
Understanding the Role of Chemerin in the Pathophysiology of Pre-Eclampsia


Abstract
1. Introduction
2. Chemerin Structure and Biological Functions
3. Key Points of PE Pathophysiology
3.1. Oxidative Stress

{kind=link}
| Oxidants—Reactive Oxygen Species (ROS) | Antioxidants |
|---|---|
| Superoxide O2●− Hydrogen peroxide H2O2 Hydroxyl radical HO● | ENZYMATIC Superoxide dismutase (SOD) Hemoxygenase (HO-1) Catalase (CAT) Glutathione peroxidase (GPX) Glutathione-S-transferase (GSX) |
| The main sources of ROS are: NO synthases (NOS) NADPH oxidases (NOX) Mitochondrial electron transport chain (ETC) Xanthine oxidase (XO) | NON-ENZYMATIC Glutathione (GSH) Vitamin C and E Nicotinamide adenine dinucleotide (NADH) Nicotinamide adenine dinucleotide phosphate (NADPH) Transferrin Selenium Melatonin (5-methoxy-N-acetyltryptamine) |
3.2. Anti-Angiogenic State
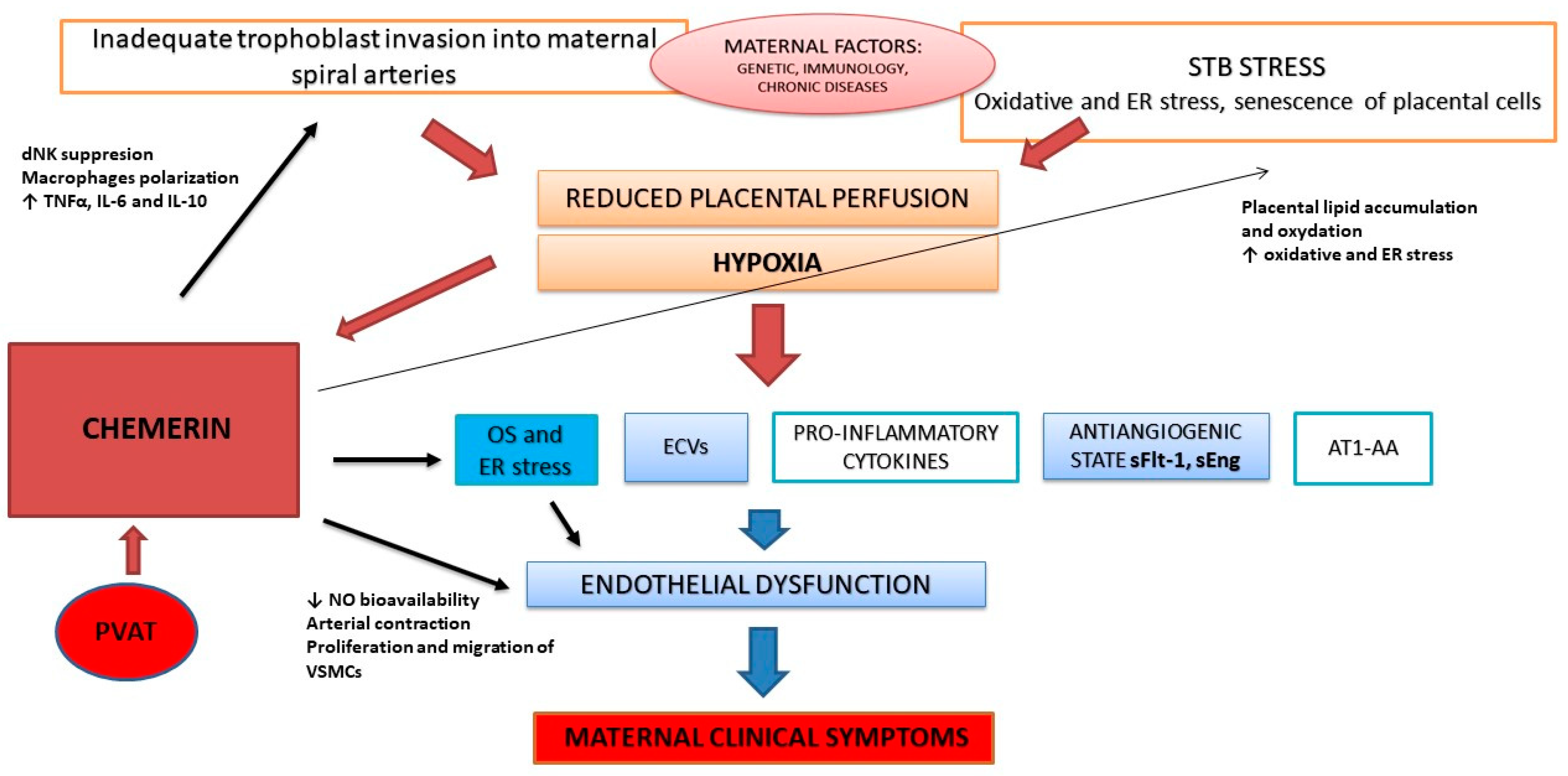
4. Potential Role of Chemerin in the Pathophysiology of PE
4.1. Chemerin during Early Pregnancy, Placentation, and Uterine Artery Remodeling
4.2. Pro-Inflammatory Effect
4.3. Angiogenesis and Imbalance between Angiogenic and Anti-Angiogenic Factors
4.4. Oxidative Stress and Endothelial Dysfunction
5. Important Implications of Chemerin Involvement in PE Pathophysiology: Early vs. Late-Onset PE and Future Cardiovascular Risk
6. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Mancuso, P. The Role of Adipokines in Chronic Inflammation. ImmunoTargets Ther. 2016, 5, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional Cloning of the Mouse Obese Gene and Its Human Homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Scotece, M.; Gómez, R.; López, V.; Gómez-Reino, J.J.; Lago, F.; Gualillo, O. Adipokines: Biofactors from White Adipose Tissue. A Complex Hub among Inflammation, Metabolism, and Immunity. BioFactors 2011, 37, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, S.; Patel, S.; Jacobe, H.; DiSepio, D.; Ghosn, C.; Malhotra, M.; Teng, M.; Duvic, M.; Chandraratna, R.A. Tazarotene-Induced Gene 2 (TIG2), a Novel Retinoid-Responsive Gene in Skin. J. Investig. Dermatol. 1997, 109, 91–95. [Google Scholar] [CrossRef]
- Helfer, G.; Wu, Q.-F. Chemerin: A Multifaceted Adipokine Involved in Metabolic Disorders. J. Endocrinol. 2018, 238, R79–R94. [Google Scholar] [CrossRef] [PubMed]
- Macvanin, M.T.; Rizzo, M.; Radovanovic, J.; Sonmez, A.; Paneni, F.; Isenovic, E.R. Role of Chemerin in Cardiovascular Diseases. Biomedicines 2022, 10, 2970. [Google Scholar] [CrossRef]
- Tan, L.; Chen, Z.; Sun, F.; Zhou, Z.; Zhang, B.; Wang, B.; Chen, J.; Li, M.; Xiao, T.; Neuman, R.I.; et al. Placental Trophoblast-Specific Overexpression of Chemerin Induces Preeclampsia-like Symptoms. Clin. Sci. 2022, 136, 257–272. [Google Scholar] [CrossRef]
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S.; et al. The Hypertensive Disorders of Pregnancy: ISSHP Classification, Diagnosis & Management Recommendations for International Practice. Pregnancy Hypertens. 2018, 13, 291–310. [Google Scholar] [CrossRef]
- Ananth, C.V.; Lavery, J.A.; Friedman, A.M.; Wapner, R.J.; Wright, J.D. Serious Maternal Complications in Relation to Severe Pre-Eclampsia: A Retrospective Cohort Study of the Impact of Hospital Volume. BJOG Int. J. Obstet. Gynaecol. 2017, 124, 1246–1253. [Google Scholar] [CrossRef]
- Bartsch, E.; Medcalf, K.E.; Park, A.L.; Ray, J.G.; High Risk of Pre-eclampsia Identification Group. Clinical Risk Factors for Pre-Eclampsia Determined in Early Pregnancy: Systematic Review and Meta-Analysis of Large Cohort Studies. BMJ 2016, 353, i1753. [Google Scholar] [CrossRef]
- Irgens, H.U.; Reisaeter, L.; Irgens, L.M.; Lie, R.T. Long Term Mortality of Mothers and Fathers after Pre-Eclampsia: Population Based Cohort Study. BMJ 2001, 323, 1213–1217. [Google Scholar] [CrossRef] [PubMed]
- Vikse, B.E. Pre-Eclampsia and the Risk of Kidney Disease. Lancet 2013, 382, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.; Peeters, L.L.; Stehouwer, C.D.A. Preeclampsia and Increased Blood Pressure in the Offspring: Meta-Analysis and Critical Review of the Evidence. J. Hypertens. 2009, 27, 1955–1959. [Google Scholar] [CrossRef] [PubMed]
- Wittamer, V.; Franssen, J.-D.; Vulcano, M.; Mirjolet, J.-F.; Le Poul, E.; Migeotte, I.; Brézillon, S.; Tyldesley, R.; Blanpain, C.; Detheux, M.; et al. Specific Recruitment of Antigen-Presenting Cells by Chemerin, a Novel Processed Ligand from Human Inflammatory Fluids. J. Exp. Med. 2003, 198, 977–985. [Google Scholar] [CrossRef]
- Meder, W.; Wendland, M.; Busmann, A.; Kutzleb, C.; Spodsberg, N.; John, H.; Richter, R.; Schleuder, D.; Meyer, M.; Forssmann, W.G. Characterization of Human Circulating TIG2 as a Ligand for the Orphan Receptor ChemR23. FEBS Lett. 2003, 555, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.J.; Davenport, A.P. International Union of Basic and Clinical Pharmacology CIII: Chemerin Receptors CMKLR1 (Chemerin1) and GPR1 (Chemerin2) Nomenclature, Pharmacology, and Function. Pharmacol. Rev. 2018, 70, 174–196. [Google Scholar] [CrossRef]
- Goralski, K.B.; McCarthy, T.C.; Hanniman, E.A.; Zabel, B.A.; Butcher, E.C.; Parlee, S.D.; Muruganandan, S.; Sinal, C.J. Chemerin, a Novel Adipokine That Regulates Adipogenesis and Adipocyte Metabolism. J. Biol. Chem. 2007, 282, 28175–28188. [Google Scholar] [CrossRef]
- Sell, H.; Laurencikiene, J.; Taube, A.; Eckardt, K.; Cramer, A.; Horrighs, A.; Arner, P.; Eckel, J. Chemerin Is a Novel Adipocyte-Derived Factor Inducing Insulin Resistance in Primary Human Skeletal Muscle Cells. Diabetes 2009, 58, 2731–2740. [Google Scholar] [CrossRef]
- Zhao, L.; Leung, L.L.; Morser, J. Chemerin Forms: Their Generation and Activity. Biomedicines 2022, 10, 2018. [Google Scholar] [CrossRef]
- Zabel, B.A.; Zuniga, L.; Ohyama, T.; Allen, S.J.; Cichy, J.; Handel, T.M.; Butcher, E.C. Chemoattractants, Extracellular Proteases, and the Integrated Host Defense Response. Exp. Hematol. 2006, 34, 1021–1032. [Google Scholar] [CrossRef]
- De Henau, O.; Degroot, G.-N.; Imbault, V.; Robert, V.; De Poorter, C.; Mcheik, S.; Galés, C.; Parmentier, M.; Springael, J.-Y. Signaling Properties of Chemerin Receptors CMKLR1, GPR1 and CCRL2. PLoS ONE 2016, 11, e0164179. [Google Scholar] [CrossRef] [PubMed]
- Serafin, D.S.; Allyn, B.; Sassano, M.F.; Timoshchenko, R.G.; Mattox, D.; Brozowski, J.M.; Siderovski, D.P.; Truong, Y.K.; Esserman, D.; Tarrant, T.K.; et al. Chemerin-Activated Functions of CMKLR1 Are Regulated by G Protein-Coupled Receptor Kinase 6 (GRK6) and β-Arrestin 2 in Inflammatory Macrophages. Mol. Immunol. 2019, 106, 12–21. [Google Scholar] [CrossRef]
- Fischer, T.F.; Czerniak, A.S.; Weiß, T.; Schoeder, C.T.; Wolf, P.; Seitz, O.; Meiler, J.; Beck-Sickinger, A.G. Ligand-Binding and -Scavenging of the Chemerin Receptor GPR1. Cell. Mol. Life Sci. 2021, 78, 6265–6281. [Google Scholar] [CrossRef]
- Degroot, G.-N.; Lepage, V.; Parmentier, M.; Springael, J.-Y. The Atypical Chemerin Receptor GPR1 Displays Different Modes of Interaction with β-Arrestins in Humans and Mice with Important Consequences on Subcellular Localization and Trafficking. Cells 2022, 11, 1037. [Google Scholar] [CrossRef]
- Yu, M.; Yang, Y.; Huang, C.; Ge, L.; Xue, L.; Xiao, Z.; Xiao, T.; Zhao, H.; Ren, P.; Zhang, J.V. Chemerin: A Functional Adipokine in Reproductive Health and Diseases. Biomedicines 2022, 10, 1910. [Google Scholar] [CrossRef] [PubMed]
- Treeck, O.; Buechler, C.; Ortmann, O. Chemerin and Cancer. Int. J. Mol. Sci. 2019, 20, 3750. [Google Scholar] [CrossRef]
- Jia, J.; Yu, F.; Xiong, Y.; Wei, W.; Ma, H.; Nisi, F.; Song, X.; Yang, L.; Wang, D.; Yuan, G.; et al. Chemerin Enhances the Adhesion and Migration of Human Endothelial Progenitor Cells and Increases Lipid Accumulation in Mice with Atherosclerosis. Lipids Health Dis. 2020, 19, 207. [Google Scholar] [CrossRef]
- Samson, M.; Edinger, A.L.; Stordeur, P.; Rucker, J.; Verhasselt, V.; Sharron, M.; Govaerts, C.; Mollereau, C.; Vassart, G.; Doms, R.W.; et al. ChemR23, a Putative Chemoattractant Receptor, Is Expressed in Monocyte-Derived Dendritic Cells and Macrophages and Is a Coreceptor for SIV and Some Primary HIV-1 Strains. Eur. J. Immunol. 1998, 28, 1689–1700. [Google Scholar] [CrossRef]
- Bozaoglu, K.; Curran, J.E.; Stocker, C.J.; Zaibi, M.S.; Segal, D.; Konstantopoulos, N.; Morrison, S.; Carless, M.; Dyer, T.D.; Cole, S.A.; et al. Chemerin, a Novel Adipokine in the Regulation of Angiogenesis. J. Clin. Endocrinol. Metab. 2010, 95, 2476–2485. [Google Scholar] [CrossRef]
- Ben Dhaou, C.; Mandi, K.; Frye, M.; Acheampong, A.; Radi, A.; De Becker, B.; Antoine, M.; Baeyens, N.; Wittamer, V.; Parmentier, M. Chemerin Regulates Normal Angiogenesis and Hypoxia-Driven Neovascularization. Angiogenesis 2022, 25, 159–179. [Google Scholar] [CrossRef]
- Muruganandan, S.; Roman, A.A.; Sinal, C.J. Role of Chemerin/CMKLR1 Signaling in Adipogenesis and Osteoblastogenesis of Bone Marrow Stem Cells. J. Bone Miner. Res. 2010, 25, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.W. Current Topic: Pre-Eclampsia and the Placenta. Placenta 1991, 12, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.W.; Sargent, I.L.; Staff, A.C. IFPA Senior Award Lecture: Making Sense of Pre-Eclampsia—Two Placental Causes of Preeclampsia? Placenta 2014, 35, S20–S25. [Google Scholar] [CrossRef]
- Staff, A.C. The Two-Stage Placental Model of Preeclampsia: An Update. J. Reprod. Immunol. 2019, 134–135, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, E.D.; Pepe, G.J. Regulation of Uterine Spiral Artery Remodeling: A Review. Reprod. Sci. 2020, 27, 1932–1942. [Google Scholar] [CrossRef]
- Redman, C.W.G.; Staff, A.C.; Roberts, J.M. Syncytiotrophoblast Stress in Preeclampsia: The Convergence Point for Multiple Pathways. Am. J. Obstet. Gynecol. 2021, 226, S907–S927. [Google Scholar] [CrossRef]
- Pankiewicz, K.; Fijałkowska, A.; Issat, T.; Maciejewski, T.M. Insight into the Key Points of Preeclampsia Pathophysiology: Uterine Artery Remodeling and the Role of MicroRNAs. Int. J. Mol. Sci. 2021, 22, 3132. [Google Scholar] [CrossRef]
- Redman, C.W.G.; Staff, A.C. Preeclampsia, Biomarkers, Syncytiotrophoblast Stress, and Placental Capacity. Am. J. Obstet. Gynecol. 2015, 213, S9.e1–S9.e4. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Burke, S.D.; Karumanchi, S.A. Imbalances in Circulating Angiogenic Factors in the Pathophysiology of Preeclampsia and Related Disorders. Am. J. Obstet. Gynecol. 2020, 226, S0002937820311960. [Google Scholar] [CrossRef]
- Chaiworapongsa, T.; Chaemsaithong, P.; Yeo, L.; Romero, R. Pre-Eclampsia Part 1: Current Understanding of Its Pathophysiology. Nat. Rev. Nephrol. 2014, 10, 466–480. [Google Scholar] [CrossRef]
- Aye, I.L.M.H.; Lager, S.; Ramirez, V.I.; Gaccioli, F.; Dudley, D.J.; Jansson, T.; Powell, T.L. Increasing Maternal Body Mass Index Is Associated with Systemic Inflammation in the Mother and the Activation of Distinct Placental Inflammatory Pathways. Biol. Reprod. 2014, 90, 129. [Google Scholar] [CrossRef] [PubMed]
- Egeland, G.M.; Klungsøyr, K.; Øyen, N.; Tell, G.S.; Næss, Ø.; Skjærven, R. Preconception Cardiovascular Risk Factor Differences Between Gestational Hypertension and Preeclampsia: Cohort Norway Study. Hypertension 2016, 67, 1173–1180. [Google Scholar] [CrossRef]
- Cadenas, E.; Sies, H. Oxidative Stress: Excited Oxygen Species and Enzyme Activity. Adv. Enzym. Regul. 1985, 23, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Freire, V.A.F.; de Melo, A.D.; de Lima Santos, H.; Barros-Pinheiro, M. Evaluation of Oxidative Stress Markers in Subtypes of Preeclampsia: A Systematic Review and Meta-Analysis. Placenta 2023, 132, 55–67. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. Oxidative Stress. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Tenório, M.B.; Ferreira, R.C.; Moura, F.A.; Bueno, N.B.; de Oliveira, A.C.M.; Goulart, M.O.F. Cross-Talk between Oxidative Stress and Inflammation in Preeclampsia. Oxid. Med. Cell. Longev. 2019, 2019, 8238727. [Google Scholar] [CrossRef]
- Aouache, R.; Biquard, L.; Vaiman, D.; Miralles, F. Oxidative Stress in Preeclampsia and Placental Diseases. Int. J. Mol. Sci. 2018, 19, 1496. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Yung, H.-W.; Cindrova-Davies, T.; Charnock-Jones, D.S. Placental Endoplasmic Reticulum Stress and Oxidative Stress in the Pathophysiology of Unexplained Intrauterine Growth Restriction and Early Onset Preeclampsia. Placenta 2009, 30 (Suppl. A), S43–S48. [Google Scholar] [CrossRef]
- Roberts, J.M. Pathophysiology of Ischemic Placental Disease. Semin. Perinatol. 2014, 38, 139–145. [Google Scholar] [CrossRef]
- Carter, A.M. Carter, A.M. Placental Oxygen Consumption. Part I: In Vivo Studies—A Review. Placenta 2000, 21 (Suppl. A), S31–S37. [Google Scholar] [CrossRef]
- Chiarello, D.I.; Abad, C.; Rojas, D.; Toledo, F.; Vázquez, C.M.; Mate, A.; Sobrevia, L.; Marín, R. Oxidative Stress: Normal Pregnancy versus Preeclampsia. Biochim. Biophys. Acta Mol. 2020, 1866, 165354. [Google Scholar] [CrossRef] [PubMed]
- Haram, K.; Mortensen, J.H.; Myking, O.; Magann, E.F.; Morrison, J.C. The Role of Oxidative Stress, Adhesion Molecules and Antioxidants in Preeclampsia. Curr. Hypertens. Rev. 2019, 15, 105–112. [Google Scholar] [CrossRef]
- Sánchez-Aranguren, L.C.; Prada, C.E.; Riaño-Medina, C.E.; Lopez, M. Endothelial Dysfunction and Preeclampsia: Role of Oxidative Stress. Front. Physiol. 2014, 5, 372. [Google Scholar] [CrossRef]
- Can, M.; Guven, B.; Bektas, S.; Arikan, I. Oxidative Stress and Apoptosis in Preeclampsia. Tissue Cell 2014, 46, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rodríguez, P.; Ramiro-Cortijo, D.; Reyes-Hernández, C.G.; López de Pablo, A.L.; González, M.C.; Arribas, S.M. Implication of Oxidative Stress in Fetal Programming of Cardiovascular Disease. Front. Physiol. 2018, 9, 602. [Google Scholar] [CrossRef] [PubMed]
- Tomimatsu, T.; Mimura, K.; Matsuzaki, S.; Endo, M.; Kumasawa, K.; Kimura, T. Preeclampsia: Maternal Systemic Vascular Disorder Caused by Generalized Endothelial Dysfunction Due to Placental Antiangiogenic Factors. Int. J. Mol. Sci. 2019, 20, 4246. [Google Scholar] [CrossRef]
- Chaiworapongsa, T.; Romero, R.; Kim, Y.M.; Kim, G.J.; Kim, M.R.; Espinoza, J.; Bujold, E.; Gonçalves, L.; Gomez, R.; Edwin, S.; et al. Plasma Soluble Vascular Endothelial Growth Factor Receptor-1 Concentration Is Elevated Prior to the Clinical Diagnosis of Pre-Eclampsia. J. Matern.-Fetal Neonatal Med. 2005, 17, 3–18. [Google Scholar] [CrossRef]
- Modzelewski, J.; Siarkowska, I.; Pajurek-Dudek, J.; Feduniw, S.; Muzyka-Placzyńska, K.; Baran, A.; Kajdy, A.; Bednarek-Jędrzejek, M.; Cymbaluk-Płoska, A.; Kwiatkowska, E.; et al. Atypical Preeclampsia before 20 Weeks of Gestation-A Systematic Review. Int. J. Mol. Sci. 2023, 24, 3752. [Google Scholar] [CrossRef]
- Dymara-Konopka, W.; Laskowska, M.; Grywalska, E.; Hymos, A.; Błażewicz, A.; Leszczyńska-Gorzelak, B. Similar Pro- and Antiangiogenic Profiles Close to Delivery in Different Clinical Presentations of Two Pregnancy Syndromes: Preeclampsia and Fetal Growth Restriction. Int. J. Mol. Sci. 2023, 24, 972. [Google Scholar] [CrossRef]
- Chau, K.; Hennessy, A.; Makris, A. Placental Growth Factor and Pre-Eclampsia. J. Hum. Hypertens. 2017, 31, 782–786. [Google Scholar] [CrossRef]
- Creswell, L.; O’Gorman, N.; Palmer, K.R.; da Silva Costa, F.; Rolnik, D.L. Perspectives on the Use of Placental Growth Factor (PlGF) in the Prediction and Diagnosis of Pre-Eclampsia: Recent Insights and Future Steps. Int. J. Womens Health 2023, 15, 255–271. [Google Scholar] [CrossRef]
- Hod, T.; Cerdeira, A.S.; Karumanchi, S.A. Molecular Mechanisms of Preeclampsia. Cold Spring Harb. Perspect. Med. 2015, 5, a023473. [Google Scholar] [CrossRef] [PubMed]
- Bdolah, Y.; Elchalal, U.; Natanson-Yaron, S.; Yechiam, H.; Bdolah-Abram, T.; Greenfield, C.; Goldman-Wohl, D.; Milwidsky, A.; Rana, S.; Karumanchi, S.A.; et al. Relationship between Nulliparity and Preeclampsia May Be Explained by Altered Circulating Soluble Fms-like Tyrosine Kinase 1. Hypertens. Pregnancy 2014, 33, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Hacker, M.R.; Modest, A.M.; Salahuddin, S.; Lim, K.-H.; Verlohren, S.; Perschel, F.H.; Karumanchi, S.A. Circulating Angiogenic Factors and Risk of Adverse Maternal and Perinatal Outcomes in Twin Pregnancies with Suspected Preeclampsia. Hypertension 2012, 60, 451–458. [Google Scholar] [CrossRef]
- Amraoui, F.; Spijkers, L.; Hassani Lahsinoui, H.; Vogt, L.; van der Post, J.; Peters, S.; Afink, G.; Ris-Stalpers, C.; van den Born, B.-J. SFlt-1 Elevates Blood Pressure by Augmenting Endothelin-1-Mediated Vasoconstriction in Mice. PLoS ONE 2014, 9, e91897. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.N.; Ratner, E.; Rutherford, T.J.; Schwartz, P.E.; Norwitz, E.R. Bevacizumab-Mediated Interference with VEGF Signaling Is Sufficient to Induce a Preeclampsia-like Syndrome in Nonpregnant Women. Rev. Obstet. Gynecol. 2012, 5, 2–8. [Google Scholar]
- Rana, S.; Rajakumar, A.; Geahchan, C.; Salahuddin, S.; Cerdeira, A.S.; Burke, S.D.; George, E.M.; Granger, J.P.; Karumanchi, S.A. Ouabain Inhibits Placental SFlt1 Production by Repressing HSP27-Dependent HIF-1α Pathway. FASEB J. 2014, 28, 4324–4334. [Google Scholar] [CrossRef]
- Tal, R.; Shaish, A.; Barshack, I.; Polak-Charcon, S.; Afek, A.; Volkov, A.; Feldman, B.; Avivi, C.; Harats, D. Effects of Hypoxia-Inducible Factor-1alpha Overexpression in Pregnant Mice: Possible Implications for Preeclampsia and Intrauterine Growth Restriction. Am. J. Pathol. 2010, 177, 2950–2962. [Google Scholar] [CrossRef] [PubMed]
- Iriyama, T.; Wang, W.; Parchim, N.F.; Song, A.; Blackwell, S.C.; Sibai, B.M.; Kellems, R.E.; Xia, Y. Hypoxia-Independent Upregulation of Placental Hypoxia Inducible Factor-1α Gene Expression Contributes to the Pathogenesis of Preeclampsia. Hypertension 2015, 65, 1307–1315. [Google Scholar] [CrossRef]
- Korkes, H.A.; De Oliveira, L.; Sass, N.; Salahuddin, S.; Karumanchi, S.A.; Rajakumar, A. Relationship between Hypoxia and Downstream Pathogenic Pathways in Preeclampsia. Hypertens. Pregnancy 2017, 36, 145–150. [Google Scholar] [CrossRef]
- Levine, R.J.; Lam, C.; Qian, C.; Yu, K.F.; Maynard, S.E.; Sachs, B.P.; Sibai, B.M.; Epstein, F.H.; Romero, R.; Thadhani, R.; et al. Soluble Endoglin and Other Circulating Antiangiogenic Factors in Preeclampsia. N. Engl. J. Med. 2006, 355, 992–1005. [Google Scholar] [CrossRef]
- Sandrim, V.C.; Palei, A.C.T.; Metzger, I.F.; Gomes, V.A.; Cavalli, R.C.; Tanus-Santos, J.E. Nitric Oxide Formation Is Inversely Related to Serum Levels of Antiangiogenic Factors Soluble Fms-like Tyrosine Kinase-1 and Soluble Endogline in Preeclampsia. Hypertension 2008, 52, 402–407. [Google Scholar] [CrossRef]
- Stepan, H.; Philipp, A.; Roth, I.; Kralisch, S.; Jank, A.; Schaarschmidt, W.; Lössner, U.; Kratzsch, J.; Blüher, M.; Stumvoll, M.; et al. Serum Levels of the Adipokine Chemerin Are Increased in Preeclampsia during and 6 Months after Pregnancy. Regul. Pept. 2011, 168, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.-M.; Niu, J.-M.; Lei, Q.; Lin, X.-H.; Chen, X. Serum Levels of the Adipokine Chemerin in Preeclampsia. J. Perinat. Med. 2011, 40, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.-L.; Zhu, M.; Jin, Y.; Wang, N.; Xu, H.-X.; Quan, L.-M.; Wang, S.-S.; Li, S.-S. The Predictive Value of the First-Trimester Maternal Serum Chemerin Level for Pre-Eclampsia. Peptides 2014, 62, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Cetin, O.; Kurdoglu, Z.; Kurdoglu, M.; Sahin, H.G. Chemerin Level in Pregnancies Complicated by Preeclampsia and Its Relation with Disease Severity and Neonatal Outcomes. J. Obstet. Gynaecol. J. Inst. Obstet. Gynaecol. 2017, 37, 195–199. [Google Scholar] [CrossRef]
- Daskalakis, G.; Bellos, I.; Nikolakea, M.; Pergialiotis, V.; Papapanagiotou, A.; Loutradis, D. The Role of Serum Adipokine Levels in Preeclampsia: A Systematic Review. Metabolism 2020, 106, 154172. [Google Scholar] [CrossRef] [PubMed]
- Turgut, A.; Ozler, A.; Goruk, N.Y.; Tunç, S.Y.; Sak, M.E.; Evsen, M.S.; Evliyaoglu, O.; Gul, T. Serum Levels of the Adipokines, Free Fatty Acids, and Oxidative Stress Markers in Obese and Non-Obese Preeclamptic Patients. Clin. Exp. Obstet. Gynecol. 2015, 42, 473–479. [Google Scholar] [CrossRef]
- Carlino, C.; Trotta, E.; Stabile, H.; Morrone, S.; Bulla, R.; Soriani, A.; Iannitto, M.L.; Agostinis, C.; Mocci, C.; Minozzi, M.; et al. Chemerin Regulates NK Cell Accumulation and Endothelial Cell Morphogenesis in the Decidua during Early Pregnancy. J. Clin. Endocrinol. Metab. 2012, 97, 3603–3612. [Google Scholar] [CrossRef]
- Garces, M.F.; Sanchez, E.; Acosta, B.J.; Angel, E.; Ruíz, A.I.; Rubio-Romero, J.A.; Diéguez, C.; Nogueiras, R.; Caminos, J.E. Expression and Regulation of Chemerin during Rat Pregnancy. Placenta 2012, 33, 373–378. [Google Scholar] [CrossRef]
- Quan, X.-Z.; Ye, J.-H.; Yang, X.-Z.; Xie, Y. HOXA9-Induced Chemerin Signals through CMKLR1/AMPK/TXNIP/NLRP3 Pathway to Induce Pyroptosis of Trophoblasts and Aggravate Preeclampsia. Exp. Cell Res. 2021, 408, 112802. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yao, J.; Wei, Q.; Ye, J.; Yin, X.; Quan, X.; Lan, Y.; Xing, H. Role of Chemerin/CMKLR1 in the Maintenance of Early Pregnancy. Front. Med. 2018, 12, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y. Endovascular Trophoblast and Spiral Artery Remodeling. Mol. Cell. Endocrinol. 2020, 503, 110699. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xiao, Z.; Lee, C.-L.; Duan, Y.-G.; Fan, X.; Yeung, W.S.B.; Chiu, P.C.N.; Zhang, J.V. The Regulatory Roles of Chemerin-Chemokine-Like Receptor 1 Axis in Placental Development and Vascular Remodeling During Early Pregnancy. Front. Cell Dev. Biol. 2022, 10, 883636. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.-S.; Jiang, H.; Xie, Y.; Wei, Q.-P.; Yin, X.-F.; Ye, J.-H.; Quan, X.-Z.; Lan, Y.-L.; Zhao, M.; Tian, X.-L.; et al. Chemerin Promotes the Pathogenesis of Preeclampsia by Activating CMKLR1/p-Akt/CEBPɑ Axis and Inducing M1 Macrophage Polarization. Cell Biol. Toxicol. 2022, 38, 611–628. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef]
- Harmon, A.C.; Cornelius, D.C.; Amaral, L.M.; Faulkner, J.L.; Cunningham, M.W.; Wallace, K.; LaMarca, B. The Role of Inflammation in the Pathology of Preeclampsia. Clin. Sci. 2016, 130, 409–419. [Google Scholar] [CrossRef]
- Laresgoiti-Servitje, E. A Leading Role for the Immune System in the Pathophysiology of Preeclampsia. J. Leukoc. Biol. 2013, 94, 247–257. [Google Scholar] [CrossRef]
- Liu, X.; Liu, X.; Liu, W.; Luo, M.; Tao, H.; Wu, D.; Zhao, Y.; Zou, L. HOXA9 Transcriptionally Regulates the EPHB4 Receptor to Modulate Trophoblast Migration and Invasion. Placenta 2017, 51, 38–48. [Google Scholar] [CrossRef]
- Brennan, M.A.; Cookson, B.T. Salmonella Induces Macrophage Death by Caspase-1-Dependent Necrosis. Mol. Microbiol. 2000, 38, 31–40. [Google Scholar] [CrossRef]
- Chen, A.; Chen, Z.; Xia, Y.; Lu, D.; Yang, X.; Sun, A.; Zou, Y.; Qian, J.; Ge, J. Liraglutide Attenuates NLRP3 Inflammasome-Dependent Pyroptosis via Regulating SIRT1/NOX4/ROS Pathway in H9c2 Cells. Biochem. Biophys. Res. Commun. 2018, 499, 267–272. [Google Scholar] [CrossRef]
- Kohli, S.; Ranjan, S.; Hoffmann, J.; Kashif, M.; Daniel, E.A.; Al-Dabet, M.M.; Bock, F.; Nazir, S.; Huebner, H.; Mertens, P.R.; et al. Maternal Extracellular Vesicles and Platelets Promote Preeclampsia via Inflammasome Activation in Trophoblasts. Blood 2016, 128, 2153–2164. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-Y.; Liu, K.; Zhang, J.-L. LINC00240/MiR-155 Axis Regulates Function of Trophoblasts and M2 Macrophage Polarization via Modulating Oxidative Stress-Induced Pyroptosis in Preeclampsia. Mol. Med. Camb. Mass 2022, 28, 119. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-B.; Nakashima, A.; Huber, W.J.; Davis, S.; Banerjee, S.; Huang, Z.; Saito, S.; Sadovsky, Y.; Sharma, S. Pyroptosis Is a Critical Inflammatory Pathway in the Placenta from Early Onset Preeclampsia and in Human Trophoblasts Exposed to Hypoxia and Endoplasmic Reticulum Stressors. Cell Death Dis. 2019, 10, 927. [Google Scholar] [CrossRef] [PubMed]
- Neves, K.B.; Lobato, N.S.; Lopes, R.A.M.; Filgueira, F.P.; Zanotto, C.Z.; Oliveira, A.M.; Tostes, R.C. Chemerin Reduces Vascular Nitric Oxide/CGMP Signalling in Rat Aorta: A Link to Vascular Dysfunction in Obesity? Clin. Sci. 2014, 127, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, K.; Friebe, D.; Ullrich, T.; Kratzsch, J.; Dittrich, K.; Herberth, G.; Adams, V.; Kiess, W.; Erbs, S.; Körner, A. Chemerin as a Mediator between Obesity and Vascular Inflammation in Children. J. Clin. Endocrinol. Metab. 2012, 97, E556–E564. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, L. Role of Chemerin/ChemR23 Axis as an Emerging Therapeutic Perspective on Obesity-Related Vascular Dysfunction. J. Transl. Med. 2022, 20, 141. [Google Scholar] [CrossRef] [PubMed]
- Szpakowicz, A.; Szpakowicz, M.; Lapinska, M.; Paniczko, M.; Lawicki, S.; Raczkowski, A.; Kondraciuk, M.; Sawicka, E.; Chlabicz, M.; Kozuch, M.; et al. Serum Chemerin Concentration Is Associated with Proinflammatory Status in Chronic Coronary Syndrome. Biomolecules 2021, 11, 1149. [Google Scholar] [CrossRef]
- Dimitriadis, G.K.; Kaur, J.; Adya, R.; Miras, A.D.; Mattu, H.S.; Hattersley, J.G.; Kaltsas, G.; Tan, B.K.; Randeva, H.S. Chemerin Induces Endothelial Cell Inflammation: Activation of Nuclear Factor-Kappa Beta and Monocyte-Endothelial Adhesion. Oncotarget 2018, 9, 16678–16690. [Google Scholar] [CrossRef]
- Socha, M.W.; Malinowski, B.; Puk, O.; Wartęga, M.; Stankiewicz, M.; Kazdepka-Ziemińska, A.; Wiciński, M. The Role of NF-ΚB in Uterine Spiral Arteries Remodeling, Insight into the Cornerstone of Preeclampsia. Int. J. Mol. Sci. 2021, 22, 704. [Google Scholar] [CrossRef]
- Kaur, J.; Adya, R.; Tan, B.K.; Chen, J.; Randeva, H.S. Identification of Chemerin Receptor (ChemR23) in Human Endothelial Cells: Chemerin-Induced Endothelial Angiogenesis. Biochem. Biophys. Res. Commun. 2010, 391, 1762–1768. [Google Scholar] [CrossRef] [PubMed]
- Corre, I.; Paris, F.; Huot, J. The P38 Pathway, a Major Pleiotropic Cascade That Transduces Stress and Metastatic Signals in Endothelial Cells. Oncotarget 2017, 8, 55684–55714. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Naruse, K.; Kobayashi, Y.; Miyabe, M.; Saiki, T.; Enomoto, A.; Takahashi, M.; Matsubara, T. Chemerin Promotes Angiogenesis In Vivo. Physiol. Rep. 2018, 6, e13962. [Google Scholar] [CrossRef] [PubMed]
- Palei, A.C.; Cruz, J.D.O.; Chaguri, J.L.; Peraçoli, J.C.; Romão-Veiga, M.; Ribeiro-Vasques, V.R.; Cavalli, R.C.; Nunes, P.R.; Luizon, M.R.; Sandrim, V.C. Circulating Levels of Tissue Inhibitor of Metalloproteinase 3, a Protein with Inhibitory Effects on Angiogenesis, Are Increased in Pre-Eclampsia. Int. J. Gynaecol. Obstet. Off. Organ Int. Fed. Gynaecol. Obstet. 2022. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Redman, C.W.; Roberts, J.M.; Moffett, A. Pre-Eclampsia: Pathophysiology and Clinical Implications. BMJ 2019, 366, l2381. [Google Scholar] [CrossRef]
- Margioula-Siarkou, G.; Margioula-Siarkou, C.; Petousis, S.; Margaritis, K.; Vavoulidis, E.; Gullo, G.; Alexandratou, M.; Dinas, K.; Sotiriadis, A.; Mavromatidis, G. The Role of Endoglin and Its Soluble Form in Pathogenesis of Preeclampsia. Mol. Cell. Biochem. 2022, 477, 479–491. [Google Scholar] [CrossRef]
- Ma, J.; Wu, H.; Yang, X.; Zheng, L.; Feng, H.; Yang, L. Identification and Validation of an Angiogenesis-Related Signature Associated with Preeclampsia by Bioinformatic Analysis. Medicine 2023, 102, e32741. [Google Scholar] [CrossRef]
- Chua, S.-K.; Shyu, K.-G.; Lin, Y.-F.; Lo, H.-M.; Wang, B.-W.; Chang, H.; Lien, L.-M. Tumor Necrosis Factor-Alpha and the ERK Pathway Drive Chemerin Expression in Response to Hypoxia in Cultured Human Coronary Artery Endothelial Cells. PLoS ONE 2016, 11, e0165613. [Google Scholar] [CrossRef]
- Watts, S.W.; Dorrance, A.M.; Penfold, M.E.; Rourke, J.L.; Sinal, C.J.; Seitz, B.; Sullivan, T.J.; Charvat, T.T.; Thompson, J.M.; Burnett, R.; et al. Chemerin Connects Fat to Arterial Contraction. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1320–1328. [Google Scholar] [CrossRef]
- Darios, E.S.; Winner, B.M.; Charvat, T.; Krasinksi, A.; Punna, S.; Watts, S.W. The Adipokine Chemerin Amplifies Electrical Field-Stimulated Contraction in the Isolated Rat Superior Mesenteric Artery. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H498–H507. [Google Scholar] [CrossRef]
- Tan, L.; Ouyang, Z.; Chen, Z.; Sun, F.; Guo, H.; Wang, F.; Mulder, M.; Sun, Y.; Lu, X.; Zhang, J.V.; et al. Adipokine Chemerin Overexpression in Trophoblasts Leads to Dyslipidemia in Pregnant Mice: Implications for Preeclampsia. Lipids Health Dis. 2023, 22, 12. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Cheng, M.; Hui, X.; Lu, B.; Jiang, W.; Shi, Z. Elevating Circulation Chemerin Level Is Associated with Endothelial Dysfunction and Early Atherosclerotic Changes in Essential Hypertensive Patients. J. Hypertens. 2015, 33, 1624–1632. [Google Scholar] [CrossRef] [PubMed]
- Joo, E.H.; Kim, Y.R.; Kim, N.; Jung, J.E.; Han, S.H.; Cho, H.Y. Effect of Endogenic and Exogenic Oxidative Stress Triggers on Adverse Pregnancy Outcomes: Preeclampsia, Fetal Growth Restriction, Gestational Diabetes Mellitus and Preterm Birth. Int. J. Mol. Sci. 2021, 22, 10122. [Google Scholar] [CrossRef]
- Shen, W.; Tian, C.; Chen, H.; Yang, Y.; Zhu, D.; Gao, P.; Liu, J. Oxidative Stress Mediates Chemerin-Induced Autophagy in Endothelial Cells. Free Radic. Biol. Med. 2013, 55, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Li, Z.; Fu, Y.; Wu, R.; Wang, Y.; Liu, C.; Yang, L.; Zhang, H. Involvement of Obesity-Associated Upregulation of Chemerin/Chemokine-like Receptor 1 in Oxidative Stress and Apoptosis in Ovaries and Granulosa Cells. Biochem. Biophys. Res. Commun. 2019, 510, 449–455. [Google Scholar] [CrossRef]
- Bulut, A.; Akca, G.; Keskin Aktan, A.; Akbulut, K.G.; Babül, A. The Significance of Blood and Salivary Oxidative Stress Markers and Chemerin in Gestational Diabetes Mellitus. Taiwan. J. Obstet. Gynecol. 2021, 60, 695–699. [Google Scholar] [CrossRef]
- Fülöp, P.; Seres, I.; Lőrincz, H.; Harangi, M.; Somodi, S.; Paragh, G. Association of Chemerin with Oxidative Stress, Inflammation and Classical Adipokines in Non-Diabetic Obese Patients. J. Cell. Mol. Med. 2014, 18, 1313–1320. [Google Scholar] [CrossRef]
- Neves, K.B.; Montezano, A.C.; Alves-Lopes, R.; Bruder-Nascimento, T.; Costa, R.M.; Costa, R.S.; Touyz, R.M.; Tostes, R.C. Upregulation of Nrf2 and Decreased Redox Signaling Contribute to Renoprotective Effects of Chemerin Receptor Blockade in Diabetic Mice. Int. J. Mol. Sci. 2018, 19, 2454. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, T.; Ding, Y.; Zhong, Y.; Yu, L.; Peng, M. Chemerin Plays a Protective Role by Regulating Human Umbilical Vein Endothelial Cell-Induced Nitric Oxide Signaling in Preeclampsia. Endocrine 2015, 48, 299–308. [Google Scholar] [CrossRef]
- Hooijschuur, M.C.E.; Ghossein-Doha, C.; Kroon, A.A.; De Leeuw, P.W.; Zandbergen, A.A.M.; Van Kuijk, S.M.J.; Spaanderman, M.E.A. Metabolic Syndrome and Pre-Eclampsia. Ultrasound Obstet. Gynecol. 2019, 54, 64–71. [Google Scholar] [CrossRef]
- Melchiorre, K.; Sharma, R.; Thilaganathan, B. Cardiovascular Implications in Preeclampsia: An Overview. Circulation 2014, 130, 703–714. [Google Scholar] [CrossRef]
- Ferland, D.J.; Darios, E.S.; Neubig, R.R.; Sjögren, B.; Truong, N.; Torres, R.; Dexheimer, T.S.; Thompson, J.M.; Watts, S.W. Chemerin-Induced Arterial Contraction Is Gi- and Calcium-Dependent. Vascul. Pharmacol. 2017, 88, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Hanthazi, A.; Jespers, P.; Vegh, G.; Degroot, G.-N.; Springael, J.-Y.; Lybaert, P.; Dewachter, L.; Mc Entee, K. Chemerin Influences Endothelin- and Serotonin-Induced Pulmonary Artery Vasoconstriction in Rats. Life Sci. 2019, 231, 116580. [Google Scholar] [CrossRef]
- Osikoya, O.; Ahmed, H.; Panahi, S.; Bourque, S.L.; Goulopoulou, S. Uterine Perivascular Adipose Tissue Is a Novel Mediator of Uterine Artery Blood Flow and Reactivity in Rat Pregnancy. J. Physiol. 2019, 597, 3833–3852. [Google Scholar] [CrossRef]
- Wen, J.; Wang, J.; Guo, L.; Cai, W.; Wu, Y.; Chen, W.; Tang, X. Chemerin Stimulates Aortic Smooth Muscle Cell Proliferation and Migration via Activation of Autophagy in VSMCs of Metabolic Hypertension Rats. Am. J. Transl. Res. 2019, 11, 1327–1342. [Google Scholar] [PubMed]
- Kunimoto, H.; Kazama, K.; Takai, M.; Oda, M.; Okada, M.; Yamawaki, H. Chemerin Promotes the Proliferation and Migration of Vascular Smooth Muscle and Increases Mouse Blood Pressure. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1017–H1028. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Chen, Y.; Li, Y.; Feng, P.; Zheng, Y.; Dong, Y.; Yang, Y.; Wang, R.; Li, A.; Yan, J.; et al. Chemerin Regulates the Proliferation and Migration of Pulmonary Arterial Smooth Muscle Cells via the ERK1/2 Signaling Pathway. Front. Pharmacol. 2022, 13, 767705. [Google Scholar] [CrossRef]
- Satoh, K.; Nigro, P.; Berk, B.C. Oxidative Stress and Vascular Smooth Muscle Cell Growth: A Mechanistic Linkage by Cyclophilin A. Antioxid. Redox Signal. 2010, 12, 675–682. [Google Scholar] [CrossRef]
- Thomas, G.D. Neural Control of the Circulation. Adv. Physiol. Educ. 2011, 35, 28–32. [Google Scholar] [CrossRef]
- Bartho, L.A.; Kandel, M.; Walker, S.P.; Cluver, C.A.; Hastie, R.; Bergman, L.; Pritchard, N.; Cannon, P.; Nguyen, T.-V.; Wong, G.P.; et al. Circulating Chemerin Is Elevated in Women with Preeclampsia. Endocrinology 2023, 164, bqad041. [Google Scholar] [CrossRef]
- Wu, P.; Haththotuwa, R.; Kwok, C.S.; Babu, A.; Kotronias, R.A.; Rushton, C.; Zaman, A.; Fryer, A.A.; Kadam, U.; Chew-Graham, C.A.; et al. Preeclampsia and Future Cardiovascular Health: A Systematic Review and Meta-Analysis. Circ. Cardiovasc. Qual. Outcomes 2017, 10, e003497. [Google Scholar] [CrossRef] [PubMed]
- Berks, D.; Hoedjes, M.; Raat, H.; Duvekot, J.J.; Steegers, E.A.P.; Habbema, J.D.F. Risk of Cardiovascular Disease after Pre-Eclampsia and the Effect of Lifestyle Interventions: A Literature-Based Study. BJOG Int. J. Obstet. Gynaecol. 2013, 120, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Pankiewicz, K.; Szczerba, E.; Maciejewski, T.; Fijałkowska, A. Non-Obstetric Complications in Preeclampsia. Prz. Menopauzalny Menopause Rev. 2019, 18, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, L.; Liu, H.; Li, Z.; Li, L.; Wu, X.; Lei, Q.; Yin, A.; Tong, J.; Liu, K.; et al. Third-Trimester Maternal Serum Chemerin and Hypertension After Preeclampsia: A Prospective Cohort Study. J. Am. Heart Assoc. 2023, 12, e027930. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pankiewicz, K.; Issat, T. Understanding the Role of Chemerin in the Pathophysiology of Pre-Eclampsia. Antioxidants 2023, 12, 830. https://doi.org/10.3390/antiox12040830
Pankiewicz K, Issat T. Understanding the Role of Chemerin in the Pathophysiology of Pre-Eclampsia. Antioxidants. 2023; 12(4):830. https://doi.org/10.3390/antiox12040830
Chicago/Turabian StylePankiewicz, Katarzyna, and Tadeusz Issat. 2023. "Understanding the Role of Chemerin in the Pathophysiology of Pre-Eclampsia" Antioxidants 12, no. 4: 830. https://doi.org/10.3390/antiox12040830
APA StylePankiewicz, K., & Issat, T. (2023). Understanding the Role of Chemerin in the Pathophysiology of Pre-Eclampsia. Antioxidants, 12(4), 830. https://doi.org/10.3390/antiox12040830