
Mitochondrial Complex I, a Possible Sensible Site of cAMP Pathway in Aging

{kind=link}
{kind=link}
Abstract
1. Introduction
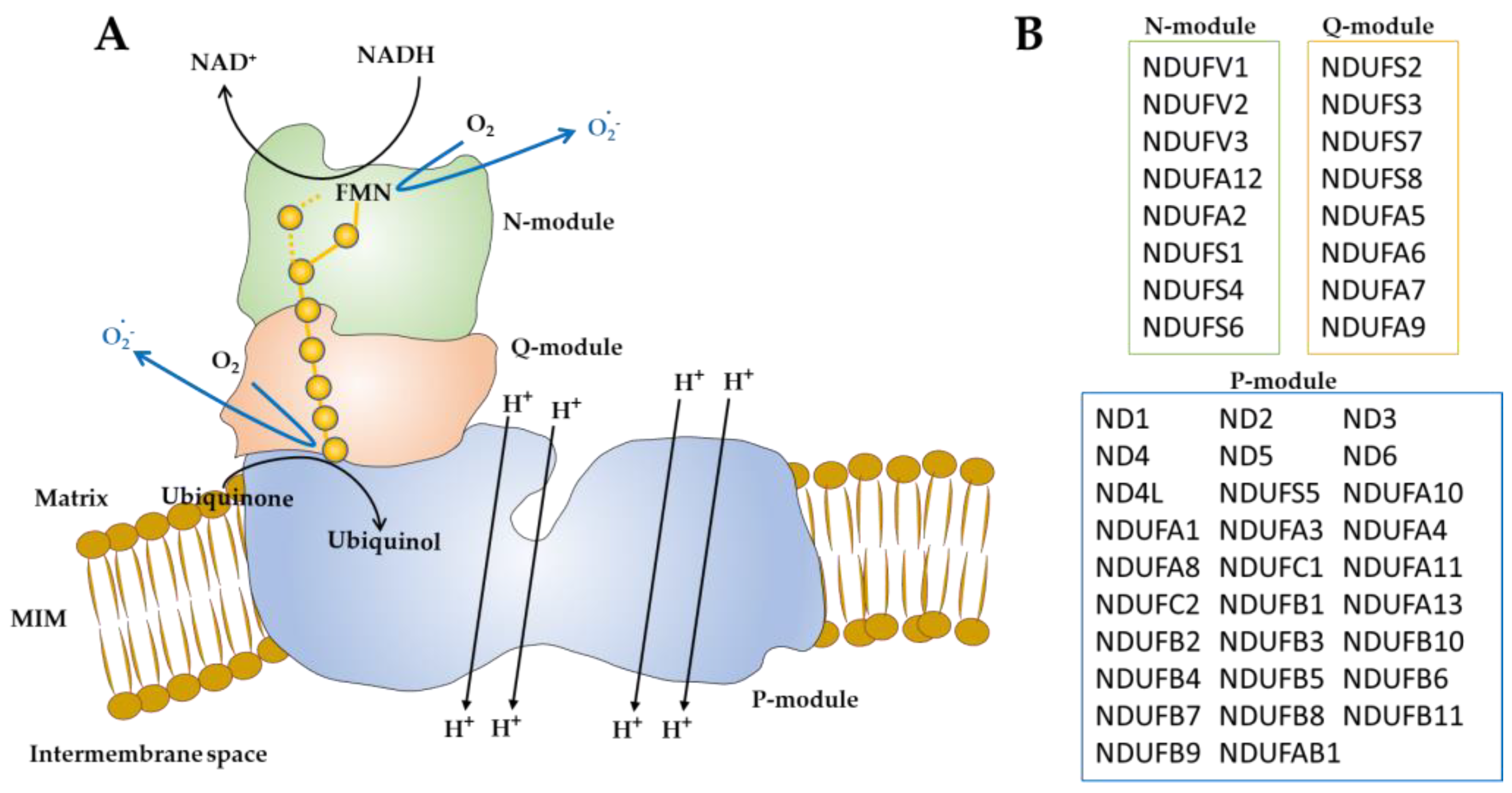
2. Mitochondrial Complex I
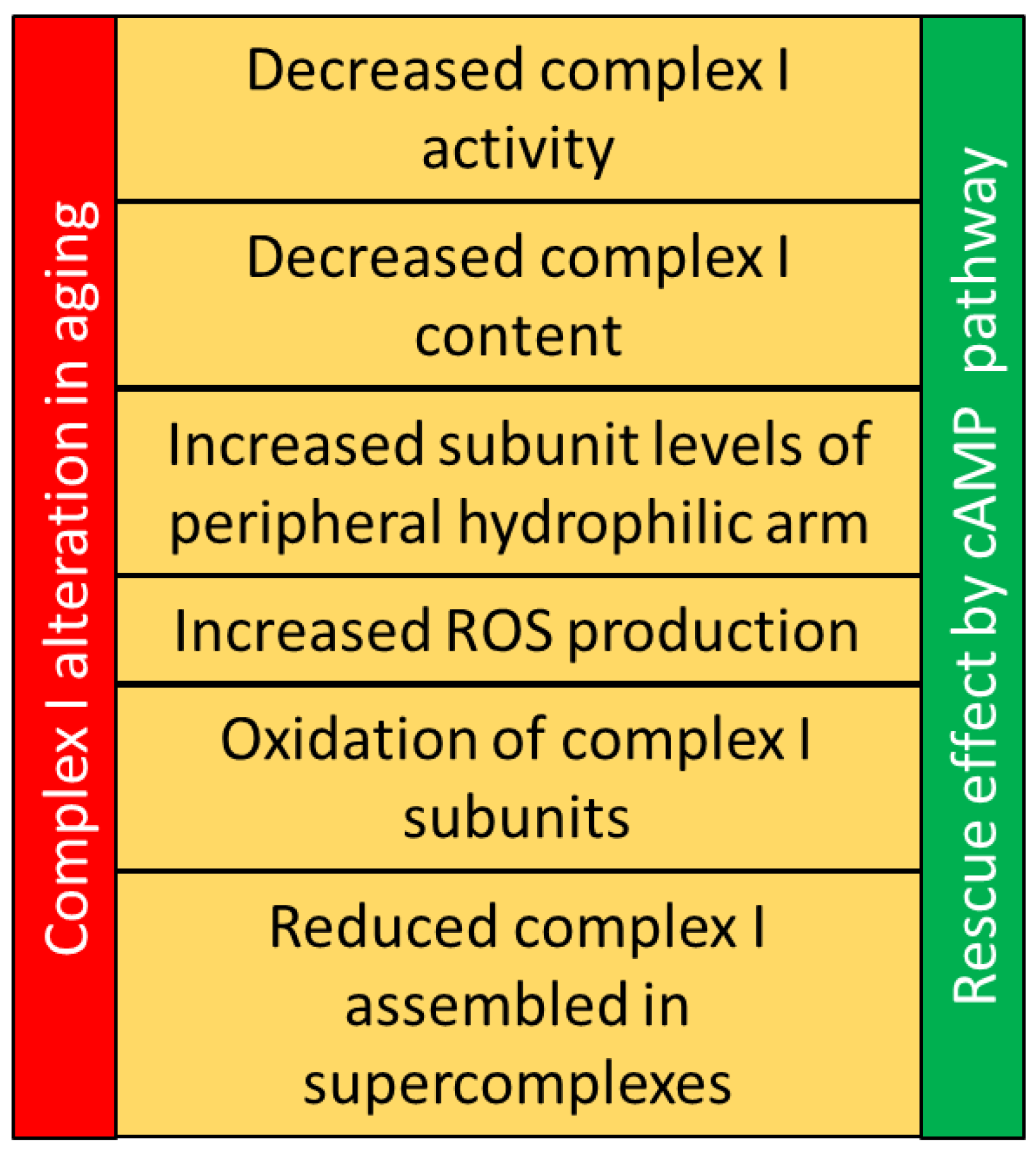
3. Mitochondrial Complex I in Aging
4. cAMP-Dependent Regulation of Mitochondria and ROS Production
5. cAMP Pathway in Aging
6. cAMP/PKA System and Mitochondria in Aging and Neurodegenerative Diseases
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Papa, S.; De Rasmo, D. Complex I Deficiencies in Neurological Disorders. Trends Mol. Med. 2013, 19, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Stefanatos, R.; Sanz, A. The Role of Mitochondrial ROS in the Aging Brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.-S.; Dighe, P.A.; Mezera, V.; Monternier, P.-A.; Brand, M.D. Production of Superoxide and Hydrogen Peroxide from Specific Mitochondrial Sites under Different Bioenergetic Conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Barja, G. Towards a Unified Mechanistic Theory of Aging. Exp. Gerontol. 2019, 124, 110627. [Google Scholar] [CrossRef]
- Ramzan, R.; Kadenbach, B.; Vogt, S. Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase. Cells 2021, 10, 514. [Google Scholar] [CrossRef]
- Módis, K.; Panopoulos, P.; Coletta, C.; Papapetropoulos, A.; Szabo, C. Hydrogen Sulfide-Mediated Stimulation of Mitochondrial Electron Transport Involves Inhibition of the Mitochondrial Phosphodiesterase 2A, Elevation of CAMP and Activation of Protein Kinase A. Biochem. Pharm. 2013, 86, 1311–1319. [Google Scholar] [CrossRef]
- Papa, S.; Scacco, S.; De Rasmo, D.; Signorile, A.; Papa, F.; Panelli, D.; Nicastro, A.; Scaringi, R.; Santeramo, A.; Roca, E.; et al. CAMP-Dependent Protein Kinase Regulates Post-Translational Processing and Expression of Complex I Subunits in Mammalian Cells. Biochim. Et Biophys. Acta 2010, 1797, 649–658. [Google Scholar] [CrossRef]
- Aslam, M.; Ladilov, Y. Emerging Role of CAMP/AMPK Signaling. Cells 2022, 11, 308. [Google Scholar] [CrossRef]
- De Rasmo, D.; Gattoni, G.; Papa, F.; Santeramo, A.; Pacelli, C.; Cocco, T.; Micelli, L.; Sardaro, N.; Larizza, M.; Scivetti, M.; et al. The β-Adrenoceptor Agonist Isoproterenol Promotes the Activity of Respiratory Chain Complex I and Lowers Cellular Reactive Oxygen Species in Fibroblasts and Heart Myoblasts. Eur. J. Pharm. 2011, 652, 15–22. [Google Scholar] [CrossRef]
- Papa, S.; Rasmo, D.D.; Technikova-Dobrova, Z.; Panelli, D.; Signorile, A.; Scacco, S.; Petruzzella, V.; Papa, F.; Palmisano, G.; Gnoni, A.; et al. Respiratory Chain Complex I, a Main Regulatory Target of the CAMP/PKA Pathway Is Defective in Different Human Diseases. FEBS Lett. 2012, 586, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Raha, S.; Myint, A.T.; Johnstone, L.; Robinson, B.H. Control of Oxygen Free Radical Formation from Mitochondrial Complex I: Roles for Protein Kinase A and Pyruvate Dehydrogenase Kinase. Free Radic. Biol. Med. 2002, 32, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, F.; Piccoli, C.; Cocco, T.; Scacco, S.; Papa, F.; Gaballo, A.; Boffoli, D.; Signorile, A.; D’Aprile, A.; Scrima, R.; et al. Regulation by the CAMP Cascade of Oxygen Free Radical Balance in Mammalian Cells. Antioxid Redox Signal. 2006, 8, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic Control through the PGC-1 Family of Transcription Coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef]
- Elkhoely, A. Liraglutide Ameliorates Gentamicin-Induced Acute Kidney Injury in Rats via PGC-1α- Mediated Mitochondrial Biogenesis: Involvement of PKA/CREB and Notch/Hes-1 Signaling Pathways. Int. Immunopharmacol. 2022, 114, 109578. [Google Scholar] [CrossRef]
- Terracina, S.; Petrella, C.; Francati, S.; Lucarelli, M.; Barbato, C.; Minni, A.; Ralli, M.; Greco, A.; Tarani, L.; Fiore, M.; et al. Antioxidant Intervention to Improve Cognition in the Aging Brain: The Example of Hydroxytyrosol and Resveratrol. Int. J. Mol. Sci. 2022, 23, 15674. [Google Scholar] [CrossRef]
- Park, S.-J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol Ameliorates Aging-Related Metabolic Phenotypes by Inhibiting CAMP Phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef]
- Signorile, A.; Micelli, L.; De Rasmo, D.; Santeramo, A.; Papa, F.; Ficarella, R.; Gattoni, G.; Scacco, S.; Papa, S. Regulation of the Biogenesis of OXPHOS Complexes in Cell Transition from Replicating to Quiescent State: Involvement of PKA and Effect of Hydroxytyrosol. Biochim. Biophys. Acta 2014, 1843, 675–684. [Google Scholar] [CrossRef]
- Li, H.; Xu, X.; Cai, M.; Qu, Y.; Ren, Z.; Ye, C.; Shen, H. The Combination of HT-Ac and HBET Improves the Cognitive and Learning Abilities of Heat-Stressed Mice by Maintaining Mitochondrial Function through the PKA-CREB-BDNF Pathway. Food Funct. 2022, 13, 6166–6179. [Google Scholar] [CrossRef]
- Dagda, R.K.; Gusdon, A.M.; Pien, I.; Strack, S.; Green, S.; Li, C.; Van Houten, B.; Cherra, S.J.; Chu, C.T. Mitochondrially Localized PKA Reverses Mitochondrial Pathology and Dysfunction in a Cellular Model of Parkinson’s Disease. Cell Death Differ. 2011, 18, 1914–1923. [Google Scholar] [CrossRef]
- Signorile, A.; Ferretta, A.; Pacelli, C.; Capitanio, N.; Tanzarella, P.; Matrella, M.L.; Valletti, A.; De Rasmo, D.; Cocco, T. Resveratrol Treatment in Human Parkin-Mutant Fibroblasts Modulates CAMP and Calcium Homeostasis Regulating the Expression of Mitochondria-Associated Membranes Resident Proteins. Biomolecules 2021, 11, 1511. [Google Scholar] [CrossRef] [PubMed]
- Sanders, O.; Rajagopal, L. Phosphodiesterase Inhibitors for Alzheimer’s Disease: A Systematic Review of Clinical Trials and Epidemiology with a Mechanistic Rationale. J. Alzheimers Dis. Rep. 2020, 4, 185–215. [Google Scholar] [CrossRef] [PubMed]
- Wiciński, M.; Socha, M.; Malinowski, B.; Wódkiewicz, E.; Walczak, M.; Górski, K.; Słupski, M.; Pawlak-Osińska, K. Liraglutide and Its Neuroprotective Properties-Focus on Possible Biochemical Mechanisms in Alzheimer’s Disease and Cerebral Ischemic Events. Int. J. Mol. Sci. 2019, 20, 1050. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; De Rasmo, D.; Signorile, A.; Rossi, L.; de Bari, L.; Scala, I.; Granese, B.; Papa, S.; Vacca, R.A. Epigallocatechin-3-Gallate Prevents Oxidative Phosphorylation Deficit and Promotes Mitochondrial Biogenesis in Human Cells from Subjects with Down’s Syndrome. Biochim. Biophys. Acta 2013, 1832, 542–552. [Google Scholar] [CrossRef]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’aquila, C.; De Mari, M.; et al. Effect of Resveratrol on Mitochondrial Function: Implications in Parkin-Associated Familiar Parkinson’s Disease. Biochim. Biophys. Acta 2014, 1842, 902–915. [Google Scholar] [CrossRef]
- Xie, Y.; Zheng, J.; Li, S.; Li, H.; Zhou, Y.; Zheng, W.; Zhang, M.; Liu, L.; Chen, Z. GLP-1 Improves the Neuronal Supportive Ability of Astrocytes in Alzheimer’s Disease by Regulating Mitochondrial Dysfunction via the CAMP/PKA Pathway. Biochem. Pharm. 2021, 188, 114578. [Google Scholar] [CrossRef]
- Zippin, J.H.; Chen, Y.; Nahirney, P.; Kamenetsky, M.; Wuttke, M.S.; Fischman, D.A.; Levin, L.R.; Buck, J. Compartmentalization of Bicarbonate-Sensitive Adenylyl Cyclase in Distinct Signaling Microdomains. FASEB J. 2003, 17, 82–84. [Google Scholar] [CrossRef]
- Signorile, A.; Sardanelli, A.M.; Nuzzi, R.; Papa, S. Serine (Threonine) Phosphatase(s) Acting on CAMP-Dependent Phosphoproteins in Mammalian Mitochondria. FEBS Lett. 2002, 512, 91–94. [Google Scholar] [CrossRef]
- Sardanelli, A.M.; Signorile, A.; Nuzzi, R.; Rasmo, D.D.; Technikova-Dobrova, Z.; Drahota, Z.; Occhiello, A.; Pica, A.; Papa, S. Occurrence of A-Kinase Anchor Protein and Associated CAMP-Dependent Protein Kinase in the Inner Compartment of Mammalian Mitochondria. FEBS Lett. 2006, 580, 5690–5696. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Russwurm, M.; Günnewig, K.; Gertz, M.; Zoidl, G.; Ramos, L.; Buck, J.; Levin, L.R.; Rassow, J.; Manfredi, G.; et al. A Phosphodiesterase 2A Isoform Localized to Mitochondria Regulates Respiration. J. Biol. Chem. 2011, 286, 30423–30432. [Google Scholar] [CrossRef]
- De Rasmo, D.; Signorile, A.; Santeramo, A.; Larizza, M.; Lattanzio, P.; Capitanio, G.; Papa, S. Intramitochondrial Adenylyl Cyclase Controls the Turnover of Nuclear-Encoded Subunits and Activity of Mammalian Complex I of the Respiratory Chain. Biochim. Biophys. Acta 2015, 1853, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP Produced inside Mitochondria Regulates Oxidative Phosphorylation. Cell Metab. 2009, 9, 265–276. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Micelli, L.; Santeramo, A.; Signorile, A.; Lattanzio, P.; Papa, S. CAMP Regulates the Functional Activity, Coupling Efficiency and Structural Organization of Mammalian FOF1 ATP Synthase. Biochim. Biophys. Acta 2016, 1857, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Scalzotto, E.; Mongillo, M.; Pozzan, T. Mitochondrial Ca2+ Uptake Induces Cyclic AMP Generation in the Matrix and Modulates Organelle ATP Levels. Cell Metab 2013, 17, 965–975. [Google Scholar] [CrossRef]
- Kumar, S.; Kostin, S.; Flacke, J.-P.; Reusch, H.P.; Ladilov, Y. Soluble Adenylyl Cyclase Controls Mitochondria-Dependent Apoptosis in Coronary Endothelial Cells. J. Biol. Chem. 2009, 284, 14760–14768. [Google Scholar] [CrossRef]
- Carroll, J.; Fearnley, I.M.; Skehel, J.M.; Shannon, R.J.; Hirst, J.; Walker, J.E. Bovine Complex I Is a Complex of 45 Different Subunits. J. Biol. Chem. 2006, 281, 32724–32727. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Thorburn, D.R.; Ryan, M.T.; McKenzie, M. Assembly of Mitochondrial Complex I and Defects in Disease. Biochim. Biophys. Acta 2009, 1793, 78–88. [Google Scholar] [CrossRef]
- Cortés-Rojo, C.; Vargas-Vargas, M.A.; Olmos-Orizaba, B.E.; Rodríguez-Orozco, A.R.; Calderón-Cortés, E. Interplay between NADH Oxidation by Complex I, Glutathione Redox State and Sirtuin-3, and Its Role in the Development of Insulin Resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165801. [Google Scholar] [CrossRef]
- Kong, D.; Zhao, L.; Du, Y.; He, P.; Zou, Y.; Yang, L.; Sun, L.; Wang, H.; Xu, D.; Meng, X.; et al. Overexpression of GRIM-19, a Mitochondrial Respiratory Chain Complex I Protein, Suppresses Hepatocellular Carcinoma Growth. Int. J. Clin. Exp. Pathol. 2014, 7, 7497–7507. [Google Scholar]
- Ricci, J.-E.; Muñoz-Pinedo, C.; Fitzgerald, P.; Bailly-Maitre, B.; Perkins, G.A.; Yadava, N.; Scheffler, I.E.; Ellisman, M.H.; Green, D.R. Disruption of Mitochondrial Function during Apoptosis Is Mediated by Caspase Cleavage of the P75 Subunit of Complex I of the Electron Transport Chain. Cell 2004, 117, 773–786. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef] [PubMed]
- Sollazzo, M.; De Luise, M.; Lemma, S.; Bressi, L.; Iorio, M.; Miglietta, S.; Milioni, S.; Kurelac, I.; Iommarini, L.; Gasparre, G.; et al. Respiratory Complex I Dysfunction in Cancer: From a Maze of Cellular Adaptive Responses to Potential Therapeutic Strategies. FEBS J. 2022, 289, 8003–8019. [Google Scholar] [CrossRef] [PubMed]
- Efremov, R.G.; Baradaran, R.; Sazanov, L.A. The Architecture of Respiratory Complex I. Nature 2010, 465, 441–445. [Google Scholar] [CrossRef]
- Brandt, U. Energy Converting NADH:Quinone Oxidoreductase (Complex I). Annu. Rev. Biochem. 2006, 75, 69–92. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial Free Radical Generation, Oxidative Stress, and Aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential Effects of Mitochondrial Complex I Inhibitors on Production of Reactive Oxygen Species. Biochim. Biophys. Acta 2009, 1787, 384–392. [Google Scholar] [CrossRef]
- Galkin, A.; Brandt, U. Superoxide Radical Formation by Pure Complex I (NADH:Ubiquinone Oxidoreductase) from Yarrowia Lipolytica. J. Biol. Chem. 2005, 280, 30129–30135. [Google Scholar] [CrossRef]
- Lambert, A.J.; Buckingham, J.A.; Boysen, H.M.; Brand, M.D. Diphenyleneiodonium Acutely Inhibits Reactive Oxygen Species Production by Mitochondrial Complex I during Reverse, but Not Forward Electron Transport. Biochim. Biophys. Acta 2008, 1777, 397–403. [Google Scholar] [CrossRef]
- Lambert, A.J.; Brand, M.D. Inhibitors of the Quinone-Binding Site Allow Rapid Superoxide Production from Mitochondrial NADH:Ubiquinone Oxidoreductase (Complex I). J. Biol. Chem. 2004, 279, 39414–39420. [Google Scholar] [CrossRef]
- Lenaz, G.; Fato, R.; Genova, M.L.; Bergamini, C.; Bianchi, C.; Biondi, A. Mitochondrial Complex I: Structural and Functional Aspects. Biochim. Biophys. Acta 2006, 1757, 1406–1420. [Google Scholar] [CrossRef] [PubMed]
- Pryde, K.R.; Hirst, J. Superoxide Is Produced by the Reduced Flavin in Mitochondrial Complex I: A Single, Unified Mechanism That Applies during Both Forward and Reverse Electron Transfer. J. Biol. Chem. 2011, 286, 18056–18065. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; De Rasmo, D.; Scacco, S.; Signorile, A.; Technikova-Dobrova, Z.; Palmisano, G.; Sardanelli, A.M.; Papa, F.; Panelli, D.; Scaringi, R.; et al. Mammalian Complex I: A Regulable and Vulnerable Pacemaker in Mitochondrial Respiratory Function. Biochim. Biophys. Acta 2008, 1777, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Cocco, T.; Pacelli, C.; Sgobbo, P.; Villani, G. Control of OXPHOS Efficiency by Complex I in Brain Mitochondria. Neurobiol. Aging 2009, 30, 622–629. [Google Scholar] [CrossRef]
- De Rasmo, D.; Signorile, A.; Larizza, M.; Pacelli, C.; Cocco, T.; Papa, S. Activation of the CAMP Cascade in Human Fibroblast Cultures Rescues the Activity of Oxidatively Damaged Complex I. Free Radic. Biol. Med. 2012, 52, 757–764. [Google Scholar] [CrossRef]
- Gonzalez-Franquesa, A.; Stocks, B.; Chubanava, S.; Hattel, H.B.; Moreno-Justicia, R.; Peijs, L.; Treebak, J.T.; Zierath, J.R.; Deshmukh, A.S. Mass-Spectrometry-Based Proteomics Reveals Mitochondrial Supercomplexome Plasticity. Cell Rep. 2021, 35, 109180. [Google Scholar] [CrossRef]
- Guarás, A.; Perales-Clemente, E.; Calvo, E.; Acín-Pérez, R.; Loureiro-Lopez, M.; Pujol, C.; Martínez-Carrascoso, I.; Nuñez, E.; García-Marqués, F.; Rodríguez-Hernández, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef]
- Signorile, A.; Pacelli, C.; Palese, L.L.; Santeramo, A.; Roca, E.; Cocco, T.; De Rasmo, D. CAMP/PKA Signaling Modulates Mitochondrial Supercomplex Organization. Int. J. Mol. Sci. 2022, 23, 9655. [Google Scholar] [CrossRef]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I Assembly into Supercomplexes Determines Differential Mitochondrial ROS Production in Neurons and Astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef]
- Boffoli, D.; Scacco, S.C.; Vergari, R.; Solarino, G.; Santacroce, G.; Papa, S. Decline with Age of the Respiratory Chain Activity in Human Skeletal Muscle. Biochim. Biophys. Acta 1994, 1226, 73–82. [Google Scholar] [CrossRef]
- Choksi, K.B.; Boylston, W.H.; Rabek, J.P.; Widger, W.R.; Papaconstantinou, J. Oxidatively Damaged Proteins of Heart Mitochondrial Electron Transport Complexes. Biochim. Biophys. Acta 2004, 1688, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Choksi, K.B.; Nuss, J.E.; Deford, J.H.; Papaconstantinou, J. Age-Related Alterations in Oxidatively Damaged Proteins of Mouse Skeletal Muscle Mitochondrial Electron Transport Chain Complexes. Free Radic. Biol. Med. 2008, 45, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s Disease Mutations in PINK1 Result in Decreased Complex I Activity and Deficient Synaptic Function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P. Parkinson’s Disease Brain Mitochondrial Complex I Has Oxidatively Damaged Subunits and Is Functionally Impaired and Misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef] [PubMed]
- Pamplona, R.; Jové, M.; Mota-Martorell, N.; Barja, G. Is the NDUFV2 Subunit of the Hydrophilic Complex I Domain a Key Determinant of Animal Longevity? FEBS J. 2021, 288, 6652–6673. [Google Scholar] [CrossRef]
- Ku, H.H.; Sohal, R.S. Comparison of Mitochondrial Pro-Oxidant Generation and Anti-Oxidant Defenses between Rat and Pigeon: Possible Basis of Variation in Longevity and Metabolic Potential. Mech. Ageing Dev. 1993, 72, 67–76. [Google Scholar] [CrossRef]
- Ku, H.H.; Brunk, U.T.; Sohal, R.S. Relationship between Mitochondrial Superoxide and Hydrogen Peroxide Production and Longevity of Mammalian Species. Free Radic. Biol. Med. 1993, 15, 621–627. [Google Scholar] [CrossRef]
- Mota-Martorell, N.; Jove, M.; Pradas, I.; Sanchez, I.; Gómez, J.; Naudi, A.; Barja, G.; Pamplona, R. Low Abundance of NDUFV2 and NDUFS4 Subunits of the Hydrophilic Complex I Domain and VDAC1 Predicts Mammalian Longevity. Redox Biol. 2020, 34, 101539. [Google Scholar] [CrossRef]
- Pamplona, R.; Barja, G. Highly Resistant Macromolecular Components and Low Rate of Generation of Endogenous Damage: Two Key Traits of Longevity. Ageing Res. Rev. 2007, 6, 189–210. [Google Scholar] [CrossRef]
- Jové, M.; Mota-Martorell, N.; Pradas, I.; Galo-Licona, J.D.; Martín-Gari, M.; Obis, È.; Sol, J.; Pamplona, R. The Lipidome Fingerprint of Longevity. Molecules 2020, 25, 4343. [Google Scholar] [CrossRef]
- Miwa, S.; Jow, H.; Baty, K.; Johnson, A.; Czapiewski, R.; Saretzki, G.; Treumann, A.; von Zglinicki, T. Low Abundance of the Matrix Arm of Complex I in Mitochondria Predicts Longevity in Mice. Nat. Commun. 2014, 5, 3837. [Google Scholar] [CrossRef] [PubMed]
- Pamplona, R.; Barja, G. Mitochondrial Oxidative Stress, Aging and Caloric Restriction: The Protein and Methionine Connection. Biochim. Biophys. Acta 2006, 1757, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Glossmann, H.H.; Lutz, O.M.D. Metformin and Aging: A Review. Gerontology 2019, 65, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cisuelo, V.; Gómez, J.; García-Junceda, I.; Naudí, A.; Cabré, R.; Mota-Martorell, N.; López-Torres, M.; González-Sánchez, M.; Pamplona, R.; Barja, G. Rapamycin Reverses Age-Related Increases in Mitochondrial ROS Production at Complex I, Oxidative Stress, Accumulation of MtDNA Fragments inside Nuclear DNA, and Lipofuscin Level, and Increases Autophagy, in the Liver of Middle-Aged Mice. Exp. Gerontol. 2016, 83, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, D.Š.; Dorđević, M.; Savković, U.; Lazarević, J. The Effect of Mitochondrial Complex I Inhibitor on Longevity of Short-Lived and Long-Lived Seed Beetles and Its Mitonuclear Hybrids. Biogerontology 2014, 15, 487–501. [Google Scholar] [CrossRef]
- Barja, G.; Herrero, A. Oxidative Damage to Mitochondrial DNA Is Inversely Related to Maximum Life Span in the Heart and Brain of Mammals. FASEB J. 2000, 14, 312–318. [Google Scholar] [CrossRef]
- Kim, T.-Y.; Wang, D.; Kim, A.K.; Lau, E.; Lin, A.J.; Liem, D.A.; Zhang, J.; Zong, N.C.; Lam, M.P.Y.; Ping, P. Metabolic Labeling Reveals Proteome Dynamics of Mouse Mitochondria. Mol. Cell Proteom. 2012, 11, 1586–1594. [Google Scholar] [CrossRef]
- Dieteren, C.E.J.; Koopman, W.J.H.; Swarts, H.G.; Peters, J.G.P.; Maczuga, P.; van Gemst, J.J.; Masereeuw, R.; Smeitink, J.A.M.; Nijtmans, L.G.J.; Willems, P.H.G.M. Subunit-Specific Incorporation Efficiency and Kinetics in Mitochondrial Complex I Homeostasis. J. Biol. Chem. 2012, 287, 41851–41860. [Google Scholar] [CrossRef]
- Lazarou, M.; McKenzie, M.; Ohtake, A.; Thorburn, D.R.; Ryan, M.T. Analysis of the Assembly Profiles for Mitochondrial- and Nuclear-DNA-Encoded Subunits into Complex I. Mol. Cell Biol. 2007, 27, 4228–4237. [Google Scholar] [CrossRef]
- Artal-Sanz, M.; Tavernarakis, N. Prohibitin Couples Diapause Signalling to Mitochondrial Metabolism during Ageing in C. elegans. Nature 2009, 461, 793–797. [Google Scholar] [CrossRef]
- McElroy, G.S.; Chakrabarty, R.P.; D’Alessandro, K.B.; Hu, Y.-S.; Vasan, K.; Tan, J.; Stoolman, J.S.; Weinberg, S.E.; Steinert, E.M.; Reyfman, P.A.; et al. Reduced Expression of Mitochondrial Complex I Subunit Ndufs2 Does Not Impact Healthspan in Mice. Sci. Rep. 2022, 12, 5196. [Google Scholar] [CrossRef] [PubMed]
- Dillin, A.; Hsu, A.-L.; Arantes-Oliveira, N.; Lehrer-Graiwer, J.; Hsin, H.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Kenyon, C. Rates of Behavior and Aging Specified by Mitochondrial Function during Development. Science 2002, 298, 2398–2401. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Lee, R.Y.N.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Ruvkun, G. A Systematic RNAi Screen Identifies a Critical Role for Mitochondria in C. elegans Longevity. Nat. Genet. 2003, 33, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.L.; Ventura, N.; Johnson, T.E. Relationship between Mitochondrial Electron Transport Chain Dysfunction, Development, and Life Extension in Caenorhabditis Elegans. PLoS Biol. 2007, 5, e259. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Hwang, A.B.; Kenyon, C. Inhibition of Respiration Extends C. elegans Life Span via Reactive Oxygen Species That Increase HIF-1 Activity. Curr. Biol. 2010, 20, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hekimi, S. A Mitochondrial Superoxide Signal Triggers Increased Longevity in Caenorhabditis Elegans. PLoS Biol. 2010, 8, e1000556. [Google Scholar] [CrossRef]
- Yee, C.; Yang, W.; Hekimi, S. The Intrinsic Apoptosis Pathway Mediates the Pro-Longevity Response to Mitochondrial ROS in C. elegans. Cell 2014, 157, 897–909. [Google Scholar] [CrossRef]
- Copeland, J.M.; Cho, J.; Lo, T.; Hur, J.H.; Bahadorani, S.; Arabyan, T.; Rabie, J.; Soh, J.; Walker, D.W. Extension of Drosophila Life Span by RNAi of the Mitochondrial Respiratory Chain. Curr. Biol. 2009, 19, 1591–1598. [Google Scholar] [CrossRef]
- Rera, M.; Monnier, V.; Tricoire, H. Mitochondrial Electron Transport Chain Dysfunction during Development Does Not Extend Lifespan in Drosophila Melanogaster. Mech. Ageing Dev. 2010, 131, 156–164. [Google Scholar] [CrossRef]
- Scialò, F.; Sriram, A.; Fernández-Ayala, D.; Gubina, N.; Lõhmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enríquez, J.A.; et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef]
- Graham, C.; Stefanatos, R.; Yek, A.E.H.; Spriggs, R.V.; Loh, S.H.Y.; Uribe, A.H.; Zhang, T.; Martins, L.M.; Maddocks, O.D.K.; Scialo, F.; et al. Mitochondrial ROS Signalling Requires Uninterrupted Electron Flow and Is Lost during Ageing in Flies. Geroscience 2022, 44, 1961–1974. [Google Scholar] [CrossRef] [PubMed]
- Calvo, E.; Cogliati, S.; Hernansanz-Agustín, P.; Loureiro-López, M.; Guarás, A.; Casuso, R.A.; García-Marqués, F.; Acín-Pérez, R.; Martí-Mateos, Y.; Silla-Castro, J.C.; et al. Functional Role of Respiratory Supercomplexes in Mice: SCAF1 Relevance and Segmentation of the Qpool. Sci. Adv. 2020, 6, eaba7509. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, M.; Rommelspacher, H.; Sugawa, M.D.; Dencher, N.A. Ageing Alters the Supramolecular Architecture of OxPhos Complexes in Rat Brain Cortex. Exp. Gerontol. 2010, 45, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Scott, J.D. AKAP Signalling Complexes: Focal Points in Space and Time. Nat. Rev. Mol. Cell Biol. 2004, 5, 959–970. [Google Scholar] [CrossRef]
- Papa, S.; Scacco, S.; Sardanelli, A.M.; Vergari, R.; Papa, F.; Budde, S.; van den Heuvel, L.; Smeitink, J. Mutation in the NDUFS4 Gene of Complex I Abolishes CAMP-Dependent Activation of the Complex in a Child with Fatal Neurological Syndrome. FEBS Lett. 2001, 489, 259–262. [Google Scholar] [CrossRef]
- Piccoli, C.; Scacco, S.; Bellomo, F.; Signorile, A.; Iuso, A.; Boffoli, D.; Scrima, R.; Capitanio, N.; Papa, S. CAMP Controls Oxygen Metabolism in Mammalian Cells. FEBS Lett. 2006, 580, 4539–4543. [Google Scholar] [CrossRef]
- De Rasmo, D.; Palmisano, G.; Scacco, S.; Technikova-Dobrova, Z.; Panelli, D.; Cocco, T.; Sardanelli, A.M.; Gnoni, A.; Micelli, L.; Trani, A.; et al. Phosphorylation Pattern of the NDUFS4 Subunit of Complex I of the Mammalian Respiratory Chain. Mitochondrion 2010, 10, 464–471. [Google Scholar] [CrossRef]
- De Rasmo, D.; Panelli, D.; Sardanelli, A.M.; Papa, S. CAMP-Dependent Protein Kinase Regulates the Mitochondrial Import of the Nuclear Encoded NDUFS4 Subunit of Complex I. Cell Signal. 2008, 20, 989–997. [Google Scholar] [CrossRef]
- Morán, M.; Rivera, H.; Sánchez-Aragó, M.; Blázquez, A.; Merinero, B.; Ugalde, C.; Arenas, J.; Cuezva, J.M.; Martín, M.A. Mitochondrial Bioenergetics and Dynamics Interplay in Complex I-Deficient Fibroblasts. Biochim. Biophys. Acta 2010, 1802, 443–453. [Google Scholar] [CrossRef]
- Valenti, D.; Rossi, L.; Marzulli, D.; Bellomo, F.; De Rasmo, D.; Signorile, A.; Vacca, R.A. Inhibition of Drp1-Mediated Mitochondrial Fission Improves Mitochondrial Dynamics and Bioenergetics Stimulating Neurogenesis in Hippocampal Progenitor Cells from a Down Syndrome Mouse Model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3117–3127. [Google Scholar] [CrossRef]
- Herzig, R.P.; Scacco, S.; Scarpulla, R.C. Sequential Serum-Dependent Activation of CREB and NRF-1 Leads to Enhanced Mitochondrial Respiration through the Induction of Cytochrome c. J. Biol. Chem. 2000, 275, 13134–13141. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, E.; Clementi, E.; Paolucci, C.; Cozzi, V.; Tonello, C.; Sciorati, C.; Bracale, R.; Valerio, A.; Francolini, M.; Moncada, S.; et al. Mitochondrial Biogenesis in Mammals: The Role of Endogenous Nitric Oxide. Science 2003, 299, 896–899. [Google Scholar] [CrossRef]
- Wu, H.; Kanatous, S.B.; Thurmond, F.A.; Gallardo, T.; Isotani, E.; Bassel-Duby, R.; Williams, R.S. Regulation of Mitochondrial Biogenesis in Skeletal Muscle by CaMK. Science 2002, 296, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Huang, X.; Feng, Y.; Handschin, C.; Feng, Y.; Gullicksen, P.S.; Bare, O.; Labow, M.; Spiegelman, B.; Stevenson, S.C. Transducer of Regulated CREB-Binding Proteins (TORCs) Induce PGC-1alpha Transcription and Mitochondrial Biogenesis in Muscle Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 14379–14384. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Signorile, A.; Papa, F.; Roca, E.; Papa, S. CAMP/Ca2+ Response Element-Binding Protein Plays a Central Role in the Biogenesis of Respiratory Chain Proteins in Mammalian Cells. IUBMB Life 2010, 62, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional Regulatory Circuits Controlling Mitochondrial Biogenesis and Function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef]
- De Rasmo, D.; Signorile, A.; Roca, E.; Papa, S. CAMP Response Element-Binding Protein (CREB) Is Imported into Mitochondria and Promotes Protein Synthesis. FEBS J. 2009, 276, 4325–4333. [Google Scholar] [CrossRef]
- Ryu, H.; Lee, J.; Impey, S.; Ratan, R.R.; Ferrante, R.J. Antioxidants Modulate Mitochondrial PKA and Increase CREB Binding to D-Loop DNA of the Mitochondrial Genome in Neurons. Proc. Natl. Acad. Sci. USA 2005, 102, 13915–13920. [Google Scholar] [CrossRef]
- Lee, J.; Kim, C.-H.; Simon, D.K.; Aminova, L.R.; Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Lonze, B.E.; Kim, K.-S.; Ginty, D.D.; et al. Mitochondrial Cyclic AMP Response Element-Binding Protein (CREB) Mediates Mitochondrial Gene Expression and Neuronal Survival. J. Biol. Chem. 2005, 280, 40398–40401. [Google Scholar] [CrossRef]
- Calabrese, V.; Dattilo, S.; Petralia, A.; Parenti, R.; Pennisi, M.; Koverech, G.; Calabrese, V.; Graziano, A.; Monte, I.; Maiolino, L.; et al. Analytical Approaches to the Diagnosis and Treatment of Aging and Aging-Related Disease: Redox Status and Proteomics. Free Radic. Res. 2015, 49, 511–524. [Google Scholar] [CrossRef]
- Barcena, M.L.; Aslam, M.; Pozdniakova, S.; Norman, K.; Ladilov, Y. Cardiovascular Inflammaging: Mechanisms and Translational Aspects. Cells 2022, 11, 1010. [Google Scholar] [CrossRef] [PubMed]
- Tengholm, A.; Gylfe, E. CAMP Signalling in Insulin and Glucagon Secretion. Diabetes Obes. Metab. 2017, 19, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Onodera, H.; Kato, H.; Kogure, K. Effects of Aging on Signal Transmission and Transduction Systems in the Gerbil Brain: Morphological and Autoradiographic Study. Neuroscience 1992, 46, 475–488. [Google Scholar] [CrossRef]
- Sugawa, M.; May, T. Age-Related Alteration in Signal Transduction: Involvement of the CAMP Cascade. Brain Res. 1993, 618, 57–62. [Google Scholar] [CrossRef]
- Puri, S.K.; Volicer, L. Age-Related Changes of Cyclic Nucleotide Levels in Rat Brain Regions. Mech. Ageing Dev. 1981, 15, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Makman, M.H.; Ahn, H.S.; Thal, L.J.; Sharpless, N.S.; Dvorkin, B.; Horowitz, S.G.; Rosenfeld, M. Evidence for Selective Loss of Brain Dopamine- and Histamine-Stimulated Adenylate Cyclase Activities in Rabbits with Aging. Brain Res. 1980, 192, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.F.; Walford, R.L. Alterations in Cyclic Nucleotides and Cyclase-Specific Activities in T Lymphocytes of Aging Normal Humans and Patients with Down’s Syndrome. J. Immunol. 1980, 125, 1665–1670. [Google Scholar] [CrossRef]
- Birkenfeld, A.; Ben Zvi, A. Age Associated Changes in Intracellular Adenosine Monophosphate. Clin. Exp. Immunol. 1984, 55, 651–654. [Google Scholar]
- Fulop, T.; Kekessy, D.; Foris, G. Altered Post-Receptorial Signal Transduction Mechanism under Various Stimulation in Polymorphonuclear Granulocytes of Alzheimer’s Disease. Mech. Ageing Dev. 1990, 52, 277–285. [Google Scholar] [CrossRef]
- Hu, R.; Yuan, B.; Wei, X.; Zhao, L.; Tang, J.; Chen, D. Enhanced CAMP/PKA Pathway by Seabuckthorn Fatty Acids in Aged Rats. J. Ethnopharmacol. 2007, 111, 248–254. [Google Scholar] [CrossRef]
- Berg, A.; Zimmerman, I.D. Effects of Electrical Stimulation and Norepinephrine on Cyclic-AMP Levels in the Cerebral Cortex of the Aging Rat. Mech. Ageing Dev. 1975, 4, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Titus, D.J.; Furones, C.; Kang, Y.; Atkins, C.M. Age-Dependent Alterations in CAMP Signaling Contribute to Synaptic Plasticity Deficits Following Traumatic Brain Injury. Neuroscience 2013, 231, 182–194. [Google Scholar] [CrossRef]
- Grammas, P.; Roher, A.E.; Ball, M.J. Increased Accumulation of CAMP in Cerebral Microvessels in Alzheimer’s Disease. Neurobiol. Aging 1994, 15, 113–116. [Google Scholar] [CrossRef]
- Martınez, M.; Hernández, A.I.; Hernanz, A. Increased CAMP Immunostaining in Cerebral Vessels in Alzheimer’s Disease. Brain Res. 2001, 922, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.J.; Thornberry, J.F. Cyclic AMP and Cyclic GMP Accumulation in Vitro in Brain Regions of Young, Old and Aged Rats. Brain Res. 1978, 139, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Karege, F.; Lambercy, C.; Schwald, M.; Steimer, T.; Cissé, M. Differential Changes of CAMP-Dependent Protein Kinase Activity and 3H-CAMP Binding Sites in Rat Hippocampus during Maturation and Aging. Neurosci. Lett. 2001, 315, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Laviada, I.D.; Galve-Roperh, I.; Malpartida, J.M.; Haro, A. CAMP Signalling Mechanisms with Aging in the Ceratitis Capitata Brain. Mech. Ageing Dev. 1997, 97, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Cashman, R.E.; Grammas, P. CAMP-Dependent Protein Kinase in Cerebral Microvessels in Aging and Alzheimer Disease. Mol. Chem. Neuropathol. 1995, 26, 247–258. [Google Scholar] [CrossRef]
- Stancheva, S.L.; Alova, L.G. Age-Related Changes of Cyclic AMP Phosphodiesterase Activity in Rat Brain Regions and a New Phosphodiesterase Inhibitor—Nootropic Agent Adafenoxate. Gen. Pharmacol. Vasc. Syst. 1991, 22, 955–958. [Google Scholar] [CrossRef]
- Chalimoniuk, M.; Strosznajder, J.B. Aging Modulates Nitric Oxide Synthesis and CGMP Levels in Hippocampus and Cerebellum: Effects of Amyloid β Peptide. Mol. Chem. Neuropathol. 1998, 35, 77–95. [Google Scholar] [CrossRef]
- Kelly, M.P.; Adamowicz, W.; Bove, S.; Hartman, A.J.; Mariga, A.; Pathak, G.; Reinhart, V.; Romegialli, A.; Kleiman, R.J. Select 3′,5′-Cyclic Nucleotide Phosphodiesterases Exhibit Altered Expression in the Aged Rodent Brain. Cell. Signal. 2014, 26, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.P. Putting Together The Pieces of Phosphodiesterase Distribution Patterns In The Brain: A Jigsaw Puzzle of Cyclic Nucleotide Regulation. In Cyclic-Nucleotide Phosphodiesterases in the Central Nervous System; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014; pp. 47–58. ISBN 978-1-118-83650-7. [Google Scholar]
- Kato, H.; Araki, T.; Chen, T.; Itoyama, Y.; Kogure, K. Effect of Rolipram on Age-Related Changes in Cyclic AMP-Selective Phosphodiesterase in the Rat Brain: An Autoradiographic Study. Methods Find Exp. Clin. Pharm. 1998, 20, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Nishiyama, S.; Ohba, H.; Sato, K.; Kakiuchi, T.; Tsukada, H. Age Differences in Phosphodiesterase Type-IV and Its Functional Response to Dopamine D1 Receptor Modulation in the Living Brain: A PET Study in Conscious Monkeys. Synapse 2002, 44, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Ramos, B.P.; Birnbaum, S.G.; Lindenmayer, I.; Newton, S.S.; Duman, R.S.; Arnsten, A.F.T. Dysregulation of Protein Kinase a Signaling in the Aged Prefrontal Cortex: New Strategy for Treating Age-Related Cognitive Decline. Neuron 2003, 40, 835–845. [Google Scholar] [CrossRef]
- Paramanik, V.; Thakur, M.K. Role of CREB Signaling in Aging Brain. Arch. Ital. Biol. 2013, 151, 33–42. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, L.; Liang, Y.; Zhang, C.; Xu, Z.; Zhang, L.; Fuji, R.; Mu, W.; Li, L.; Jiang, J.; et al. Cyclic AMP Mimics the Anti-Ageing Effects of Calorie Restriction by Up-Regulating Sirtuin. Sci. Rep. 2015, 5, 12012. [Google Scholar] [CrossRef]
- Mathieu, L.; Lopes Costa, A.; Le Bachelier, C.; Slama, A.; Lebre, A.-S.; Taylor, R.W.; Bastin, J.; Djouadi, F. Resveratrol Attenuates Oxidative Stress in Mitochondrial Complex I Deficiency: Involvement of SIRT3. Free Radic. Biol. Med. 2016, 96, 190–198. [Google Scholar] [CrossRef]
- Signorile, A.; Santeramo, A.; Tamma, G.; Pellegrino, T.; D’Oria, S.; Lattanzio, P.; De Rasmo, D. Mitochondrial CAMP Prevents Apoptosis Modulating Sirt3 Protein Level and OPA1 Processing in Cardiac Myoblast Cells. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 355–366. [Google Scholar] [CrossRef]
- Valenti, D.; Manente, G.A.; Moro, L.; Marra, E.; Vacca, R.A. Deficit of Complex I Activity in Human Skin Fibroblasts with Chromosome 21 Trisomy and Overproduction of Reactive Oxygen Species by Mitochondria: Involvement of the CAMP/PKA Signalling Pathway. Biochem. J. 2011, 435, 679–688. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Tolosa, E. Molecular and Clinical Prodrome of Parkinson Disease: Implications for Treatment. Nat. Rev. Neurol. 2010, 6, 309–317. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in Humans Due to a Product of Meperidine-Analog Synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.-S.; Kruse, S.E.; Palmiter, R.D.; Xia, Z. Mitochondrial Complex I Inhibition Is Not Required for Dopaminergic Neuron Death Induced by Rotenone, MPP+, or Paraquat. Proc. Natl. Acad. Sci. USA 2008, 105, 15136–15141. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.-S.; Palmiter, R.D.; Xia, Z. Loss of Mitochondrial Complex I Activity Potentiates Dopamine Neuron Death Induced by Microtubule Dysfunction in a Parkinson’s Disease Model. J. Cell Biol. 2011, 192, 873–882. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging Themes in Mitochondrial Homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Zanellati, M.C.; Monti, V.; Barzaghi, C.; Reale, C.; Nardocci, N.; Albanese, A.; Valente, E.M.; Ghezzi, D.; Garavaglia, B. Mitochondrial Dysfunction in Parkinson Disease: Evidence in Mutant PARK2 Fibroblasts. Front. Genet. 2015, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, C.; De Rasmo, D.; Signorile, A.; Grattagliano, I.; di Tullio, G.; D’Orazio, A.; Nico, B.; Comi, G.P.; Ronchi, D.; Ferranini, E.; et al. Mitochondrial Defect and PGC-1α Dysfunction in Parkin-Associated Familial Parkinson’s Disease. Biochim. Biophys. Acta 2011, 1812, 1041–1053. [Google Scholar] [CrossRef]
- Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting Mitochondrial Impairment in Parkinson’s Disease: Challenges and Opportunities. Front. Cell Dev. Biol. 2021, 8, 615461. [Google Scholar] [CrossRef]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New Insights into the Complex Role of Mitochondria in Parkinson’s Disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.-g.; Casadesus, G.; Perry, G.; Zhu, X. Impaired Mitochondrial Biogenesis Contributes to Mitochondrial Dysfunction in Alzheimer’s Disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Vrettou, S.; Wirth, B. S-Glutathionylation and S-Nitrosylation in Mitochondria: Focus on Homeostasis and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 15849. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Ferretta, A.; Russo, S.; Ruggieri, M.; Lasorella, P.; Paolicelli, D.; Trojano, M.; Signorile, A. PBMC of Multiple Sclerosis Patients Show Deregulation of OPA1 Processing Associated with Increased ROS and PHB2 Protein Levels. Biomedicines 2020, 8, 85. [Google Scholar] [CrossRef]
- Signorile, A.; Ferretta, A.; Ruggieri, M.; Paolicelli, D.; Lattanzio, P.; Trojano, M.; De Rasmo, D. Mitochondria, Oxidative Stress, CAMP Signalling and Apoptosis: A Crossroads in Lymphocytes of Multiple Sclerosis, a Possible Role of Nutraceutics. Antioxidants 2020, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Pyrgelis, E.-S.; Piperi, C. Emerging Potential of the Phosphodiesterase (PDE) Inhibitor Ibudilast for Neurodegenerative Diseases: An Update on Preclinical and Clinical Evidence. Molecules 2022, 27, 8448. [Google Scholar] [CrossRef]
- Ribaudo, G.; Landucci, E.; Giannangeli, M.; Mazzantini, C.; Maccarinelli, G.; Mastinu, A.; Bonini, S.A.; Memo, M.; Pellegrini-Giampietro, D.E.; Gianoncelli, A. Virtual Screening and in Vitro Experiments Highlight Cannabidiol as a Drug-like Phosphodiesterase 9 Inhibitor. Eur. J. Neurosci. 2022; Online ahead of print. [Google Scholar] [CrossRef]
- Sheng, J.; Zhang, S.; Wu, L.; Kumar, G.; Liao, Y.; Gk, P.; Fan, H. Inhibition of Phosphodiesterase: A Novel Therapeutic Target for the Treatment of Mild Cognitive Impairment and Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 1019187. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Signorile, A.; De Rasmo, D. Mitochondrial Complex I, a Possible Sensible Site of cAMP Pathway in Aging. Antioxidants 2023, 12, 221. https://doi.org/10.3390/antiox12020221
Signorile A, De Rasmo D. Mitochondrial Complex I, a Possible Sensible Site of cAMP Pathway in Aging. Antioxidants. 2023; 12(2):221. https://doi.org/10.3390/antiox12020221
Chicago/Turabian StyleSignorile, Anna, and Domenico De Rasmo. 2023. "Mitochondrial Complex I, a Possible Sensible Site of cAMP Pathway in Aging" Antioxidants 12, no. 2: 221. https://doi.org/10.3390/antiox12020221
APA StyleSignorile, A., & De Rasmo, D. (2023). Mitochondrial Complex I, a Possible Sensible Site of cAMP Pathway in Aging. Antioxidants, 12(2), 221. https://doi.org/10.3390/antiox12020221