
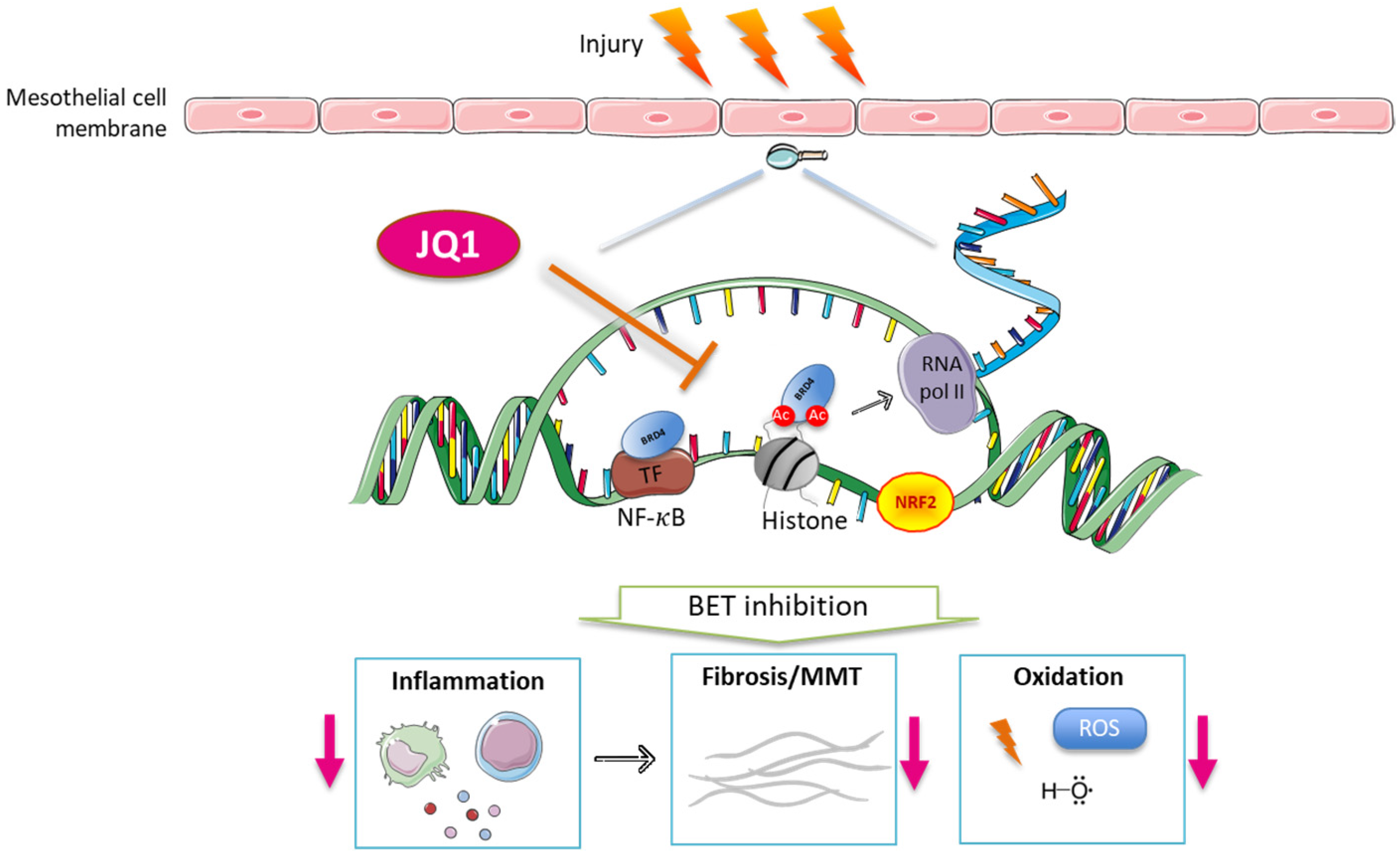
BET Protein Inhibitor JQ1 Ameliorates Experimental Peritoneal Damage by Inhibition of Inflammation and Oxidative Stress

 , , , , , , , , and
, , , , , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Chlorhexidine Gluconate-Induced Peritoneal Damage in Mice
2.3. Cell Culture
2.4. Histological and Immunohistochemical Analyses
2.5. Protein Level Studies
2.6. Gene Expression Assays
2.7. Statistical Analysis
3. Results
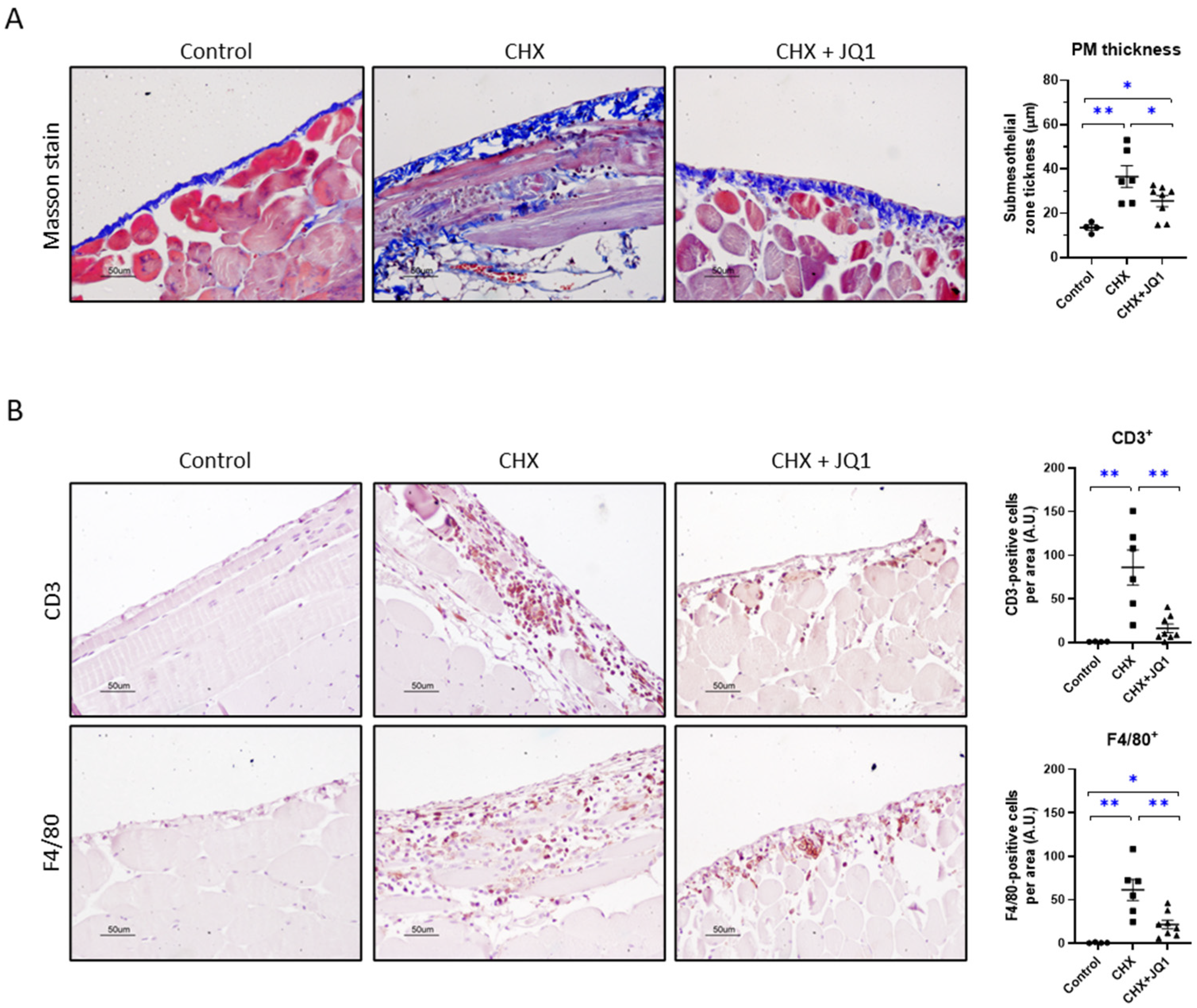
3.1. BET Inhibition Ameliorated Experimental Peritoneal Damage in Mice
3.2. BET Inhibition Improved CHX-Induced Peritoneal Fibrosis in Mice
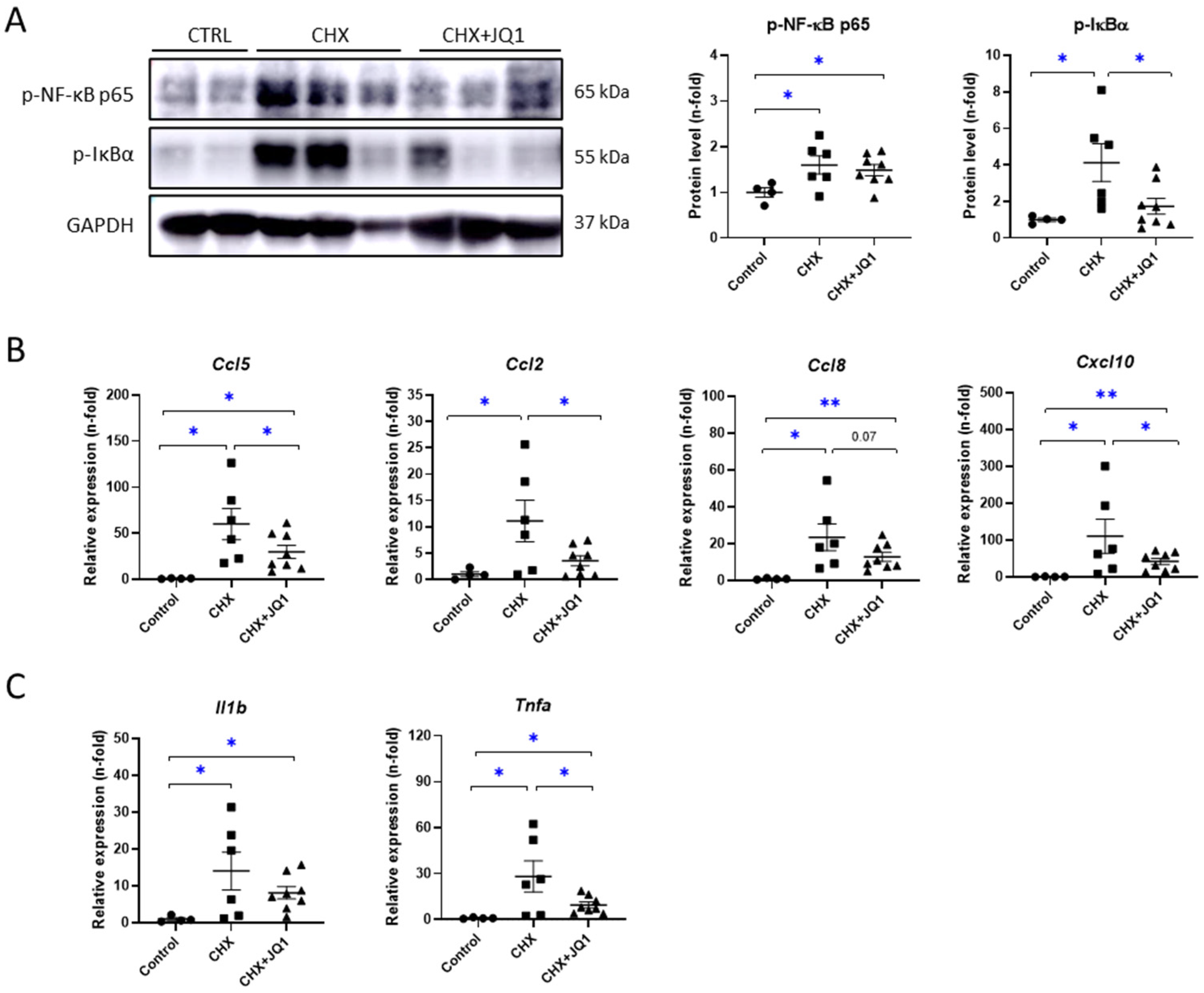
3.3. BET Inhibition Decreased CHX-Induced Peritoneal Inflammation in Mice
3.4. BET Inhibition Modulated Oxidative-Stress-Related Factors in CHX-Treated Mice
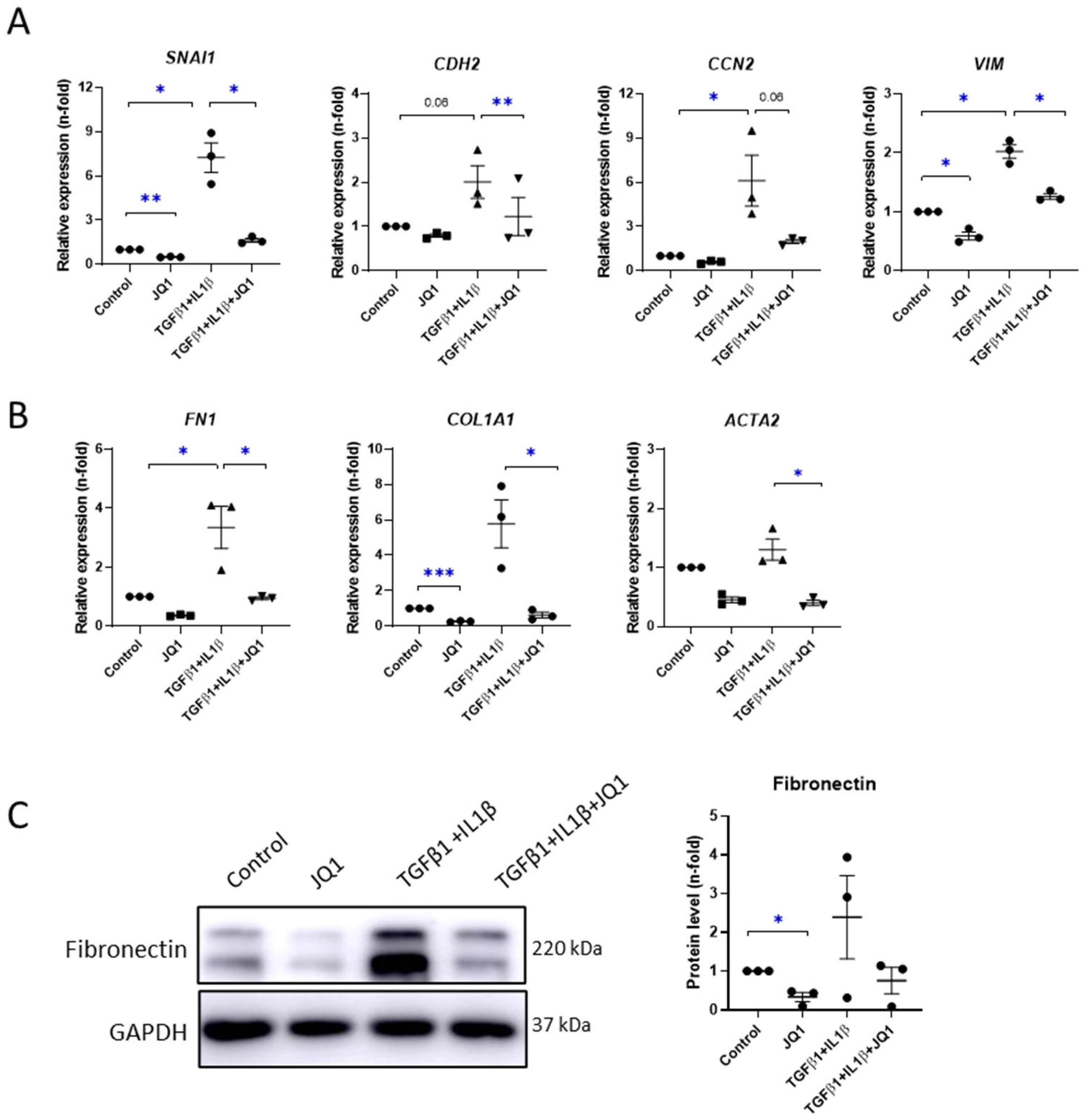
3.5. BET Inhibition Diminished Profibrotic- and Proinflammatory-Related Responses in Cultured Mesothelial Cells
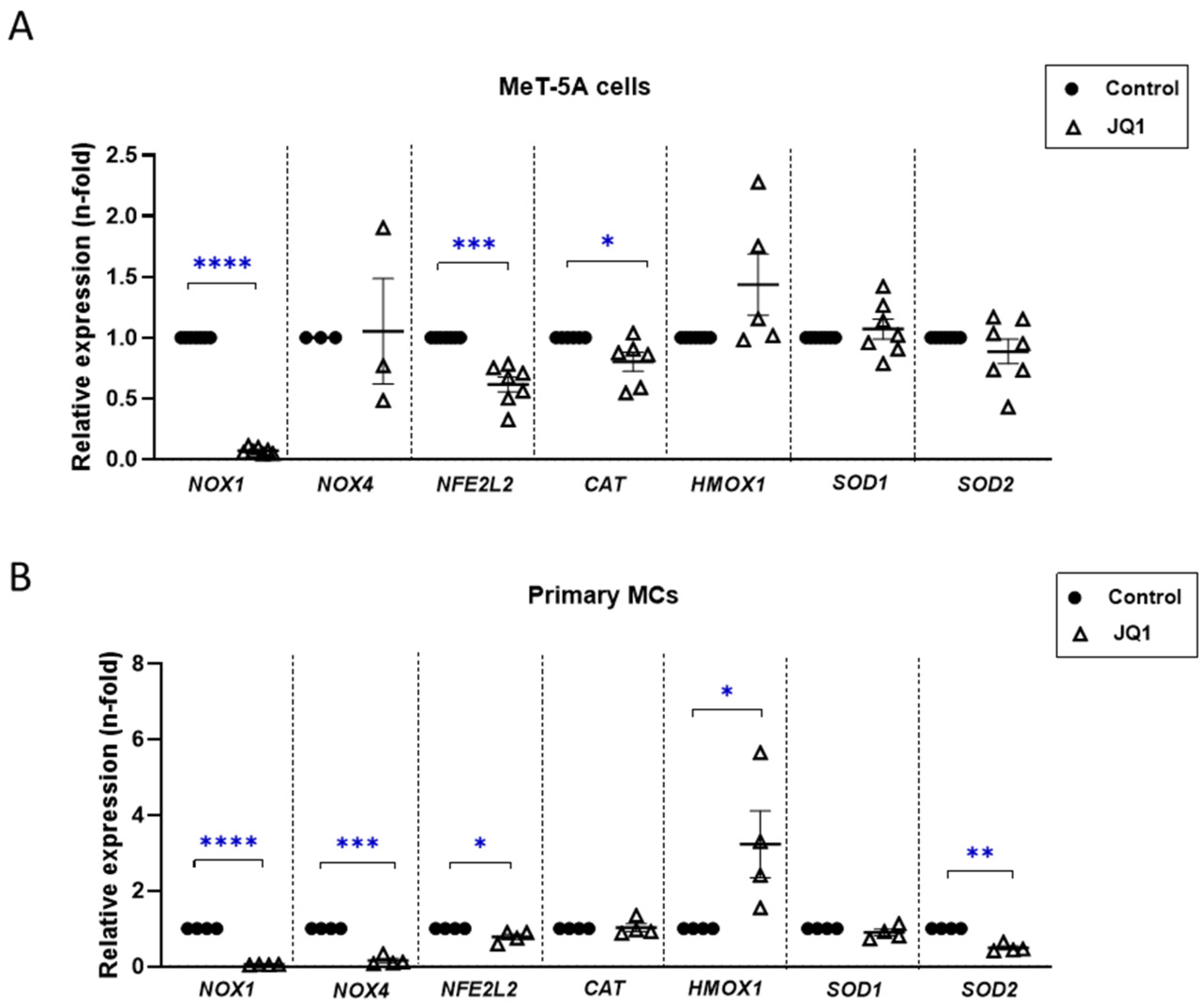
3.6. BET Inhibition Reduced Oxidative Stress in Cultured Mesothelial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ortiz, A.; Roger, M.; Jiménez, V.M.; Perez, J.C.R.; Furlano, M.; Atxer, L.S.; Zurro, D.G.; Casabona, C.M.R.; Gómez, C.G.; Bermúdez, P.P.; et al. RICORS2040: The Need for Collaborative Research in Chronic Kidney Disease. Clin. Kidney J. 2022, 15, 372–387. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Covic, A.; Fliser, D.; Fouque, D.; Goldsmith, D.; Kanbay, M.; Mallamaci, F.; Massy, Z.A.; Rossignol, P.; Vanholder, R.; et al. Epidemiology, Contributors to, and Clinical Trials of Mortality Risk in Chronic Kidney Failure. Lancet 2014, 383, 1831–1843. [Google Scholar] [CrossRef] [PubMed]
- Trionfetti, F.; Marchant, V.; González-Mateo, G.T.; Kawka, E.; Márquez-Expósito, L.; Ortiz, A.; López-Cabrera, M.; Ruiz-Ortega, M.; Strippoli, R. Novel Aspects of the Immune Response Involved in the Peritoneal Damage in Chronic Kidney Disease Patients under Dialysis. Int. J. Mol. Sci. 2023, 24, 5763. [Google Scholar] [CrossRef] [PubMed]
- López-Cabrera, M. Mesenchymal Conversion of Mesothelial Cells Is a Key Event in the Pathophysiology of the Peritoneum during Peritoneal Dialysis. Adv. Med. 2014, 2014, 473134. [Google Scholar] [CrossRef] [PubMed]
- Strippoli, R.; Moreno-Vicente, R.; Battistelli, C.; Cicchini, C.; Noce, V.; Amicone, L.; Marchetti, A.; Del Pozo, M.A.; Tripodi, M. Molecular Mechanisms Underlying Peritoneal EMT and Fibrosis. Stem Cells Int. 2016, 2016, 3543678. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, Y.; Tao, M.; Zhuang, S.; Liu, N. Peritoneal Fibrosis and Epigenetic Modulation. Perit. Dial. Int. 2021, 41, 168–178. [Google Scholar] [CrossRef]
- Shi, Y.; Tao, M.; Wang, Y.; Zang, X.; Ma, X.; Qiu, A.; Zhuang, S.; Liu, N. Genetic or Pharmacologic Blockade of Enhancer of Zeste Homolog 2 Inhibits the Progression of Peritoneal Fibrosis. J. Pathol. 2020, 250, 79–94. [Google Scholar] [CrossRef]
- Maeda, K.; Doi, S.; Nakashima, A.; Nagai, T.; Irifuku, T.; Ueno, T.; Masaki, T. Inhibition of H3K9 Methyltransferase G9a Ameliorates Methylglyoxal-Induced Peritoneal Fibrosis. PLoS ONE 2017, 12, e0173706. [Google Scholar] [CrossRef]
- Xu, L.; Liu, N.; Gu, H.; Wang, H.; Shi, Y.; Ma, X.; Ma, S.; Ni, J.; Tao, M.; Qiu, A.; et al. Histone Deacetylase 6 Inhibition Counteracts the Epithelial-Mesenchymal Transition of Peritoneal Mesothelial Cells and Prevents Peritoneal Fibrosis. Oncotarget 2017, 8, 88730. [Google Scholar] [CrossRef]
- Rossi, L.; Battistelli, C.; Turris, V.D.; Noce, V.; Zwergel, C.; Valente, S.; Moioli, A.; Manzione, A.; Palladino, M.; Bordoni, V.; et al. HDAC1 Inhibition by MS-275 in Mesothelial Cells Limits Cellular Invasion and Promotes MMT Reversal. Sci. Rep. 2018, 8, 8492. [Google Scholar] [CrossRef]
- Bontempi, G.; Terri, M.; Garbo, S.; Montaldo, C.; Mariotti, D.; Bordoni, V.; Valente, S.; Zwergel, C.; Mai, A.; Marchetti, A.; et al. Restoration of WT1/MiR-769-5p Axis by HDAC1 Inhibition Promotes MMT Reversal in Mesenchymal-like Mesothelial Cells. Cell Death Dis. 2022, 13, 965. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.L.; Kim, C.; Zhou, M.M. The Functions of BET Proteins in Gene Transcription of Biology and Diseases. Front. Mol. Biosci. 2021, 8, 728777. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, D.C.; Horng, T.; Medzhitov, R. Control of Inducible Gene Expression by Signal-Dependent Transcriptional Elongation. Cell 2009, 138, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 152, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Gajjela, B.K.; Zhou, M.M. Bromodomain Inhibitors and Therapeutic Applications. Curr. Opin. Chem. Biol. 2023, 75, 102323. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Alvarez, B.; Morgado-Pascual, J.L.; Rayego-Mateos, S.; Rodriguez, R.M.; Rodrigues-Diez, R.; Cannata-Ortiz, P.; Sanz, A.B.; Egido, J.; Tharaux, P.L.; Ortiz, A.; et al. Inhibition of Bromodomain and Extraterminal Domain Family Proteins Ameliorates Experimental Renal Damage. J. Am. Soc. Nephrol. 2017, 28, 504–519. [Google Scholar] [CrossRef] [PubMed]
- Chaidos, A.; Caputo, V.; Karadimitris, A. Inhibition of Bromodomain and Extra-Terminal Proteins (BET) as a Potential Therapeutic Approach in Haematological Malignancies: Emerging Preclinical and Clinical Evidence. Ther. Adv. Hematol. 2015, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Ogawa, K.; Kawaguchi, M.; Fumoto, S.; Mukai, H.; Kawakami, S. Suppression of Peritoneal Fibrosis by Sonoporation of Hepatocyte Growth Factor Gene-Encoding Plasmid DNA in Mice. Pharmaceutics 2021, 13, 115. [Google Scholar] [CrossRef]
- Duan, Q.; Wu, P.; Liu, Z.; Xia, F.; Zhu, L.; Zheng, Z.; Yang, T.; Qi, J. BET Bromodomain Inhibition Suppresses Adipogenesis in Mice. Endocrine 2020, 67, 264. [Google Scholar] [CrossRef]
- López-Cabrera, M.; Aguilera, A.; Aroeira, L.S.; Ramírez-Huesca, M.; Pérez-Lozano, M.L.; Jiménez-Heffernan, J.A.; Bajo, M.A.; del Peso, G.; Sánchez-Tomero, J.A.; Selgas, R. Ex Vivo Analysis of Dialysis Effluent-Derived Mesothelial Cells as an Approach to Unveiling the Mechanism of Peritoneal Membrane Failure. Perit. Dial. Int. 2006, 26, 26–34. [Google Scholar] [CrossRef]
- Chang, M.Y.; Wang, H.H.; Chen, L.H.; Gao, J.; Hung, S.Y.; Chiou, Y.Y.; Lee, Y.C. A Mice Model of Chlorhexidine Gluconate-Induced Peritoneal Damage. J. Vis. Exp. 2022, 2022, 63903. [Google Scholar] [CrossRef]
- Shi, Y.; Hu, Y.; Wang, Y.; Ma, X.; Tang, L.; Tao, M.; Qiu, A.; Zhuang, S.; Liu, N. Blockade of Autophagy Prevents the Development and Progression of Peritoneal Fibrosis. Front. Pharmacol. 2021, 12, 724141. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Li, J.; Chen, H.; Hu, Y.; Tang, L.; Zhou, X.; Tao, M.; Lv, Z.; Chen, S.; Qiu, A.; et al. Pharmacologic Inhibition of Histone Deacetylase 6 Prevents the Progression of Chlorhexidine Gluconate-Induced Peritoneal Fibrosis by Blockade of M2 Macrophage Polarization. Front. Immunol. 2022, 13, 899140. [Google Scholar] [CrossRef] [PubMed]
- Io, K.; Nishino, T.; Obata, Y.; Kitamura, M.; Koji, T.; Kohno, S. Saha Suppresses Peritoneal Fibrosis in Mice. Perit. Dial. Int. 2015, 35, 246. [Google Scholar] [CrossRef]
- Stergar, J.; Dolenec, R.; Kojc, N.; Lakota, K.; Perše, M.; Tomšič, M.; Milanic, M. Hyperspectral Evaluation of Peritoneal Fibrosis in Mouse Models. Biomed. Opt. Express 2020, 11, 1991. [Google Scholar] [CrossRef]
- Zhou, F.; Yao, L.; Lu, X.; Li, Y.; Han, X.; Wang, P. Therapeutic Targeting of GSK3β-Regulated Nrf2 and NFκB Signaling Pathways by Salvianolic Acid A Ameliorates Peritoneal Fibrosis. Front. Med. 2022, 9, 804899. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, W.; Peng, X.; Zhu, L.; Wang, Z.; Shen, H.; Chen, C.; Xiao, L.; Li, S.; Zhao, Y.; et al. Parthenolide Alleviates Peritoneal Fibrosis by Inhibiting Inflammation via the NF-ΚB/ TGF-β/Smad Signaling Axis. Lab. Investig. 2022, 102, 1346. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S.; Sandhu, M.A. PGC-1 α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef]
- To, K.K.W.; Xing, E.; Larue, R.C.; Li, P.K. BET Bromodomain Inhibitors: Novel Design Strategies and Therapeutic Applications. Molecules 2023, 28, 3043. [Google Scholar] [CrossRef]
- Schooling, C.M.; Zhao, J.V. How Might Bromodomain and Extra-Terminal (BET) Inhibitors Operate in Cardiovascular Disease? Am. J. Cardiovasc. Drugs 2019, 19, 107. [Google Scholar] [CrossRef]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond Transcriptional Regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of Inflammation by a Synthetic Histone Mimic. Nature 2010, 468, 1119. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Pascual, J.L.; Rayego-Mateos, S.; Tejedor, L.; Suarez-Alvarez, B.; Ruiz-Ortega, M. Bromodomain and Extraterminal Proteins as Novel Epigenetic Targets for Renal Diseases. Front. Pharmacol. 2019, 10, 1315. [Google Scholar] [CrossRef] [PubMed]
- Jörres, A.; Ludat, K.; Lang, J.; Sander, K.; Gahl, G.M.; Frei, U.; DeJonge, K.; Williams, J.D.; Topley, N. Establishment and Functional Characterization of Human Peritoneal Fibroblasts in Culture: Regulation of Interleukin-6 Production by Proinflammatory Cytokines. J. Am. Soc. Nephrol. 1996, 7, 2192. [Google Scholar] [CrossRef]
- Kang, D.H.; Hong, Y.S.; Lim, H.J.; Choi, J.H.; Han, D.S.; Yoon, K.L. High Glucose Solution and Spent Dialysate Stimulate the Synthesis of Transforming Growth Factor-Β1 of Human Peritoneal Mesothelial Cells: Effect of Cytokine Costimulation. Perit. Dial. Int. 1999, 19, 221. [Google Scholar] [CrossRef] [PubMed]
- Helmke, A.; Nordlohne, J.; Balzer, M.S.; Dong, L.; Rong, S.; Hiss, M.; Shushakova, N.; Haller, H.; von Vietinghoff, S. CX3CL1–CX3CR1 Interaction Mediates Macrophage-Mesothelial Cross Talk and Promotes Peritoneal Fibrosis. Kidney Int. 2019, 95, 1405. [Google Scholar] [CrossRef]
- Catar, R.A.; Bartosova, M.; Kawka, E.; Chen, L.; Marinovic, I.; Zhang, C.; Zhao, H.; Wu, D.; Zickler, D.; Stadnik, H.; et al. Angiogenic Role of Mesothelium-Derived Chemokine CXCL1 During Unfavorable Peritoneal Tissue Remodeling in Patients Receiving Peritoneal Dialysis as Renal Replacement Therapy. Front. Immunol. 2022, 13, 821681. [Google Scholar] [CrossRef]
- Li, Q.; Zheng, M.; Liu, Y.; Sun, W.; Shi, J.; Ni, J.; Wang, Q. A Pathogenetic Role for M1 Macrophages in Peritoneal Dialysis-Associated Fibrosis. Mol. Immunol. 2018, 94, 131. [Google Scholar] [CrossRef]
- Zhou, Q.; Bajo, M.A.; del Peso, G.; Yu, X.; Selgas, R. Preventing Peritoneal Membrane Fibrosis in Peritoneal Dialysis Patients. Kidney Int. 2016, 90, 515. [Google Scholar] [CrossRef]
- Cantero-Navarro, E.; Rayego-Mateos, S.; Orejudo, M.; Tejedor-Santamaria, L.; Tejera-Muñoz, A.; Sanz, A.B.; Marquez-Exposito, L.; Marchant, V.; Santos-Sanchez, L.; Egido, J.; et al. Role of Macrophages and Related Cytokines in Kidney Disease. Front. Med. 2021, 8, 688060. [Google Scholar] [CrossRef]
- Herzog, R.; Sacnun, J.M.; González-Mateo, G.; Bartosova, M.; Bialas, K.; Wagner, A.; Unterwurzacher, M.; Sobieszek, I.J.; Daniel-Fischer, L.; Rusai, K.; et al. Lithium Preserves Peritoneal Membrane Integrity by Suppressing Mesothelial Cell AB-Crystallin. Sci. Transl. Med. 2021, 13, eaaz9705. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, J.; Park, M.; Seo, A.; Kim, K.H.; Kim, S.; Kang, M.; Kang, E.; Yoo, K.D.; Lee, S.; et al. Inflammatory Chemokine (C-C Motif) Ligand 8 Inhibition Ameliorates Peritoneal Fibrosis. FASEB J. 2023, 37, e22632. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jiang, Q.; Ren, L.; Ren, H.; Xu, H.; Wang, J.; Wang, P.; Chen, S.; Hua, Y.; Ren, S.; et al. BET Proteins Inhibitor JQ1 Impairs GM-CSF-Promoted Peritoneal Macrophage Self-Renewal and IL-4-Induced Alternative Polarization. Int. Immunopharmacol. 2023, 124, 110942. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Hsu, H.; Lin, C.C.; Pan, S.Y.; Liu, S.Y.; Wu, C.F.; Tsai, P.Z.; Liao, C.T.; Cheng, H.T.; Chiang, W.C.; et al. Inflammatory Macrophages Switch to CCL17-Expressing Phenotype and Promote Peritoneal Fibrosis. J. Pathol. 2020, 250, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Tieu, B.; Recinos, A.; Tilton, R.G.; Brasier, A.R. RhoA Mediates Angiotensin II-Induced Phospho-Ser536 Nuclear Factor ΚB/RelA Subunit Exchange on the Interleukin-6 Promoter in VSMCs. Circ. Res. 2006, 99, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Yang, X.-D.; Zhou, M.-M.; Ozato, K.; Chen, L.-F. Brd4 Coactivates Transcriptional Activation of NF-ΚB via Specific Binding to Acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Qi, J.; Bradner, J.E.; Xiao, G.; Chen, L.F. Bromodomain and Extraterminal (BET) Protein Inhibition Suppresses Human T Cell Leukemia Virus 1 (HTLV-1) Tax Protein-Mediated Tumorigenesis by Inhibiting Nuclear Factor ΚB (NF-ΚB) Signaling. J. Biol. Chem. 2013, 288, 36094. [Google Scholar] [CrossRef] [PubMed]
- Lüdemann, W.M.; Heide, D.; Kihm, L.; Zeier, M.; Scheurich, P.; Schwenger, V.; Ranzinger, J. TNF Signaling in Peritoneal Mesothelial Cells: Pivotal Role of CFLIPL. Perit. Dial. Int. 2017, 37, 250–258. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, Q.; Chen, Y.; Peng, X.; Wang, Y.; Li, S.; Wu, J.; Luo, C.; Gong, W.; Yin, B.; et al. Parthenolide, an NF-ΚB Inhibitor, Alleviates Peritoneal Fibrosis by Suppressing the TGF-β/Smad Pathway. Int. Immunopharmacol. 2020, 78, 106064. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Lara-Pezzi, E.; Selgas, R.; Ramírez-Huesca, M.; Domínguez-Jiménez, C.; Jiménez-Heffernan, J.A.; Aguilera, A.; Sánchez-Tomero, J.A.; Bajo, M.A.; Álvarez, V.; et al. Peritoneal Dialysis and Epithelial-to-Mesenchymal Transition of Mesothelial Cells. N. Engl. J. Med. 2003, 348, 403. [Google Scholar] [CrossRef]
- Stratton, M.S.; Bagchi, R.A.; Felisbino, M.B.; Hirsch, R.A.; Smith, H.E.; Riching, A.S.; Enyart, B.Y.; Koch, K.A.; Cavasin, M.A.; Alexanian, M.; et al. Dynamic Chromatin Targeting of BRD4 Stimulates Cardiac Fibroblast Activation. Circ. Res. 2019, 125, 662. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A.; Stratton, M.S.; Haldar, S.M. BRD4 Inhibition for the Treatment of Pathological Organ Fibrosis. F1000Research 2017, 6, 1015. [Google Scholar] [CrossRef]
- Hao, K.; Jiang, W.; Zhou, M.; Li, H.; Chen, Y.; Jiang, F.; Hu, Q. Targeting BRD4 Prevents Acute Gouty Arthritis by Regulating Pyroptosis. Int. J. Biol. Sci. 2020, 16, 3163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, R.; Xiao, H.; Liu, W.; Zeng, X.; Xie, G.; Yang, W.; Shi, L.; Yin, Y.; Tao, K. BRD4 Inhibitor AZD5153 Suppresses the Proliferation of Colorectal Cancer Cells and Sensitizes the Anticancer Effect of PARP Inhibitor. Int. J. Biol. Sci. 2019, 15, 1942. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, L.; Weng, X.; Chen, H.; Du, Y.; Diao, C.; Chen, Z.; Liu, X. Inhibition of Brd4 Alleviates Renal Ischemia/Reperfusion Injury-Induced Apoptosis and Endoplasmic Reticulum Stress by Blocking FoxO4-Mediated Oxidative Stress. Redox Biol. 2019, 24, 101195. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Mu, J.; Gong, Y.; Lu, C.; Zhao, Y.; He, T.; Qin, Z. Brd4 Inhibition Attenuates Unilateral Ureteral Obstruction-Induced Fibrosis by Blocking TGF-β-Mediated Nox4 Expression. Redox Biol. 2017, 11, 390. [Google Scholar] [CrossRef]
- Morgado-Pascual, J.L.; Suarez-Alvarez, B.; Marchant, V.; Basantes, P.; Tharaux, P.L.; Ortiz, A.; Lopez-Larrea, C.; Ruiz-Ortega, M.; Rayego-Mateos, S. Type IV Collagen and SOX9 Are Molecular Targets of BET Inhibition in Experimental Glomerulosclerosis. Int. J. Mol. Sci. 2023, 24, 468. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Peng, Y.; Huang, T.; Xia, F.; Yang, T.; Duan, Q.; Zhang, W. Bromodomain-Containing Protein 4 Contributes to Renal Fibrosis through the Induction of Epithelial-Mesenchymal Transition. Exp. Cell Res. 2019, 383, 111507. [Google Scholar] [CrossRef]
- Andrieu, G.P.; Denis, G.V. BET Proteins Exhibit Transcriptional and Functional Opposition in the Epithelial-to-Mesenchymal Transition. Mol. Cancer Res. 2018, 16, 580. [Google Scholar] [CrossRef]
- Qu, M.; Zhang, X.; Hu, X.; Dong, M.; Pan, X.; Bian, J.; Zhou, Q. BRD4 Inhibitor JQ1 Inhibits and Reverses Mechanical Injury-Induced Corneal Scarring. Cell Death Discov. 2018, 4, 64. [Google Scholar] [CrossRef]
- Tan, Y.F.; Wang, M.; Chen, Z.Y.; Wang, L.; Liu, X.H. Inhibition of BRD4 Prevents Proliferation and Epithelial–Mesenchymal Transition in Renal Cell Carcinoma via NLRP3 Inflammasome-Induced Pyroptosis. Cell Death Dis. 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Doi, S.; Nakashima, A.; Sasaki, K.; Maeda, K.; Ueno, T.; Masaki, T. Inhibition of the H3K4 Methyltransferase SET7/9 Ameliorates Peritoneal Fibrosis. PLoS ONE 2018, 13, e0196844. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, K.; Liang, Y.; Chen, Y.; Chen, Y.; Gong, Y. Histone Acetyltransferase Inhibitor C646 Reverses Epithelial to Mesenchymal Transition of Human Peritoneal Mesothelial Cells via Blocking TGF-Β1/Smad3 Signaling Pathway in Vitro. Int. J. Clin. Exp. Pathol. 2015, 8, 2746. [Google Scholar] [PubMed]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-ΚB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81. [Google Scholar] [CrossRef]
- Shin, H.S.; Ko, J.; Kim, D.A.; Ryu, E.S.; Ryu, H.M.; Park, S.H.; Kim, Y.L.; Oh, E.S.; Kang, D.H. Metformin Ameliorates the Phenotype Transition of Peritoneal Mesothelial Cells and Peritoneal Fibrosis via a Modulation of Oxidative Stress. Sci. Rep. 2017, 7, 5690. [Google Scholar] [CrossRef]
- Książek, K. Mesothelial Cell: A Multifaceted Model of Aging. Ageing Res. Rev. 2013, 12, 595. [Google Scholar] [CrossRef]
- Duan, S.; Yu, J.; Liu, Q.; Wang, Y.; Pan, P.; Xiao, L.; Ling, G.; Liu, F. Epithelial-to-Mesenchymal Transdifferentiation of Peritoneal Mesothelial Cells Mediated by Oxidative Stress in Peritoneal Fibrosis Rats. J. Cent. South Univ. 2011, 36, 34. [Google Scholar] [CrossRef]
- Xie, M.; Xia, B.; Xiao, L.; Yang, D.; Li, Z.; Wang, H.; Wang, X.; Zhang, X.; Peng, Q. Astragaloside IV Ameliorates Peritoneal Fibrosis by Promoting PGC-1α to Reduce Apoptosis in Vitro and in Vivo. J. Cell. Mol. Med. 2023, 27, 2945. [Google Scholar] [CrossRef]
- Huang, Q.; Sun, Y.; Sun, J.; Peng, L.; Shang, H.; Wei, D.; Li, C.; Hu, Z.; Peng, H. Proteomic Characterization of Peritoneal Extracellular Vesicles in a Mouse Model of Peritoneal Fibrosis. J. Proteome Res. 2023, 22, 908. [Google Scholar] [CrossRef]
- Hung, K.Y.; Liu, S.Y.; Yang, T.C.; Liao, T.L.; Kao, S.H. High-Dialysate-Glucose-Induced Oxidative Stress and Mitochondrial-Mediated Apoptosis in Human Peritoneal Mesothelial Cells. Oxid. Med. Cell Longev. 2014, 2014, 642793. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Ke, Q.; Nie, Q.; Qi, R.; Zhu, X.; Liu, W.; Hu, X.; Sun, Q.; Fu, J.L.; Tang, X.; et al. Inhibition of CGAS-STING by JQ1 Alleviates Oxidative Stress-Induced Retina Inflammation and Degeneration. Cell Death Differ. 2022, 29, 1816. [Google Scholar] [CrossRef] [PubMed]
- Younis, S.S.; Ghafil, F.A.A.; Majeed, S.; Hadi, N.R. The Effect of JQ1 Systemic Administration on Oxidative Stress and Apoptotic Markers in Renal Ischemic Reperfusion Injury in a Rat Model. J. Med. Life 2023, 16, 682. [Google Scholar] [CrossRef] [PubMed]
- Rayego-Mateos, S.; Basantes, P.; Morgado-Pascual, J.L.; Prieto, B.B.; Suarez-Alvarez, B.; Ortiz, A.; Lopez-Larrea, C.; Ruiz-Ortega, M. BET Protein Inhibitor JQ1 Modulates Mitochondrial Dysfunction and Oxidative Stress Induced by Chronic Kidney Disease. Antioxidants 2023, 12, 1130. [Google Scholar] [CrossRef] [PubMed]
- An, Q.D.; Li, Y.Y.; Zhang, H.X.; Lu, J.; Yu, X.D.; Lin, Q.; Zhang, Y.G. Inhibition of Bromodomain-Containing Protein 4 Ameliorates Oxidative Stress–Mediated Apoptosis and Cartilage Matrix Degeneration through Activation of NF-E2–Related Factor 2-Heme Oxygenase-1 Signaling in Rat Chondrocytes. J. Cell Biochem. 2018, 119, 7719. [Google Scholar] [CrossRef] [PubMed]
- Hussong, M.; Börno, S.T.; Kerick, M.; Wunderlich, A.; Franz, A.; Sültmann, H.; Timmermann, B.; Lehrach, H.; Hirsch-Kauffmann, M.; Schweiger, M.R. The Bromodomain Protein BRD4 Regulates the KEAP1/NRF2-Dependent Oxidative Stress Response. Cell Death Dis. 2014, 5, e1195. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liu, J.; Yuan, Y.; Zhang, I.; Dong, Z. Protective Effect of the BET Protein Inhibitor JQ1 in Cisplatin-Induced Nephrotoxicity. Am. J. Physiol. Renal Physiol. 2018, 315, F469. [Google Scholar] [CrossRef]
- Fahey, J.M.; Stancill, J.S.; Smith, B.C.; Girotti, A.W. Nitric Oxide Antagonism to Glioblastoma Photodynamic Therapy and Mitigation Thereof by BET Bromodomain Inhibitor JQ1. J. Biol. Chem. 2018, 293, 5345. [Google Scholar] [CrossRef]
- Li, L.; Meng, Y.; Wu, X.; Li, J.; Sun, Y. Bromodomain-Containing Protein 4 Inhibitor JQ1 Promotes Melanoma Cell Apoptosis by Regulating Mitochondrial Dynamics. Cancer Sci. 2021, 112, 4013. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Lyv, X.; Zhou, Q.J.; Xiang, Z.; Stanford, D.; Bodduluri, S.; Rowe, S.M.; Thannickal, V.J. Brd4-P300 Inhibition Downregulates Nox4 and Accelerates Lung Fibrosis Resolution in Aged Mice. JCI Insight 2020, 5, 14. [Google Scholar] [CrossRef]
- Guerrero-Hue, M.; Rayego-Mateos, S.; Vázquez-Carballo, C.; Palomino-Antolín, A.; García-Caballero, C.; Opazo-Rios, L.; Morgado-Pascual, J.L.; Herencia, C.; Mas, S.; Ortiz, A.; et al. Protective Role of Nrf2 in Renal Disease. Antioxidants 2021, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Debray-García, Y.; Sánchez, E.I.; Rodríguez-Muñoz, R.; Venegas, M.A.; Velazquez, J.; Reyes, J.L. Diabetes and Exposure to Peritoneal Dialysis Solutions Alter Tight Junction Proteins and Glucose Transporters of Rat Peritoneal Mesothelial Cells. Life Sci. 2016, 161, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Shi, J.; Shan, Y.; Yu, M.; Zhu, X.; Zhu, Y.; Liu, L.; Sheng, M. Asiaticoside Inhibits TGF-Β1-Induced Mesothelial-Mesenchymal Transition and Oxidative Stress via the Nrf2/HO-1 Signaling Pathway in the Human Peritoneal Mesothelial Cell Line HMrSV5. Cell Mol. Biol. Lett. 2020, 25, 33. [Google Scholar] [CrossRef] [PubMed]
- Zaza, G.; Granata, S.; Masola, V.; Rugiu, C.; Fantin, F.; Gesualdo, L.; Schena, F.P.; Lupo, A. Downregulation of Nuclear-Encoded Genes of Oxidative Metabolism in Dialyzed Chronic Kidney Disease Patients. PLoS ONE 2013, 8, e77847. [Google Scholar] [CrossRef] [PubMed]
- Michaeloudes, C.; Mercado, N.; Clarke, C.; Bhavsar, P.K.; Adcock, I.M.; Barnes, P.J.; Chung, K.F. Bromodomain and Extraterminal Proteins Suppress NF-E2–Related Factor 2–Mediated Antioxidant Gene Expression. J. Immunol. 2014, 192, 4913. [Google Scholar] [CrossRef]
- Jin, N.; George, T.L.; Otterson, G.A.; Verschraegen, C.; Wen, H.; Carbone, D.; Herman, J.; Bertino, E.M.; He, K. Advances in Epigenetic Therapeutics with Focus on Solid Tumors. Clin. Epigenet. 2021, 13, 83. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Zhang, Z.C.; Wu, Y.Y.; Pi, Y.N.; Lou, S.H.; Liu, T.B.; Lou, G.; Yang, C. Bromodomain and extraterminal (BET) proteins: Biological functions, diseases, and targeted therapy. Signal Transduct. Target. Ther. 2023, 8, 420. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchant, V.; Trionfetti, F.; Tejedor-Santamaria, L.; Rayego-Mateos, S.; Rotili, D.; Bontempi, G.; Domenici, A.; Menè, P.; Mai, A.; Martín-Cleary, C.; et al. BET Protein Inhibitor JQ1 Ameliorates Experimental Peritoneal Damage by Inhibition of Inflammation and Oxidative Stress. Antioxidants 2023, 12, 2055. https://doi.org/10.3390/antiox12122055
Marchant V, Trionfetti F, Tejedor-Santamaria L, Rayego-Mateos S, Rotili D, Bontempi G, Domenici A, Menè P, Mai A, Martín-Cleary C, et al. BET Protein Inhibitor JQ1 Ameliorates Experimental Peritoneal Damage by Inhibition of Inflammation and Oxidative Stress. Antioxidants. 2023; 12(12):2055. https://doi.org/10.3390/antiox12122055
Chicago/Turabian StyleMarchant, Vanessa, Flavia Trionfetti, Lucia Tejedor-Santamaria, Sandra Rayego-Mateos, Dante Rotili, Giulio Bontempi, Alessandro Domenici, Paolo Menè, Antonello Mai, Catalina Martín-Cleary, and et al. 2023. "BET Protein Inhibitor JQ1 Ameliorates Experimental Peritoneal Damage by Inhibition of Inflammation and Oxidative Stress" Antioxidants 12, no. 12: 2055. https://doi.org/10.3390/antiox12122055
APA StyleMarchant, V., Trionfetti, F., Tejedor-Santamaria, L., Rayego-Mateos, S., Rotili, D., Bontempi, G., Domenici, A., Menè, P., Mai, A., Martín-Cleary, C., Ortiz, A., Ramos, A. M., Strippoli, R., & Ruiz-Ortega, M. (2023). BET Protein Inhibitor JQ1 Ameliorates Experimental Peritoneal Damage by Inhibition of Inflammation and Oxidative Stress. Antioxidants, 12(12), 2055. https://doi.org/10.3390/antiox12122055