
Ether Lipid-Mediated Antioxidant Defense in Alzheimer’s Disease

 , , , , ,
, , , , ,  , and
, and
Abstract
1. Introduction
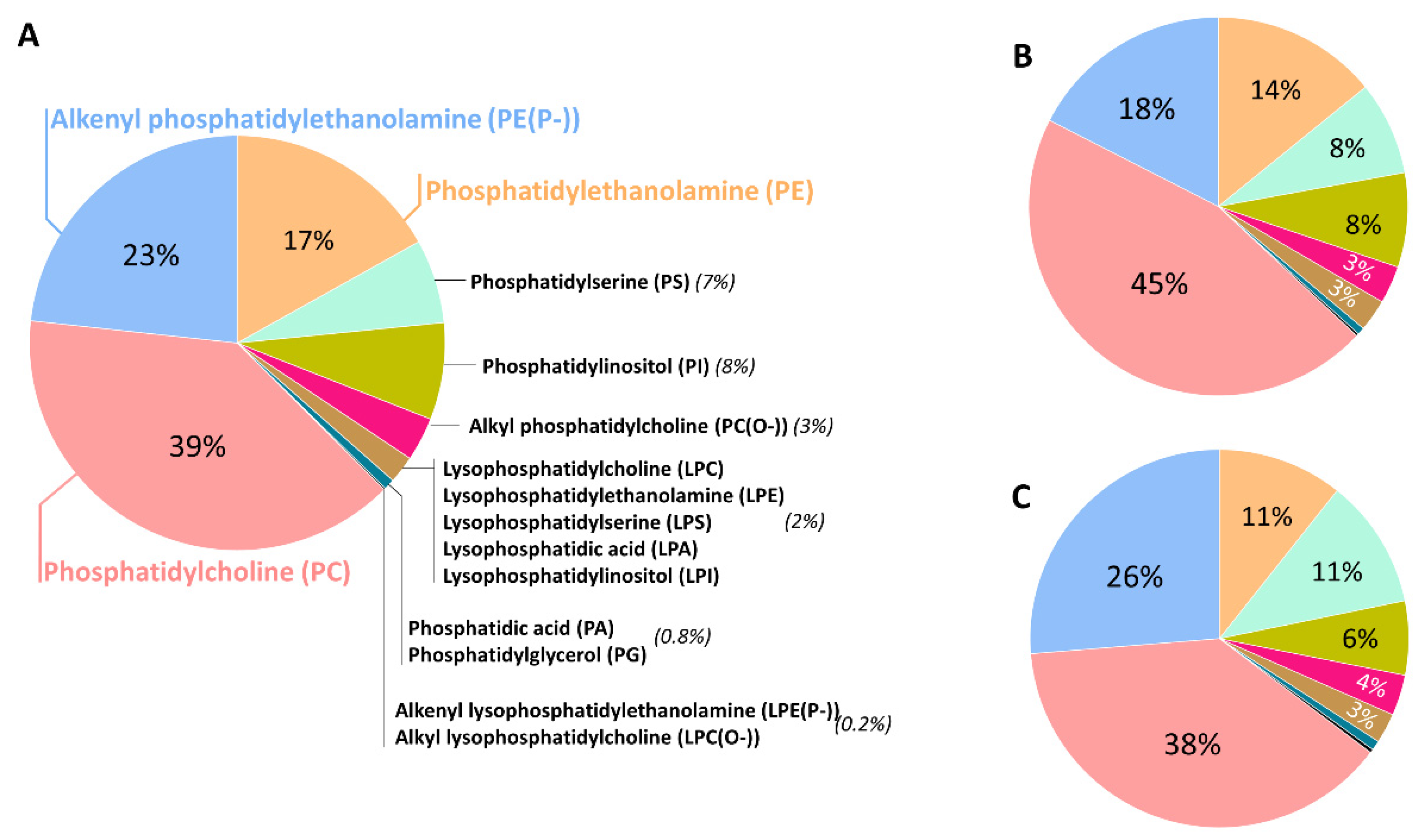
2. Lipid Species and the Human Brain
3. Ether Lipids and the Human Brain Evolution
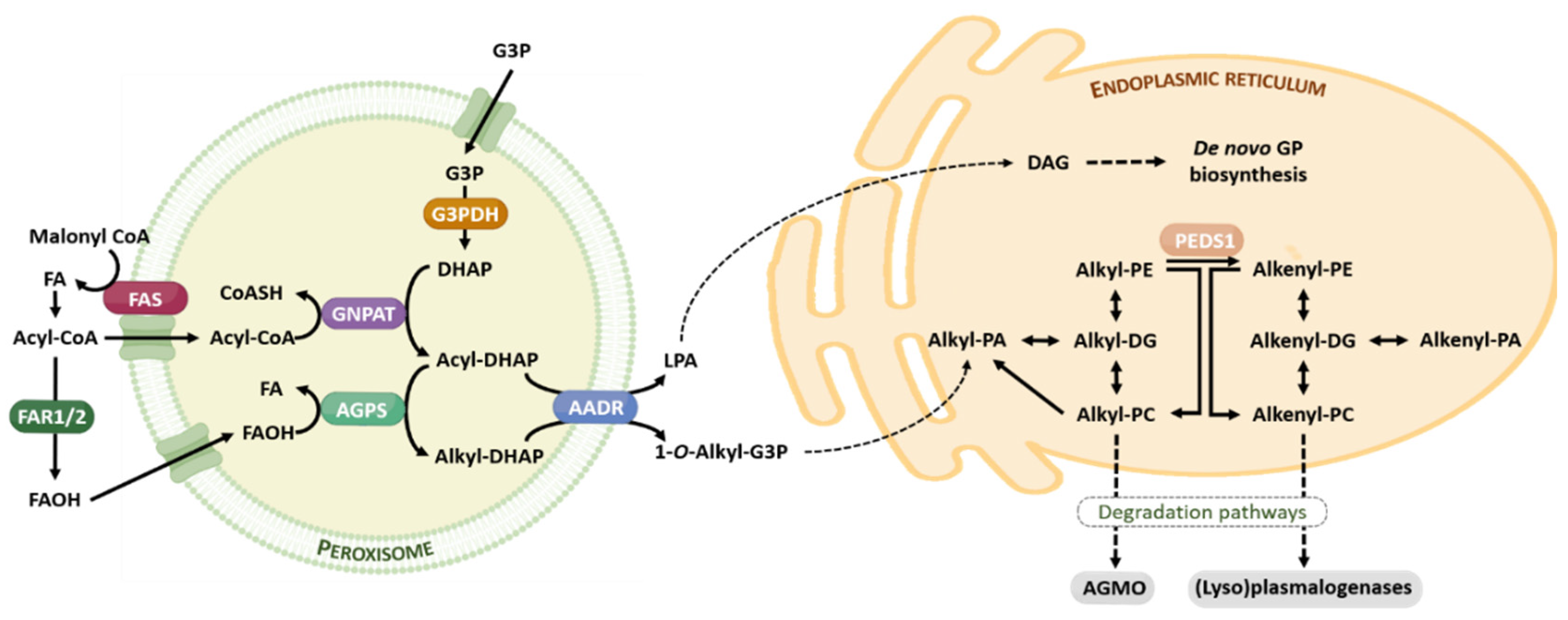
4. Basic Traits of Ether Lipids: Structure, Metabolism, and Function
4.1. Structural Roles
4.2. Membrane Trafficking
4.3. Cell Signaling
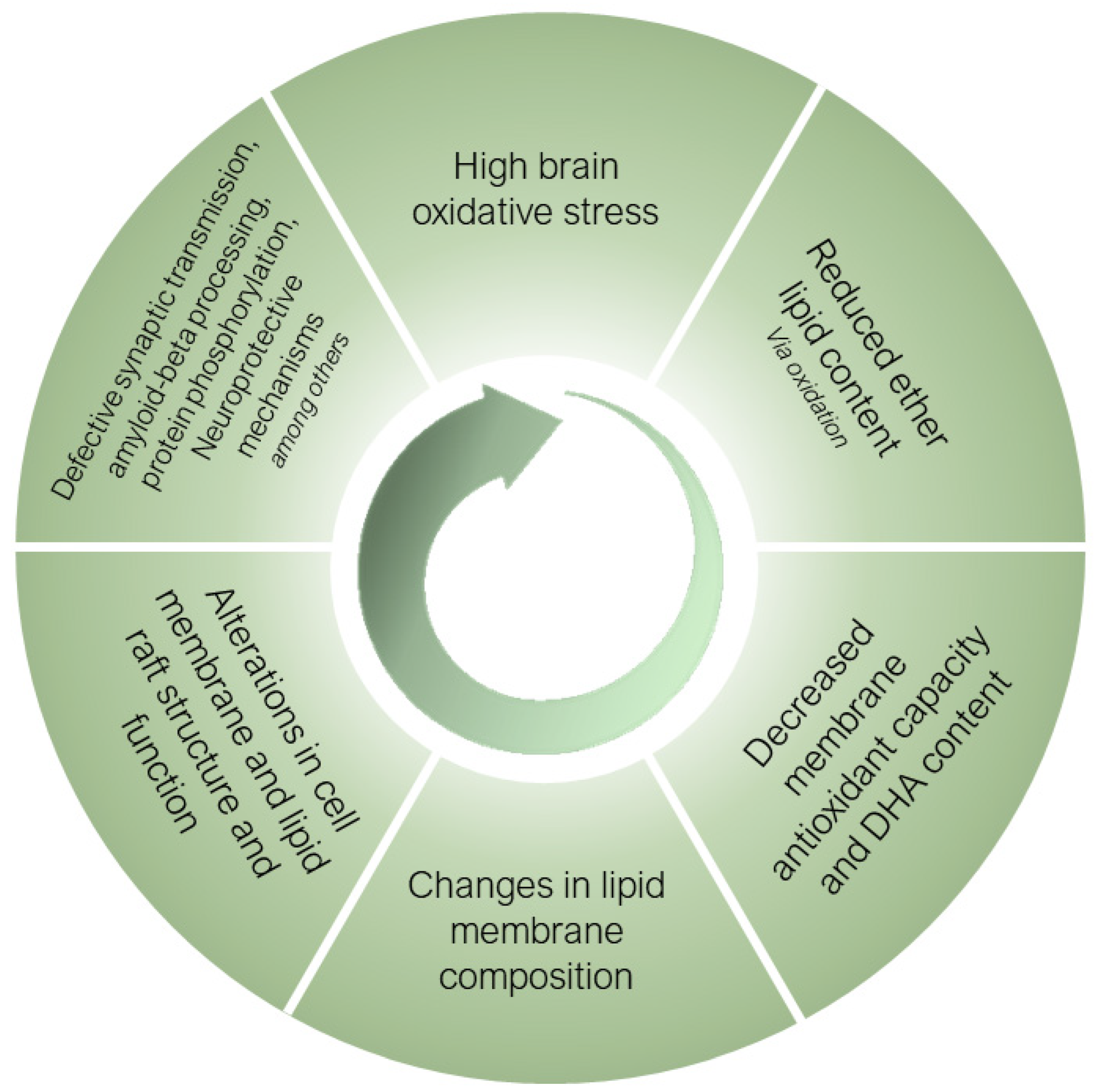
5. Plasmalogens as Endogenous Antioxidants in the Human Brain
6. Ether Lipids in Alzheimer’s Disease
6.1. Ether Lipids and Alzheimer’s Disease
6.2. Potential Interventions to Ameliorate the Ether Lipid Content in the Brain Tissue
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sastry, P.S. Lipids of Nervous Tissue: Composition and Metabolism. Prog. Lipid Res. 1985, 24, 69–176. [Google Scholar] [CrossRef] [PubMed]
- Thudichum, J.L. A Treatise on the Chemical Constitution of the Brain; Archon Books: Hamden, CT, USA, 1962. [Google Scholar]
- Piomelli, D.; Astarita, G.; Rapaka, R. A Neuroscientist’s Guide to Lipidomics. Nat. Rev. Neurosci. 2007, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Naudí, A.; Cabré, R.; Jové, M.; Ayala, V.; Gonzalo, H.; Portero-Otín, M.; Ferrer, I.; Pamplona, R. Lipidomics of Human Brain Aging and Alzheimer’s Disease Pathology. Int. Rev. Neurobiol. 2015, 122, 133–189. [Google Scholar] [CrossRef] [PubMed]
- Bozek, K.; Wei, Y.; Yan, Z.; Liu, X.; Xiong, J.; Sugimoto, M.; Tomita, M.; Pääbo, S.; Sherwood, C.C.; Hof, P.R.; et al. Organization and Evolution of Brain Lipidome Revealed by Large-Scale Analysis of Human, Chimpanzee, Macaque, and Mouse Tissues. Neuron 2015, 85, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Naudí, A.; Cabré, R.; Ayala, V.; Jové, M.; Mota-Martorell, N.; Portero-Otín, M.; Pamplona, R. Region-Specific Vulnerability to Lipid Peroxidation and Evidence of Neuronal Mechanisms for Polyunsaturated Fatty Acid Biosynthesis in the Healthy Adult Human Central Nervous System. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2017, 1862, 485–495. [Google Scholar] [CrossRef]
- Mota-Martorell, N.; Andrés-Benito, P.; Martín-Gari, M.; Galo-Licona, J.D.; Sol, J.; Fernández-Bernal, A.; Portero-Otín, M.; Ferrer, I.; Jove, M.; Pamplona, R. Selective Brain Regional Changes in Lipid Profile with Human Aging. GeroScience 2022, 44, 763–783. [Google Scholar] [CrossRef]
- Mattson, M.P.; Magnus, T. Ageing and Neuronal Vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef]
- Stack, C.; Jainuddin, S.; Elipenahli, C.; Gerges, M.; Starkova, N.; Starkov, A.A.; Jové, M.; Portero-Otin, M.; Launay, N.; Pujol, A.; et al. Methylene Blue Upregulates Nrf2/ARE Genes and Prevents Tau-Related Neurotoxicity. Hum. Mol. Genet. 2014, 23, 3716–3732. [Google Scholar] [CrossRef]
- Merrill, A.H.; Sullards, M.C.; Allegood, J.C.; Kelly, S.; Wang, E. Sphingolipidomics: High-Throughput, Structure-Specific, and Quantitative Analysis of Sphingolipids by Liquid Chromatography Tandem Mass Spectrometry. Methods 2005, 36, 207–224. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D. Cholesterol Metabolism in the Brain. Curr. Opin. Lipidol. 2001, 12, 105–112. [Google Scholar] [CrossRef]
- Rouser, G.; Yamamoto, A. Curvilinear Regression Course of Human Brain Lipid Composition Changes with Age. Lipids 1968, 3, 284–287. [Google Scholar] [CrossRef]
- Rouser, G.; Galli, C.; Kritchevsky, G. Lipid Class Composition of Normal Human Brain and Variations in Metachromatic Leucodystrophy, Tay-Sachs, Niemann-Pick, Chronic Gaucher’s and Alzheimer’s Diseases. J. Am. Oil Chem. Soc. 1965, 42, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Rouser, G.; Feldman, G.; Galli, C. Fatty Acid Compositions of Human Brain Lecithin and Sphingomyelin in Normal Individuals, Senile Cerebral Cortical Atrophy, Alzheimer’s Disease, Metachromatic Leucodystrophy, Tay-Sachs and Niemann-Pick Diseases. J. Am. Oil Chem. Soc. 1965, 42, 411–412. [Google Scholar] [CrossRef]
- O’brien, J.S.; Sampson, E.L.; Brien, O.; Fillerup, D.L.; Mead, J.F.; Lz, J. Lipid Composition of the Normal Human Brain: Gray Matter, White Matter, and Myelin. J. Lipid Res. 1965, 5, 329. [Google Scholar] [CrossRef]
- Panganamala, R.V.; Horrocks, L.A.; Geer, J.C.; Cornwell, D.G. Positions of Double Bonds in the Monounsaturated Alk-1-Enyl Groups from the Plasmalogens of Human Heart and Brain. Chem. Phys. Lipids 1971, 6, 97–102. [Google Scholar] [CrossRef]
- Kahma, K.; Brotherus, J.; Haltia, M.; Renkonen, O. Low and Moderate Concentrations of Lysobisphosphatidic Acid in Brain and Liver of Patients Affected by Some Storage Diseases. Lipids 1976, 11, 539–544. [Google Scholar] [CrossRef]
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paolo, G. Comparative Lipidomic Analysis of Mouse and Human Brain with Alzheimer Disease. J. Biol. Chem. 2012, 287, 2678–2688. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.M.; Lodhi, I.J. Structural and Functional Roles of Ether Lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.; Watschinger, K.; Werner, E.R.; Keller, M.A. Tricky Isomers—The Evolution of Analytical Strategies to Characterize Plasmalogens and Plasmanyl Ether Lipids. Front. Cell Dev. Biol. 2022, 10, 768. [Google Scholar] [CrossRef] [PubMed]
- Snyder, F. The Ether Lipid Trail: A Historical Perspective. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 1999, 1436, 265–278. [Google Scholar] [CrossRef]
- Honsho, M.; Fujiki, Y. Plasmalogen Homeostasis—Regulation of Plasmalogen Biosynthesis and Its Physiological Consequence in Mammals. FEBS Lett. 2017, 591, 2720–2729. [Google Scholar] [CrossRef] [PubMed]
- Dorninger, F.; Forss-Petter, S.; Wimmer, I.; Berger, J. Plasmalogens, Platelet-Activating Factor and beyond–Ether Lipids in Signaling and Neurodegeneration. Neurobiol. Dis. 2020, 145, 105061. [Google Scholar] [CrossRef] [PubMed]
- Lohner, K. Is the High Propensity of Ethanolamine Plasmalogens to Form Non-Lamellar Lipid Structures Manifested in the Properties of Biomembranes? Chem. Phys. Lipids 1996, 81, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Gross, R.W. Plasmenylcholine and Phosphatidylcholine Membrane Bilayers Possess Distinct Conformational Motifs. Biochemistry 1990, 29, 4992–4996. [Google Scholar] [CrossRef]
- Paltauf, F. Ether Lipids in Biomembranes. Chem. Phys. Lipids 1994, 74, 101–139. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Horrocks, L.A. Book Review: Plasmalogens: Workhorse Lipids of Membranes in Normal and Injured Neurons and Glia. Neuroscientist 2016, 7, 232–245. [Google Scholar] [CrossRef]
- Ferreira Da Silva, T.; Eira, J.; Lopes, A.T.; Malheiro, A.R.; Sousa, V.; Luoma, A.; Avila, R.L.; Wanders, R.J.A.; Just, W.W.; Kirschner, D.A.; et al. Peripheral Nervous System Plasmalogens Regulate Schwann Cell Differentiation and Myelination. J. Clin. Investig. 2014, 124, 2560–2570. [Google Scholar] [CrossRef]
- Pike, L.J.; Han, X.; Chung, K.N.; Gross, R.W. Lipid Rafts Are Enriched in Arachidonic Acid and Plasmenylethanolamine and Their Composition Is Independent of Caveolin-1 Expression: A Quantitative Electrospray Ionization/Mass Spectrometric Analysis†. Biochemistry 2002, 41, 2075–2088. [Google Scholar] [CrossRef]
- Rodemer, C.; Thai, T.P.; Brugger, B.; Kaercher, T.; Werner, H.; Nave, K.A.; Wieland, F.; Gorgas, K.; Just, W.W. Inactivation of Ether Lipid Biosynthesis Causes Male Infertility, Defects in Eye Development and Optic Nerve Hypoplasia in Mice. Hum. Mol. Genet. 2003, 12, 1881–1895. [Google Scholar] [CrossRef]
- Marrink, S.J.; Mark, A.E. Molecular View of Hexagonal Phase Formation in Phospholipid Membranes. Biophys. J. 2004, 87, 3894. [Google Scholar] [CrossRef]
- Glaser, P.E.; Gross, R.W. Plasmenylethanolamine Facilitates Rapid Membrane Fusion: A Stopped-Flow Kinetic Investigation Correlating the Propensity of a Major Plasma Membrane Constituent To Adopt an HII Phase with Its Ability To Promote Membrane Fusion. Biochemistry 1994, 33, 5805–5812. [Google Scholar] [CrossRef] [PubMed]
- Brodde, A.; Teigler, A.; Brugger, B.; Lehmann, W.D.; Wieland, F.; Berger, J.; Just, W.W. Impaired Neurotransmission in Ether Lipid-Deficient Nerve Terminals. Hum. Mol. Genet. 2012, 21, 2713. [Google Scholar] [CrossRef] [PubMed]
- Nagan, N.; Zoeller, R.A. Plasmalogens: Biosynthesis and Functions. Prog. Lipid Res. 2001, 40, 199–229. [Google Scholar] [CrossRef] [PubMed]
- Dorninger, F.; Forss-Petter, S.; Berger, J. From Peroxisomal Disorders to Common Neurodegenerative Diseases–the Role of Ether Phospholipids in the Nervous System. FEBS Lett. 2017, 591, 2761–2788. [Google Scholar] [CrossRef] [PubMed]
- Díaz, M.; Mesa-Herrera, F.; Marín, R. DHA and Its Elaborated Modulation of Antioxidant Defenses of the Brain: Implications in Aging and AD Neurodegeneration. Antioxidants 2021, 10, 907. [Google Scholar] [CrossRef]
- Katsuki, H.; Okuda, S. Arachidonic Acid as a Neurotoxic and Neurotrophic Substance. Prog. Neurobiol. 1995, 46, 607–636. [Google Scholar] [CrossRef] [PubMed]
- Goldfine, H. The Appearance, Disappearance and Reappearance of Plasmalogens in Evolution. Prog. Lipid Res. 2010, 49, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Yavin, E.; Gatt, S. Oxygen-Dependent Cleavage of the Vinyl-Ether Linkage of Plasmalogens. Eur. J. Biochem. 1972, 25, 437–446. [Google Scholar] [CrossRef]
- Morand, O.H.; Zoeller, R.A.; Raetz, C.R.H. Disappearance of Plasmalogens from Membranes of Animal Cells Subjected to Photosensitized Oxidation. J. Biol. Chem. 1988, 263, 11597–11606. [Google Scholar] [CrossRef]
- Khaselev, N.; Murphy, R.C. Susceptibility of Plasmenyl Glycerophosphoethanolamine Lipids Containing Arachidonate to Oxidative Degradation. Free Radic. Biol. Med. 1999, 26, 275–284. [Google Scholar] [CrossRef]
- Maeba, R.; Sawada, Y.; Shimasaki, H.; Takahashi, I.; Ueta, N. Ethanolamine Plasmalogens Protect Cholesterol-Rich Liposomal Membranes from Oxidation Caused by Free Radicals. Chem. Phys. Lipids 2002, 120, 145–151. [Google Scholar] [CrossRef]
- Skaff, O.; Pattison, D.I.; Davies, M.J. The Vinyl Ether Linkages of Plasmalogens Are Favored Targets for Myeloperoxidase-Derived Oxidants: A Kinetic Study. Biochemistry 2008, 47, 8237–8245. [Google Scholar] [CrossRef]
- Broniec, A.; Klosinski, R.; Pawlak, A.; Wrona-Krol, M.; Thompson, D.; Sarna, T. Interactions of Plasmalogens and Their Diacyl Analogs with Singlet Oxygen in Selected Model Systems. Free Radic. Biol. Med. 2011, 50, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive Oxygen Species and the Central Nervous System. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Miles Bailey, D. 13 Reasons Why the Brain Is Susceptible to Oxidative Stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Zoeller, R.A.; Morand, O.H.; Raetz, C.R.H. A Possible Role for Plasmalogens in Protecting Animal Cells against Photosensitized Killing. J. Biol. Chem. 1988, 263, 11590–11596. [Google Scholar] [CrossRef]
- Reiss, D.; Beyer, K.; Engelmann, B. Delayed Oxidative Degradation of Polyunsaturated Diacyl Phospholipids in the Presence of Plasmalogen Phospholipids in Vitro. Biochem. J. 1997, 323, 807–814. [Google Scholar] [CrossRef]
- Luoma, A.M.; Kuo, F.; Cakici, O.; Crowther, M.N.; Denninger, A.R.; Avila, R.L.; Brites, P.; Kirschner, D.A. Plasmalogen Phospholipids Protect Internodal Myelin from Oxidative Damage. Free Radic. Biol. Med. 2015, 84, 296–310. [Google Scholar] [CrossRef]
- Ferrer, I. Hypothesis Review: Alzheimer’s Overture Guidelines. Brain Pathol. 2022, e13122. [Google Scholar] [CrossRef]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The Global Prevalence of Dementia: A Systematic Review and Metaanalysis. Alzheimer’s Dement. 2013, 9, 63–75.e2. [Google Scholar] [CrossRef]
- Nichols, E.; Steinmetz, J.D.; Vollset, S.E.; Fukutaki, K.; Chalek, J.; Abd-Allah, F.; Abdoli, A.; Abualhasan, A.; Abu-Gharbieh, E.; Akram, T.T.; et al. Estimation of the Global Prevalence of Dementia in 2019 and Forecasted Prevalence in 2050: An Analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef] [PubMed]
- Weisser, M.; Vieth, M.; Stolte, M.; Riederer, P.; Pfeuffer, R.; Leblhuber, F.; Spiteller, G. Dramatic Increase of α-Hydroxyaldehydes Derived from Plasmalogens in the Aged Human Brain. Chem. Phys. Lipids 1997, 90, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, L.; Rafique, S.; Xuereb, J.H.; Rapoport, S.I.; Gershfeld, N.L. Disease and Anatomic Specificity of Ethanolamine Plasmalogen Deficiency in Alzheimer’s Disease Brain. Brain Res. 1995, 698, 223–226. [Google Scholar] [CrossRef]
- Guan, Z.; Wang, Y.; Cairns, N.J.; Lantos, P.L.; Dallner, G.; Sindelar, P.J. Decrease and Structural Modifications of Phosphatidylethanolamine Plasmalogen in the Brain with Alzheimer Disease. J. Neuropathol. Exp. Neurol. 1999, 58, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Holtzman, D.M.; McKeel, D.W. Plasmalogen Deficiency in Early Alzheimer’s Disease Subjects and in Animal Models: Molecular Characterization Using Electrospray Ionization Mass Spectrometry. J. Neurochem. 2001, 77, 1168–1180. [Google Scholar] [CrossRef]
- Pettegrew, J.W.; Panchalingam, K.; Hamilton, R.L.; Mcclure, R.J. Brain Membrane Phospholipid Alterations in Alzheimer’s Disease. Neurochem. Res. 2001, 26, 771–782. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Grösgen, S.; Riemenschneider, M.; Tanila, H.; Grimm, H.S.; Hartmann, T. From Brain to Food: Analysis of Phosphatidylcholins, Lyso-Phosphatidylcholins and Phosphatidylcholin–Plasmalogens Derivates in Alzheimer’s Disease Human Post Mortem Brains and Mice Model via Mass Spectrometry. J. Chromatogr. A 2011, 1218, 7713–7722. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Kuchenbecker, J.; Rothhaar, T.L.; Grösgen, S.; Hundsdörfer, B.; Burg, V.K.; Friess, P.; Müller, U.; Grimm, H.S.; Riemenschneider, M.; et al. Plasmalogen Synthesis Is Regulated via Alkyl-Dihydroxyacetonephosphate-Synthase by Amyloid Precursor Protein Processing and Is Affected in Alzheimer’s Disease. J. Neurochem. 2011, 116, 916–925. [Google Scholar] [CrossRef]
- Igarashi, M.; Ma, K.; Gao, F.; Kim, H.W.; Rapoport, S.I.; Rao, J.S. Disturbed Choline Plasmalogen and Phospholipid Fatty Acid Concentrations in Alzheimer Disease Prefrontal Cortex. J. Alzheimers. Dis. 2011, 24, 507. [Google Scholar] [CrossRef]
- Kou, J.; Kovacs, G.G.; Höftberger, R.; Kulik, W.; Brodde, A.; Forss-Petter, S.; Hönigschnabl, S.; Gleiss, A.; Brügger, B.; Wanders, R.; et al. Peroxisomal Alterations in Alzheimer’s Disease. Acta Neuropathol. 2011, 122, 271. [Google Scholar] [CrossRef]
- Wood, P.L.; Barnette, B.L.; Kaye, J.A.; Quinn, J.F.; Woltjer, R.L. Non-Targeted Lipidomics of CSF and Frontal Cortex Grey and White Matter in Control, Mild Cognitive Impairment, and Alzheimer’s Disease Subjects. Acta Neuropsychiatr. 2015, 27, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Otoki, Y.; Kato, S.; Nakagawa, K.; Harvey, D.J.; Jin, L.W.; Dugger, B.N.; Taha, A.Y. Lipidomic Analysis of Postmortem Prefrontal Cortex Phospholipids Reveals Changes in Choline Plasmalogen Containing Docosahexaenoic Acid and Stearic Acid Between Cases with and Without Alzheimer’s Disease. NeuroMolecular Med. 2021, 23, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Rustam, Y.H.; Masters, C.L.; Makalic, E.; McLean, C.A.; Hill, A.F.; Barnham, K.J.; Reid, G.E.; Vella, L.J. Characterization of Brain-Derived Extracellular Vesicle Lipids in Alzheimer’s Disease. J. Extracell. Vesicles 2021, 10, e12089. [Google Scholar] [CrossRef]
- Obis, E.; Sol, J.; Andres-Benito, P.; Martín-Gari, M.; Mota-Martorell, N.; Daniel Galo-Licona, J.; Piñol-Ripoll, G.; Portero-Otin, M.; Ferrer, I.; Jové, M.; et al. Lipidomic Alterations in the Cerebral Cortex and White Matter in Sporadic Alzheimer’s Disease. bioRxiv 2022. [Google Scholar] [CrossRef]
- Wood, P.L.; Woltjer, R.L. Serine Ether Glycerophospholipids: Decrements in the Frontal Cortex Associated with Mild Cognitive Impairment and Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 870. [Google Scholar] [CrossRef]
- Goodenowe, D.B.; Cook, L.L.; Liu, J.; Lu, Y.; Jayasinghe, D.A.; Ahiahonu, P.W.K.; Heath, D.; Yamazaki, Y.; Flax, J.; Krenitsky, K.F.; et al. Peripheral Ethanolamine Plasmalogen Deficiency: A Logical Causative Factor in Alzheimer’s Disease and Dementia. J. Lipid Res. 2007, 48, 2485–2498. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Mankidy, R.; Ritchie, S.; Heath, D.; Wood, J.A.; Flax, J.; Goodenowe, D.B. Circulating Plasmalogen Levels and Alzheimer Disease Assessment Scale–Cognitive Scores in Alzheimer Patients. J. Psychiatry Neurosci. 2010, 35, 59. [Google Scholar] [CrossRef]
- Orešič, M.; Hyötyläinen, T.; Herukka, S.-K.; Sysi-Aho, M.; Mattila, I.; Seppänan-Laakso, T.; Julkunen, V.; Gopalacharyulu, P.V.; Hallikainen, M.; Koikkalainen, J.; et al. Metabolome in Progression to Alzheimer’s Disease. Transl. Psychiatry 2011, 1, e57. [Google Scholar] [CrossRef]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; MacArthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma Phospholipids Identify Antecedent Memory Impairment in Older Adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Zhong, X.; Cheema, A.K.; Orquiza, M.H.; Chidambaram, S.; Tan, M.T.; Gresenz, C.R.; FitzGerald, K.T.; Nalls, M.A.; Singleton, A.B.; et al. Plasma 24-Metabolite Panel Predicts Preclinical Transition to Clinical Stages of Alzheimer’s Disease. Front. Neurol. 2015, 6, 12. [Google Scholar] [CrossRef]
- Maeba, R.; Maeda, T.; Kinoshita, M.; Takao, K.; Takenaka, H.; Kusano, J.; Yoshimura, N.; Takeoka, Y.; Yasuda, D.; Okazaki, T.; et al. Plasmalogens in Human Serum Positively Correlate with High- Density Lipoprotein and Decrease with Aging. J. Atheroscler. Thromb. 2007, 14, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Kiko, T.; Fujiwara, H.; Hashimoto, M.; Nakagawa, K.; Kinoshita, M.; Furukawa, K.; Arai, H.; Miyazawa, T. Alterations in the Levels of Amyloid-β, Phospholipid Hydroperoxide, and Plasmalogen in the Blood of Patients with Alzheimer’s Disease: Possible Interactions between Amyloid-β and These Lipids. J. Alzheimer’s Dis. 2016, 50, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Toledo, J.B.; Arnold, M.; Kastenmüller, G.; Chang, R.; Baillie, R.A.; Han, X.; Thambisetty, M.; Tenenbaum, J.D.; Suhre, K.; Thompson, J.W.; et al. Metabolic Network Failures in Alzheimer’s Disease: A Biochemical Road Map. Alzheimer’s Dement. 2017, 13, 965–984. [Google Scholar] [CrossRef] [PubMed]
- Dorninger, F.; Moser, A.B.; Kou, J.; Wiesinger, C.; Forss-Petter, S.; Gleiss, A.; Hinterberger, M.; Jungwirth, S.; Fischer, P.; Berger, J. Alterations in the Plasma Levels of Specific Choline Phospholipids in Alzheimer’s Disease Mimic Accelerated Aging. J. Alzheimer’s Dis. 2018, 62, 841. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Cheong, L.-Z.; Man, Q.-Q.; Pang, S.-J.; Li, Y.-Q.; Ren, B.; Zhang, J. Characterization of Potential Plasma Biomarkers Related to Cognitive Impairment by Untargeted Profiling of Phospholipids Using the HILIC-ESI-IT-TOF-MS System. Anal. Bioanal. Chem. 2018, 410, 2937–2948. [Google Scholar] [CrossRef]
- Huynh, K.; Lim, W.L.F.; Giles, C.; Jayawardana, K.S.; Salim, A.; Mellett, N.A.; Smith, A.A.T.; Olshansky, G.; Drew, B.G.; Chatterjee, P.; et al. Concordant Peripheral Lipidome Signatures in Two Large Clinical Studies of Alzheimer’s Disease. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Sakr, F.; Dyrba, M.; Bräuer, A.; Teipel, S. Association of Lipidomics Signatures in Blood with Clinical Progression in Preclinical and Prodromal Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 85, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Han, X. Lipid Alterations in the Earliest Clinically Recognizable Stage of Alzheimers Disease: Implication of the Role of Lipids in the Pathogenesis of Alzheimers Disease. Curr. Alzheimer Res. 2005, 2, 65–77. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria Dysfunction in the Pathogenesis of Alzheimer’s Disease: Recent Advances. Mol. Neurodegener. 2020, 15, 1–22. [Google Scholar] [CrossRef]
- Lizard, G.; Rouaud, O.; Demarquoy, J.; Cherkaoui-Malki, M.; Iuliano, L. Potential Roles of Peroxisomes in Alzheimer’s Disease and in Dementia of the Alzheimer’s Type. J. Alzheimer’s Dis. 2012, 29, 241–254. [Google Scholar] [CrossRef]
- Farooqui, A.A. Studies on Plasmalogen-Selective Phospholipase A2 in Brain. Mol. Neurobiol. 2010, 41, 267–273. [Google Scholar] [CrossRef]
- Bennett, S.A.L.; Valenzuela, N.; Xu, H.; Franko, B.; Fai, S.; Figeys, D. Using Neurolipidomics to Identify Phospholipid Mediators of Synaptic (Dys)Function in Alzheimer’s Disease. Front. Physiol. 2013, 4, 168. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Abe, Y.; Ali, F.; Youssef, M.; Honsho, M.; Fujiki, Y.; Katafuchi, T. Reduction of Ether-Type Glycerophospholipids, Plasmalogens, by NF-ΚB Signal Leading to Microglial Activation. J. Neurosci. 2017, 37, 4074. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Andrés-Benito, P.; Ausín, K.; Pamplona, R.; del Rio, J.A.; Fernández-Irigoyen, J.; Santamaría, E. Dysregulated Protein Phosphorylation: A Determining Condition in the Continuum of Brain Aging and Alzheimer’s Disease. Brain Pathol. 2021, 31, e12996. [Google Scholar] [CrossRef]
- Hernández, F.; Ferrer, I.; Pérez, M.; Zabala, J.C.; del Rio, J.A.; Avila, J. Tau Aggregation. Neuroscience 2022. [Google Scholar] [CrossRef]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic Processing of the Alzheimer β-Amyloid Precursor Protein Depends on Lipid Rafts. J. Cell Biol. 2003, 160, 113. [Google Scholar] [CrossRef]
- Bazinet, R.P.; Layé, S. Polyunsaturated Fatty Acids and Their Metabolites in Brain Function and Disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Dyall, S.C. Long-Chain Omega-3 Fatty Acids and the Brain: A Review of the Independent and Shared Effects of EPA, DPA and DHA. Front. Aging Neurosci. 2015, 7, 52. [Google Scholar] [CrossRef]
- Udagawa, J.; Hino, K. Plasmalogen in the Brain: Effects on Cognitive Functions and Behaviors Attributable to Its Properties. Brain Res. Bull. 2022, 188, 197–202. [Google Scholar] [CrossRef]
- Bieri, G.; Schroer, A.B.; Villeda, S.A. Blood-to-Brain Communication in Aging and Rejuvenation. Nat. Neurosci. 2023, 1–15. [Google Scholar] [CrossRef]
- Assogna, M.; Di Lorenzo, F.; Martorana, A.; Koch, G. Synaptic Effects of Palmitoylethanolamide in Neurodegenerative Disorders. Biomolecules 2022, 12, 1161. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Tissue | Analytical Approach | Cases | Effect of AD on Ether Lipid Content | Reference |
|---|---|---|---|---|
| Human Brain Tissue | ||||
| Temporal cortex, cerebellum, caudate nuclei, substantia nigra | TLC | Ctl (9) vs. AD (9). | Decreased plasmalogens in TC, not in cerebellum, caudate nuclei and substantia nigra. | [54] |
| Frontal cortex, hippocampus, white matter | HPLC and GC | Ctl (13) vs. AD (15). | Decreased PE-plasmalogens (PE(P-)). | [55] |
| Frontal cortex, parietal cortex, temporal cortex and cerebellum (grey matter and white matter) | ESI/MS | Subjects with a spectrum of AD clinical dementia rating (CDR from 0 to 3) (n = 6 per group). | Decreased PE(P-) with increase in CDR, except in cerebellum. | [56] |
| Superior/middle frontal, Inferior parietal occipital, superior temporal, cerebellum | NMR | Ctl (6–16) vs. AD (37–43) Most of the control subjects presented concomitant neuropathological disorders. | Increased PE(P-) in frontal and temporal cortex. | [57] |
| Frontal cortex | TLC | Ctl (26) vs. AD (39). | Decreased PE(P-). | [58] |
| Frontal cortex, temporal cortex, cerebellum | LC-MS | Ctl (14) vs. AD (30). | Decreased PC(P-). | [59] |
| Prefrontal cortex | TLC | Ctl (9) vs. AD (10). | Decreased PE(P-). | [60] |
| Gyrus frontalis | GC/MS | Human AD grouped by Braak staging of neuropathology. | Decreased PE(P-). | [61] |
| Prefrontal cortex, entorhinal cortex, cerebellum | HPLC-QQQ/Ion Trap-MS | Ctl (10) vs. AD (10) per region. AD was defined as “high probability of AD” based on NIA_RI (National Institutes of Health-Reagan Institute) criteria. | No significant differences in any brain region for plasmalogen content. | [18] |
| Grey matter, white matter | HR-ESI-MS | Young dementia, old dementia and MCI cohort. | Decreased PE(P-) in grey matter from young dementia and old dementia. | [62] |
| Prefrontal cortex | UPLC-MS/MS | Ctl (20) vs AD (21). | Decreased PC plasmalogens (PC(P-)) species containing 18:0 and 22:6. | [63] |
| Brain-derived extracellular vesicles (BDEV) from frontal cortex | nESI-UHRAMS and HCD-MS/MS | Ctl (8) vs AD (8). | Increased specific species of PE(P-). | [64] |
| Frontal cortex (white matter and grey matter) | UPLC-ESI-QTOF-MS/MS | Middle-aged cases (Ctl group, 6) vs. sAD cases (lacking co-morbidities and concomitant brain pathologies) categorized according to Braak and Braak neurofibrillary tangle (NFT) and β-amyloid stages as ADI–II/0-A (n = 7); ADIII– IV/0-C (n = 5), and ADV–VI/B-C (n = 6) | Decreased specific PC(O-), PC(P-) and PE(P-) species in grey matter and white matter with AD progression. | [65] |
| Frontal cortex | HR-ESI-MS | Ctl (20) vs. MCI (19), EOAD (17), and LOAD (17). | Decreased serine ether GPs (PS(O-), PS(P-)). | [66] |
| Human Plasma/Serum Tissue | ||||
| Serum | LC-MS/MS | >350 non-demented subjects vs. >400 demented subjects (dementia of the Alzheimer’s type). | Decreased PE(P-) 16:0/22:6 with severity pf dementia. | [67] |
| Serum | LC-MS | Ctl (66) vs. AD patients with an ADS-Cog score between 20 and 46 (40). | Decreased PE(P-) and there is a correlation between plasmalogen depletion and cognitive decline. | [68] |
| Serum | UPLC-MS | Ctl (46), MCI (143), AD (47). | Decreased ether GPs (PC(O-)). | [69] |
| Plasma | UPLC-ESI-QTOF-MS and LC-MS/MS | Five hundred and twenty-five community-dwelling participants aged 70 and older and otherwise healthy into this 5 years observational study. Three groups were defined: normal control, aMCI/AD, and converted. | Decreased PC(O-40:6) predict phenoconvertion to either MCI or AD within a 2–3-year timeframe. | [70] |
| Plasma | MRM-SID-MS | Ctl (73) vs. phenoconverters (28). | Decreased specific PC(P-) species allow to determine the risk of phenoconversion from normal cognition to aMCI or AD. | [71] |
| Serum | LC using radioactive iodine | NE (cognitively normal elderly) (107) vs. MCOs (memory clinic outpatients) (55). | Decreased PE(P-) | [72] |
| Plasma | LC-MS | Ctl vs AD. | Decreased PE(P-), bearing the DHA moiety. | [73] |
| Serum | UPLC-QQQ-MS/MS | Ctl (199), MCI (356), AD (175). | Increased specific ether-linked PCs in earliest AD. | [74] |
| Plasma | UFLC-MS/MS | From 606 participants, two groups are generated: aged controls and probable AD after 30, 60 and 90 months (ctl vs. aging and phenoconversion to AD). | Increased lysoPAF and PC(P-). | [75] |
| Plasma | HILIC-ESI-IT-TOF-MS | Ctl (41) vs. Cognitive Impairment (41). | Decreased PE(P-), especially with PUFAs. | [76] |
| Plasma | HPLC-QQQ-MS | Two cohorts: 1) Ctl (768), MCI (131), AD (211); 2) Ctl (200), MCI (400), mild AD (200). | Decreased PE(O-) and PE(P-) species. | [77] |
| Plasma | LC ESI-QQQ MS/MS | Preclinical and prodromal AD cases from the ADNI cohort (529 participants). | Decreased content of ether-linked GPs. | [78] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jové, M.; Mota-Martorell, N.; Obis, È.; Sol, J.; Martín-Garí, M.; Ferrer, I.; Portero-Otin, M.; Pamplona, R. Ether Lipid-Mediated Antioxidant Defense in Alzheimer’s Disease. Antioxidants 2023, 12, 293. https://doi.org/10.3390/antiox12020293
Jové M, Mota-Martorell N, Obis È, Sol J, Martín-Garí M, Ferrer I, Portero-Otin M, Pamplona R. Ether Lipid-Mediated Antioxidant Defense in Alzheimer’s Disease. Antioxidants. 2023; 12(2):293. https://doi.org/10.3390/antiox12020293
Chicago/Turabian StyleJové, Mariona, Natàlia Mota-Martorell, Èlia Obis, Joaquim Sol, Meritxell Martín-Garí, Isidre Ferrer, Manuel Portero-Otin, and Reinald Pamplona. 2023. "Ether Lipid-Mediated Antioxidant Defense in Alzheimer’s Disease" Antioxidants 12, no. 2: 293. https://doi.org/10.3390/antiox12020293
APA StyleJové, M., Mota-Martorell, N., Obis, È., Sol, J., Martín-Garí, M., Ferrer, I., Portero-Otin, M., & Pamplona, R. (2023). Ether Lipid-Mediated Antioxidant Defense in Alzheimer’s Disease. Antioxidants, 12(2), 293. https://doi.org/10.3390/antiox12020293