stopper, H.; Heidland, A. DNA Damage in Chronic Kidney Disease: Evaluation of Clinical Biomarkers. Oxid. Med. Cell. Longev. 2016, 2016, 3592042. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, G.; Mehta, T.; Contractor, P.; Tung, G. Genotoxic damage in end-stage renal disease. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2018, 835, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Lopez, A.; Paniagua-Medina, M.E.; Urban-Reyes, M.; Cortes-Arredondo, M.; Alvarez-Aguilar, C.; Lopez-Meza, J.; Ochoa-Zarzosa, A.; Lindholm, B.; Garcia-Lopez, E.; Paniagua, J.R. Genetic damage in patients with chronic kidney disease, peritoneal dialysis and haemodialysis: A comparative study. Mutagenesis 2013, 28, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Tin, A.; Scharpf, R.; Estrella, M.M.; Yu, B.; Grove, M.L.; Chang, P.P.; Matsushita, K.; Kottgen, A.; Arking, D.E.; Boerwinkle, E.; et al. The Loss of GSTM1 Associates with Kidney Failure and Heart Failure. J. Am. Soc. Nephrol. 2017, 28, 3345–3352. [Google Scholar] [CrossRef] [PubMed]
- Schaub, J.A.; Venkatachalam, M.A.; Weinberg, J.M. Proximal Tubular Oxidative Metabolism in Acute Kidney Injury and the Transition to CKD. Kidney360 2021, 2, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Gee, H.Y.; Otto, E.A.; Hurd, T.W.; Ashraf, S.; Chaki, M.; Cluckey, A.; Vega-Warner, V.; Saisawat, P.; Diaz, K.A.; Fang, H.; et al. Whole-exome resequencing distinguishes cystic kidney diseases from phenocopies in renal ciliopathies. Kidney Int. 2014, 85, 880–887. [Google Scholar] [CrossRef]
- Lyakhovich, A. Damaged mitochondria and overproduction of ROS in Fanconi anemia cells. Rare Dis. 2013, 1, e24048. [Google Scholar] [CrossRef]
- Kumari, U.; Ya Jun, W.; Huat Bay, B.; Lyakhovich, A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi anemia cells. Oncogene 2014, 33, 165–172. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- Airik, M.; Phua, Y.L.; Huynh, A.B.; McCourt, B.T.; Rush, B.M.; Tan, R.J.; Vockley, J.; Murray, S.L.; Dorman, A.; Conlon, P.J.; et al. Persistent DNA damage underlies tubular cell polyploidization and progression to chronic kidney disease in kidneys deficient in the DNA repair protein FAN1. Kidney Int. 2022, 102, 1042–1056. [Google Scholar] [CrossRef]
- Frantz, M.C.; Pierce, J.G.; Pierce, J.M.; Kangying, L.; Qingwei, W.; Johnson, M.; Wipf, P. Large-scale asymmetric synthesis of the bioprotective agent JP4-039 and analogs. Org. Lett. 2011, 13, 2318–2321. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chen, C.W.; Buj, R.; Tangudu, N.K.; Fang, R.S.; Leon, K.E.; Dahl, E.S.; Varner, E.L.; von Krusenstiern, E.; Cole, A.R.; et al. ATM inhibition drives metabolic adaptation via induction of macropinocytosis. J. Cell Biol. 2023, 222, e202007026. [Google Scholar] [CrossRef]
- Zhang, Y.; Bharathi, S.S.; Beck, M.E.; Goetzman, E.S. The fatty acid oxidation enzyme long-chain acyl-CoA dehydrogenase can be a source of mitochondrial hydrogen peroxide. Redox Biol. 2019, 26, 101253. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, D.; Hamasaki, Y.; Doi, K.; Negishi, K.; Sugaya, T.; Nangaku, M.; Noiri, E. Interstitial renal fibrosis due to multiple cisplatin treatments is ameliorated by semicarbazide-sensitive amine oxidase inhibition. Kidney Int. 2016, 89, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Ogrunc, M.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gorgoulis, V.G.; d’Adda di Fagagna, F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014, 21, 998–1012. [Google Scholar] [CrossRef]
- Czerwinska, J.; Nowak, M.; Wojtczak, P.; Dziuban-Lech, D.; Ciesla, J.M.; Kolata, D.; Gajewska, B.; Baranczyk-Kuzma, A.; Robinson, A.R.; Shane, H.L.; et al. ERCC1-deficient cells and mice are hypersensitive to lipid peroxidation. Free Radic. Biol. Med. 2018, 124, 79–96. [Google Scholar] [CrossRef]
- Wilmes, A.; Bielow, C.; Ranninger, C.; Bellwon, P.; Aschauer, L.; Limonciel, A.; Chassaigne, H.; Kristl, T.; Aiche, S.; Huber, C.G.; et al. Mechanism of cisplatin proximal tubule toxicity revealed by integrating transcriptomics, proteomics, metabolomics and biokinetics. Toxicol. Vitr. 2015, 30, 117–127. [Google Scholar] [CrossRef]
- Kruidering, M.; Van de Water, B.; de Heer, E.; Mulder, G.J.; Nagelkerke, J.F. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: Mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J. Pharmacol. Exp. Ther. 1997, 280, 638–649. [Google Scholar] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Kagan, V.E.; Wipf, P.; Stoyanovsky, D.; Greenberger, J.S.; Borisenko, G.; Belikova, N.A.; Yanamala, N.; Samhan Arias, A.K.; Tungekar, M.A.; Jiang, J.; et al. Mitochondrial targeting of electron scavenging antioxidants: Regulation of selective oxidation vs random chain reactions. Adv. Drug Deliv. Rev. 2009, 61, 1375–1385. [Google Scholar] [CrossRef]
- Epperly, M.W.; Fisher, R.; Zhang, X.; Hou, W.; Shields, D.; Wipf, P.; Wang, H.; Thermozier, S.; Greenberger, J.S. Fanconi Anemia Mouse Genotype-specific Mitigation of Total Body Irradiation by GS-Nitroxide JP4-039. In Vivo 2020, 34, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Glanzel, N.M.; Grings, M.; da Rosa-Junior, N.T.; de Carvalho, L.M.C.; Mohsen, A.W.; Wipf, P.; Wajner, M.; Vockley, J.; Leipnitz, G. The mitochondrial-targeted reactive species scavenger JP4-039 prevents sulfite-induced alterations in antioxidant defenses, energy transfer, and cell death signaling in striatum of rats. J. Inherit. Metab. Dis. 2021, 44, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Pabla, N.; Tang, C.; He, L.; Dong, Z. DNA damage response in cisplatin-induced nephrotoxicity. Arch. Toxicol. 2015, 89, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Thongthip, S.; Bellani, M.; Gregg, S.Q.; Sridhar, S.; Conti, B.A.; Chen, Y.; Seidman, M.M.; Smogorzewska, A. Fan1 deficiency results in DNA interstrand cross-link repair defects, enhanced tissue karyomegaly, and organ dysfunction. Genes Dev. 2016, 30, 645–659. [Google Scholar] [CrossRef]
- Lachaud, C.; Slean, M.; Marchesi, F.; Lock, C.; Odell, E.; Castor, D.; Toth, R.; Rouse, J. Karyomegalic interstitial nephritis and DNA damage-induced polyploidy in Fan1 nuclease-defective knock-in mice. Genes Dev. 2016, 30, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Yoshikiyo, K.; Kratz, K.; Hirota, K.; Nishihara, K.; Takata, M.; Kurumizaka, H.; Horimoto, S.; Takeda, S.; Jiricny, J. KIAA1018/FAN1 nuclease protects cells against genomic instability induced by interstrand cross-linking agents. Proc. Natl. Acad. Sci. USA 2010, 107, 21553–21557. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Xu, S.; Jia, P.; Fang, Y.; Jin, J.; Sun, Z.; Zhou, W.; Li, J.; Zhang, Y.; Wang, X.; Ren, T.; et al. Nuclear farnesoid X receptor attenuates acute kidney injury through fatty acid oxidation. Kidney Int. 2022, 101, 987–1002. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Mapuskar, K.A.; Wen, H.; Holanda, D.G.; Rastogi, P.; Steinbach, E.; Han, R.; Coleman, M.C.; Attanasio, M.; Riley, D.P.; Spitz, D.R.; et al. Persistent increase in mitochondrial superoxide mediates cisplatin-induced chronic kidney disease. Redox Biol. 2019, 20, 98–106. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Reyes-Fermin, L.M.; Briones-Herrera, A.; Tapia, E.; Leon-Contreras, J.C.; Hernandez-Pando, R.; Sanchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective effects of N-acetyl-cysteine in mitochondria bioenergetics, oxidative stress, dynamics and S-glutathionylation alterations in acute kidney damage induced by folic acid. Free Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Oberg, B.P.; McMenamin, E.; Lucas, F.L.; McMonagle, E.; Morrow, J.; Ikizler, T.A.; Himmelfarb, J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004, 65, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Himmelfarb, J.; McMonagle, E.; Freedman, S.; Klenzak, J.; McMenamin, E.; Le, P.; Pupim, L.B.; Ikizler, T.A.; The PICARD Group. Oxidative stress is increased in critically ill patients with acute renal failure. J. Am. Soc. Nephrol. 2004, 15, 2449–2456. [Google Scholar] [CrossRef]
- Joenje, H.; Oostra, A.B. Effect of oxygen tension on chromosomal aberrations in Fanconi anaemia. Hum. Genet. 1983, 65, 99–101. [Google Scholar] [CrossRef]
- Saito, H.; Hammond, A.T.; Moses, R.E. Hypersensitivity to oxygen is a uniform and secondary defect in Fanconi anemia cells. Mutat. Res. 1993, 294, 255–262. [Google Scholar] [CrossRef]
- Schindler, D.; Hoehn, H. Fanconi anemia mutation causes cellular susceptibility to ambient oxygen. Am. J. Hum. Genet. 1988, 43, 429–435. [Google Scholar] [PubMed]
- Walter, D.; Lier, A.; Geiselhart, A.; Thalheimer, F.B.; Huntscha, S.; Sobotta, M.C.; Moehrle, B.; Brocks, D.; Bayindir, I.; Kaschutnig, P.; et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 2015, 520, 549–552. [Google Scholar] [CrossRef]
- Robinson, A.R.; Yousefzadeh, M.J.; Rozgaja, T.A.; Wang, J.; Li, X.; Tilstra, J.S.; Feldman, C.H.; Gregg, S.Q.; Johnson, C.H.; Skoda, E.M.; et al. Spontaneous DNA damage to the nuclear genome promotes senescence, redox imbalance and aging. Redox Biol. 2018, 17, 259–273. [Google Scholar] [CrossRef]
- Zhang, Q.S.; Eaton, L.; Snyder, E.R.; Houghtaling, S.; Mitchell, J.B.; Finegold, M.; Van Waes, C.; Grompe, M. Tempol protects against oxidative damage and delays epithelial tumor onset in Fanconi anemia mice. Cancer Res. 2008, 68, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Koshy, P.J.; Sudhakar, D.V.S.; Anupama, S.H.; Mathew, M.; Parthasarthy, R.; Thangaraj, K.; Yaqoob, M.M.; Abraham, G. Novel Homozygous FAN1 Mutation in a Familial Case of Karyomegalic Interstitial Nephritis. Indian J. Nephrol. 2020, 30, 283–285. [Google Scholar] [CrossRef]
- Pontel, L.B.; Rosado, I.V.; Burgos-Barragan, G.; Garaycoechea, J.I.; Yu, R.; Arends, M.J.; Chandrasekaran, G.; Broecker, V.; Wei, W.; Liu, L.; et al. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol. Cell 2015, 60, 177–188. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Airik, M.; Arbore, H.; Childs, E.; Huynh, A.B.; Phua, Y.L.; Chen, C.W.; Aird, K.; Bharathi, S.; Zhang, B.; Conlon, P.; et al. Mitochondrial ROS Triggers KIN Pathogenesis in FAN1-Deficient Kidneys. Antioxidants 2023, 12, 900. https://doi.org/10.3390/antiox12040900
Airik M, Arbore H, Childs E, Huynh AB, Phua YL, Chen CW, Aird K, Bharathi S, Zhang B, Conlon P, et al. Mitochondrial ROS Triggers KIN Pathogenesis in FAN1-Deficient Kidneys. Antioxidants. 2023; 12(4):900. https://doi.org/10.3390/antiox12040900
Chicago/Turabian StyleAirik, Merlin, Haley Arbore, Elizabeth Childs, Amy B. Huynh, Yu Leng Phua, Chi Wei Chen, Katherine Aird, Sivakama Bharathi, Bob Zhang, Peter Conlon, and et al. 2023. "Mitochondrial ROS Triggers KIN Pathogenesis in FAN1-Deficient Kidneys" Antioxidants 12, no. 4: 900. https://doi.org/10.3390/antiox12040900
APA StyleAirik, M., Arbore, H., Childs, E., Huynh, A. B., Phua, Y. L., Chen, C. W., Aird, K., Bharathi, S., Zhang, B., Conlon, P., Kmoch, S., Kidd, K., Bleyer, A. J., Vockley, J., Goetzman, E., Wipf, P., & Airik, R. (2023). Mitochondrial ROS Triggers KIN Pathogenesis in FAN1-Deficient Kidneys. Antioxidants, 12(4), 900. https://doi.org/10.3390/antiox12040900