
Metabolic Syndrome: A Narrative Review from the Oxidative Stress to the Management of Related Diseases

 ,
,  ,
,  , and
, and
Abstract
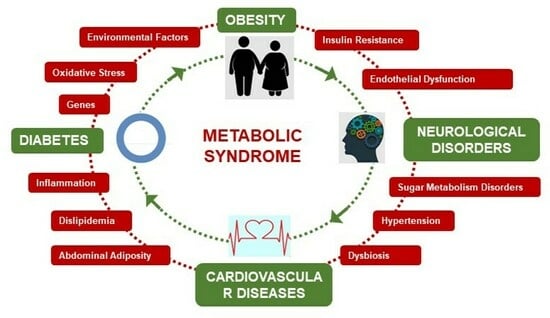
1. Introduction
2. Free Radicals, Oxidative Stress and Metabolic Syndrome
3. Metabolic Syndrome and Diabesity
3.1. Diabetes Mellitus
3.2. Diabesity
3.3. Gut Microbiota Interactions
4. Metabolic Syndrome and Cardiovascular Diseases
4.1. Dyslipidaemia
4.2. Hypertension
4.3. Hyperglycaemia and Vascular Complications
5. Metabolic Syndrome and Neurological Diseases
5.1. Stroke
5.2. Alzheimer
5.3. Depression
6. Management of Metabolic Syndrome
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engin, A. The Definition and Prevalence of Obesity and Metabolic Syndrome. Adv. Exp. Med. Biol. 2017, 960, 1–17. [Google Scholar] [CrossRef]
- Clodoveo, M.L.; Muraglia, M.; Fino, V.; Curci, F.; Fracchiolla, G.; Corbo, F.F.R. Overview on Innovative Packaging Methods Aimed to Increase the Shelf-Life of Cook-Chill Foods. Foods 2021, 10, 2086. [Google Scholar] [CrossRef]
- Ford, E.S.; Li, C.; Zhao, G. Prevalence and correlates of metabolic syndrome based on a harmonious definition among adults in the US. J. Diabetes 2010, 2, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Riediger, N.D.; Clara, I. Prevalence of metabolic syndrome in the Canadian adult population. CMAJ 2011, 183, E1127–E1134. [Google Scholar] [CrossRef] [PubMed]
- Beltrán-Sánchez, H.; Harhay, M.O.; Harhay, M.M.; McElligott, S. Prevalence and trends of metabolic syndrome in the adult U.S. population, 1999–2010. J. Am. Coll. Cardiol. 2013, 62, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, P.; Mathangasinghe, Y.; Jayawardena, R.; Hills, A.P.; Misra, A. Prevalence and trends of metabolic syndrome among adults in the asia-pacific region: A systematic review. BMC Public Health 2017, 17, 101. [Google Scholar] [CrossRef]
- Sarafidis, P.A.; Nilsson, P.M. The metabolic syndrome: A glance at its history. J. Hypertens. 2006, 24, 621–626. [Google Scholar] [CrossRef]
- Wilson, P.W.; D’Agostino, R.B.; Levy, D.; Belanger, A.M.; Silbershatz, H.; Kannel, W.B. Prediction of coronary heart disease using risk factor categories. Circulation 1998, 97, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
- Sigit, F.S.; Tahapary, D.L.; Trompet, S.; Sartono, E.; Willems van Dijk, K.; Rosendaal, F.R.; de Mutsert, R. The prevalence of metabolic syndrome and its association with body fat distribution in middle-aged individuals from Indonesia and the Netherlands: A cross-sectional analysis of two population-based studies. Diabetol. Metabol. Syndr. 2020, 12, 2. [Google Scholar] [CrossRef]
- Reaven, G.M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef]
- Kassi, E.; Pervanidou, P.; Kaltsas, G.; Chrousos, G. Metabolic syndrome: Definitions and controversies. BMC Med. 2011, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Martemucci, G.; Portincasa, P.; Di Ciaula, A.; Mariano, M.; Centonze, V.; D’Alessandro, A.G. Oxidative stress, aging, antioxidant supplementation and their impact on human health: An overview. Mech. Ageing Dev. 2022, 206, 111707. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Brewer, H.B., Jr.; Cleeman, J.I.; Smith, S.C., Jr.; Lenfant, C. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 2004, 109, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- van den Brink, W.; van Bilsen, J.; Salic, K.; Hoevenaars, F.P.M.; Verschuren, L.; Kleemann, R.; Bouwman, J.; Ronnett, G.V.; van Ommen, B.; Wopereis, S. Current and Future Nutritional Strategies to Modulate Inflammatory Dynamics in Metabolic Disorders. Front. Nutr. 2019, 6, 129. [Google Scholar] [CrossRef] [PubMed]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Farooqui, T.; Panza, F.; Frisardi, V. Metabolic syndrome as a risk factor for neurological disorders. Cell. Mol. Life Sci. 2012, 69, 741–762. [Google Scholar] [CrossRef]
- López-Jiménez, T.; Duarte-Salles, T.; Plana-Ripoll, O.; Recalde, M.; Xavier-Cos, F.; Puente, D. Association between metabolic syndrome and 13 types of cancer in Catalonia: A matched case-control study. PLoS ONE 2022, 17, e0264634. [Google Scholar] [CrossRef]
- Zhu, L.; Spence, C.; Yang, J.W.; Ma, G.X. The IDF Definition Is Better Suited for Screening Metabolic Syndrome and Estimating Risks of Diabetes in Asian American Adults: Evidence from NHANES 2011–2016. J. Clin. Med. 2020, 9, 3871. [Google Scholar] [CrossRef]
- Yamagishi, K.; Iso, H. The criteria for metabolic syndrome and the national health screening and education system in Japan. Epidemiol. Health 2017, 39, e2017003. [Google Scholar] [CrossRef]
- Desroches, S.; Lamarche, B. The evolving definitions and increasing prevalence of the metabolic syndrome. Appl. Physiol. Nutr. Metab. 2007, 32, 23–32. [Google Scholar] [CrossRef]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar] [PubMed]
- Giles, G.I.; Jacob, C. Reactive sulfur species: An emerging concept in oxidative stress. Biol. Chem. 2002, 383, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Martemucci, G.; Costagliola, C.; Mariano, M.; D’andrea, L.; Napolitano, P.; D’Alessandro, A.G. Free radical properties, source and targets, antioxidant consumption and health. Oxygen 2022, 2, 48–78. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 4th ed.; Oxford Press: Oxford, UK, 2020; p. 26. [Google Scholar]
- Min, B.; Ahn, D.U. Mechanism of lipid peroxidation in meat and meat products—A review. Food Sci. Biotechnol. 2005, 14, 152–163. [Google Scholar]
- Vijayalaxmi Reiter, R.J.; Tan, D.X.; Herman, T.S.; Thomas, C.R., Jr. Melatonin as a radioprotective agent: A review. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, B.; Pretorius, E. Hydroxyl radical-modified fibrinogen as a marker of thrombosis: The role of iron. Hematology 2012, 17, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012, 46, 382–419. [Google Scholar] [CrossRef]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar]
- Martemucci, G.; Portincasa, P.; Centonze, V.; Mariano, M.; Khalil, M.; D’Alessandro, A.G. Prevention of Oxidative Stress and Diseases by Antioxidant Supplementation. Med. Chem. 2023, 19, 509–537. [Google Scholar] [CrossRef]
- Repetto, M.; Semprine, J.; Boveris, A. Lipid peroxidation: Chemical mechanism, biological implications and analytical determination. In Lipid Peroxidation; Catala, D.A., Ed.; InTech: London, UK, 2012. [Google Scholar] [CrossRef]
- Poljsak, B.; Šuput, D.; Milisav, I. Achieving the balance between ROS and antioxidants: When to use the synthetic antioxidants. Oxid. Med. Cell. Longev. 2013, 2013, 956792. [Google Scholar] [CrossRef] [PubMed]
- Petersen, D.R.; Doorn, J.A. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic. Biol. Med. 2004, 37, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Giustarini, D.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation in human diseases. Trends Mol. Med. 2003, 9, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Fraley, A.E.; Tsimikas, S. Clinical applications of circulating oxidized low-density lipoprotein biomarkers in cardiovascular disease. Curr. Opin. Lipidol. 2006, 17, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Makita, Z.; Yanagisawa, K.; Kuwajima, S.; Bucala, R.; Vlassara, H.; Koike, T. The role of advanced glycosylation end-products in the pathogenesis of atherosclerosis. Nephrol. Dial. Transplant. 1996, 11 (Suppl. S5), 31–33. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Vannucci, S.J.; Yan, S.S.; Herold, K.; Yan, S.F.; Schmidt, A.M. Advanced glycation end products and RAGE: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005, 15, 16R–28R. [Google Scholar] [CrossRef] [PubMed]
- Grimsrud, P.A.; Picklo MJSr Griffin, T.J.; Bernlohr, D.A. Carbonylation of adipose proteins in obesity and insulin resistance: Identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Mol. Cell. Proteom. 2007, 6, 624–637. [Google Scholar] [CrossRef]
- Ceriello, A.; Motz, E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 816–823. [Google Scholar] [CrossRef]
- Carrier, A. Metabolic Syndrome and Oxidative Stress: A Complex Relationship. Antioxid. Redox Signal. 2017, 26, 429–431. [Google Scholar] [CrossRef]
- Urakawa, H.; Katsuki, A.; Sumida, Y.; Gabazza, E.C.; Murashima, S.; Morioka, K.; Maruyama, N.; Kitagawa, N.; Tanaka, T.; Hori, Y.; et al. Oxidative stress is associated with adiposity and insulin resistance in men. J. Clin. Endocrinol. Metab. 2003, 88, 4673–4676. [Google Scholar] [CrossRef]
- Ford, E.S.; Mokdad, A.H.; Giles, W.H.; Brown, D.W. The metabolic syndrome and antioxidant concentrations: Findings from the Third National Health and Nutrition Examination Survey. Diabetes 2003, 52, 2346–2352. [Google Scholar] [CrossRef]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2013, 36 (Suppl. S1), S67–S74. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States; U.S. Department of Health and Human Services, Centers for Disease Control and Prevention: Atlanta, GA, USA, 2014.
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, A.; Gudbjörnsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, Å. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Cruzat, V.; Arfuso, F.; Keane, K. Nutrient regulation of insulin secretion and action. J. Endocrinol. 2014, 221, R105–R120. [Google Scholar] [CrossRef]
- Polonsky, K.S. The past 200 years in diabetes. N. Engl. J. Med. 2012, 367, 1332–1340. [Google Scholar] [CrossRef]
- Weir, G.C.; Bonner-Weir, S. Islet β cell mass in diabetes and how it relates to function, birth, and death. Ann. N. Y. Acad. Sci. 2013, 1281, 92–105. [Google Scholar] [CrossRef]
- American Diabetes Association Professional Practice Committee. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022, 45 (Suppl. S1), S17–S38. [Google Scholar] [CrossRef]
- Bellamy, L.; Casas, J.P.; Hingorani, A.D.; Williams, D. Type 2 diabetes mellitus after gestational diabetes: A systematic review and meta-analysis. Lancet 2009, 373, 1773–1779. [Google Scholar] [CrossRef]
- Vounzoulaki, E.; Khunti, K.; Abner, S.C.; Tan, B.K.; Davies, M.J.; Gillies, C.L. Progression to type 2 diabetes in women with a known history of gestational diabetes: Systematic review and meta-analysis. BMJ 2020, 369, m1361. [Google Scholar] [CrossRef]
- World Health Organization. Use of Glycated Haemoglobin (HbA1c) in the Diagnosis of Diabetes Mellitus: Abbreviated Report of a WHO Consultation; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Sørgjerd, E.P. Type 1 Diabetes-related Autoantibodies in Different Forms of Diabetes. Curr. Diabetes Rev. 2019, 15, 199–204. [Google Scholar] [CrossRef]
- Kawasaki, M.; Arata, N.; Sakamoto, N.; Osamura, A.; Sato, S.; Ogawa, Y.; Yasuhi, I.; Waguri, M.; Hiramatsu, Y. Risk factors during the early postpartum period for type 2 diabetes mellitus in women with gestational diabetes. Endocr. J. 2020, 67, 427–437. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Bloch-Damti, A.; Bashan, N. Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxid. Redox Signal. 2005, 7, 1553–1567. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.-I.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.J.; Oates, P.J.; Hammes, H.-P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Al Ghouleh, I.; Khoo, N.K.; Knaus, U.G.; Griendling, K.K.; Touyz, R.M.; Thannickal, V.J.; Barchowsky, A.; Nauseef, W.M.; Kelley, E.E.; Bauer, P.M.; et al. Oxidases and peroxidases in cardiovascular and lung disease: New concepts in reactive oxygen species signaling. Free Radic. Biol. Med. 2011, 51, 1271–1288. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Shah, A.M. ROS generation by nonphagocytic NADPH oxidase: Potential relevance in diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14 (Suppl. S3), S221–S226. [Google Scholar] [CrossRef]
- Dal, S.; Jeandidier, N.; Schaschkow, A.; Spizzo, A.; Seyfritz, E.; Sookhareea, C.; Bietiger, W.; Péronet, C.; Moreau, F.; Pinget, M.; et al. Portal or subcutaneous insulin infusion: Efficacy and impact on liver inflammation. Fundam. Clin. Pharmacol. 2015, 29, 488–498. [Google Scholar] [CrossRef]
- Newsholme, P.; Morgan, D.; Rebelato, E.; Oliveira-Emilio, H.C.; Procopio, J.; Curi, R.; Carpinelli, A. Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell. Diabetologia 2009, 52, 2489–2498. [Google Scholar] [CrossRef]
- Kaneto, H.; Fujii, J.; Myint, T.; Miyazawa, N.; Islam, K.N.; Kawasaki, Y.; Suzuki, K.; Nakamura, M.; Tatsumi, H.; Yamasaki, Y.; et al. Reducing sugars trigger oxidative modification and apoptosis in pancreatic beta-cells by provoking oxidative stress through the glycation reaction. Biochem. J. 1996, 320 Pt 3, 855–863. [Google Scholar] [CrossRef]
- Gkogkolou, P.; Böhm, M. Advanced glycation end products: Key players in skin aging? Dermato-Endocrinology 2012, 4, 259–270. [Google Scholar] [CrossRef]
- Medzhitov, R.; Horng, T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 2009, 9, 692–703. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Defraigne, J.O. Un mécanisme physiopathologique central à l’origine des complications du diabète? [A central pathological mechanism explaining diabetic complications?]. Rev. Med. Liege 2005, 60, 472–478. [Google Scholar]
- Zhao, Z.; Zhao, C.; Zhang, X.H.; Zheng, F.; Cai, W.; Vlassara, H.; Ma, Z.A. Advanced glycation end products inhibit glucose-stimulated insulin secretion through nitric oxide-dependent inhibition of cytochrome c oxidase and adenosine triphosphate synthesis. Endocrinology 2009, 150, 2569–2576. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; He, J.C.; Zhu, L.; Chen, X.; Zheng, F.; Striker, G.E.; Vlassara, H. Oral glycotoxins determine the effects of calorie restriction on oxidant stress, age-related diseases, and lifespan. Am. J. Pathol. 2008, 173, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Beyan, H.; Riese, H.; Hawa, M.I.; Beretta, G.; Davidson, H.W.; Hutton, J.C.; Burger, H.; Schlosser, M.; Snieder, H.; Boehm, B.O.; et al. Glycotoxin and autoantibodies are additive environmentally determined predictors of type 1 diabetes: A twin and population study. Diabetes 2012, 61, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Peppa, M.; Goodman, S.; Ferrucci, L.; Striker, G.; Vlassara, H. Circulating glycotoxins and dietary advanced glycation endproducts: Two links to inflammatory response, oxidative stress, and aging. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 427–433. [Google Scholar] [CrossRef]
- Krhač, M.; Lovrenčić, M.V. Update on biomarkers of glycemic control. World J. Diabetes 2019, 10, 1. [Google Scholar] [CrossRef]
- Marcovecchio, M.L.; Dalton, R.N.; Chiarelli, F.; Dunger, D.B. A1C variability as an independent risk factor for microalbuminuria in young people with type 1 diabetes. Diabetes Care 2011, 34, 1011–1013. [Google Scholar] [CrossRef] [PubMed]
- The ADVANCE Collaborative Group; Patel, A.; MacMahon, S.; Chalmers, J.; Neal, B.; Billot, L.; Woodward, M.; Marre, M.; Cooper, M.; Glasziou, P.; et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008, 358, 2560–2572. [Google Scholar] [CrossRef]
- Schulman, I.H.; Zhou, M.S. Vascular insulin resistance: A potential link between cardiovascular and metabolic diseases. Curr. Hypertens. Rep. 2009, 11, 48–55. [Google Scholar] [CrossRef]
- Soeters, M.R.; Soeters, P.B.; Schooneman, M.G.; Houten, S.M.; Romijn, J.A. Adaptive reciprocity of lipid and glucose metabolism in human short-term starvation. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1397–E1407. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.R.; Milner, J.J.; Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 2012, 249, 218–238. [Google Scholar] [CrossRef] [PubMed]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Pratipanawatr, T.; Berria, R.; Wang, E.; DeFronzo, R.A.; Sullards, M.C.; Mandarino, L.J. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 2004, 53, 25–31. [Google Scholar] [CrossRef]
- Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Gerber, P.A.; Rutter, G.A. The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid. Redox Signal. 2017, 26, 501–518. [Google Scholar] [CrossRef]
- Newsholme, P.; Haber, E.P.; Hirabara, S.M.; Rebelato, E.L.O.; Procopio, J.; Morgan, D.; Oliveira-Emilio, H.C.; Carpinelli, A.R.; Curi, R. Diabetes associated cell stress and dysfunction: Role of mitochondrial and non-mitochondrial ROS production and activity. J. Physiol. 2007, 583 Pt 1, 9–24. [Google Scholar] [CrossRef]
- Morgan, D.; Oliveira-Emilio, H.R.; Keane, D.; Hirata, A.E.; Da Rocha, M.S.; Bordin, S.; Curi, R.; Newsholme, P.; Carpinelli, A.R. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 2007, 50, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Maechler, P.; Jornot, L.; Wollheim, C.B. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J. Biol. Chem. 1999, 274, 27905–27913. [Google Scholar] [CrossRef] [PubMed]
- Boitard, C. Pancreatic islet autoimmunity. Presse Med. 2012, 41 Pt 2, e636–e650. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Sammeth, M.; Bouckenooghe, T.; Bottu, G.; Sisino, G.; Igoillo-Esteve, M.; Ortis, F.; Santin, I.; Colli, M.L.; Barthson, J.; et al. The human pancreatic islet transcriptome: Expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012, 8, e1002552. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Ley, R.E.; Volchkov, P.Y.; Stranges, P.B.; Avanesyan, L.; Stonebraker, A.C.; Hu, C.; Wong, F.S.; Szot, G.L.; Bluestone, J.A.; et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008, 455, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.Y.; Vidal-Puig, A. Adipose tissue expandability: The metabolic problems of obesity may arise from the inability to become more obese. Biochem. Soc. Trans. 2008, 36 Pt 5, 935–940. [Google Scholar] [CrossRef]
- Cruzat, V.F.; Keane, K.N.; Scheinpflug, A.L.; Cordeiro, R.; Soares, M.J.; Newsholme, P. Alanyl-glutamine improves pancreatic β-cell function following ex vivo inflammatory challenge. J. Endocrinol. 2015, 224, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Colli, M.L.; Ortis, F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat. Rev. Endocrinol. 2009, 5, 219–226. [Google Scholar] [CrossRef]
- Araki, E.; Nishikawa, T. Oxidative stress: A cause and therapeutic target of diabetic complications. J. Diabetes Investig. 2010, 1, 90–96. [Google Scholar] [CrossRef]
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and Oxidative Stress: The Molecular Connectivity between Insulin Resistance, Obesity, and Alzheimer’s Disease. Mediat. Inflamm. 2015, 2015, 105828. [Google Scholar] [CrossRef]
- Bastard, J.-P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Emanuela, F.; Grazia, M.; de Marco, R.; Maria Paola, L.; Giorgio, F.; Marco, B. Inflammation as a Link between Obesity and Metabolic Syndrome. J. Nutr. Metab. 2012, 2012, 476380. [Google Scholar] [CrossRef] [PubMed]
- Murdolo, G.; Piroddi, M.; Luchetti, F.; Tortoioli, C.; Canonico, B.; Zerbinati, C.; Galli, F.; Iuliano, L. Oxidative stress and lipid peroxidation by-products at the crossroad between adipose organ dysregulation and obesity-linked insulin resistance. Biochimie 2013, 95, 585–594. [Google Scholar] [CrossRef]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Deyoung, S.M.; Bodzin, J.L.; Saltiel, A.R. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 2007, 56, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Koppaka, S.; Kehlenbrink, S.; Carey, M.; Li, W.; Sanchez, E.; Lee, D.-E.; Lee, H.; Chen, J.; Carrasco, E.; Kishore, P.; et al. Reduced adipose tissue macrophage content is associated with improved insulin sensitivity in thiazolidinedione-treated diabetic humans. Diabetes 2013, 62, 1843–1854. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Moro, C.; Klimcakova, E.; Lolmède, K.; Berlan, M.; Lafontan, M.; Stich, V.; Bouloumié, A.; Galitzky, J.; Arner, P.; Langin, D. Atrial natriuretic peptide inhibits the production of adipokines and cytokines linked to inflammation and insulin resistance in human subcutaneous adipose tissue. Diabetologia 2007, 50, 1038–1047. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Lastra, G.; Dhuper, S.; Johnson, M.S.; Sowers, J.R. Salt, aldosterone, and insulin resistance: Impact on the cardiovascular system. Nat. Rev. Cardiol. 2010, 7, 577–584. [Google Scholar] [CrossRef]
- Zhou, M.S.; Schulman, I.H.; Zeng, Q. Link between the renin-angiotensin system and insulin resistance: Implications for cardiovascular disease. Vasc. Med. 2012, 17, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M.; Lithell, H.; Landsberg, L. Hypertension and associated metabolic abnormalities—The role of insulin resistance and the sympathoadrenal system. N. Engl. J. Med. 1996, 334, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H. Evolutionary medicine and chronic inflammatory state—Known and new concepts in pathophysiology. J. Mol. Med. 2012, 90, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.A.; Habibi, J.; Wei, Y.; Lastra, G.; Manrique, C.; Stas, S.; Sowers, J.R.; Boese, A.C.; Kim, S.C.; Yin, K.-J.; et al. Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2009–H2023. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.S.; Schulman, I.H.; Raij, L. Vascular inflammation, insulin resistance, and endothelial dysfunction in salt-sensitive hypertension: Role of nuclear factor kappa B activation. J. Hypertens. 2010, 28, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.V.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef] [PubMed]
- Halpern, A.; Mancini, M.C. Diabesity: Are weight loss medications effective? Treat. Endocrinol. 2005, 4, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, T.M.; Jakupović, H.; Carrasquilla, G.D.; Ängquist, L.; Grarup, N.; Sørensen, T.I.A.; Tjønneland, A.; Overvad, K.; Pedersen, O.; Hansen, T.; et al. Obesity, unfavourable lifestyle and genetic risk of type 2 diabetes: A case-cohort study. Diabetologia 2020, 63, 1324–1332. [Google Scholar] [CrossRef]
- Ford, E.S.; Mokdad, A.H. Fruit and vegetable consumption and diabetes mellitus incidence among U.S. adults. Prev. Med. 2001, 32, 33–39. [Google Scholar] [CrossRef]
- Nolan, P.B.; Carrick-Ranson, G.; Stinear, J.W.; Reading, S.A.; Dalleck, L.C. Prevalence of metabolic syndrome and metabolic syndrome components in young adults: A pooled analysis. Prev. Med. Rep. 2017, 7, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Lois, K.; Kumar, S. Obesity and diabetes. Endocrinol. Nutr. 2009, 56 (Suppl. S4), 38–42. [Google Scholar] [CrossRef] [PubMed]
- Baldeweg, S.E.; Golay, A.; Natali, A.; Balkau, B.; Del Prato, S.; Coppack, S.W. Insulin resistance, lipid and fatty acid concentrations in 867 healthy Europeans. European Group for the Study of Insulin Resistance (EGIR). Eur. J. Clin. Investig. 2000, 30, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Felber, J.P.; Golay, A. Pathways from obesity to diabetes. Int. J. Obes. Relat. Metab. Disord. 2002, 26 (Suppl. S2), S39–S45. [Google Scholar] [CrossRef] [PubMed]
- Lönnqvist, F.; Thöme, A.; Nilsell, K.; Hoffstedt, J.; Arner, P. A pathogenic role of visceral fat beta 3-adrenoceptors in obesity. J. Clin. Investig. 1995, 95, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Shulman, G.I.; Rothman, D.L.; Jue, T.; Stein, P.; DeFronzo, R.A.; Shulman, R.G. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N. Engl. J. Med. 1990, 322, 223–228. [Google Scholar] [CrossRef]
- Golay, A.; Ybarra, J. Link between obesity and type 2 diabetes. Best Pract. Res. Clin. Endocrinol. Metab. 2005, 19, 649–663. [Google Scholar] [CrossRef]
- Ouchi, N.; PDer, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef]
- Groop, L.; Forsblom, C.; Lehtovirta, M.; Tuomi, T.; Karanko, S.; Nissén, M.; Ehrnström, B.-O.; Forsén, B.; Isomaa, B.; Snickars, B.; et al. Metabolic consequences of a family history of NIDDM (the Botnia study): Evidence for sex-specific parental effects. Diabetes 1996, 45, 1585–1593. [Google Scholar] [CrossRef]
- Maes, H.H.; Neale, M.C.; Eaves, L.J. Genetic and environmental factors in relative body weight and human adiposity. Behav. Genet. 1997, 27, 325–351. [Google Scholar] [CrossRef]
- Stunkard, A.J.; Harris, J.R.; Pedersen, N.L.; McClearn, G.E. The body-mass index of twins who have been reared apart. N. Engl. J. Med. 1990, 322, 1483–1487. [Google Scholar] [CrossRef]
- Whitaker, R.C.; Wright, J.A.; Pepe, M.S.; Seidel, K.D.; Dietz, W.H. Predicting obesity in young adulthood from childhood and parental obesity. N. Engl. J. Med. 1997, 337, 869–873. [Google Scholar] [CrossRef]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef]
- Bray, M.S.; Loos, R.J.F.; McCaffery, J.M.; Ling, C.; Franks, P.W.; Weinstock, G.M.; Snyder, M.P.; Vassy, J.L.; Agurs-Collins, T.; The The Conference Working Group. NIH working group report—Using genomic information to guide weight management: From universal to precision treatment. Obesity 2016, 24, 14–22, Correction in Obesity 2016, 24, 757. [Google Scholar] [CrossRef] [PubMed]
- Bogardus, C. Missing heritability and GWAS utility. Obesity 2009, 17, 209–210. [Google Scholar] [CrossRef]
- Lander, E.S. Initial impact of the sequencing of the human genome. Nature 2011, 470, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.G.; Pluzhnikov, A.; Miyake, K.; Sun, Y.; Ng, M.C.; Roe, C.A.; Below, J.E.; Nicolae, R.I.; Konkashbaev, A.; Bell, G.I.; et al. Identification of type 2 diabetes genes in Mexican Americans through genome-wide association studies. Diabetes 2007, 56, 3033–3044. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, C.M.; Heid, I.M.; Randall, J.C.; Lamina, C.; Steinthorsdottir, V.; Qi, L.; Speliotes, E.K.; Thorleifsson, G.; Willer, C.J.; Herrera, B.M.; et al. Genome-wide association scan meta-analysis identifies three Loci influencing adiposity and fat distribution. PLoS Genet. 2009, 5, e1000508, Correction in PLoS Genet. 2009, 5. [Google Scholar] [CrossRef]
- Spiegelman, B.M.; Flier, J.S. Obesity and the regulation of energy balance. Cell 2001, 104, 531–543. [Google Scholar] [CrossRef]
- Seeley, R.J.; Woods, S.C. Monitoring of stored and available fuel by the CNS: Implications for obesity. Nat. Rev. Neurosci. 2003, 4, 901–909, Correction in Nat. Rev. Neurosci. 2006, 7, 167. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Chawla, A. Connecting type 1 and type 2 diabetes through innate immunity. Cold Spring Harb. Perspect. Med. 2012, 2, a007724. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Obin, M.S. Obesity and the role of adipose tissue in inflammation and metabolism. Am. J. Clin. Nutr. 2006, 83, 461S–465S. [Google Scholar] [CrossRef] [PubMed]
- Van Gaal, L.F.; Mertens, I.L.; De Block, C.E. Mechanisms linking obesity with cardiovascular disease. Nature 2006, 444, 875–880. [Google Scholar] [CrossRef]
- Te Morenga, L.; Mallard, S.; Mann, J. Dietary sugars and body weight: Systematic review and meta-analyses of randomised controlled trials and cohort studies. BMJ 2012, 346, e7492. [Google Scholar] [CrossRef]
- Chen, L.; Appel, L.J.; Loria, C.; Lin, P.-H.; Champagne, C.M.; Elmer, P.J.; Ard, J.D.; Mitchell, D.; Batch, B.C.; Svetkey, L.P.; et al. Reduction in consumption of sugar-sweetened beverages is associated with weight loss: The PREMIER trial. Am. J. Clin. Nutr. 2009, 89, 1299–1306. [Google Scholar] [CrossRef]
- PDs, E.J.; Skokan, L.E.; Timlin, M.T.; Dingfelder, C.S. Dietary sugars stimulate fatty acid synthesis in adults. J. Nutr. 2008, 138, 1039–1046. [Google Scholar] [CrossRef]
- Taskinen, M.R.; Packard, C.J.; Borén, J. Dietary Fructose and the Metabolic Syndrome. Nutrients 2019, 11, 1987. [Google Scholar] [CrossRef]
- Lecoultre, V.; Egli, L.; Carrel, G.; Theytaz, F.; Kreis, R.; Schneiter, P.; Boss, A.; Zwygart, K.; Lê, K.; Bortolotti, M.; et al. Effects of fructose and glucose overfeeding on hepatic insulin sensitivity and intrahepatic lipids in healthy humans. Obesity 2013, 21, 782–785. [Google Scholar] [CrossRef]
- Tappy, L. Fructose-containing caloric sweeteners as a cause of obesity and metabolic disorders. J. Exp. Biol. 2018, 221 (Suppl. S1), jeb164202. [Google Scholar] [CrossRef]
- Sattar, N.; Forrest, E.; Preiss, D. Non-alcoholic fatty liver disease. BMJ 2014, 349, g4596. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; Montezinho, L.P.; Oliveira, P.J.; Frühbeck, G.; Gómez-Ambrosi, J.; Montecucco, F.; Carbone, F.; Wieckowski, M.R.; Wang, D.Q.-H.; Portincasa, P. Targeting mitochondria to oppose the progression of nonalcoholic fatty liver disease. Biochem. Pharmacol. 2019, 160, 34–45. [Google Scholar] [CrossRef]
- Molina-Molina, E.; Krawczyk, M.; Stachowska, E.; Lammert, F.; Portincasa, P. Non-Alcoholic Fatty Liver Disease in Non-Obese Individuals: Prevalence, Pathogenesis and Treatment. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, E.; Lunardi Baccetto, R.; Wang, D.Q.; de Bari, O.; Krawczyk, M.; Portincasa, P. Exercising the hepatobiliary-gut axis. The impact of physical activity performance. Eur. J. Clin. Investig. 2018, 48, e12958. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Pardee, P.E.; Lavine, J.E.; Blumkin, A.K.; Cook, S. Cardiovascular risk factors and the metabolic syndrome in pediatric nonalcoholic fatty liver disease. Circulation 2008, 118, 277–283. [Google Scholar] [CrossRef]
- Bellentani, S.; Scaglioni, F.; Marino, M.; Bedogni, G. Epidemiology of non-alcoholic fatty liver disease. Dig. Dis. 2010, 28, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Mendrick, D.L.; Diehl, A.M.; Topor, L.S.; Dietert, R.R.; Will, Y.; A La Merrill, M.; Bouret, S.; Varma, V.; Hastings, K.L.; Schug, T.T.; et al. Metabolic Syndrome and Associated Diseases: From the Bench to the Clinic. Toxicol. Sci. 2018, 162, 36–42. [Google Scholar] [CrossRef]
- Surowska, A.; Jegatheesan, P.; Campos, V.; Marques, A.-S.; Egli, L.; Cros, J.; Rosset, R.; Lecoultre, V.; Kreis, R.; Boesch, C.; et al. Effects of Dietary Protein and Fat Content on Intrahepatocellular and Intramyocellular Lipids during a 6-Day Hypercaloric, High Sucrose Diet: A Randomized Controlled Trial in Normal Weight Healthy Subjects. Nutrients 2019, 11, 209. [Google Scholar] [CrossRef]
- Johnston, R.D.; Stephenson, M.C.; Crossland, H.; Cordon, S.M.; Palcidi, E.; Cox, E.F.; Taylor, M.A.; Aithal, G.P.; Macdonald, I.A. No difference between high-fructose and high-glucose diets on liver triacylglycerol or biochemistry in healthy overweight men. Gastroenterology 2013, 145, 1016–1025.e2. [Google Scholar] [CrossRef] [PubMed]
- Campos, V.; Despland, C.; Brandejsky, V.; Kreis, R.; Schneiter, P.; Chiolero, A.; Boesch, C.; Tappy, L. Sugar- and artificially sweetened beverages and intrahepatic fat: A randomized controlled trial. Obesity 2015, 23, 2335–2339. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Ugalde-Nicalo, P.; Welsh, J.A.; Angeles, J.E.; Cordero, M.; Harlow, K.E.; Alazraki, A.; Durelle, J.; Knight-Scott, J.; Newton, K.P.; et al. Effect of a Low Free Sugar Diet vs Usual Diet on Nonalcoholic Fatty Liver Disease in Adolescent Boys: A Randomized Clinical Trial. JAMA 2019, 321, 256–265, Correction in JAMA 2019, 322, 469. [Google Scholar] [CrossRef]
- Imamura, F.; O’Connor, L.; Ye, Z.; Mursu, J.; Hayashino, Y.; Bhupathiraju, S.N.; Forouhi, N.G. Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: Systematic review, meta-analysis, and estimation of population attributable fraction. BMJ 2015, 351, h3576. [Google Scholar] [CrossRef]
- Kelishadi, R.; Mansourian, M.; Heidari-Beni, M. Association of fructose consumption and components of metabolic syndrome in human studies: A systematic review and meta-analysis. Nutrition 2014, 30, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Wijarnpreecha, K.; Thongprayoon, C.; Edmonds, P.J.; Cheungpasitporn, W. Associations of sugar- and artificially sweetened soda with nonalcoholic fatty liver disease: A systematic review and meta-analysis. QJM 2016, 109, 461–466. [Google Scholar] [CrossRef]
- Johnson, R.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Shafiu, M.; Sundaram, S.; Le, M.; Ishimoto, T.; Sautin, Y.Y.; Lanaspa, M.A. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 2013, 62, 3307–3315. [Google Scholar] [CrossRef]
- Cheungpasitporn, W.; Thongprayoon, C.; Edmonds, P.J.; Srivali, N.; Ungprasert, P.; Kittanamongkolchai, W.; Erickson, S.B. Sugar and artificially sweetened soda consumption linked to hypertension: A systematic review and meta-analysis. Clin. Exp. Hypertens. 2015, 37, 587–593. [Google Scholar] [CrossRef]
- Xi, B.; Huang, Y.; Reilly, K.H.; Li, S.; Zheng, R.; Barrio-Lopez, M.T.; Martinez-Gonzalez, M.A.; Zhou, D. Sugar-sweetened beverages and risk of hypertension and CVD: A dose-response meta-analysis. Br. J. Nutr. 2015, 113, 709–717. [Google Scholar] [CrossRef]
- Sung, H.; Siegel, R.L.; Torre, L.A.; Pearson-Stuttard, J.; Islami, F.; Fedewa, S.A.; Sauer, A.G.; Shuval, K.; Gapstur, S.M.; Jacobs, E.J.; et al. Global patterns in excess body weight and the associated cancer burden. CA Cancer J. Clin. 2019, 69, 88–112. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martinez, P.; Garcia-Quintana, J.M.; Yubero-Serrano, E.M.; Tasset-Cuevas, I.; Tunez, I.; Garcia-Rios, A.; Delgado-Lista, J.; Marin, C.; Perez-Jimenez, F.; Roche, H.M.; et al. Postprandial oxidative stress is modified by dietary fat: Evidence from a human intervention study. Clin. Sci. 2010, 119, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Cardona, F.; Túnez, I.; Tasset, I.; Montilla, P.; Collantes, E.; Tinahones, F.J. Fat overload aggravates oxidative stress in patients with the metabolic syndrome. Eur. J. Clin. Investig. 2008, 38, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Yen, C.H.; Huang, Y.C.; Lee, B.J.; Hsia, S.; Lin, P.T. Relationships between inflammation, adiponectin, and oxidative stress in metabolic syndrome. PLoS ONE 2012, 7, e45693. [Google Scholar] [CrossRef] [PubMed]
- Smith, U. Abdominal obesity: A marker of ectopic fat accumulation. J. Clin. Investig. 2015, 125, 1790–1792. [Google Scholar] [CrossRef]
- Krotkiewski, M.; Björntorp, P.; Sjöström, L.; Smith, U. Impact of obesity on metabolism in men and women. Importance of regional adipose tissue distribution. J. Clin. Investig. 1983, 72, 1150–1162. [Google Scholar] [CrossRef]
- Lönn, M.; Mehlig, K.; Bengtsson, C.; Lissner, L. Adipocyte size predicts incidence of type 2 diabetes in women. FASEB J. 2010, 24, 326–331. [Google Scholar] [CrossRef]
- Perry, R.J.; Camporez, J.-P.G.; Kursawe, R.; Titchenell, P.M.; Zhang, D.; Perry, C.J.; Jurczak, M.J.; Abudukadier, A.; Han, M.S.; Zhang, X.-M.; et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015, 160, 745–758. [Google Scholar] [CrossRef]
- Summers, S.A. Ceramides in insulin resistance and lipotoxicity. Prog. Lipid Res. 2006, 45, 42–72. [Google Scholar] [CrossRef]
- van Herpen, N.A.; Schrauwen-Hinderling, V.B. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol. Behav. 2008, 94, 231–241. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol. Sci. 2017, 38, 649–665. [Google Scholar] [CrossRef]
- Nedergaard, J.; Bengtsson, T.; Cannon, B. New powers of brown fat: Fighting the metabolic syndrome. Cell Metab. 2011, 13, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef] [PubMed]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar] [CrossRef]
- Tchoukalova, Y.D.; Votruba, S.B.; Tchkonia, T.; Giorgadze, N.; Kirkland, J.L.; Jensen, M.D. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc. Natl. Acad. Sci. USA 2010, 107, 18226–18231. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.; Westermark, P.O.; Spalding, K.L.; Britton, T.; Rydén, M.; Frisén, J.; Bernard, S.; Arner, P. Adipocyte turnover: Relevance to human adipose tissue morphology. Diabetes 2010, 59, 105–109. [Google Scholar] [CrossRef]
- Hoffstedt, J.; Arner, E.; Wahrenberg, H.; Andersson, D.P.; Qvisth, V.; Löfgren, P.; Rydén, M.; Thörne, A.; Wirén, M.; Palmér, M.; et al. Regional impact of adipose tissue morphology on the metabolic profile in morbid obesity. Diabetologia 2010, 53, 2496–2503. [Google Scholar] [CrossRef] [PubMed]
- Isakson, P.; Hammarstedt, A.; Gustafson, B.; Smith, U. Impaired preadipocyte differentiation in human abdominal obesity: Role of Wnt, tumor necrosis factor-alpha, and inflammation. Diabetes 2009, 58, 1550–1557. [Google Scholar] [CrossRef]
- Langin, D. In and out: Adipose tissue lipid turnover in obesity and dyslipidemia. Cell Metab. 2011, 14, 569–570. [Google Scholar] [CrossRef]
- Arner, P.; Bernard, S.; Salehpour, M.; Possnert, G.; Liebl, J.; Steier, P.; Buchholz, B.A.; Eriksson, M.; Arner, E.; Hauner, H.; et al. Dynamics of human adipose lipid turnover in health and metabolic disease. Nature 2011, 478, 110–113. [Google Scholar] [CrossRef]
- Spalding, K.L.; Bernard, S.; Näslund, E.; Salehpour, M.; Possnert, G.; Appelsved, L.; Fu, K.-Y.; Alkass, K.; Druid, H.; Thorell, A.; et al. Impact of fat mass and distribution on lipid turnover in human adipose tissue. Nat. Commun. 2017, 8, 15253. [Google Scholar] [CrossRef]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef]
- Matsuzawa, Y.; Funahashi, T.; Nakamura, T. The concept of metabolic syndrome: Contribution of visceral fat accumulation and its molecular mechanism. J. Atheroscler. Thromb. 2011, 18, 629–639. [Google Scholar] [CrossRef]
- Rasouli, N.; Kern, P.A. Adipocytokines and the metabolic complications of obesity. J. Clin. Endocrinol. Metab. 2008, 93 (Suppl. S1), S64–S73. [Google Scholar] [CrossRef]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef]
- Tilg, H.; Kaser, A. Gut microbiome, obesity, and metabolic dysfunction. J. Clin. Investig. 2011, 121, 2126–2132. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef]
- Malagon, M.M.; Díaz-Ruiz, A.; Guzman-Ruiz, R.; Jimenez-Gomez, Y.; Moreno, N.R.; Garcia-Navarro, S.; Vazquez-Martinez, R.; Peinado, J.R. Adipobiology for novel therapeutic approaches in metabolic syndrome. Curr. Vasc. Pharmacol. 2013, 11, 954–967. [Google Scholar] [CrossRef] [PubMed]
- Toubal, A.; Treuter, E.; Clément, K.; Venteclef, N. Genomic and epigenomic regulation of adipose tissue inflammation in obesity. Trends Endocrinol. Metab. 2013, 24, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Kraja, A.T.; Chasman, D.I.; North, K.E.; Reiner, A.P.; Yanek, L.R.; Kilpeläinen, T.O.; Smith, J.A.; Dehghan, A.; Dupuis, J.; Johnson, A.D.; et al. Pleiotropic genes for metabolic syndrome and inflammation. Mol. Genet. Metab. 2014, 112, 317–338. [Google Scholar] [CrossRef] [PubMed]
- Sell, H.; Habich, C.; Eckel, J. Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 2012, 8, 709–716. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801, Correction in J. Clin. Investig. 2006, 116, 2308. [Google Scholar] [CrossRef]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef]
- Donath, M.Y. Targeting inflammation in the treatment of type 2 diabetes. Diabetes Obes. Metab. 2013, 15 (Suppl. S3), 193–196. [Google Scholar] [CrossRef]
- Ortega, F.B.; Lavie, C.J.; Blair, S.N. Obesity and Cardiovascular Disease. Circ. Res. 2016, 118, 1752–1770. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.A.; Spencer, S.J. Obesity and neuroinflammation: A pathway to cognitive impairment. Brain Behav. Immun. 2014, 42, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.F.; Feng, D.D.; Chen, C. Contribution of adipocyte-derived factors to beta-cell dysfunction in diabetes. Int. J. Biochem. Cell Biol. 2006, 38, 804–819. [Google Scholar] [CrossRef] [PubMed]
- Booth, A.; Magnuson, A.; Fouts, J.; Foster, M.T. Adipose tissue: An endocrine organ playing a role in metabolic regulation. Horm. Mol. Biol. Clin. Investig. 2016, 26, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Osei, S.Y. Adipokines in obesity. Front. Horm. Res. 2008, 36, 182–197. [Google Scholar] [CrossRef]
- Martí, A.; Berraondo, B.; Martínez, J.A. Leptin: Physiological actions. J. Physiol. Biochem. 1999, 55, 43–49. [Google Scholar]
- Fehmann, H.-C.; Peiser, C.; Bode, H.-P.; Stamm, M.; Staats, P.; Hedetoft, C.; Lang, R.E.; Göke, B. Leptin: A potent inhibitor of insulin secretion. Peptides 1997, 18, 1267–1273. [Google Scholar] [CrossRef]
- Hill, J.W.; Elias, C.F.; Fukuda, M.; Williams, K.W.; Berglund, E.D.; Holland, W.L.; Cho, Y.-R.; Chuang, J.-C.; Xu, Y.; Choi, M.; et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010, 11, 286–297. [Google Scholar] [CrossRef]
- Caron, A.; Lemko, H.M.D.; Castorena, C.M.; Fujikawa, T.; Lee, S.; Lord, C.C.; Ahmed, N.; E Lee, C.; Holland, W.L.; Liu, C.; et al. POMC neurons expressing leptin receptors coordinate metabolic responses to fasting via suppression of leptin levels. Elife 2018, 7, e33710. [Google Scholar] [CrossRef] [PubMed]
- Kamohara, S.; Burcelin, R.; Halaas, J.L.; Friedman, J.M.; Charron, M.J. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature 1997, 389, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Kim, Y.-B.; Peroni, O.D.; Fryer, L.G.D.; Müller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Al Maskari, M.Y.; Alnaqdy, A.A. Correlation between Serum Leptin Levels, Body Mass Index and Obesity in Omanis. Sultan. Qaboos. Univ. Med. J. 2006, 6, 27–31. [Google Scholar] [PubMed]
- Zuo, H.; Shi, Z.; Yuan, B.; Dai, Y.; Wu, G.; Hussain, A. Association between serum leptin concentrations and insulin resistance: A population-based study from China. PLoS ONE 2013, 8, e54615. [Google Scholar] [CrossRef] [PubMed]
- Weyer, C.; Funahashi, T.; Tanaka, S.; Hotta, K.; Matsuzawa, Y.; Pratley, R.E.; Tataranni, P.A. Hypoadiponectinemia in obesity and type 2 diabetes: Close association with insulin resistance and hyperinsulinemia. J. Clin. Endocrinol. Metab. 2001, 86, 1930–1935. [Google Scholar] [CrossRef] [PubMed]
- Kazumi, T.; Kawaguchi, A.; Hirano, T.; Yoshino, G. Serum adiponectin is associated with high-density lipoprotein cholesterol, triglycerides, and low-density lipoprotein particle size in young healthy men. Metabolism 2004, 53, 589–593. [Google Scholar] [CrossRef]
- Kim, K.Y.; Kim, J.K.; Jeon, J.H.; Yoon, S.R.; Choi, I.; Yang, Y. c-Jun N-terminal kinase is involved in the suppression of adiponectin expression by TNF-alpha in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2005, 327, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Kamigaki, M.; Sakaue, S.; Tsujino, I.; Ohira, H.; Ikeda, D.; Itoh, N.; Ishimaru, S.; Ohtsuka, Y.; Nishimura, M. Oxidative stress provokes atherogenic changes in adipokine gene expression in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2006, 339, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef]
- Madsen, E.L.; Rissanen, A.; Bruun, J.M.; Skogstrand, K.; Tonstad, S.; Hougaard, D.M.; Richelsen, B. Weight loss larger than 10% is needed for general improvement of levels of circulating adiponectin and markers of inflammation in obese subjects: A 3-year weight loss study. Eur. J. Endocrinol. 2008, 158, 179–187. [Google Scholar] [CrossRef]
- Pischon, T.; Girman, C.J.; Hotamisligil, G.S.; Rifai, N.; Hu, F.B.; Rimm, E.B. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA 2004, 291, 1730–1737. [Google Scholar] [CrossRef]
- Hotta, K.; Funahashi, T.; Arita, Y.; Takahashi, M.; Matsuda, M.; Okamoto, Y.; Iwahashi, H.; Kuriyama, H.; Ouchi, N.; Maeda, K.; et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1595–1599. [Google Scholar] [CrossRef]
- Putz, D.M.; Goldner, W.S.; Bar, R.S.; Haynes, W.G.; Sivitz, W.I. Adiponectin and C-reactive protein in obesity, type 2 diabetes, and monodrug therapy. Metabolism 2004, 53, 1454–1461. [Google Scholar] [CrossRef]
- Bahceci, M.; Gokalp, D.; Bahceci, S.; Tuzcu, A.; Atmaca, S.; Arikan, S. The correlation between adiposity and adiponectin, tumor necrosis factor alpha, interleukin-6 and high sensitivity C-reactive protein levels. Is adipocyte size associated with inflammation in adults? J. Endocrinol. Investig. 2007, 30, 210–214. [Google Scholar] [CrossRef]
- Kim, C.; Park, J.; Park, J.; Kang, E.; Ahn, C.; Cha, B.; Lim, S.; Kim, K.; Lee, H. Comparison of body fat composition and serum adiponectin levels in diabetic obesity and non-diabetic obesity. Obesity 2006, 14, 1164–1171. [Google Scholar] [CrossRef]
- Derosa, G.; Catena, G.; Gaudio, G.; D’Angelo, A.; Maffioli, P. Adipose tissue dysfunction and metabolic disorders: Is it possible to predict who will develop type 2 diabetes mellitus? Role of markErs in the progreSsion of dIabeteS in obese paTIeNts (The RESISTIN trial). Cytokine 2020, 127, 154947. [Google Scholar] [CrossRef]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef]
- Sandeep, S.; Velmurugan, K.; Deepa, R.; Mohan, V. Serum visfatin in relation to visceral fat, obesity, and type 2 diabetes mellitus in Asian Indians. Metabolism 2007, 56, 565–570. [Google Scholar] [CrossRef]
- Boucher, J.; Masri, B.; Daviaud, D.; Gesta, S.; Guigné, C.; Mazzucotelli, A.; Castan-Laurell, I.; Tack, I.; Knibiehler, B.; Carpéné, C.; et al. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology 2005, 146, 1764–1771. [Google Scholar] [CrossRef]
- Xu, A.; Wang, Y.; Xu, J.Y.; Stejskal, D.; Tam, S.; Zhang, J.; Wat, N.M.; Wong, W.K.; Lam, K.S. Adipocyte fatty acid-binding protein is a plasma biomarker closely associated with obesity and metabolic syndrome. Clin. Chem. 2006, 52, 405–413. [Google Scholar] [CrossRef]
- Guo, D.; Liu, J.; Zhang, P.; Yang, X.; Liu, D.; Lin, J.; Wei, X.; Xu, B.; Huang, C.; Zhou, X.; et al. Adiposity Measurements and Metabolic Syndrome Are Linked Through Circulating Neuregulin 4 and Adipsin Levels in Obese Adults. Front. Physiol. 2021, 12, 667330. [Google Scholar] [CrossRef]
- Shoukry, A.; Shalaby, S.M.; El-Arabi Bdeer, S.; Mahmoud, A.A.; Mousa, M.M.; Khalifa, A. Circulating serum irisin levels in obesity and type 2 diabetes mellitus. IUBMB Life 2016, 68, 544–556. [Google Scholar] [CrossRef]
- Gil-Campos, M.; Aguilera, C.M.; Cañete, R.; Gil, A. Ghrelin: A hormone regulating food intake and energy homeostasis. Br. J. Nutr. 2006, 96, 201–226. [Google Scholar] [CrossRef]
- Colldén, G.; Tschöp, M.H.; Müller, T.D. Therapeutic Potential of Targeting the Ghrelin Pathway. Int. J. Mol. Sci. 2017, 18, 798. [Google Scholar] [CrossRef]
- Katsuki, A.; Urakawa, H.; Gabazza, E.; Murashima, S.; Nakatani, K.; Togashi, K.; Yano, Y.; Adachi, Y.; Sumida, Y. Circulating levels of active ghrelin is associated with abdominal adiposity, hyperinsulinemia and insulin resistance in patients with type 2 diabetes mellitus. Eur. J. Endocrinol. 2004, 151, 573–577. [Google Scholar] [CrossRef]
- Cuevas-Sierra, A.; Ramos-Lopez, O.; Riezu-Boj, J.I.; Milagro, F.I.; Martinez, J.A. Diet, Gut Microbiota, and Obesity: Links with Host Genetics and Epigenetics and Potential Applications. Adv. Nutr. 2019, 10 (Suppl. S1), S17–S30. [Google Scholar] [CrossRef]
- Amabebe, E.; Robert, F.O.; Agbalalah, T.; Orubu, E.S.F. Microbial dysbiosis-induced obesity: Role of gut microbiota in homoeostasis of energy metabolism. Br. J. Nutr. 2020, 123, 1127–1137. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martínez, O.; Naya, I.; García-Honduvilla, N.; Álvarez-Mon, M.; Buján, J.; Asúnsolo, Á.; de la Torre, B. Type 2 Diabetes Mellitus Associated with Obesity (Diabesity). The Central Role of Gut Microbiota and Its Translational Applications. Nutrients 2020, 12, 2749. [Google Scholar] [CrossRef]
- Bauer, P.V.; Hamr, S.C.; Duca, F.A. Regulation of energy balance by a gut-brain axis and involvement of the gut microbiota. Cell. Mol. Life Sci. 2016, 73, 737–755. [Google Scholar] [CrossRef]
- Barrea, L.; Muscogiuri, G.; Annunziata, G.; Laudisio, D.; Pugliese, G.; Salzano, C.; Colao, A.; Savastano, S. From gut microbiota dysfunction to obesity: Could short-chain fatty acids stop this dangerous course? Hormones 2019, 18, 245–250. [Google Scholar] [CrossRef]
- Wang, S.Z.; Yu, Y.J.; Adeli, K. Role of Gut Microbiota in Neuroendocrine Regulation of Carbohydrate and Lipid Metabolism via the Microbiota-Gut-Brain-Liver Axis. Microorganisms 2020, 8, 527. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Cerf-Bensussan, N.; Gaboriau-Routhiau, V. The immune system and the gut microbiota: Friends or foes? Nat. Rev. Immunol. 2010, 10, 735–744. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef]
- Hugon, P.; Lagier, J.C.; Colson, P.; Bittar, F.; Raoult, D. Repertoire of human gut microbes. Microb. Pathog. 2017, 106, 103–112. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Lepage, P.; Leclerc, M.C.; Joossens, M.; Mondot, S.; Blottière, H.M.; Raes, J.; Ehrlich, D.; Doré, J. A metagenomic insight into our gut’s microbiome. Gut 2013, 62, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi-Man, O.; Davenport, E.R.; Gilad, Y. Taxonomic classification of bacterial 16S rRNA genes using short sequencing reads: Evaluation of effective study designs. PLoS ONE 2013, 8, e53608. [Google Scholar] [CrossRef]
- Poretsky, R.; Rodriguez-R, L.M.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef]
- Kommineni, S.; Bretl, D.J.; Lam, V.; Chakraborty, R.; Hayward, M.; Simpson, P.; Cao, Y.; Bousounis, P.; Kristich, C.J.; Salzman, N.H. Bacteriocin production augments niche competition by enterococci in the mammalian gastrointestinal tract. Nature 2015, 526, 719–722. [Google Scholar] [CrossRef]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef]
- Mshvildadze, M.; Neu, J. The infant intestinal microbiome: Friend or foe? Early Hum. Dev. 2010, 86 (Suppl. S1), 67–71, Correction in Early Hum. Dev. 2014, 90, 163–164. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef]
- Harmsen, H.J.M.; Wildeboer–Veloo, A.C.M.; Raangs, G.C.; Wagendorp, A.A.; Klijn, N.; Bindels, J.G.; Welling, G.W. Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J. Pediatr. Gastroenterol. Nutr. 2000, 30, 61–67. [Google Scholar] [CrossRef]
- Biasucci, G.; Benenati, B.; Morelli, L.; Bessi, E.; Boehm, G. Cesarean delivery may affect the early biodiversity of intestinal bacteria. J. Nutr. 2008, 138, 1796S–1800S. [Google Scholar] [CrossRef]
- Dedrick, S.; Sundaresh, B.; Huang, Q.; Brady, C.; Yoo, T.; Cronin, C.; Rudnicki, C.; Flood, M.; Momeni, B.; Ludvigsson, J.; et al. The Role of Gut Microbiota and Environmental Factors in Type 1 Diabetes Pathogenesis. Front. Endocrinol 2020, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Power, S.E.; O’Toole, P.W.; Stanton, C.; Ross, R.P.; Fitzgerald, G.F. Intestinal microbiota, diet and health. Br. J. Nutr. 2014, 111, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Mika, A.; Van Treuren, W.; González, A.; Herrera, J.J.; Knight, R.; Fleshner, M. Exercise is More Effective at Altering Gut Microbial Composition and Producing Stable Changes in Lean Mass in Juvenile versus Adult Male F344 Rats. PLoS ONE 2015, 10, e0125889. [Google Scholar] [CrossRef]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Bäckhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef]
- Rodríguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 2015, 26, 26050. [Google Scholar] [CrossRef]
- Kleessen, B.; Bezirtzoglou, E.; Maettoe, J. Culture-based knowledge on biodiversity, development and stability of human gastrointestinal microflora. Microb. Ecol. Health Dis. 2000, 12, 53–63. [Google Scholar] [CrossRef]
- Leser, T.D.; Mølbak, L. Better living through microbial action: The benefits of the mammalian gastrointestinal microbiota on the host. Environ. Microbiol. 2009, 11, 2194–2206. [Google Scholar] [CrossRef]
- Wang, M.; Ahrné, S.; Jeppsson, B.; Molin, G. Comparison of bacterial diversity along the human intestinal tract by direct cloning and sequencing of 16S rRNA genes. FEMS Microbiol. Ecol. 2005, 54, 219–231. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; Raes, J.; van den Bogert, B.; Arumugam, M.; Booijink, C.C.G.M.; Troost, F.J.; Bork, P.; Wels, M.; De Vos, W.M.; Kleerebezem, M. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012, 6, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Yankovsky, D.S.; Shyrokobokov, V.P.; Dyment, G.S. Integral Role of Symbiotic Microflora in Human Physiology; Ltd. Chervona Ruta–Turs: Kyiv, Ukraine, 2011. [Google Scholar]
- Kobyliak, N.; Virchenko, O.; Falalyeyeva, T. Pathophysiological role of host microbiota in the development of obesity. Nutr. J. 2016, 15, 43. [Google Scholar] [CrossRef] [PubMed]
- Brestoff, J.R.; Artis, D. Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 2013, 14, 676–684. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef]
- Flint, H.J.; Duncan, S.H.; Scott, K.P.; Louis, P. Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc. 2015, 74, 13–22. [Google Scholar] [CrossRef]
- Adak, A.; Khan, M.R. An insight into gut microbiota and its functionalities. Cell. Mol. Life Sci. 2019, 76, 473–493. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, N.; Duncan, S.H.; Young, P.; Belenguer, A.; McWilliam Leitch, C.; Scott, K.P.; Flint, H.J.; Louis, P. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 2014, 8, 1323–1335, Correction in ISME J. 2014, 8, 1352. [Google Scholar] [CrossRef]
- Delzenne, N.M.; Cani, P.D.; Everard, A.; Neyrinck, A.M.; Bindels, L.B. Gut microorganisms as promising targets for the management of type 2 diabetes. Diabetologia 2015, 58, 2206–2217. [Google Scholar] [CrossRef]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [CrossRef]
- Schwiertz, A.; Jacobi, M.; Frick, J.S.; Richter, M.; Rusch, K.; Köhler, H. Microbiota in pediatric inflammatory bowel disease. J. Pediatr. 2010, 157, 240–244.e1. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.K.; Muir, J.G.; Gibson, P.R. Review article: Insights into colonic protein fermentation, its modulation and potential health implications. Aliment. Pharmacol. Ther. 2016, 43, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Yamaoka, M.; Takei, M.; Ando, T.; Taniguchi, S.; Ishii, I.; Tohya, K.; Ishizaki, T.; Niki, I.; Kimura, T. Endogenous hydrogen sulfide protects pancreatic beta-cells from a high-fat diet-induced glucotoxicity and prevents the development of type 2 diabetes. Biochem. Biophys. Res. Commun. 2013, 442, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Blaut, M. Gut microbiota and energy balance: Role in obesity. Proc. Nutr. Soc. 2015, 74, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Demigné, C.; Morand, C.; Levrat, M.A.; Besson, C.; Moundras, C.; Rémésy, C. Effect of propionate on fatty acid and cholesterol synthesis and on acetate metabolism in isolated rat hepatocytes. Br. J. Nutr. 1995, 74, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Louis, P.; Thomson, J.M.; Flint, H.J. The role of pH in determining the species composition of the human colonic microbiota. Environ. Microbiol. 2009, 11, 2112–2122. [Google Scholar] [CrossRef]
- Macfarlane, G.T.; Macfarlane, S. Bacteria, colonic fermentation, and gastrointestinal health. J. AOAC Int. 2012, 95, 50–60. [Google Scholar] [CrossRef]
- Wang, H.B.; Wang, P.Y.; Wang, X.; Wan, Y.L.; Liu, Y.C. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Dig. Dis. Sci. 2012, 57, 3126–3135. [Google Scholar] [CrossRef]
- Khosravi, A.; Mazmanian, S.K. Disruption of the gut microbiome as a risk factor for microbial infections. Curr. Opin. Microbiol. 2013, 16, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.T.; Davis-Richardson, A.G.; Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE 2011, 6, e25792. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Emoto, T.; Yamashita, T.; Watanabe, H.; Hayashi, T.; Tabata, T.; Hoshi, N.; Hatano, N.; Ozawa, G.; Sasaki, N.; et al. Bacteroides vulgatus and Bacteroides dorei Reduce Gut Microbial Lipopolysaccharide Production and Inhibit Atherosclerosis. Circulation 2018, 138, 2486–2498. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.; Suzuki, Y.; Saito, Y. Butyrate reduces colonic paracellular permeability by enhancing PPARgamma activation. Biochem. Biophys. Res. Commun. 2002, 293, 827–831. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed]
- Reimann, F.; Tolhurst, G.; Gribble, F.M. G-protein-coupled receptors in intestinal chemosensation. Cell Metab. 2012, 15, 421–431. [Google Scholar] [CrossRef]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef]
- Bindels, L.B.; Dewulf, E.M.; Delzenne, N.M. GPR43/FFA2: Physiopathological relevance and therapeutic prospects. Trends Pharmacol. Sci. 2013, 34, 226–232. [Google Scholar] [CrossRef]
- Reimer, R.A.; Darimont, C.; Gremlich, S.; Nicolas-Métral, V.; Rüegg, U.T.; Macé, K. A human cellular model for studying the regulation of glucagon-like peptide-1 secretion. Endocrinology 2001, 142, 4522–4528. [Google Scholar] [CrossRef]
- Larraufie, P.; Martin-Gallausiaux, C.; Lapaque, N.; Dore, J.; Gribble, F.M.; Reimann, F.; Blottiere, H.M. SCFAs strongly stimulate PYY production in human enteroendocrine cells. Sci. Rep. 2018, 8, 74. [Google Scholar] [CrossRef]
- Al-Lahham, S.H.; Roelofsen, H.; Priebe, M.; Weening, D.; Dijkstra, M.; Hoek, A.; Rezaee, F.; Venema, K.; Vonk, R.J. Regulation of adipokine production in human adipose tissue by propionic acid. Eur. J. Clin. Investig. 2010, 40, 401–407. [Google Scholar] [CrossRef]
- Everard, A.; Cani, P.D. Gut microbiota and GLP-1. Rev. Endocr. Metab. Disord. 2014, 15, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef] [PubMed]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Ilyés, T.; Silaghi, C.N.; Crăciun, A.M. Diet-Related Changes of Short-Chain Fatty Acids in Blood and Feces in Obesity and Metabolic Syndrome. Biology 2022, 11, 1556. [Google Scholar] [CrossRef] [PubMed]
- May, K.S.; den Hartigh, L.J. Gut Microbial-Derived Short Chain Fatty Acids: Impact on Adipose Tissue Physiology. Nutrients 2023, 15, 272. [Google Scholar] [CrossRef] [PubMed]
- Benahmed, A.G.; Gasmi, A.; Doşa, A.; Chirumbolo, S.; Mujawdiya, P.K.; Aaseth, J.; Dadar, M.; Bjørklund, G. Association between the gut and oral microbiome with obesity. Anaerobe 2021, 70, 102248. [Google Scholar] [CrossRef]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 2014, 5, 3611. [Google Scholar] [CrossRef]
- Chambers, E.S.; Byrne, C.S.; Aspey, K.; Chen, Y.; Khan, S.; Morrison, D.J.; Frost, G. Acute oral sodium propionate supplementation raises resting energy expenditure and lipid oxidation in fasted humans. Diabetes Obes. Metab. 2018, 20, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Baothman, O.A.; Zamzami, M.A.; Taher, I.; Abubaker, J.; Abu-Farha, M. The role of Gut Microbiota in the development of obesity and Diabetes. Lipids Health Dis. 2016, 15, 108. [Google Scholar] [CrossRef]
- Lefort, C.; Cani, P.D. The Liver under the Spotlight: Bile Acids and Oxysterols as Pivotal Actors Controlling Metabolism. Cells 2021, 10, 400. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acids as Metabolic Regulators and Nutrient Sensors. Annu. Rev. Nutr. 2019, 39, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.R.; Want, E.J.; Geier, F.M.; Spagou, K.; Wilson, I.D.; Sidaway, J.E.; Nicholson, J.K.; Holmes, E. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4523–4530. [Google Scholar] [CrossRef] [PubMed]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.-U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Karpen, S.J. Intestinal transport and metabolism of bile acids. J. Lipid Res. 2015, 56, 1085–1099. [Google Scholar] [CrossRef]
- Kato, M.; Muto, Y.; Tanaka-Bandoh, K.; Watanabe, K.; Ueno, K. Sphingolipid composition in Bacteroides species. Anaerobe 1995, 1, 135–139. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Allin, K.H.; Nielsen, T.; Pedersen, O. Mechanisms in endocrinology: Gut microbiota in patients with type 2 diabetes mellitus. Eur. J. Endocrinol. 2015, 172, R167–R177. [Google Scholar] [CrossRef] [PubMed]
- Trabelsi, M.-S.; Daoudi, M.; Prawitt, J.; Ducastel, S.; Touche, V.; Sayin, S.I.; Perino, A.; Brighton, C.A.; Sebti, Y.; Kluza, J.; et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat. Commun. 2015, 6, 7629. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap—bile acids in metabolic control. Nat. Rev. Endocrinol. 2014, 10, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.R.; Haeusler, R.A. Bile acids in glucose metabolism and insulin signalling—Mechanisms and research needs. Nat. Rev. Endocrinol. 2019, 15, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.C.; Chickering, T.W.; Rosen, E.D.; Dussault, B.; Qin, Y.; Soukas, A.; Friedman, J.M.; Holmes, W.E.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma target gene encoding a novel angiopoietin-related protein associated with adipose differentiation. Mol. Cell. Biol. 2000, 20, 5343–5349. [Google Scholar] [CrossRef]
- El Aidy, S.; A Merrifield, C.; Derrien, M.; van Baarlen, P.; Hooiveld, G.; Levenez, F.; Doré, J.; Dekker, J.; Holmes, E.; Claus, S.P.; et al. The gut microbiota elicits a profound metabolic reorientation in the mouse jejunal mucosa during conventionalisation. Gut 2013, 62, 1306–1314. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef]
- Wu, H.J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef]
- Lathrop, S.K.; Bloom, S.M.; Rao, S.M.; Nutsch, K.; Lio, C.-W.; Santacruz, N.; Peterson, D.A.; Stappenbeck, T.S.; Hsieh, C.-S. Peripheral education of the immune system by colonic commensal microbiota. Nature 2011, 478, 250–254. [Google Scholar] [CrossRef]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; Van Der Veeken, J.; DeRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Rosenstiel, P. Stories of love and hate: Innate immunity and host-microbe crosstalk in the intestine. Curr. Opin. Gastroenterol. 2013, 29, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010, 328, 228–231. [Google Scholar] [CrossRef]
- Sonnenburg, E.D.; Smits, S.A.; Tikhonov, M.; Higginbottom, S.K.; Wingreen, N.S.; Sonnenburg, J.L. Diet-induced extinctions in the gut microbiota compound over generations. Nature 2016, 529, 212–215. [Google Scholar] [CrossRef]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.-Z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 90. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, K.; Rebholz, C.M.; Kim, J. Plant-based diets and incident metabolic syndrome: Results from a South Korean prospective cohort study. PLoS Med. 2020, 17, e1003371. [Google Scholar] [CrossRef]
- Suzuki, T.A.; Worobey, M. Geographical variation of human gut microbial composition. Biol. Lett. 2014, 10, 20131037. [Google Scholar] [CrossRef] [PubMed]
- Heiman, M.L.; Greenway, F.L. A healthy gastrointestinal microbiome is dependent on dietary diversity. Mol. Metab. 2016, 5, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Healey, G.R.; Murphy, R.; Brough, L.; Butts, C.A.; Coad, J. Interindividual variability in gut microbiota and host response to dietary interventions. Nutr. Rev. 2017, 75, 1059–1080. [Google Scholar] [CrossRef] [PubMed]
- Pu, S.; Khazanehei, H.; Jones, P.J.; Khafipour, E. Interactions between Obesity Status and Dietary Intake of Monounsaturated and Polyunsaturated Oils on Human Gut Microbiome Profiles in the Canola Oil Multicenter Intervention Trial (COMIT). Front. Microbiol. 2016, 7, 1612. [Google Scholar] [CrossRef]
- Grosicki, G.J.; Durk, R.P.; Bagley, J.R. Rapid gut microbiome changes in a world-class ultramarathon runner. Physiol. Rep. 2019, 7, e14313. [Google Scholar] [CrossRef]
- Rogers, M.A.M.; Aronoff, D.M. The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin. Microbiol. Infect. 2016, 22, e1–e178. [Google Scholar] [CrossRef]
- Bender, J.M.; Li, F.; Purswani, H.; Capretz, T.; Cerini, C.; Zabih, S.; Hung, L.; Francis, N.; Chin, S.; Pannaraj, P.S.; et al. Early exposure to antibiotics in the neonatal intensive care unit alters the taxonomic and functional infant gut microbiome. J. Matern. Fetal. Neonatal. Med. 2021, 34, 3335–3343. [Google Scholar] [CrossRef]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, B.S. Role of the gut microbiota in human nutrition and metabolism. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. S4), 9–17. [Google Scholar] [CrossRef] [PubMed]
- Tagliabue, A.; Elli, M. The role of gut microbiota in human obesity: Recent findings and future perspectives. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 160–168. [Google Scholar] [CrossRef]
- Bervoets, L.; Van Hoorenbeeck, K.; Kortleven, I.; Van Noten, C.; Hens, N.; Vael, C.; Goossens, H.; Desager, K.N.; Vankerckhoven, V. Differences in gut microbiota composition between obese and lean children: A cross-sectional study. Gut Pathog. 2013, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Riva, A.; Borgo, F.; Lassandro, C.; Verduci, E.; Morace, G.; Borghi, E.; Berry, D. Pediatric obesity is associated with an altered gut microbiota and discordant shifts in Firmicutes populations. Environ. Microbiol. 2017, 19, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Munukka, E.; Wiklund, P.; Pekkala, S.; Völgyi, E.; Xu, L.; Cheng, S.; Lyytikäinen, A.; Marjomäki, V.; Alen, M.; Vaahtovuo, J.; et al. Women with and without metabolic disorder differ in their gut microbiota composition. Obesity 2012, 20, 1082–1087. [Google Scholar] [CrossRef]
- Kasai, C.; Sugimoto, K.; Moritani, I.; Tanaka, J.; Oya, Y.; Inoue, H.; Tameda, M.; Shiraki, K.; Ito, M.; Takei, Y.; et al. Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. BMC Gastroenterol. 2015, 15, 100. [Google Scholar] [CrossRef]
- Remely, M.; Aumueller, E.; Jahn, D.; Hippe, B.; Brath, H.; Haslberger, A.G. Microbiota and epigenetic regulation of inflammatory mediators in type 2 diabetes and obesity. Benef. Microbes 2014, 5, 33–43. [Google Scholar] [CrossRef]
- Moreno-Indias, I.; Cardona, F.; Tinahones, F.J.; Queipo-Ortuño, M.I. Impact of the gut microbiota on the development of obesity and type 2 diabetes mellitus. Front. Microbiol. 2014, 5, 190. [Google Scholar] [CrossRef]
- Mai, V.; McCrary, Q.M.; Sinha, R.; Glei, M. Associations between dietary habits and body mass index with gut microbiota composition and fecal water genotoxicity: An observational study in African American and Caucasian American volunteers. Nutr. J. 2009, 8, 49. [Google Scholar] [CrossRef]
- Jumpertz, R.; Le, D.S.; Turnbaugh, P.J.; Trinidad, C.; Bogardus, C.; Gordon, J.I.; Krakoff, J. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am. J. Clin. Nutr. 2011, 94, 58–65. [Google Scholar] [CrossRef]
- Kocełak, P.; Żak-Gołąb, A.; Zahorska-Markiewicz, B.; Aptekorz, M.; Zientara, M.; Martirosian, G.; Chudek, J.; Olszanecka-Glinianowicz, M. Resting energy expenditure and gut microbiota in obese and normal weight subjects. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2816–2821. [Google Scholar]
- Cândido, F.G.; Valente, F.X.; Grześkowiak, Ł.M.; Moreira, A.P.B.; Rocha, D.M.U.P.; Alfenas, R.C.G. Impact of dietary fat on gut microbiota and low-grade systemic inflammation: Mechanisms and clinical implications on obesity. Int. J. Food Sci. Nutr. 2018, 69, 125–143. [Google Scholar] [CrossRef]
- Gomes, A.C.; Hoffmann, C.; Mota, J.F. The human gut microbiota: Metabolism and perspective in obesity. Gut Microbes 2018, 9, 308–325. [Google Scholar] [CrossRef]
- Cani, P.D.; Osto, M.; Geurts, L.; Everard, A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 2012, 3, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Kaessmann, H.; Pääbo, S. The genetical history of humans and the great apes. J. Intern. Med. 2002, 251, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ramadass, B.; Rani, B.S.; Pugazhendhi, S.; John, K.R.; Ramakrishna, B.S. Faecal microbiota of healthy adults in south India: Comparison of a tribal & a rural population. Indian J. Med. Res. 2017, 145, 237–246. [Google Scholar]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The role of short-chain fatty acids in health and disease. Adv. Immunol. 2014, 121, 91–119. [Google Scholar] [CrossRef] [PubMed]
- Segata, N. Gut Microbiome: Westernization and the Disappearance of Intestinal Diversity. Curr. Biol. 2015, 25, R611–R613. [Google Scholar] [CrossRef]
- Simões, C.D.; Maukonen, J.; Kaprio, J.; Rissanen, A.; Pietiläinen, K.H.; Saarela, M. Habitual dietary intake is associated with stool microbiota composition in monozygotic twins. J. Nutr. 2013, 143, 417–423. [Google Scholar] [CrossRef]
- Patterson, E.; O’Doherty, R.M.; Murphy, E.F.; Wall, R.; O’Sullivan, O.; Nilaweera, K.; Fitzgerald, G.F.; Cotter, P.D.; Ross, R.P.; Stanton, C. Impact of dietary fatty acids on metabolic activity and host intestinal microbiota composition in C57BL/6J mice. Br. J. Nutr. 2014, 111, 1905–1917. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.F.; Cotter, P.D.; Healy, S.; Marques, T.M.; O’Sullivan, O.; Fouhy, F.; Clarke, S.F.; O’Toole, P.W.; Quigley, E.M.; Stanton, C.; et al. Composition and energy harvesting capacity of the gut microbiota: Relationship to diet, obesity and time in mouse models. Gut 2010, 59, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cui, Y.; Zhu, S.; Feng, F.; Zheng, X. Characterization and antimicrobial activity of a pharmaceutical microemulsion. Int. J. Pharm. 2010, 395, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Li, X.; Weiszmann, J.; Wang, P.; Baribault, H.; Chen, J.-L.; Tian, H.; Li, Y. Activation of G protein-coupled receptor 43 in adipocytes leads to inhibition of lipolysis and suppression of plasma free fatty acids. Endocrinology 2008, 149, 4519–4526. [Google Scholar] [CrossRef] [PubMed]
- Bjursell, M.; Admyre, T.; Göransson, M.; Marley, A.E.; Smith, D.M.; Oscarsson, J.; Bohlooly, Y.M. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E211–E220. [Google Scholar] [CrossRef] [PubMed]
- Blaut, M.; Klaus, S. Intestinal microbiota and obesity. Handb. Exp. Pharmacol. 2012, 209, 251–273. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Park, J.-H.; Seok, S.-H.; Baek, M.-W.; Kim, D.-J.; Lee, K.-E.; Paek, K.-S.; Lee, Y.; Park, J.-H. Human originated bacteria, Lactobacillus rhamnosus PL60, produce conjugated linoleic acid and show anti-obesity effects in diet-induced obese mice. Biochim. Biophys. Acta 2006, 1761, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, T.; Kamei, A.; Watanabe, Y.; Koga, J.; Abe, K. Short-chain fructooligosaccharide regulates hepatic peroxisome proliferator-activated receptor alpha and farnesoid X receptor target gene expression in rats. J. Agric. Food Chem. 2010, 58, 7007–7012. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; DuGar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.-P.; Michel, M.-L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694.e3. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef]
- Conlon, M.A.; Bird, A.R. The impact of diet and lifestyle on gut microbiota and human health. Nutrients 2014, 7, 17–44. [Google Scholar] [CrossRef] [PubMed]
- Mayerhofer, C.C.; Ueland, T.; Broch, K.; Vincent, R.P.; Cross, G.F.; Dahl, C.P.; Aukrust, P.; Gullestad, L.; Hov, J.R.; Trøseid, M. Increased Secondary/Primary Bile Acid Ratio in Chronic Heart Failure. J. Card. Fail. 2017, 23, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Carbonero, F.; Zoetendal, E.G.; DeLany, J.P.; Wang, M.; Newton, K.; Gaskins, H.R.; O’keefe, S.J. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am. J. Clin. Nutr. 2013, 98, 111–120. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101, Correction in Nature 2014, 506, 396. [Google Scholar] [CrossRef]
- O’Keefe, S.J.D.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E.; et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 2015, 6, 6342. [Google Scholar] [CrossRef]
- Vaarala, O. Gut microbiota and type 1 diabetes. Rev. Diabet. Stud. 2012, 9, 251–259. [Google Scholar] [CrossRef]
- Alkanani, A.K.; Hara, N.; Gottlieb, P.A.; Ir, D.; Robertson, C.E.; Wagner, B.D.; Frank, D.N.; Zipris, D. Alterations in Intestinal Microbiota Correlate With Susceptibility to Type 1 Diabetes. Diabetes 2015, 64, 3510–3520. [Google Scholar] [CrossRef]
- Murri, M.; Leiva, I.; Gomez-Zumaquero, J.M.; Tinahones, F.J.; Cardona, F.; Soriguer, F.; Queipo-Ortuño, M.I. Gut microbiota in children with type 1 diabetes differs from that in healthy children: A case-control study. BMC Med. 2013, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- SEARCH for Diabetes in Youth Study Group; Liese, A.D.; D’Agostino, R.B., Jr.; Hamman, R.F.; Kilgo, P.D.; Lawrence, L.M.; Liu, L.L.; Loots, B.; Linder, B.; Marcovina, S.; et al. The burden of diabetes mellitus among US youth: Prevalence estimates from the SEARCH for Diabetes in Youth Study. Pediatrics 2006, 118, 1510–1518. [Google Scholar] [CrossRef]
- Gale, E.A. Declassifying diabetes. Diabetologia 2006, 49, 1989–1995. [Google Scholar] [CrossRef] [PubMed]
- Ludvigsson, J. Increasing Incidence but Decreasing Awareness of Type 1 Diabetes in Sweden. Diabetes Care 2017, 40, e143–e144. [Google Scholar] [CrossRef] [PubMed]
- Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Novelo, L.L.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; Hyoty, H.; et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011, 5, 82–91. [Google Scholar] [CrossRef]
- Mejía-León, M.E.; Barca, A.M. Diet, Microbiota and Immune System in Type 1 Diabetes Development and Evolution. Nutrients 2015, 7, 9171–9184. [Google Scholar] [CrossRef]
- Wen, L.; Duffy, A. Factors Influencing the Gut Microbiota, Inflammation, and Type 2 Diabetes. J. Nutr. 2017, 147, 1468S–1475S. [Google Scholar] [CrossRef]
- Trigwell, S.M.; Radford, P.M.; Page, S.R.; Loweth, A.C.; James, R.F.L.; Morgan, N.G.; Todd, I. Islet glutamic acid decarboxylase modified by reactive oxygen species is recognized by antibodies from patients with type 1 diabetes mellitus. Clin. Exp. Immunol. 2001, 126, 242–249. [Google Scholar] [CrossRef]
- Culina, S.; Brezar, V.; Mallone, R. Insulin and type 1 diabetes: Immune connections. Eur. J. Endocrinol. 2013, 168, R19–R31. [Google Scholar] [CrossRef]
- Atkinson, M.; Gale, E.A. Infant diets and type 1 diabetes: Too early, too late, or just too complicated? JAMA 2003, 290, 1771–1772. [Google Scholar] [CrossRef]
- Stewart, C.J.; Ajami, N.J.; O’brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef]
- Alves, J.G.; Figueiroa, J.N.; Meneses, J.; Alves, G.V. Breastfeeding protects against type 1 diabetes mellitus: A case-sibling study. Breastfeed. Med. 2012, 7, 25–28. [Google Scholar] [CrossRef]
- Mueller, N.T.; Bakacs, E.; Combellick, J.; Grigoryan, Z.; Dominguez-Bello, M.G. The infant microbiome development: Mom matters. Trends Mol. Med. 2015, 21, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Solís, G.; de Los Reyes-Gavilan, C.G.; Fernández, N.; Margolles, A.; Gueimonde, M. Establishment and development of lactic acid bacteria and bifidobacteria microbiota in breast-milk and the infant gut. Anaerobe 2010, 16, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Fioramonti, J.; Francois, A.; Robinson, T.; Neant, F.; Matuchansky, C. Review article: Bifidobacteria as probiotic agents—physiological effects and clinical benefits. Aliment. Pharmacol. Ther. 2005, 22, 495–512. [Google Scholar] [CrossRef] [PubMed]
- Insel, R.; Knip, M. Prospects for primary prevention of type 1 diabetes by restoring a disappearing microbe. Pediatr. Diabetes 2018, 19, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Vatanen, T.; Kostic, A.D.; D’hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hämäläinen, A.-M.; et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 2016, 165, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Gülden, E.; Ihira, M.; Ohashi, A.; Reinbeck, A.L.; Freudenberg, M.A.; Kolb, H.; Burkart, V. Toll-like receptor 4 deficiency accelerates the development of insulin-deficient diabetes in non-obese diabetic mice. PLoS ONE 2013, 8, e75385. [Google Scholar] [CrossRef] [PubMed]
- Benus, R.F.J.; van der Werf, T.S.; Welling, G.W.; Judd, P.A.; Taylor, M.A.; Harmsen, H.J.M.; Whelan, K. Association between Faecalibacterium prausnitzii and dietary fibre in colonic fermentation in healthy human subjects. Br. J. Nutr. 2010, 104, 693–700. [Google Scholar] [CrossRef]
- Aw, W.; Fukuda, S. Toward the comprehensive understanding of the gut ecosystem via metabolomics-based integrated omics approach. Semin. Immunopathol. 2015, 37, 5–16. [Google Scholar] [CrossRef]
- de Goffau, M.C.; Luopajärvi, K.; Knip, M.; Ilonen, J.; Ruohtula, T.; Härkönen, T.; Orivuori, L.; Hakala, S.; Welling, G.W.; Harmsen, H.J.; et al. Fecal microbiota composition differs between children with β-cell autoimmunity and those without. Diabetes 2013, 62, 1238–1244. [Google Scholar] [CrossRef]
- Dietert, R.R. The microbiome in early life: Self-completion and microbiota protection as health priorities. Birth Defects Res. B Dev. Reprod. Toxicol. 2014, 101, 333–340. [Google Scholar] [CrossRef]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E. Obesity and the human microbiome. Curr. Opin. Gastroenterol. 2010, 26, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.; Vogensen, F.K.; Van Den Berg, F.W.J.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef]
- Furet, J.-P.; Kong, L.-C.; Tap, J.; Poitou, C.; Basdevant, A.; Bouillot, J.-L.; Mariat, D.; Corthier, G.; Doré, J.; Henegar, C.; et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: Links with metabolic and low-grade inflammation markers. Diabetes 2010, 59, 3049–3057. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Sepp, E.; Kolk, H.; Lõivukene, K.; Mikelsaar, M. Higher blood glucose level associated with body mass index and gut microbiota in elderly people. Microb. Ecol. Health Dis. 2014, 25, 22857. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Everard, A.; Lazarevic, V.; Derrien, M.; Girard, M.; Muccioli, G.G.; Neyrinck, A.M.; Possemiers, S.; Van Holle, A.; François, P.; de Vos, W.M.; et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 2011, 60, 2775–2786, Correction in Diabetes 2011, 60, 3307. [Google Scholar] [CrossRef]
- Moreira, A.P.; Texeira, T.F.; Ferreira, A.B.; Peluzio Mdo, C.; Alfenas Rde, C. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br. J. Nutr. 2012, 108, 801–809. [Google Scholar] [CrossRef]
- Santos, N.C.; Silva, A.C.; Castanho, M.A.; Martins-Silva, J.; Saldanha, C. Evaluation of lipopolysaccharide aggregation by light scattering spectroscopy. Chembiochem 2003, 4, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Laugerette, F.; Vors, C.; Géloën, A.; Chauvin, M.-A.; Soulage, C.; Lambert-Porcheron, S.; Peretti, N.; Alligier, M.; Burcelin, R.; Laville, M.; et al. Emulsified lipids increase endotoxemia: Possible role in early postprandial low-grade inflammation. J. Nutr. Biochem. 2011, 22, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Louis, S.; Tappu, R.M.; Damms-Machado, A.; Huson, D.H.; Bischoff, S.C. Characterization of the Gut Microbial Community of Obese Patients Following a Weight-Loss Intervention Using Whole Metagenome Shotgun Sequencing. PLoS ONE 2016, 11, e0149564. [Google Scholar] [CrossRef] [PubMed]
- Gérard, C.; Brown, K.A. Obesity and breast cancer—Role of estrogens and the molecular underpinnings of aromatase regulation in breast adipose tissue. Mol. Cell Endocrinol. 2018, 466, 15–30. [Google Scholar] [CrossRef]
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Rose, D.J. Long-term dietary pattern of fecal donor correlates with butyrate production and markers of protein fermentation during in vitro fecal fermentation. Nutr. Res. 2014, 34, 749–759. [Google Scholar] [CrossRef]
- Neal, M.D.; Leaphart, C.; Levy, R.; Prince, J.; Billiar, T.R.; Watkins, S.; Li, J.; Cetin, S.; Ford, H.; Schreiber, A.; et al. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J. Immunol. 2006, 176, 3070–3079. [Google Scholar] [CrossRef]
- Radilla-Vázquez, R.B.; Parra-Rojas, I.; Martínez-Hernández, N.E.; Márquez-Sandoval, Y.F.; Illades-Aguiar, B.; Castro-Alarcón, N. Gut Microbiota and Metabolic Endotoxemia in Young Obese Mexican Subjects. Obes. Facts 2016, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.M.G.; Costa, J.A.; Alfenas, R.C.G. Metabolic endotoxemia and diabetes mellitus: A systematic review. Metabolism 2017, 68, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, W.C.; He, C.Y.; Han, Z.; Jin, D.Y.; Wang, L. Change of intestinal mucosa barrier function in the progress of non-alcoholic steatohepatitis in rats. World J. Gastroenterol. 2008, 14, 3254–3258. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Plöger, S.; Stumpff, F.; Penner, G.B.; Schulzke, J.; Gäbel, G.; Martens, H.; Shen, Z.; Günzel, D.; Aschenbach, J.R. Microbial butyrate and its role for barrier function in the gastrointestinal tract. Ann. N. Y. Acad. Sci. 2012, 1258, 52–59. [Google Scholar] [CrossRef]
- Muccioli, G.G.; Naslain, D.; Bäckhed, F.; Reigstad, C.S.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D. The endocannabinoid system links gut microbiota to adipogenesis. Mol. Syst. Biol. 2010, 6, 392. [Google Scholar] [CrossRef]
- Farhadi, A.; Gundlapalli, S.; Shaikh, M.; Frantzides, C.; Harrell, L.; Kwasny, M.M.; Keshavarzian, A. Susceptibility to gut leakiness: A possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int. 2008, 28, 1026–1033. [Google Scholar] [CrossRef]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Osbak, P.S.; Bindslev, N.; Hansen, M.B. Relationships between body mass index and short-circuit current in human duodenal and colonic mucosal biopsies. Acta Physiol. 2011, 201, 47–53. [Google Scholar] [CrossRef]
- Vors, C.; Pineau, G.; Drai, J.; Meugnier, E.; Pesenti, S.; Laville, M.; Laugerette, F.; Malpuech-Brugère, C.; Vidal, H.; Michalski, M.-C. Postprandial Endotoxemia Linked With Chylomicrons and Lipopolysaccharides Handling in Obese Versus Lean Men: A Lipid Dose-Effect Trial. J. Clin. Endocrinol. Metab. 2015, 100, 3427–3435. [Google Scholar] [CrossRef]
- Kim, S.H.; Plutzky, J. Brown Fat and Browning for the Treatment of Obesity and Related Metabolic Disorders. Diabetes Metab. J. 2016, 40, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; De Nigris, V.; Spiga, R.; Mancuso, E.; La Sala, L.; Antonicelli, R.; Testa, R.; Procopio, A.D.; Olivieri, F.; Ceriello, A. Inflammageing and metaflammation: The yin and yang of type 2 diabetes. Ageing Res. Rev. 2018, 41, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Iacob, S.; Iacob, D.G. Infectious Threats, the Intestinal Barrier, and Its Trojan Horse: Dysbiosis. Front. Microbiol. 2019, 10, 1676. [Google Scholar] [CrossRef] [PubMed]
- Stolfi, C.; Maresca, C.; Monteleone, G.; Laudisi, F. Implication of Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases. Biomedicines 2022, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.F. The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 2002, 347, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Alpizar-Rodriguez, D.; Lesker, T.R.; Gronow, A.; Gilbert, B.; Raemy, E.; Lamacchia, C.; Gabay, C.; Finckh, A.; Strowig, T. Prevotella copri in individuals at risk for rheumatoid arthritis. Ann. Rheum. Dis. 2019, 78, 590–593. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Huerta, O.D.; Pastor-Villaescusa, B.; Aguilera, C.M.; Gil, A. A Systematic Review of the Efficacy of Bioactive Compounds in Cardiovascular Disease: Phenolic Compounds. Nutrients 2015, 7, 5177–5216. [Google Scholar] [CrossRef]
- Panda, P.; Verma, H.K.; Lakkakula, S.; Merchant, N.; Kadir, F.; Rahman, S.; Jeffree, M.S.; Lakkakula, B.V.K.S.; Rao, P.V. Biomarkers of Oxidative Stress Tethered to Cardiovascular Diseases. Oxid. Med. Cell Longev. 2022, 2022, 9154295. [Google Scholar] [CrossRef] [PubMed]
- Silveira Rossi, J.L.; Barbalho, S.M.; Reverete de Araujo, R.; Bechara, M.D.; Sloan, K.P.; Sloan, L.A. Metabolic syndrome and cardiovascular diseases: Going beyond traditional risk factors. Diabetes Metab. Res. Rev. 2022, 38, e3502. [Google Scholar] [CrossRef]
- Münzel, T.; Daiber, A.; Ullrich, V.; Mülsch, A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Quagliaro, L.; D’amico, M.; Di Filippo, C.; Marfella, R.; Nappo, F.; Berrino, L.; Rossi, F.; Giugliano, D. Acute hyperglycemia induces nitrotyrosine formation and apoptosis in perfused heart from rat. Diabetes 2002, 51, 1076–1082. [Google Scholar] [CrossRef]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef]
- Turkbey, E.B.; McClelland, R.L.; Kronmal, R.A.; Burke, G.L.; Bild, D.E.; Tracy, R.P.; Arai, A.E.; Lima, J.A.; Bluemke, D.A. The impact of obesity on the left ventricle: The Multi-Ethnic Study of Atherosclerosis (MESA). JACC Cardiovasc. Imaging 2010, 3, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Shulman, G.I. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N. Engl. J. Med. 2014, 371, 1131–1141, Correction in N. Engl. J. Med. 2014, 371, 2241. [Google Scholar] [CrossRef]
- Marsh, J.B. Lipoprotein metabolism in obesity and diabetes: Insights from stable isotope kinetic studies in humans. Nutr. Rev. 2003, 61, 363–375. [Google Scholar] [CrossRef]
- Barter, P.J.; Hopkins, G.J.; Calvert, G.D. Transfers and exchanges of esterified cholesterol between plasma lipoproteins. Biochem. J. 1982, 208, 1–7. [Google Scholar] [CrossRef]
- Santamarina-Fojo, S.; Haudenschild, C.; Amar, M. The role of hepatic lipase in lipoprotein metabolism and atherosclerosis. Curr. Opin. Lipidol. 1998, 9, 211–219. [Google Scholar] [CrossRef]
- Khirfan, G.; Tejwani, V.; Wang, X.; Li, M.; DiDonato, J.; Dweik, R.A.; Smedira, N.; Heresi, G.A. Plasma levels of high density lipoprotein cholesterol and outcomes in chronic thromboembolic pulmonary hypertension. PLoS ONE 2018, 13, e0197700. [Google Scholar] [CrossRef] [PubMed]
- Kopeć, G.; Waligóra, M.; Tyrka, A.; Jonas, K.; Pencina, M.J.; Zdrojewski, T.; Moertl, D.; Stokwiszewski, J.; Zagożdżon, P.; Podolec, P. Low-density lipoprotein cholesterol and survival in pulmonary arterial hypertension. Sci. Rep. 2017, 7, 41650. [Google Scholar] [CrossRef]
- Hurt-Camejo, E.; Camejo, G.; Rosengren, B.; Lopez, F.; Wiklund, O.; Bondjers, G. Differential uptake of proteoglycan-selected subfractions of low density lipoprotein by human macrophages. J. Lipid Res. 1990, 31, 1387–1398. [Google Scholar] [CrossRef]
- Tribble, D.L.; Rizzo, M.; Chait, A.; Lewis, D.M.; Blanche, P.J.; Krauss, R.M. Enhanced oxidative susceptibility and reduced antioxidant content of metabolic precursors of small, dense low-density lipoproteins. Am. J. Med. 2001, 110, 103–110. [Google Scholar] [CrossRef]
- Zmysłowski, A.; Szterk, A. Current knowledge on the mechanism of atherosclerosis and pro-atherosclerotic properties of oxysterols. Lipids Health Dis. 2017, 16, 188. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.K.; Barnard, R.J.; Sindhu, R.K.; Jurczak, M.; Ehdaie, A.; Vaziri, N.D. A high-fat, refined-carbohydrate diet induces endothelial dysfunction and oxidant/antioxidant imbalance and depresses NOS protein expression. J. Appl. Physiol. 2005, 98, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Czernichow, S.; Greenfield, J.R.; Galan, P.; Jellouli, F.; E Safar, M.; Blacher, J.; Hercberg, S.; I Levy, B. Macrovascular and microvascular dysfunction in the metabolic syndrome. Hypertens. Res. 2010, 33, 293–297. [Google Scholar] [CrossRef]
- Greenstein, A.S.; Khavandi, K.; Withers, S.B.; Sonoyama, K.; Clancy, O.; Jeziorska, M.; Laing, I.; Yates, A.P.; Pemberton, P.W.; Malik, R.A.; et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation 2009, 119, 1661–1670. [Google Scholar] [CrossRef]
- Lteif, A.A.; Han, K.; Mather, K.J. Obesity, insulin resistance, and the metabolic syndrome: Determinants of endothelial dysfunction in whites and blacks. Circulation 2005, 112, 32–38. [Google Scholar] [CrossRef]
- Rutter, M.K.; Meigs, J.B.; Sullivan, L.M.; D’Agostino RBSr Wilson, P.W. C-reactive protein, the metabolic syndrome, and prediction of cardiovascular events in the Framingham Offspring Study. Circulation 2004, 110, 380–385. [Google Scholar] [CrossRef]
- Prenner, S.B.; Chirinos, J.A. Arterial stiffness in diabetes mellitus. Atherosclerosis 2015, 238, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, A.H.; Oommen, A.T.; Bridges, L.R. Endothelial cells and human cerebral small vessel disease. Brain Pathol. 2015, 25, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Moorhouse, P.; Rockwood, K. Vascular cognitive impairment: Current concepts and clinical developments. Lancet Neurol. 2008, 7, 246–255. [Google Scholar] [CrossRef]
- van Norden, A.G.; van Dijk, E.J.; de Laat, K.F.; Scheltens, P.; Olderikkert, M.G.; de Leeuw, F.E. Dementia: Alzheimer pathology and vascular factors: From mutually exclusive to interaction. Biochim. Biophys. Acta 2012, 1822, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Fowler, M.J. Microvascular and macrovascular complications of diabetes. Clin. Diabetes 2008, 26, 77–82. [Google Scholar] [CrossRef]
- Pugh, M.E.; Hemnes, A.R. Metabolic and hormonal derangements in pulmonary hypertension: From mouse to man. Int. J. Clin. Pract. Suppl. 2010, 64, 5–13. [Google Scholar] [CrossRef]
- Magliano, D.J.; Islam, R.M.; Barr, E.L.M.; Gregg, E.W.; E Pavkov, M.; Harding, J.L.; Tabesh, M.; Koye, D.N.; E Shaw, J. Trends in incidence of total or type 2 diabetes: Systematic review. BMJ 2019, 366, l5003. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Bir, S.C.; Kevil, C.G. Endothelial dysfunction and diabetes: Effects on angiogenesis, vascular remodeling, and wound healing. Int. J. Vasc. Med. 2012, 2012, 918267. [Google Scholar] [CrossRef]
- Hamilton, S.J.; Watts, G.F. Endothelial dysfunction in diabetes: Pathogenesis, significance, and treatment. Rev. Diabet. Stud. 2013, 10, 133–156. [Google Scholar] [CrossRef]
- El-Najjar, N.; Kulkarni, R.P.; Nader, N.; Hodeify, R.; Machaca, K. Effects of Hyperglycemia on Vascular Smooth Muscle Ca2+ Signaling. BioMed Res. Int. 2017, 2017, 3691349. [Google Scholar] [CrossRef]
- Makino, A.; Kamata, K. Time-course changes in plasma endothelin-1 and its effects on the mesenteric arterial bed in streptozotocin-induced diabetic rats. Diabetes Obes. Metab. 2000, 2, 47–55. [Google Scholar] [CrossRef]
- Kizub, I.V.; Klymenko, K.I.; Soloviev, A.I. Protein kinase C in enhanced vascular tone in diabetes mellitus. Int. J. Cardiol. 2014, 174, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, D.L.; Hutsell, T.C.; Keefer, L.K. Nitric oxide (NO) donor molecules: Effect of NO release rate on vascular smooth muscle cell proliferation in vitro. J. Cardiovasc. Pharmacol. 1995, 25, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Radomski, M.W.; Palmer, R.M.; Moncada, S. Modulation of platelet aggregation by an L-arginine-nitric oxide pathway. Trends Pharmacol. Sci. 1991, 12, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhou, Z.; Zhang, M.; Fan, L.; Forudi, F.; Zhou, X.; Qu, W.; Lincoff, A.M.; Schmidt, A.M.; Topol, E.J.; et al. Peroxisome proliferator-activated receptor gamma down-regulates receptor for advanced glycation end products and inhibits smooth muscle cell proliferation in a diabetic and nondiabetic rat carotid artery injury model. J. Pharmacol. Exp. Ther. 2006, 317, 37–43. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 2021, 11, 613780. [Google Scholar] [CrossRef]
- Scheen, A.J. De l’athérosclérose à l’athérothrombose: D’une pathologie chronique silencieuse à un accident aigu critique [From atherosclerosis to atherothrombosis: From a silent chronic pathology to an acute critical event]. Rev. Med. Liege 2018, 73, 224–228. [Google Scholar]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Otsuka, F.; Yasuda, S.; Noguchi, T.; Ishibashi-Ueda, H. Pathology of coronary atherosclerosis and thrombosis. Cardiovasc. Diagn. Ther. 2016, 6, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Hafiane, A. Vulnerable Plaque, Characteristics, Detection, and Potential Therapies. J. Cardiovasc. Dev Dis. 2019, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Bello, B.D.; Morbini, P.; Burke, A.P.; Bocciarelli, M.; Specchia, G.; Virmani, R. Plaque erosion is a major substrate for coronary thrombosis in acute myocardial infarction. Heart 1999, 82, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Burke, A.P.; Farb, A.; Kolodgie, F.D. Pathology of the vulnerable plaque. J. Am. Coll. Cardiol. 2006, 47 (Suppl. S8), C13–C18. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Kunju, S.U.; Badarudeen, S.; Schwarz, E.R. Impact of obesity in patients with congestive heart failure. Rev. Cardiovasc. Med. 2009, 10, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Cermenati, G.; Mitro, N.; Audano, M.; Melcangi, R.C.; Crestani, M.; De Fabiani, E.; Caruso, D. Lipids in the nervous system: From biochemistry and molecular biology to patho-physiology. Biochim. Biophys. Acta 2015, 1851, 51–60. [Google Scholar] [CrossRef]
- Penke, B.; Paragi, G.; Gera, J.; Berkecz, R.; Kovács, Z.; Crul, T.; Vígh, L. The Role of Lipids and Membranes in the Pathogenesis of Alzheimer’s Disease: A Comprehensive View. Curr. Alzheimer Res. 2018, 15, 1191–1212. [Google Scholar] [CrossRef]
- Hooijmans, C.R.; Kiliaan, A.J. Fatty acids, lipid metabolism and Alzheimer pathology. Eur. J. Pharmacol. 2008, 585, 176–196. [Google Scholar] [CrossRef]
- Berg, D.; Youdim, M.B.; Riederer, P. Redox imbalance. Cell Tissue Res. 2004, 318, 201–213. [Google Scholar] [CrossRef] [PubMed]
- McQuillen, P.S.; Ferriero, D.M. Selective vulnerability in the developing central nervous system. Pediatr. Neurol. 2004, 30, 227–235. [Google Scholar] [CrossRef] [PubMed]
- von Arnim, C.A.; Gola, U.; Biesalski, H.K. More than the sum of its parts? Nutrition in Alzheimer’s disease. Nutrition 2010, 26, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Grünblatt, E.; Riederer, P.; Gerlach, M.; Levites, Y.; Youdim, M.B. Neuroprotective strategies in PDinson’s disease: An update on progress. CNS Drugs 2003, 17, 729–762. [Google Scholar] [CrossRef] [PubMed]
- Stack, E.C.; Matson, W.R.; Ferrante, R.J. Evidence of oxidant damage in Huntington’s disease: Translational strategies using antioxidants. Ann. N. Y. Acad. Sci. 2008, 1147, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.C.; Kuo, C.L.; Cheng, W.L.; Liu, C.S.; Hsieh, M. Decreased antioxidant enzyme activity and increased mitochondrial DNA damage in cellular models of Machado-Joseph disease. J. Neurosci. Res. 2009, 87, 1884–1891. [Google Scholar] [CrossRef] [PubMed]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem. 1998, 71, 2034–2040. [Google Scholar] [CrossRef]
- Wong, M.W.; Braidy, N.; Poljak, A.; Pickford, R.; Thambisetty, M.; Sachdev, P.S. Dysregulation of lipids in Alzheimer’s disease and their role as potential biomarkers. Alzheimers Dement. 2017, 13, 810–827. [Google Scholar] [CrossRef]
- Sonnino, S.; Prinetti, A. Membrane domains and the “lipid raft” concept. Curr. Med. Chem. 2013, 20, 4–21. [Google Scholar]
- Díaz, M.; Fabelo, N.; Martín, V.; Ferrer, I.; Gómez, T.; Marín, R. Biophysical alterations in lipid rafts from human cerebral cortex associate with increased BACE1/AβPP interaction in early stages of Alzheimer’s disease. J. Alzheimers Dis. 2015, 43, 1185–1198. [Google Scholar] [CrossRef]
- Grassi, S.; Giussani, P.; Mauri, L.; Prioni, S.; Sonnino, S.; Prinetti, A. Lipid rafts and neurodegeneration: Structural and functional roles in physiologic aging and neurodegenerative diseases. J. Lipid Res. 2020, 61, 636–654. [Google Scholar] [CrossRef]
- Marin, R.; Fabelo, N.; Fernández-Echevarría, C.; Canerina-Amaro, A.; Rodríguez-Barreto, D.; Quinto-Alemany, D.; Mesa-Herrera, F.; Díaz, M. Lipid Raft Alterations in Aged-Associated Neuropathologies. Curr. Alzheimer Res. 2016, 13, 973–984. [Google Scholar] [CrossRef]
- Frisardi, V.; Solfrizzi, V.; Seripa, D.; Capurso, C.; Santamato, A.; Sancarlo, D.; Vendemiale, G.; Pilotto, A.; Panza, F. Metabolic-cognitive syndrome: A cross-talk between metabolic syndrome and Alzheimer’s disease. Ageing Res. Rev. 2010, 9, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Gustafson, D.R. Adiposity, type 2 diabetes, and Alzheimer’s disease. J. Alzheimers Dis. 2009, 16, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.S.; Beiser, A.S.; Fox, C.S.; Au, R.; Himali, J.J.; Debette, S.; DeCarli, C.; Vasan, R.S.; Wolf, P.A.; Seshadri, S. Association of metabolic dysregulation with volumetric brain magnetic resonance imaging and cognitive markers of subclinical brain aging in middle-aged adults: The Framingham Offspring Study. Diabetes Care 2011, 34, 1766–1770. [Google Scholar] [CrossRef]
- Debette, S.; Beiser, A.; Hoffmann, U.; DeCarli, C.; O’Donnell, C.J.; Massaro, J.M.; Au, R.; Himali, J.J.; Wolf, P.A.; Fox, C.S.; et al. Visceral fat is associated with lower brain volume in healthy middle-aged adults. Ann. Neurol. 2010, 68, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Yates, K.F.; Sweat, V.; Yau, P.L.; Turchiano, M.M.; Convit, A. Impact of metabolic syndrome on cognition and brain: A selected review of the literature. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2060–2067. [Google Scholar] [CrossRef]
- Burns, J.M.; Honea, R.A.; Vidoni, E.D.; Hutfles, L.J.; Brooks, W.M.; Swerdlow, R.H. Insulin is differentially related to cognitive decline and atrophy in Alzheimer’s disease and aging. Biochim. Biophys. Acta 2012, 1822, 333–339. [Google Scholar] [CrossRef]
- Knopman, D.; Boland, L.; Mosley, T.; Howard, G.; Liao, D.; Szklo, M.; McGovern, P.; Folsom, A.; Atherosclerosis Risk in Communities (ARIC) Study Investigators. Cardiovascular risk factors and cognitive decline in middle-aged adults. Neurology 2001, 56, 42–48. [Google Scholar] [CrossRef]
- Akomolafe, A.; Beiser, A.; Meigs, J.B.; Au, R.; Green, R.C.; Farrer, L.A.; Wolf, P.A.; Seshadri, S. Diabetes mellitus and risk of developing Alzheimer disease: Results from the Framingham Study. Arch. Neurol. 2006, 63, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kåreholt, I.; Winblad, B.; Helkala, E.-L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [CrossRef]
- Gorelick, P.B.; Scuteri, A.; Black, S.E.; DeCarli, C.; Greenberg, S.M.; Iadecola, C.; Launer, L.J.; Laurent, S.; Lopez, O.L.; Nyenhuis, D.; et al. Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the american heart association/american stroke association. Stroke 2011, 42, 2672–2713. [Google Scholar] [CrossRef]
- Grammas, P. Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflamm. 2011, 8, 26. [Google Scholar] [CrossRef]
- Marchesi, V.T. Alzheimer’s disease and CADASIL are heritable, adult-onset dementias that both involve damaged small blood vessels. Cell. Mol. Life Sci. 2014, 71, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Parikh, N.S.; Merkler, A.E.; Iadecola, C. Inflammation, Autoimmunity, Infection, and Stroke: Epidemiology and Lessons From Therapeutic Intervention. Stroke 2020, 51, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Hart, R.G.; Pearce, L.A.; Bakheet, M.F.; Benavente, O.R.; Conwit, R.A.; McClure, L.A.; Talbert, R.L.; Anderson, D.C. Predictors of stroke recurrence in patients with recent lacunar stroke and response to interventions according to risk status: Secondary prevention of small subcortical strokes trial. J. Stroke Cerebrovasc. Dis. 2014, 23, 618–624. [Google Scholar] [CrossRef] [PubMed][Green Version]
- PD, Y.K.; Yi, H.J.; Lee, Y.J.; Cho, H.; Chun, H.J.; Oh, S.J. The relationship between metabolic syndrome (MetS) and spontaneous intracerebral hemorrhage (ICH). Neurol. Sci. 2013, 34, 1523–1528. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Fang, F.; Fu, X.; Lin, H.; Gao, Q. Is Metabolic Syndrome Associated with the Risk of Recurrent Stroke: A Meta-Analysis of Cohort Studies. J. Stroke Cerebrovasc. Dis. 2017, 26, 2700–2705. [Google Scholar] [CrossRef]
- Peters, S.A.; Huxley, R.R.; Woodward, M. Diabetes as a risk factor for stroke in women compared with men: A systematic review and meta-analysis of 64 cohorts, including 775,385 individuals and 12,539 strokes. Lancet 2014, 383, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Folstein, M.F. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: Review and hypothesis. Neurobiol. Aging 2006, 27, 190–198. [Google Scholar] [CrossRef]
- Levine, D.A.; Galecki, A.T.; Langa, K.M.; Unverzagt, F.W.; Kabeto, M.U.; Giordani, B.; Wadley, V.G. Trajectory of Cognitive Decline After Incident Stroke. JAMA 2015, 314, 41–51. [Google Scholar] [CrossRef]
- Thiel, A.; Cechetto, D.F.; Heiss, W.D.; Hachinski, V.; Whitehead, S.N. Amyloid burden, neuroinflammation, and links to cognitive decline after ischemic stroke. Stroke 2014, 45, 2825–2829. [Google Scholar] [CrossRef] [PubMed]
- Pendlebury, S.T.; Rothwell, P.M. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: A systematic review and meta-analysis. Lancet Neurol. 2009, 8, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Sposato, L.A.; Kapral, M.K.; Fang, J.; Gill, S.S.; Hackam, D.G.; Cipriano, L.E.; Hachinski, V. Declining Incidence of Stroke and Dementia: Coincidence or Prevention Opportunity? JAMA Neurol. 2015, 72, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Cerasuolo, J.O.; Cipriano, L.E.; Sposato, L.A.; Kapral, M.K.; Fang, J.; Gill, S.S.; Hackam, D.G.; Hachinski, V. Population-based stroke and dementia incidence trends: Age and sex variations. Alzheimers Dement. 2017, 13, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Castro, D.M.; Dillon, C.; Machnicki, G.; Allegri, R.F. The economic cost of Alzheimer’s disease: Family or public health burden? Dement. Neuropsychol. 2010, 4, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Tóth, P.; Gavurová, B.; Barták, M. Alzheimer’s Disease Mortality according to Socioeconomic Factors: Country Study. Int. J. Alzheimers Dis. 2018, 2018, 8137464. [Google Scholar] [CrossRef]
- Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J. Alzheimers Dis. 2001, 3, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.E.; Green, K.N. Inflammation in Alzheimer’s disease: Lessons learned from microglia-depletion models. Brain Behav. Immun. 2017, 61, 1–11. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Verdile, G.; Fuller, S.; Atwood, C.S.; Laws, S.M.; Gandy, S.E.; Martins, R.N. The role of beta amyloid in Alzheimer’s disease: Still a cause of everything or the only one who got caught? Pharmacol. Res. 2004, 50, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Gouras, G.K.; Wang, R.; Gross, R.S.; Beal, M.F.; Greengard, P.; Xu, H. Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J. Neurosci. 2001, 21, 2561–2570. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: Implications for therapy. Acta Neuropathol. 2013, 126, 479–497. [Google Scholar] [CrossRef] [PubMed]
- McCaulley, M.E.; Grush, K.A. Seeking a New Paradigm for Alzheimer’s Disease: Considering the Roles of Inflammation, Blood-Brain Barrier Dysfunction, and Prion Disease. Int. J. Alzheimers Dis. 2017, 2017, 2438901. [Google Scholar] [CrossRef]
- Le Page, A.; Dupuis, G.; Frost, E.H.; Larbi, A.; Pawelec, G.; Witkowski, J.M.; Fulop, T. Role of the peripheral innate immune system in the development of Alzheimer’s disease. Exp. Gerontol. 2018, 107, 59–66. [Google Scholar] [CrossRef]
- Dansokho, C.; Heneka, M.T. Neuroinflammatory responses in Alzheimer’s disease. J. Neural Transm. 2018, 125, 771–779. [Google Scholar] [CrossRef]
- Tan, M.S.; Yu, J.T.; Jiang, T.; Zhu, X.C.; Tan, L. The NLRP3 inflammasome in Alzheimer’s disease. Mol. Neurobiol. 2013, 48, 875–882. [Google Scholar] [CrossRef]
- Zhou, K.; Shi, L.; Wang, Y.; Chen, S.; Zhang, J. Recent Advances of the NLRP3 Inflammasome in Central Nervous System Disorders. J. Immunol. Res. 2016, 2016, 9238290. [Google Scholar] [CrossRef] [PubMed]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-beta: A crucial factor in Alzheimer’s disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513. [Google Scholar] [CrossRef]
- McArthur, S.; Loiola, R.A.; Maggioli, E.; Errede, M.; Virgintino, D.; Solito, E. The restorative role of annexin A1 at the blood-brain barrier. Fluids Barriers CNS 2016, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Kinder, L.S.; Carnethon, M.R.; Palaniappan, L.P.; King, A.C.; Fortmann, S.P. Depression and the metabolic syndrome in young adults: Findings from the Third National Health and Nutrition Examination Survey. Psychosom. Med. 2004, 66, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Skilton, M.R.; Moulin, P.; Terra, J.L.; Bonnet, F. Associations between anxiety, depression, and the metabolic syndrome. Biol. Psychiatry 2007, 62, 1251–1257. [Google Scholar] [CrossRef]
- Ghanei Gheshlagh, R.; Parizad, N.; Sayehmiri, K. The Relationship Between Depression and Metabolic Syndrome: Systematic Review and Meta-Analysis Study. Iran Red. Crescent. Med. J. 2016, 18, e26523. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Cuijpers, P.; Penninx, B.W. Is depression associated with increased oxidative stress? A systematic review and meta-analysis. Psychoneuroendocrinology 2015, 51, 164–175. [Google Scholar] [CrossRef]
- Stetler, C.; Miller, G.E. Depression and hypothalamic-pituitary-adrenal activation: A quantitative summary of four decades of research. Psychosom. Med. 2011, 73, 114–126. [Google Scholar] [CrossRef]
- Fisher, A.J.; Newman, M.G. Heart rate and autonomic response to stress after experimental induction of worry versus relaxation in healthy, high-worry, and generalized anxiety disorder individuals. Biol. Psychol. 2013, 93, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Penninx, B.W.; Milaneschi, Y.; Lamers, F.; Vogelzangs, N. Understanding the somatic consequences of depression: Biological mechanisms and the role of depression symptom profile. BMC Med. 2013, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Alexopoulos, G.S.; Lopez, O.L.; Williamson, J.D.; Yaffe, K. Depressive symptoms, vascular disease, and mild cognitive impairment: Findings from the Cardiovascular Health Study. Arch. Gen. Psychiatry 2006, 63, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.E.; Myint, A. The psychoneuroimmunology of depression. Hum. Psychopharmacol. 2009, 24, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Swaab, D.F.; Bao, A.M.; Lucassen, P.J. The stress system in the human brain in depression and neurodegeneration. Ageing Res. Rev. 2005, 4, 141–194. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, S.R.; Schuppenies, A.; Wong, M.L.; Licinio, J. Approaching the shared biology of obesity and depression: The stress axis as the locus of gene-environment interactions. Mol. Psychiatry 2006, 11, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Eddouks, M.; Chattopadhyay, D.; De Feo, V.; Cho, W.C. Medicinal plants in the prevention and treatment of chronic diseases. Evid. Based Complement. Alternat. Med. 2012, 2012, 458274. [Google Scholar] [CrossRef]
- Tabatabaei-Malazy, O.; Larijani, B.; Abdollahi, M. Targeting metabolic disorders by natural products. J. Diabetes Metab. Disord. 2015, 14, 57. [Google Scholar] [CrossRef]
- Höhn, A.; Weber, D.; Jung, T.; Ott, C.; Hugo, M.; Kochlik, B.; Kehm, R.; König, J.; Grune, T.; Castro, J.P. Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 2017, 11, 482–501. [Google Scholar] [CrossRef]
- Halliwell, B. Free radicals and antioxidants—Quo vadis? Trends Pharmacol. Sci. 2011, 32, 125–130. [Google Scholar] [CrossRef]
- Bast, A.; Haenen, G.R. Ten misconceptions about antioxidants. Trends Pharmacol. Sci. 2013, 34, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Papas, A.M. Diet and antioxidant status. Food Chem. Toxicol. 1999, 37, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Akbarirad, H.; Ardabili, A.G.; Kazemeini, S.M.; Khaneghah, A.M. An overview on some of important sources of natural antioxidants. Int. Food Res. J. 2016, 23, 928–933. [Google Scholar]
- Pandey, K.B.; Rizvi, S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxid. Med. Cell. Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef]
- Parasuraman, S.; Thing, G.S.; Dhanaraj, S.A. Polyherbal formulation: Concept of ayurveda. Pharmacogn. Rev. 2014, 8, 73–80. [Google Scholar] [CrossRef]
- Saxena MS, J.; Nema, R.; Sigh, D.; Gupta, A. Phytochemsitry of Medical Plants. J. Pharm. Phytochem. 2013, 1, 168–182. [Google Scholar]
- Kumar, S. The importance of antioxidant and their role in pharmaceutical science—A review. Asian J. Res. Chem. Pharm. Sci. 2014, 1, 27–44. [Google Scholar]
- Rochlani, Y.; Pothineni, N.V.; Kovelamudi, S.; Mehta, J.L. Metabolic syndrome: Pathophysiology, management, and modulation by natural compounds. Ther. Adv. Cardiovasc. Dis. 2017, 11, 215–225. [Google Scholar] [CrossRef]
- Alesci, A.; Miller, A.; Tardugno, R.; Pergolizzi, S. Chemical analysis, biological and therapeutic activities of Olea europaea L. extracts. Nat. Prod. Res. 2022, 36, 2932–2945. [Google Scholar] [CrossRef]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (poly)phenolics in human health: Structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, S.E.; Nguyen, M.; Malik, V.S. Association between adherence to plant-based dietary patterns and obesity risk: A systematic review of prospective cohort studies. Appl. Physiol. Nutr. Metab. 2022, 47, 1115–1133. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Huang, J.; Song, D.; Deng, R.; Wei, J.; Zhang, Z. Increased Consumption of Fruit and Vegetables Is Related to a Reduced Risk of Cognitive Impairment and Dementia: Meta-Analysis. Front. Aging Neurosci. 2017, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Mazo, N.A.; Echeverria, V.; Cabezas, R.; Avila-Rodríguez, M.; Tarasov, V.V.; Yarla, N.S.; Aliev, G.; Barreto, G.E. Medicinal Plants as Protective Strategies Against Parkinson’s Disease. Curr. Pharm. Des. 2017, 23, 4180–4188. [Google Scholar] [CrossRef] [PubMed]
- Stefaniak, O.; Dobrzyńska, M.; Drzymała-Czyż, S.; Przysławski, J. Diet in the Prevention of Alzheimer’s Disease: Current Knowledge and Future Research Requirements. Nutrients 2022, 14, 4564. [Google Scholar] [CrossRef] [PubMed]
- Abourashed, E.A. Bioavailability of Plant-Derived Antioxidants. Antioxidants 2013, 2, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Margaritelis, N.V. Antioxidants as therapeutics in the intensive care unit: Have we ticked the redox boxes? Pharmacol. Res. 2016, 111, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Martins, N.; Barros, L.; Ferreira, I.C. In vivo antioxidant activity of phenolic compounds: Facts and gaps. Trends Food Sci. Technol. 2016, 48, 1–2. [Google Scholar] [CrossRef]
- Altenhöfer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef]
- Fields, M.; Marcuzzi, A.; Gonelli, A.; Celeghini, C.; Maximova, N.; Rimondi, E. Mitochondria-Targeted Antioxidants, an Innovative Class of Antioxidant Compounds for Neurodegenerative Diseases: Perspectives and Limitations. Int. J. Mol. Sci. 2023, 24, 3739. [Google Scholar] [CrossRef]
- Grundy, S.M. Metabolic syndrome update. Trends Cardiovasc. Med. 2016, 26, 364–373. [Google Scholar] [CrossRef]
- Sjöholm, K.; Sjöström, E.; Carlsson, L.M.; Peltonen, M. Weight Change-Adjusted Effects of Gastric Bypass Surgery on Glucose Metabolism: 2- and 10-Year Results From the Swedish Obese Subjects (SOS) Study. Diabetes Care 2016, 39, 625–631. [Google Scholar] [CrossRef]
- Lean, M.E.J.; Leslie, W.S.; Barnes, A.C.; Brosnahan, N.; Thom, G.; McCombie, L.; Peters, C.; Zhyzhneuskaya, S.; Al-Mrabeh, A.; Hollingsworth, K.G.; et al. Durability of a primary care-led weight-management intervention for remission of type 2 diabetes: 2-year results of the DiRECT open-label, cluster-randomised trial. Lancet Diabetes Endocrinol. 2019, 7, 344–355. [Google Scholar] [CrossRef]
- Lingvay, I.; Sumithran, P.; Cohen, R.V.; le Roux, C.W. Obesity management as a primary treatment goal for type 2 diabetes: Time to reframe the conversation. Lancet 2022, 399, 394–405, Correction in Lancet. 2022, 399, 358. [Google Scholar] [CrossRef] [PubMed]
- Clamp, L.D.; Hume, D.J.; Lambert, E.V.; Kroff, J. Enhanced insulin sensitivity in successful, long-term weight loss maintainers compared with matched controls with no weight loss history. Nutr. Diabetes 2017, 7, e282. [Google Scholar] [CrossRef] [PubMed]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Moazzeni, S.S.; Arani, R.H.; Deravi, N.; Hasheminia, M.; Khalili, D.; Azizi, F.; Hadaegh, F. Weight change and risk of cardiovascular disease among adults with type 2 diabetes: More than 14 years of follow-up in the Tehran Lipid and Glucose Study. Cardiovasc. Diabetol. 2021, 20, 141. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Suarez, J.A.; Longo, V.D. Protein and amino acid restriction, aging and disease: From yeast to humans. Trends Endocrinol. Metab. 2014, 25, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Most, J.; Tosti, V.; Redman, L.M.; Fontana, L. Calorie restriction in humans: An update. Ageing Res. Rev. 2017, 39, 36–45. [Google Scholar] [CrossRef]
- Montefusco, L.; D’addio, F.; Loretelli, C.; Ben Nasr, M.; Garziano, M.; Rossi, A.; Pastore, I.; Plebani, L.; Lunati, M.E.; Bolla, A.M.; et al. Anti-inflammatory effects of diet and caloric restriction in metabolic syndrome. J. Endocrinol. Investig. 2021, 44, 2407–2415. [Google Scholar] [CrossRef]
- Boden, G.; Sargrad, K.; Homko, C.; Mozzoli, M.; Stein, T.P. Effect of a low-carbohydrate diet on appetite, blood glucose levels, and insulin resistance in obese patients with type 2 diabetes. Ann. Intern. Med. 2005, 142, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.S.; Tjonn, S.L.; Swan, P.D.; White, A.; Hutchins, H.; Sears, B. Ketogenic low-carbohydrate diets have no metabolic advantage over nonketogenic low-carbohydrate diets. Am. J. Clin. Nutr. 2006, 83, 1055–1061. [Google Scholar] [CrossRef]
- Ferraz-Bannitz, R.; Beraldo, R.A.; Peluso, A.A.; Dall, M.; Babaei, P.; Foglietti, R.C.; Martins, L.M.; Gomes, P.M.; Marchini, J.S.; Suen, V.M.M.; et al. Dietary Protein Restriction Improves Metabolic Dysfunction in Patients with Metabolic Syndrome in a Randomized, Controlled Trial. Nutrients 2022, 14, 2670. [Google Scholar] [CrossRef]
- Schübel, R.; Nattenmüller, J.; Sookthai, D.; Nonnenmacher, T.; E Graf, M.; Riedl, L.; Schlett, C.L.; von Stackelberg, O.; Johnson, T.; Nabers, D.; et al. Effects of intermittent and continuous calorie restriction on body weight and metabolism over 50 wk: A randomized controlled trial. Am. J. Clin. Nutr. 2018, 108, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Vasim, I.; Majeed, C.N.; DeBoer, M.D. Intermittent Fasting and Metabolic Health. Nutrients 2022, 14, 631. [Google Scholar] [CrossRef]
- Saxton, S.N.; Clark, B.J.; Withers, S.B.; Eringa, E.C.; Heagerty, A.M. Mechanistic Links between Obesity, Diabetes, and Blood Pressure: Role of Perivascular Adipose Tissue. Physiol. Rev. 2019, 99, 1701–1763. [Google Scholar] [CrossRef] [PubMed]
- García-Fernández, E.; Rico-Cabanas, L.; Rosgaard, N.; Estruch, R.; Bach-Faig, A. Mediterranean diet and cardiodiabesity: A review. Nutrients 2014, 6, 3474–3500. [Google Scholar] [CrossRef]
- Ruiz-Canela, M.; Zazpe, I.; Shivappa, N.; Hébert, J.R.; Sánchez-Tainta, A.; Corella, D.; Salas-Salvadó, J.; Fitó, M.; Lamuela-Raventós, R.M.; Rekondo, J.; et al. Dietary inflammatory index and anthropometric measures of obesity in a population sample at high cardiovascular risk from the PREDIMED (PREvención con DIeta MEDiterránea) trial. Br. J. Nutr. 2015, 113, 984–995. [Google Scholar] [CrossRef]
- Pappachan, J.M.; Viswanath, A.K. Medical Management of Diabesity: Do We Have Realistic Targets? Curr. Diab. Rep. 2017, 17, 4. [Google Scholar] [CrossRef]
- Metro, D.; Tardugno, R.; Papa, M.; Bisignano, C.; Manasseri, L.; Calabrese, G.; Gervasi, T.; Dugo, G.; Cicero, N. Adherence to the Mediterranean diet in a Sicilian student population. Nat. Prod. Res. 2018, 2, 1775–1781. [Google Scholar] [CrossRef]
- Metro, D.; Papa, M.; Manasseri, L.; Gervasi, T.; Campone, L.; Pellizzeri, V.; Tardugno, R.; Dugo, G. Mediterranean diet in a Sicilian student population. Second part: Breakfast and its nutritional profile. Nat. Prod. Res. 2020, 34, 2255–2261. [Google Scholar] [CrossRef] [PubMed]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef]
- Bianchi, F.; Cappella, A.; Gagliano, N.; Sfondrini, L.; Stacchiotti, A. Polyphenols-Gut-Heart: An Impactful Relationship to Improve Cardiovascular Diseases. Antioxidants 2022, 11, 1700. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Dash, R.; Nishan, A.A.; Habiba, S.U.; Moon, I.S. Brain modulation by the gut microbiota: From disease to therapy. Adv. Res. 2022, 53, 153–173. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
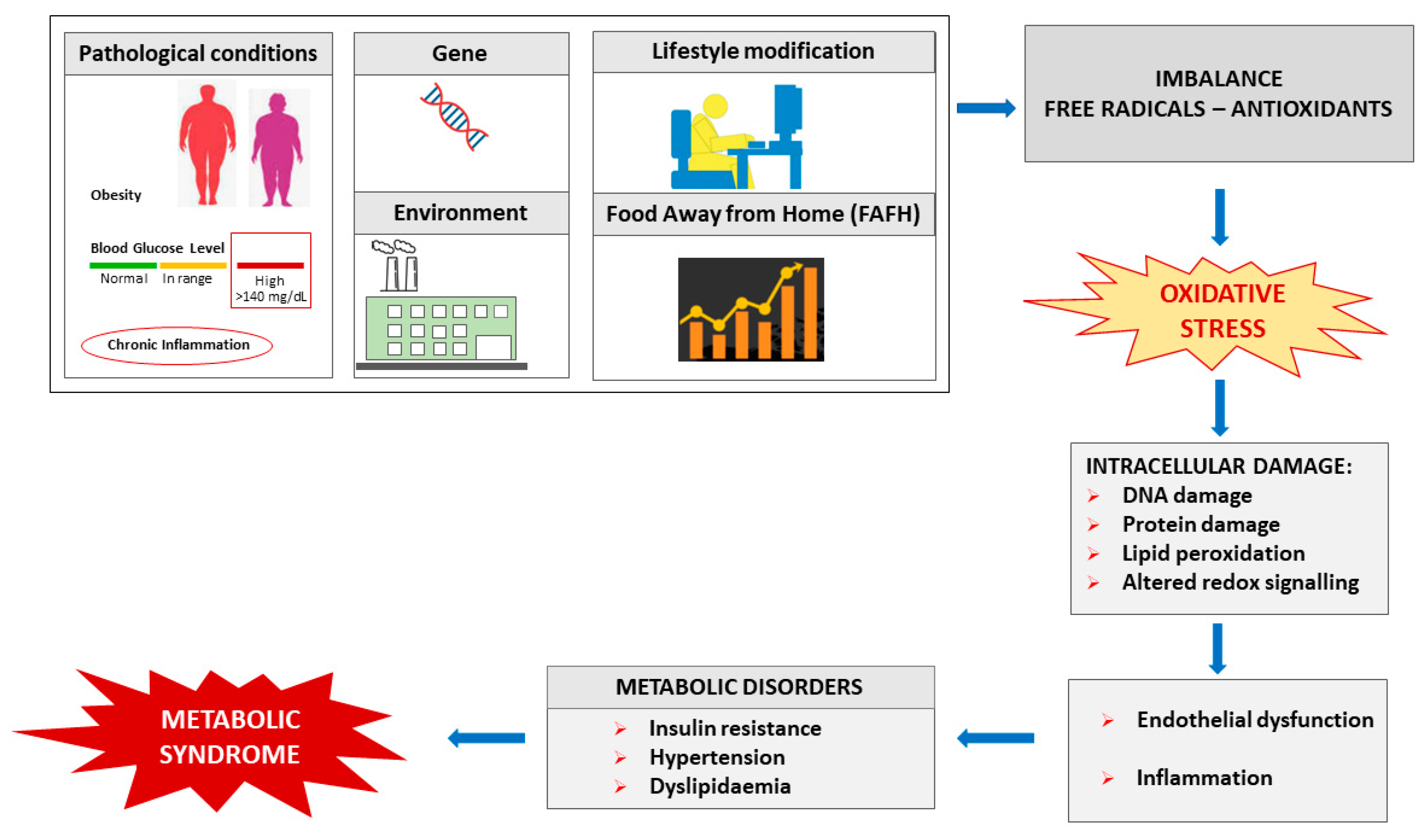{kind=link}
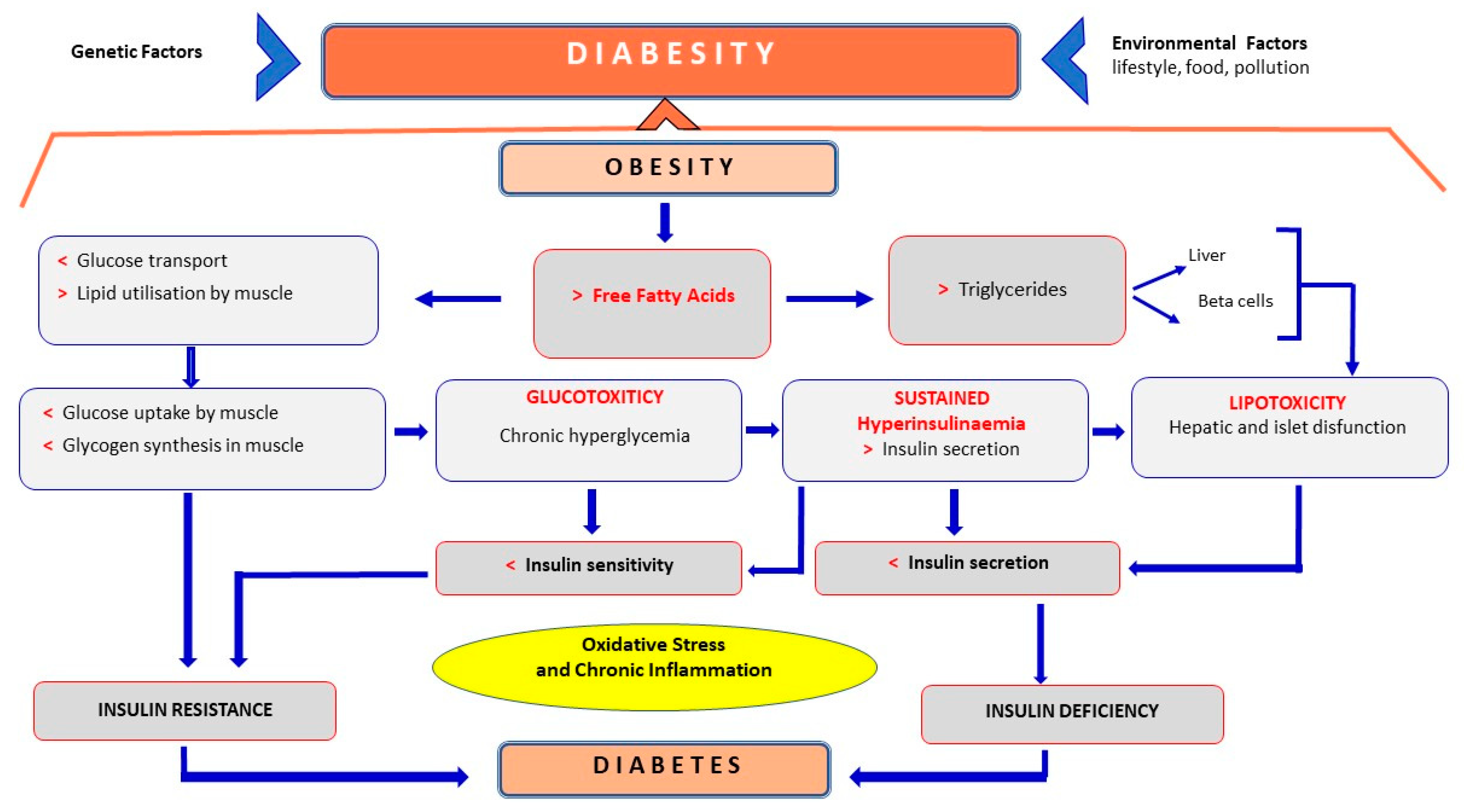{kind=link}
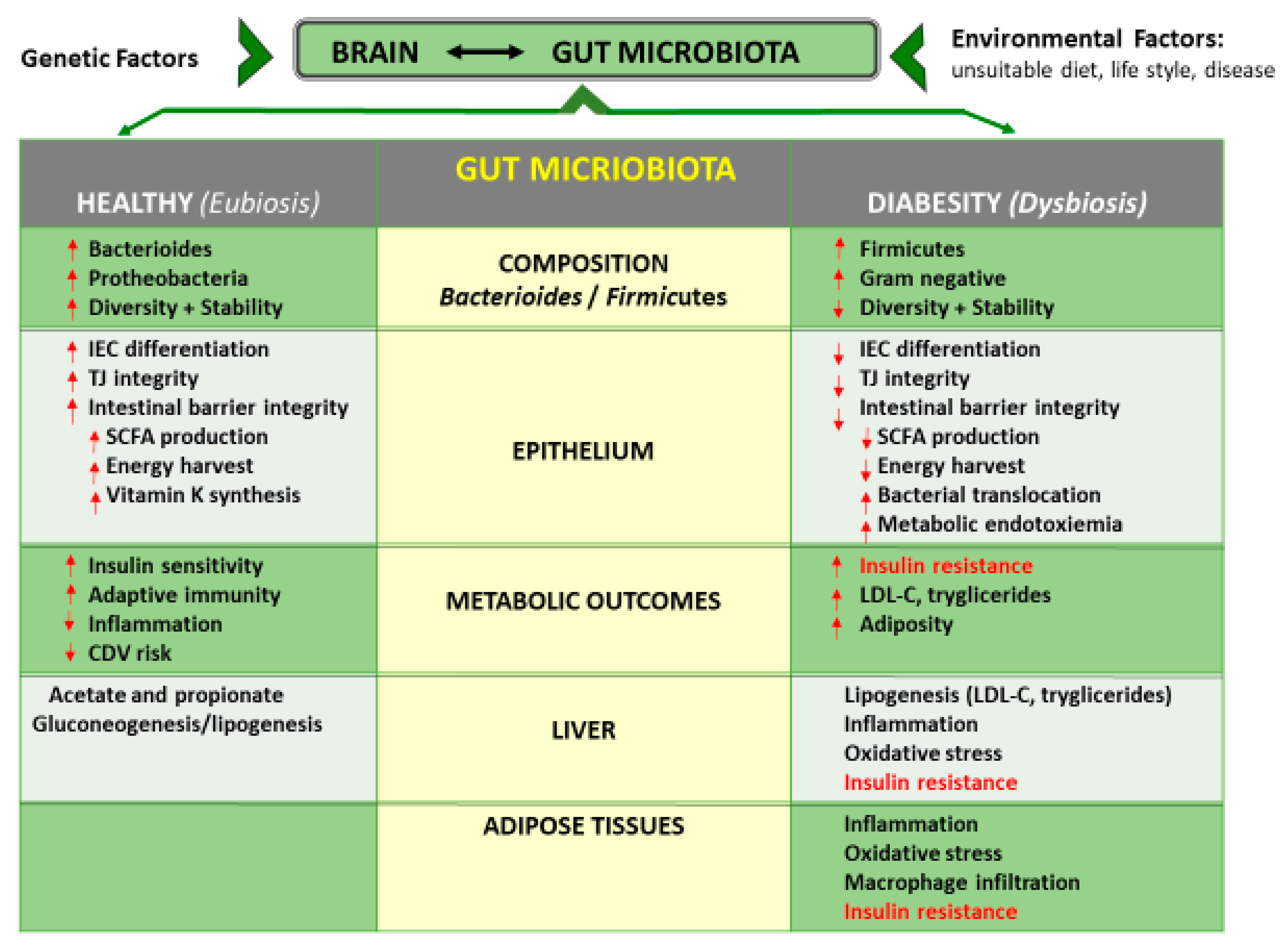{kind=link}
| Clinical Parameters | Criteria | |||||
|---|---|---|---|---|---|---|
| Central Obesity | Fasting Blood Sugar | ↑ Triglycerides | ↓ HDL-Cholesterol | ↑ Blood Pressure | Diagnosed as MS | |
| IDF (2005) [19] | Waist circumference defined in terms of ethnicity-specific values | ≥100 mg/dL or on medication | ≥150 mg/dL or on medication | Male: <40 mg/dL Female: <50 mg/dL | Diastolic ≥130 and/or systolic ≥85 mmHg or on medication | Absolutely required central obesity plus ≥2 criteria |
| AHA/NHLBI (2005) [20] | Waist circumference Male: ≥102 cm Female: ≥88 cm | ≥100 mg/dL or on medication | ≥150 mg/dL or on medication | Male: <40 mg/dL Female: <50 mg/dL | Diastolic ≥130 and/or systolic ≥85 mmHg or on medication | ≥3 criteria |
| AHA/NHLBI and IDF:2009 [21] | Waist circumference defined in terms of population- and country-based-specific definition | ≥100 mg/dL or on medication | ≥150 mg/dL or on medication | Male: <40 mg/dL Female: <50 mg/dL | Diastolic ≥130 and/or systolic ≥85 mmHg or on medication | ≥3 criteria |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martemucci, G.; Fracchiolla, G.; Muraglia, M.; Tardugno, R.; Dibenedetto, R.S.; D’Alessandro, A.G. Metabolic Syndrome: A Narrative Review from the Oxidative Stress to the Management of Related Diseases. Antioxidants 2023, 12, 2091. https://doi.org/10.3390/antiox12122091
Martemucci G, Fracchiolla G, Muraglia M, Tardugno R, Dibenedetto RS, D’Alessandro AG. Metabolic Syndrome: A Narrative Review from the Oxidative Stress to the Management of Related Diseases. Antioxidants. 2023; 12(12):2091. https://doi.org/10.3390/antiox12122091
Chicago/Turabian StyleMartemucci, Giovanni, Giuseppe Fracchiolla, Marilena Muraglia, Roberta Tardugno, Roberta Savina Dibenedetto, and Angela Gabriella D’Alessandro. 2023. "Metabolic Syndrome: A Narrative Review from the Oxidative Stress to the Management of Related Diseases" Antioxidants 12, no. 12: 2091. https://doi.org/10.3390/antiox12122091
APA StyleMartemucci, G., Fracchiolla, G., Muraglia, M., Tardugno, R., Dibenedetto, R. S., & D’Alessandro, A. G. (2023). Metabolic Syndrome: A Narrative Review from the Oxidative Stress to the Management of Related Diseases. Antioxidants, 12(12), 2091. https://doi.org/10.3390/antiox12122091