

Effects of Acetyl-L-Carnitine on Oxidative Stress in Amyotrophic Lateral Sclerosis Patients: Evaluation on Plasma Markers and Members of the Neurovascular Unit

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Collection of Blood Samples
2.3. Measurements of the Plasma Oxidants/Antioxidants and NO
2.3.1. Quantification of Thiobarbituric Acid Reactive Substances (TBARS) in Plasma Samples
2.3.2. Quantification of the 4-Hydroxy Nonenal (4-HNE) in Plasma Samples
2.3.3. Quantification of Glutathione (GSH) in Plasma Samples
2.3.4. Quantification of the Glutathione Peroxidase Activity (GPx) in Plasma Samples
2.3.5. Evaluation of Plasma NO
2.4. In Vitro Experiments
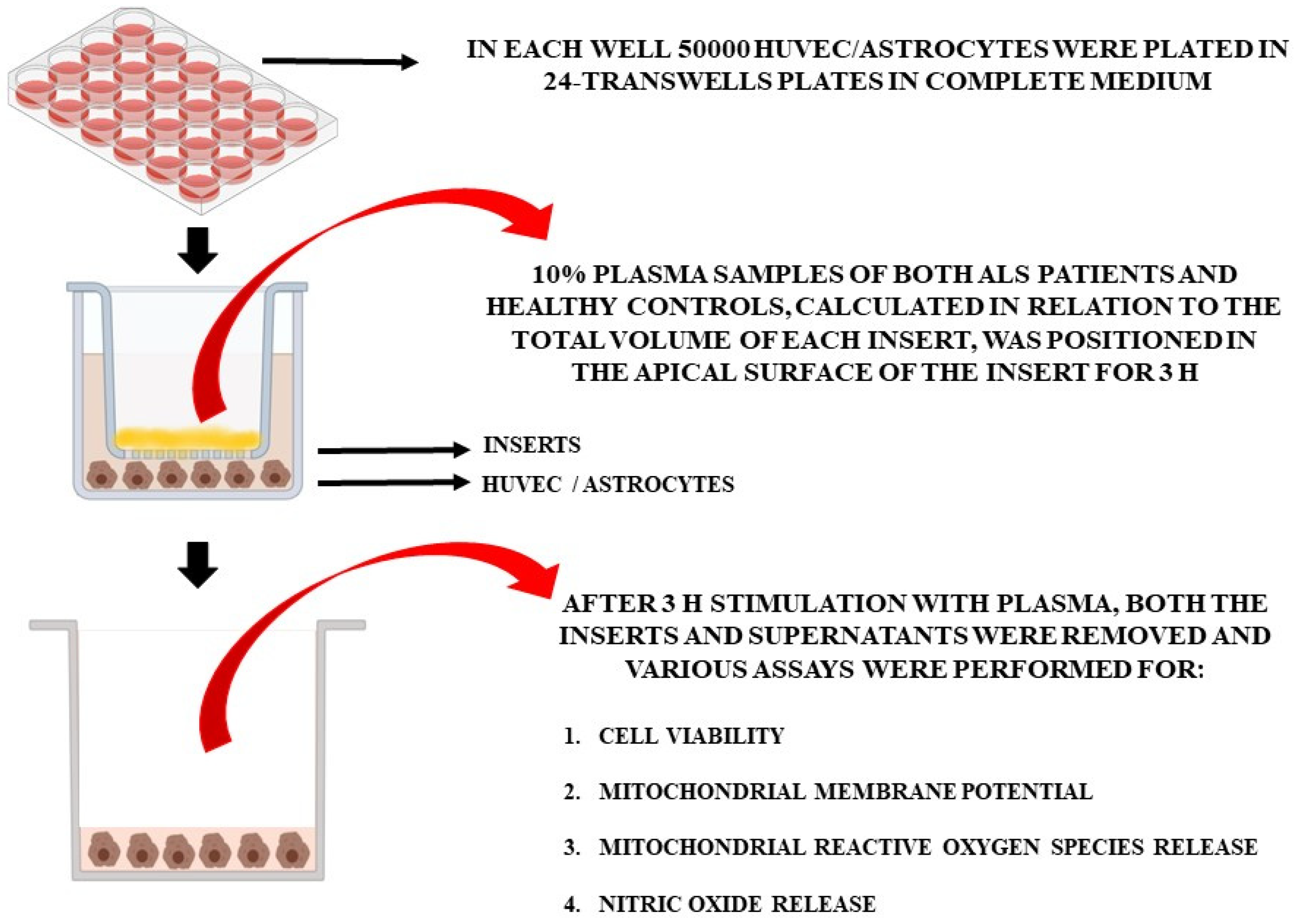
2.4.1. Effects of Plasma Samples on HUVEC and Astrocytes
2.4.2. Cell Cultures
2.4.3. Cell Viability
2.4.4. Mitochondrial Membrane Potential Measurement
2.4.5. MitoROS Release
2.4.6. NO Release
2.5. Statistical Analysis
3. Results
3.1. Clinical Data
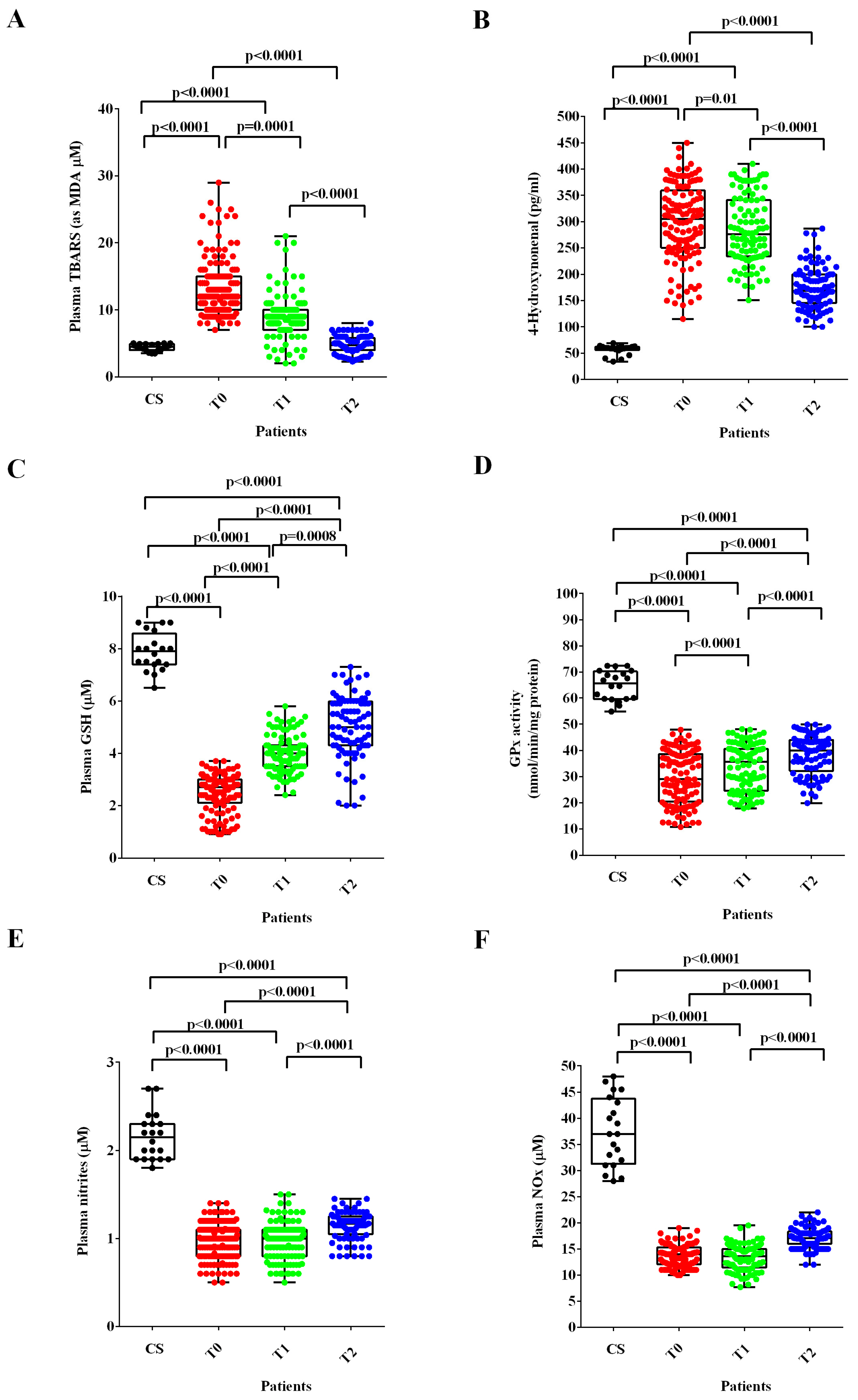
3.2. Plasmatic Quantifications
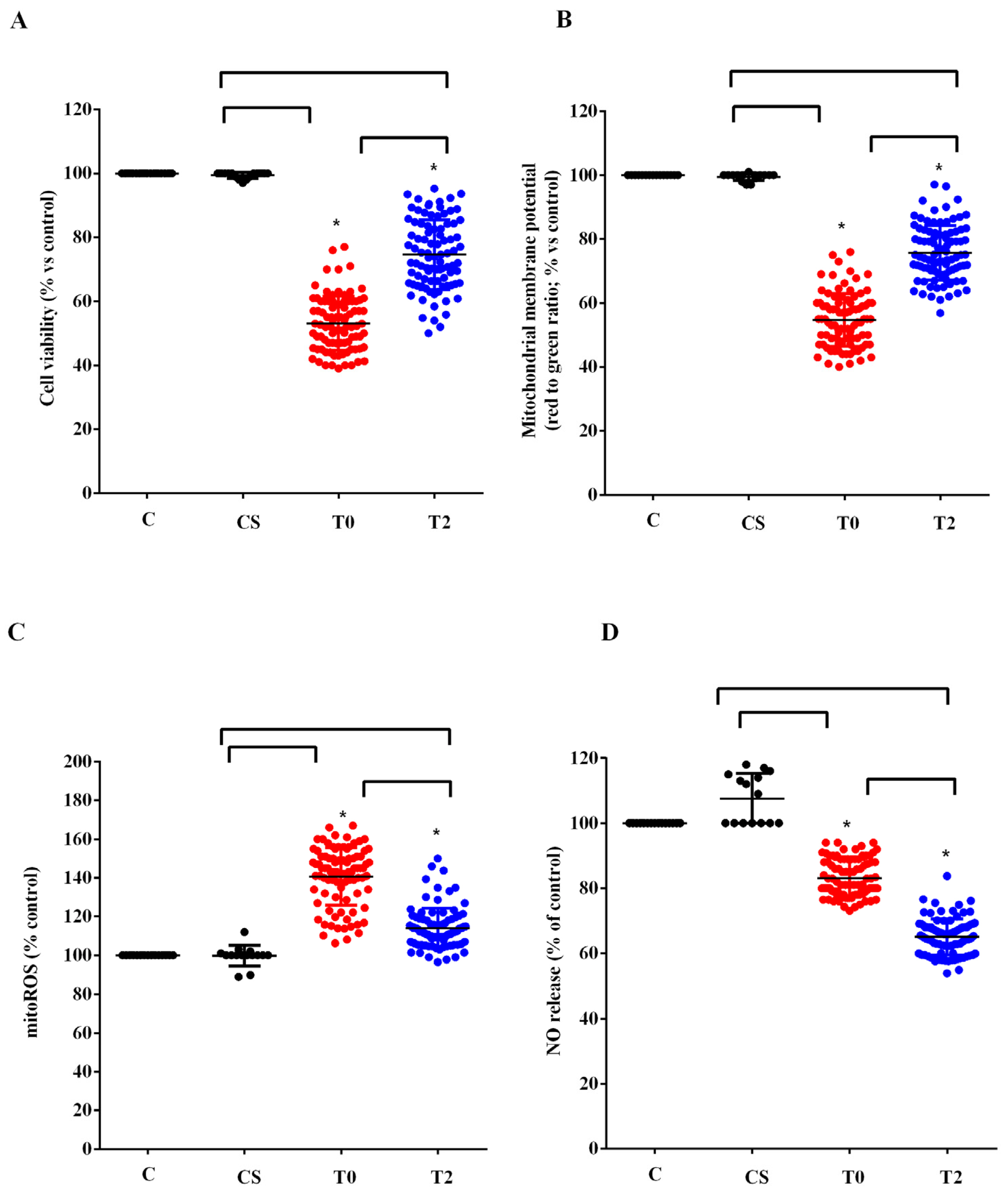
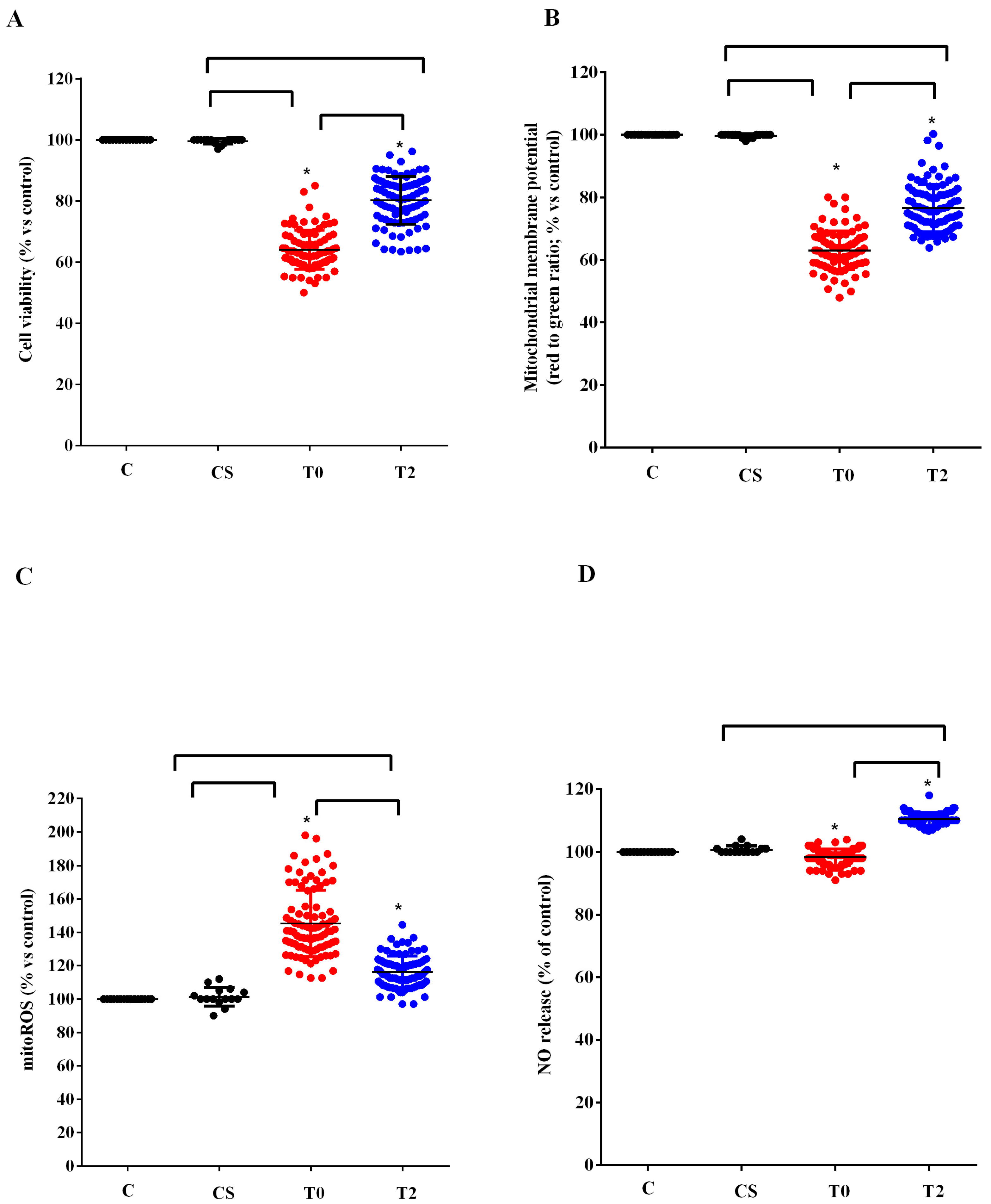
3.3. In Vitro Experiments
3.4. Clinical Correlations
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.A.; Fang, T.; De Marchi, F.; Neel, D.; Van Weehaeghe, D.; Berry, J.D.; Paganoni, S. Pharmacotherapy for Amyotrophic Lateral Sclerosis: A Review of Approved and Upcoming Agents. Drugs 2022, 82, 1367–1388. [Google Scholar] [CrossRef]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chió, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Recent advances in the diagnosis and prognosis of amyotrophic lateral sclerosis. Lancet Neurol. 2022, 21, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Rothstein, J.D.; Kiernan, M.C. Advances in treating amyotrophic lateral sclerosis: Insights from pathophysiological studies. Trends Neurosci. 2014, 37, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Shatunov, A.; Al-Chalabi, A. The genetic architecture of ALS. Neurobiol. Dis. 2021, 147, 105156. [Google Scholar] [CrossRef]
- Oskarsson, B.; Horton, D.K.; Mitsumoto, H. Potential Environmental Factors in Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 877–888. [Google Scholar] [CrossRef]
- Alonso, A.; Logroscino, G.; Hernán, M.A. Smoking and the risk of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1249–1252. [Google Scholar] [CrossRef]
- Amyotrophic Lateral Sclerosis/Riluzole Study Group II; Lacomblez, L.; Bensimon, G.; Meininger, V.; Leigh, P.N.; Guillet, P. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Lancet 1996, 347, 1425–1431. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V.; the ALS/Riluzole Study Group. A controlled trial of riluzole in amyotrophic lateral sclerosis. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Dash, R.P.; Babu, R.J.; Srinivas, N.R. Two Decades-Long Journey from Riluzole to Edaravone: Revisiting the Clinical Pharmacokinetics of the Only Two Amyotrophic Lateral Sclerosis Therapeutics. Clin. Pharmacokinet. 2018, 57, 1385–1398. [Google Scholar] [CrossRef] [PubMed]
- Group WG on Behalf of the E (MCI-186) ALS 18 S. Exploratory double-blind, parallel-group, placebo-controlled study of edaravone (MCI-186) in amyotrophic lateral sclerosis (Japan ALS severity classification: Grade 3, requiring assistance for eating, excretion or ambulation). Amyotroph. Lateral Scler. Front. Degener. 2017, 18 (Suppl. 1), 40–48. [Google Scholar] [CrossRef] [PubMed]
- Bedlack, R.S.; Joyce, N.; Carter, G.T.; Paganoni, S.; Karam, C. Complementary and alternative therapies in amyotrophic lateral sclerosis. Neurol. Clin. 2015, 33, 909–936. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.L.; McDonald, D.A.; Borum, P.R. Acylcarnitines: Role in brain. Prog. Lipid Res. 2010, 49, 61–75. [Google Scholar] [CrossRef]
- De Marchi, F.; Venkatesan, S.; Saraceno, M.; Mazzini, L.; Grossini, E. Acetyl-L-carnitine and Amyotrophic Lateral Sclerosis: Current evidence and potential use. CNS Neurol. Disord. Drug Targets 2023. [Google Scholar] [CrossRef]
- Zanelli, S.A.; Solenski, N.J.; Rosenthal, R.E.; Fiskum, G. Mechanisms of ischemic neuroprotection by acetyl-L-carnitine. Ann. N. Y. Acad. Sci. 2005, 1053, 153–161. [Google Scholar]
- White, H.L.; Scates, P.W. Acetyl-L-carnitine as a precursor of acetylcholine. Neurochem. Res. 1990, 15, 597–601. [Google Scholar] [CrossRef]
- Kidd, P.M. Neurodegeneration from mitochondrial insufficiency: Nutrients, stem cells, growth factors, and prospects for brain rebuilding using integrative management. Altern. Med. Rev. 2005, 10, 268. [Google Scholar]
- Hota, K.B.; Hota, S.K.; Chaurasia, O.P.; Singh, S.B. Acetyl-L-carnitine-mediated neuroprotection during hypoxia is attributed to ERK1/2-Nrf2-regulated mitochondrial biosynthesis. Hippocampus 2012, 22, 723–736. [Google Scholar] [CrossRef]
- Patel, S.P.; Sullivan, P.G.; Lyttle, T.S.; Magnuson, D.S.K.; Rabchevsky, A.G. Acetyl-L-carnitine treatment following spinal cord injury improves mitochondrial function correlated with remarkable tissue sparing and functional recovery. Neuroscience 2012, 210, 296–307. [Google Scholar] [CrossRef]
- Bigini, P.; Larini, S.; Pasquali, C.; Muzio, V.; Mennini, T. Acetyl-L-carnitine shows neuroprotective and neurotrophic activity in primary culture of rat embryo motoneurons. Neurosci. Lett. 2002, 329, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Kira, Y.; Nishikawa, M.; Ochi, A.; Sato, E.; Inoue, M. L-carnitine suppresses the onset of neuromuscular degeneration and increases the life span of mice with familial amyotrophic lateral sclerosis. Brain Res. 2006, 1070, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Beghi, E.; Pupillo, E.; Bonito, V.; Buzzi, P.; Caponnetto, C.; Chiò, A.; Corbo, M.; Giannini, F.; Inghilleri, M.; Bella, V.L.; et al. Randomized double-blind placebo-controlled trial of acetyl-L-carnitine for ALS. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Saporta, S.; Sanberg, P.R. Implications of blood-brain barrier disruption in ALS. Amyotroph. Lateral Scler. 2008, 9, 375–376. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Ehrhart, J.; Mustafa, H.; Llauget, A.; Boccio, K.J.; Sanberg, P.R.; Appel, S.H.; Borlongan, C.V. Phenotypic characteristics of human bone marrow-derived endothelial progenitor cells in vitro support cell effectiveness for repair of the blood-spinal cord barrier in ALS. Brain Res. 2019, 1724, 146428. [Google Scholar] [CrossRef]
- Yu, X.; Ji, C.; Shao, A. Neurovascular Unit Dysfunction and Neurodegenerative Disorders. Front. Neurosci. 2020, 14, 334. [Google Scholar] [CrossRef]
- Grossini, E.; Garhwal, D.; Venkatesan, S.; Ferrante, D.; Mele, A.; Saraceno, M.; Scognamiglio, A.; Mandrioli, J.; Amedei, A.; De Marchi, F.; et al. The Potential Role of Peripheral Oxidative Stress on the Neurovascular Unit in Amyotrophic Lateral Sclerosis Pathogenesis: A Preliminary Report from Human and In Vitro Evaluations. Biomedicines 2022, 10, 691. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Ludolph, A.; Drory, V.; Hardiman, O.; Nakano, I.; Ravits, J.; Robberecht, W.; Shefner, J. A revision of the El Escorial criteria-2015. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 291–292. [Google Scholar] [CrossRef]
- Galiniak, S.; Mołoń, M.; Biesiadecki, M.; Bożek, A.; Rachel, M. The Role of Oxidative Stress in Atopic Dermatitis and Chronic Urticaria. Antioxidants 2022, 11, 1590. [Google Scholar] [CrossRef]
- Grossini, E.; Farruggio, S.; Pierelli, D.; Bolzani, V.; Rossi, L.; Pollesello, P.; Monaco, C. Levosimendan Improves Oxidative Balance in Cardiogenic Shock/Low Cardiac Output Patients. J. Clin. Med. 2020, 9, 373. [Google Scholar] [CrossRef] [PubMed]
- Bilgic, H.A.; Kilic, B.; Kockaya, B.D.; Sarac, B.E.; Suloglu, A.K.; Kalayci, O.; Karaaslan, C. Oxidative stress stimulation leads to cell-specific oxidant and antioxidant responses in airway resident and inflammatory cells. Life Sci. 2023, 315, 121358. [Google Scholar] [CrossRef] [PubMed]
- Dematteis, G.; Vydmantaitė, G.; Ruffinatti, F.A.; Chahin, M.; Farruggio, S.; Barberis, E.; Ferrari, E.; Marengo, E.; Distasi, C.; Morkūnienė, R.; et al. Proteomic analysis links alterations of bioenergetics, mitochondria-ER interactions and proteostasis in hippocampal astrocytes from 3xTg-AD mice. Cell Death Dis. 2020, 11, 645. [Google Scholar] [CrossRef] [PubMed]
- De Cillà, S.; Farruggio, S.; Cocomazzi, G.; Mary, D.; Alkabes, M.; Rossetti, L.; Vujosevic, S.; Grossini, E. Aflibercept and Ranibizumab Modulate Retinal Pigment Epithelial Cells Function by Acting on Their Cross Talk with Vascular Endothelial Cells. Cell. Physiol. Biochem. 2020, 54, 161–179. [Google Scholar] [PubMed]
- Grossini, E.; Venkatesan, S.; Alkabes, M.; Toma, C.; de Cillà, S. Membrane Blue Dual Protects Retinal Pigment Epithelium Cells/Ganglion Cells-Like through Modulation of Mitochondria Function. Biomedicines 2022, 10, 2854. [Google Scholar] [CrossRef]
- Roos, N.J.; Duthaler, U.; Bouitbir, J.; Krähenbühl, S. The uricosuric benzbromarone disturbs the mitochondrial redox homeostasis and activates the NRF2 signaling pathway in HepG2 cells. Free Radic. Biol. Med. 2020, 152, 216–226. [Google Scholar] [CrossRef]
- Stufano, A.; Isgrò, C.; Palese, L.L.; Caretta, P.; De Maria, L.; Lovreglio, P.; Sardanelli, A.M. Oxidative Damage and Post-COVID Syndrome: A Cross-Sectional Study in a Cohort of Italian Workers. Int. J. Mol. Sci. 2023, 24, 7445. [Google Scholar] [CrossRef]
- Bahrami, A.; Nikoomanesh, F.; Khorasanchi, Z.; Mohamadian, M.; Ferns, G.A. The relationship between food quality score with inflammatory biomarkers, and antioxidant capacity in young women. Physiol. Rep. 2023, 11, e15590. [Google Scholar] [CrossRef]
- Dos Santos, J.M.; Taiar, R.; Ribeiro, V.G.C.; da Silva Lage, V.K.; Scheidt Figueiredo, P.H.; Costa, H.S.; Pereira Lima, V.; Sañudo, B.; Bernardo-Filho, M.; Sá-Caputo, D.D.C.D.; et al. Whole-Body Vibration Training on Oxidative Stress Markers, Irisin Levels, and Body Composition in Women with Fibromyalgia: A Randomized Controlled Trial. Bioengineering 2023, 10, 260. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Kyheng, M.; Garçon, G.; Rolland, A.S.; Blasco, H.; Gelé, P.; Timothée Lenglet, T.; Veyrat-Durebex, C.; Corcia, P.; et al. A ferroptosis-based panel of prognostic biomarkers for Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 2918. [Google Scholar] [CrossRef]
- Lan, J.; Zhou, Y.; Wang, H.; Tang, J.; Kang, Y.; Wang, P.; Liu, X.; Peng, Y. Protective effect of human umbilical cord mesenchymal stem cell derived conditioned medium in a mutant TDP-43 induced motoneuron-like cellular model of ALS. Brain Res. Bull. 2023, 193, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Dzik, K.P.; Flis, D.J.; Bytowska, Z.K.; Karnia, M.J.; Ziolkowski, W.; Kaczor, J.J. Swim Training Ameliorates Hyperlocomotion of ALS Mice and Increases Glutathione Peroxidase Activity in the Spinal Cord. Int. J. Mol. Sci. 2021, 22, 11614. [Google Scholar] [CrossRef] [PubMed]
- Aguilar Diaz De Leon, J.; Borges, C.R. Evaluation of Oxidative Stress in Biological Samples Using the Thiobarbituric Acid Reactive Substances Assay. J. Vis. Exp. 2020, 159, e61122. [Google Scholar]
- Grossini, E.; Concina, D.; Rinaldi, C.; Russotto, S.; Garhwal, D.; Zeppegno, P.; Gramaglia, C.; Kul, S.; Panella, M. Association Between Plasma Redox State/Mitochondria Function and a Flu-Like Syndrome/COVID-19 in the Elderly Admitted to a Long-Term Care Unit. Front. Physiol. 2021, 12, 707587. [Google Scholar] [CrossRef]
- Zeppegno, P.; Krengli, M.; Ferrante, D.; Bagnati, M.; Burgio, V.; Farruggio, S.; Rolla, R.; Gramaglia, C.; Grossini, E. Psychotherapy with Music Intervention Improves Anxiety, Depression and the Redox Status in Breast Cancer Patients Undergoing Radiotherapy: A Randomized Controlled Clinical Trial. Cancers 2021, 13, 1752. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Fanti, P.; Matteucci, E.; Rossi, R. Micro-method for the determination of glutathione in human blood. J. Chromatogr. B 2014, 964, 191–194. [Google Scholar] [CrossRef]
- Kalita, J.; Shukla, R.; Pandey, P.C.; Misra, U.K. Balancing between apoptosis and survival biomarkers in the patients with tuberculous meningitis. Cytokine 2022, 157, 155960. [Google Scholar] [CrossRef] [PubMed]
- Safe, I.P.; Amaral, E.P.; Araújo-Pereira, M.; Lacerda, M.V.G.; Printes, V.S.; Souza, A.B.; Beraldi-Magalhães, F.; Monteiro, W.M.; Sampaio, V.S.; Barreto-Duarte, B.; et al. Adjunct N-Acetylcysteine Treatment in Hospitalized Patients With HIV-Associated Tuberculosis Dampens the Oxidative Stress in Peripheral Blood: Results from the RIPENACTB Study Trial. Front. Immunol. 2020, 11, 602589. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef]
- Kim, K. Glutathione in the Nervous System as a Potential Therapeutic Target to Control the Development and Progression of Amyotrophic Lateral Sclerosis. Antioxidants 2021, 10, 1011. [Google Scholar] [CrossRef]
- Babu, G.N.; Kumar, A.; Chandra, R.; Puri, S.; Singh, R.; Kalita, J.; Misra, U. Oxidant-antioxidant imbalance in the erythrocytes of sporadic amyotrophic lateral sclerosis patients correlates with the progression of disease. Neurochem. Int. 2008, 52, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Cova, E.; Bongioanni, P.; Cereda, C.; Metelli, M.R.; Salvaneschi, L.; Bernuzzi, S.; Guareschi, S.; Rossi, B.; Ceroni, M. Time course of oxidant markers and antioxidant defenses in subgroups of amyotrophic lateral sclerosis patients. Neurochem. Int. 2010, 56, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Kuźma, M.; Jamrozik, Z.; Barańczyk-Kuźma, A. Activity and expression of glutathione S-transferase pi in patients with amyotrophic lateral sclerosis. Clin. Chim. Acta 2006, 364, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Abe, T.; Yamazaki, K.; Murata, T.; Ishizaki, E.; Isobe, C. Increase in oxidized NO products and reduction in oxidized glutathione in cerebrospinal fluid from patients with sporadic form of amyotrophic lateral sclerosis. Neurosci. Lett. 1999, 260, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Hyun, D.; Jenner, P.; Halliwell, B. Effect of overexpression of wild-type and mutant Cu/Zn-superoxide dismutases on oxidative damage and antioxidant defences: Relevance to Down’s syndrome and familial amyotrophic lateral sclerosis. J. Neurochem. 2001, 76, 957–965. [Google Scholar] [CrossRef]
- Przedborski, S.; Donaldson, D.; Jakowec, M.; Kish, S.J.; Guttman, M.; Rosoklija, G.; Hays, A.P. Brain superoxide dismutase, catalase, and glutathione peroxidase activities in amyotrophic lateral sclerosis. Ann. Neurol. 1996, 39, 158–165. [Google Scholar] [CrossRef]
- Boillée, S.; Velde CVande Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef]
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; Van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Bonconpagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef]
- Magrané, J.; Cortez, C.; Gan, W.-B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Abdul Muneer, P.M.; Alikunju, S.; Szlachetka, A.M.; Mercer, A.J.; Haorah, J. Ethanol impairs glucose uptake by human astrocytes and neurons: Protective effects of acetyl-L-carnitine. Int. J. Physiol. Pathophysiol. Pharmacol. 2011, 3, 48–56. [Google Scholar] [PubMed]
- Karalija, A.; Novikova, L.N.; Kingham, P.J.; Wiberg, M.; Novikov, L.N. Neuroprotective effects of N-acetyl-cysteine and acetyl-L-carnitine after spinal cord injury in adult rats. PLoS ONE 2012, 7, e41086. [Google Scholar] [CrossRef]
- Sorrentino, S.; Polini, A.; Arima, V.; Romano, A.; Quattrini, A.; Gigli, G.; Mozetic, P.; Moroni, L. Neurovascular signals in amyotrophic lateral sclerosis. Curr. Opin. Biotechnol. 2022, 74, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Haller, E.; Saporta, S.; Kolomey, I.; Nicosia, S.V.; Sanberg, P.R. Ultrastructure of blood–brain barrier and blood–spinal cord barrier in SOD1 mice modeling ALS. Brain Res. 2007, 1157, 126–137. [Google Scholar] [CrossRef]
- Miyazaki, K.; Ohta, Y.; Nagai, M.; Morimoto, N.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Matsuura, T.; Abe, K. Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J. Neurosci. Res. 2011, 89, 718–728. [Google Scholar] [CrossRef]
- Zhong, Z.; Deane, R.; Ali, Z.; Parisi, M.; Shapovalov, Y.; O’Banion, M.K.; Stojanovic, K.; Sagare, A.; Boillee, S.; Cleveland, D.W.; et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat. Neurosci. 2008, 11, 420–422. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Woods, R.L., III; Louis, M.K.; Zesiewicz, T.A.; Kuzmin-Nichols, N.; Sullivan, K.L.; Miller, A.M.; Hernandez-Ontiveros, D.G.; Sanberg, P.R. Reduction of circulating endothelial cells in peripheral blood of ALS patients. PLoS ONE 2010, 5, e10614. [Google Scholar] [CrossRef]
- Zhong, Z.; Ilieva, H.; Hallagan, L.; Bell, R.; Singh, I.; Paquette, N.; Thiyagarajan, M.; Deane, R.; Fernandez, J.A.; Lane, S.; et al. Activated protein C therapy slows ALS-like disease in mice by transcriptionally inhibiting SOD1 in motor neurons and microglia cells. J. Clin. Investig. 2009, 119, 3437–3449. [Google Scholar] [CrossRef]
- Levine, J.B.; Kong, J.; Nadler, M.; Xu, Z. Astrocytes interact intimately with degenerating motor neurons in mouse amyotrophic lateral sclerosis (ALS). Glia 1999, 28, 215–224. [Google Scholar] [CrossRef]
- Alexianu, M.E.; Kozovska, M.; Appel, S.H. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 2001, 57, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.I.; Tajti, J.; Appel, S.H. Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch. Neurol. 1993, 50, 30–36. [Google Scholar] [CrossRef]
- Nakamura, T.; Lipton, S.A. Protein S-Nitrosylation as a Therapeutic Target for Neurodegenerative Diseases. Trends Pharmacol. Sci. 2016, 37, 73–84. [Google Scholar] [CrossRef]
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M.G. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775. [Google Scholar] [CrossRef]
- Contestabile, A.; Monti, B.; Polazzi, E. Neuronal-glial Interactions Define the Role of Nitric Oxide in Neural Functional Processes. Curr. Neuropharmacol. 2012, 10, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Fatemi, A. Pathologic role of glial nitric oxide in adult and pediatric neuroinflammatory diseases. Neurosci. Biobehav. Rev. 2014, 45, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Ros-Bernal, F. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef]
- Ferreira, G.C.; McKenna, M.C. L-Carnitine and acetyl-L-carnitine roles and neuroprotection in developing brain. Neurochem. Res. 2017, 42, 1661–1675. [Google Scholar] [CrossRef]
- Virmani, M.A.; Cirulli, M. The Role of l-Carnitine in Mitochondria, Prevention of Metabolic Inflexibility and Disease Initiation. Int. J. Mol. Sci. 2022, 23, 2717. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients’ Features (n = 32) | |
|---|---|
| Sex (male/female) | 19 (59%)/13 (41%) |
| Age at onset (median, IQR) | 67 (58–70) |
| Phenotype (spinal/bulbar, %) | 22 (69%)/10 (31%) |
| ALSFRS-R baseline (median, IQR) | 39 (35–43) |
| FVC% baseline (median, IQR) | 72 (49–94) |
| BMI baseline (median, IQR) | 23.2 (20.0–26.0) |
| Rate of progression (fast/slow, %) | 19 (59%)/13 (41%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grossini, E.; De Marchi, F.; Venkatesan, S.; Mele, A.; Ferrante, D.; Mazzini, L. Effects of Acetyl-L-Carnitine on Oxidative Stress in Amyotrophic Lateral Sclerosis Patients: Evaluation on Plasma Markers and Members of the Neurovascular Unit. Antioxidants 2023, 12, 1887. https://doi.org/10.3390/antiox12101887
Grossini E, De Marchi F, Venkatesan S, Mele A, Ferrante D, Mazzini L. Effects of Acetyl-L-Carnitine on Oxidative Stress in Amyotrophic Lateral Sclerosis Patients: Evaluation on Plasma Markers and Members of the Neurovascular Unit. Antioxidants. 2023; 12(10):1887. https://doi.org/10.3390/antiox12101887
Chicago/Turabian StyleGrossini, Elena, Fabiola De Marchi, Sakthipriyan Venkatesan, Angelica Mele, Daniela Ferrante, and Letizia Mazzini. 2023. "Effects of Acetyl-L-Carnitine on Oxidative Stress in Amyotrophic Lateral Sclerosis Patients: Evaluation on Plasma Markers and Members of the Neurovascular Unit" Antioxidants 12, no. 10: 1887. https://doi.org/10.3390/antiox12101887
APA StyleGrossini, E., De Marchi, F., Venkatesan, S., Mele, A., Ferrante, D., & Mazzini, L. (2023). Effects of Acetyl-L-Carnitine on Oxidative Stress in Amyotrophic Lateral Sclerosis Patients: Evaluation on Plasma Markers and Members of the Neurovascular Unit. Antioxidants, 12(10), 1887. https://doi.org/10.3390/antiox12101887