
Fundamental Role of Pentose Phosphate Pathway within the Endoplasmic Reticulum in Glutamine Addiction of Triple-Negative Breast Cancer Cells

 , ,
, ,  , ,
, ,  ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability and Proliferation Assays
2.3. Seahorse Analysis
2.4. Metabolites Extraction
2.5. Liquid Chromatography with Tandem Mass Spectrometry Metabolic Profiling
2.6. NADP/NADPH Ratio and Malondialdehyde Assay
2.7. Radioactive Glucose Analog Uptake and its Relationship with Glycolytic Flux
2.8. Western Blot Analysis
2.9. 2-NBDG/ER Colocalization
2.10. Mitochondrial–ER Colocalization and Mitochondrial Morphology
2.11. Ca2+ Experiments
2.12. Real-Time Polymerase Chain Reaction
2.13. Statistical Analysis
3. Results
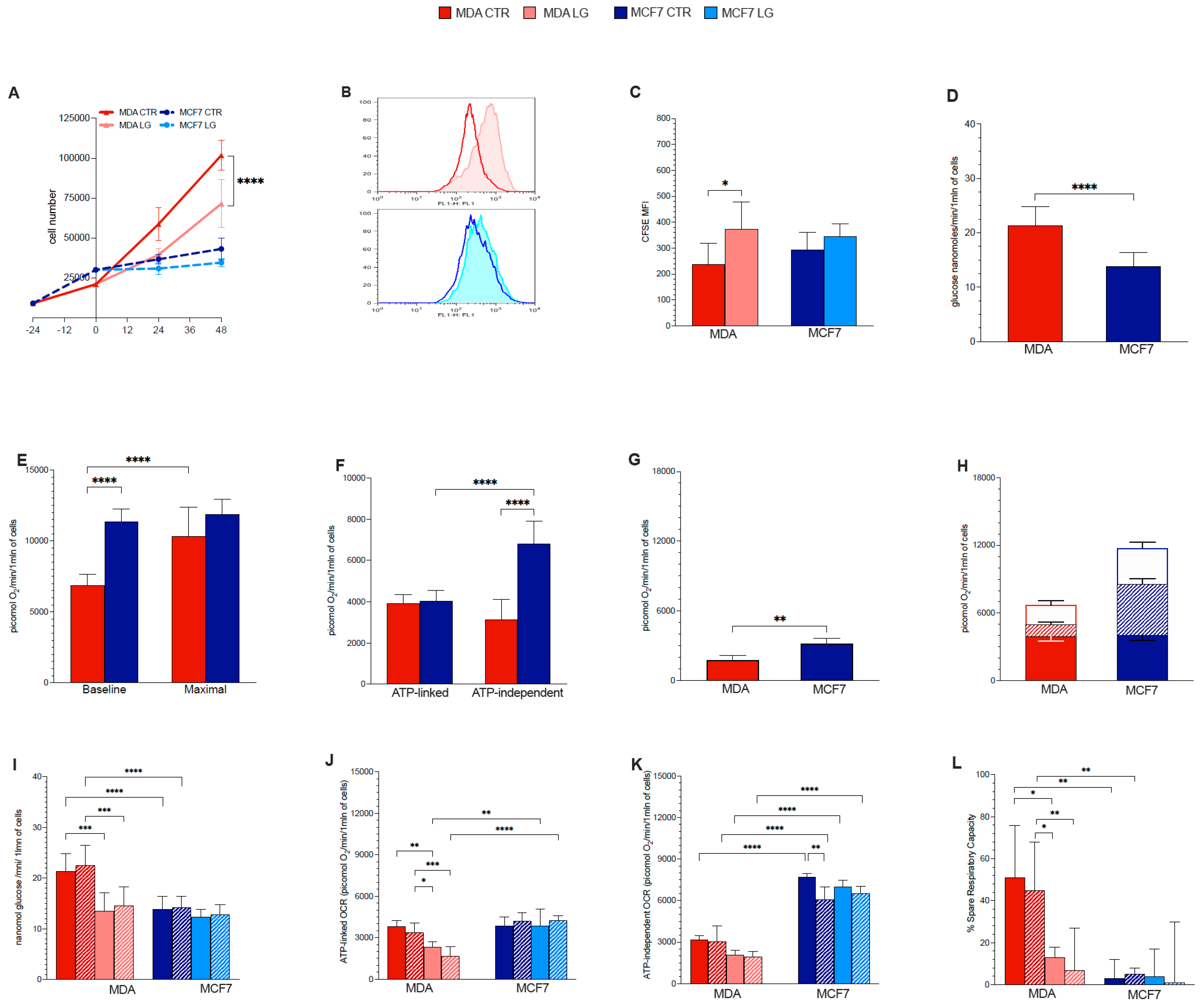
3.1. Proliferation Rate
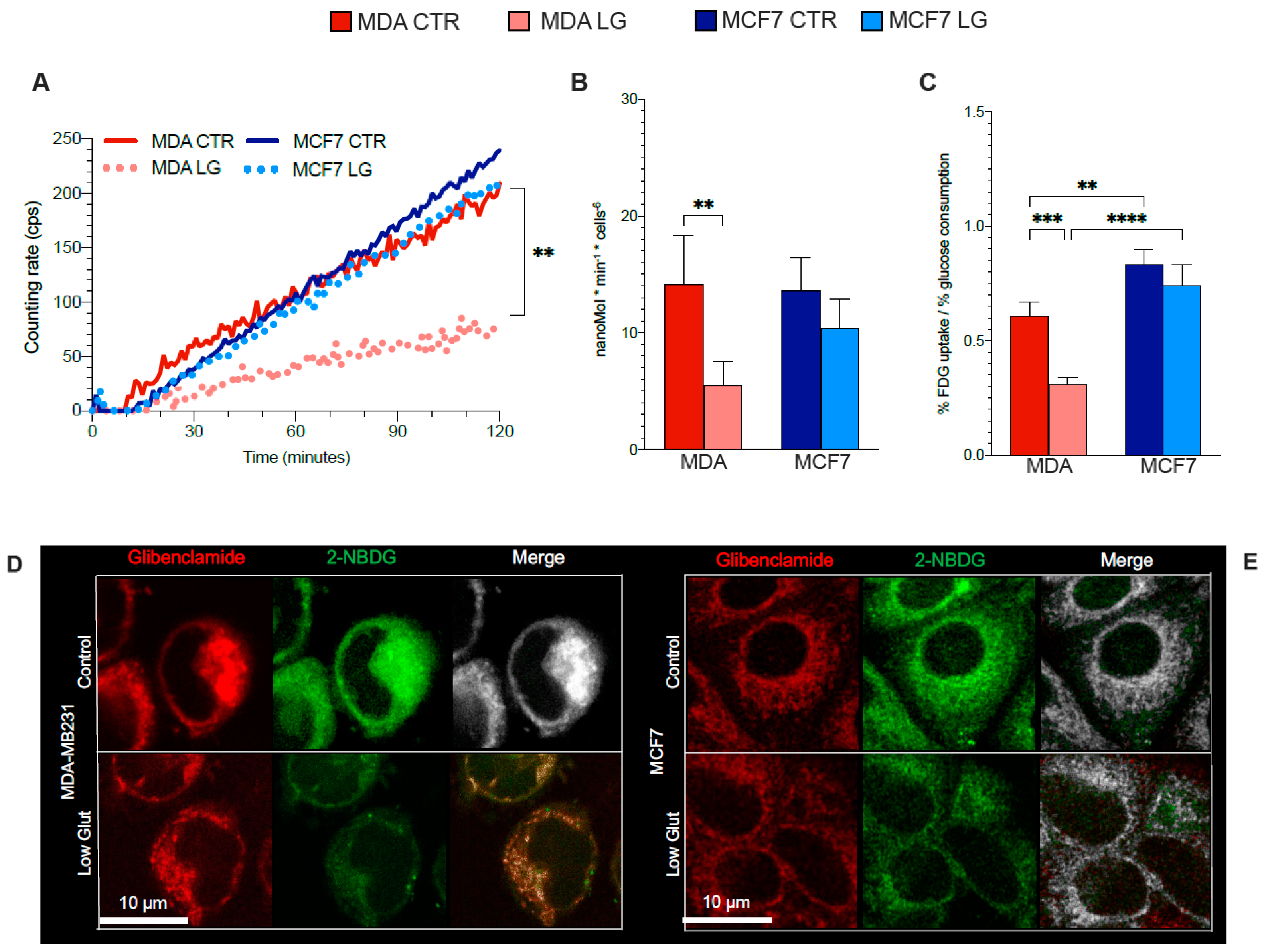
3.2. Warburg Effect Is Independent of Energy Needs
3.3. Glutamine Availability and Non-Respiratory Mitochondrial Function
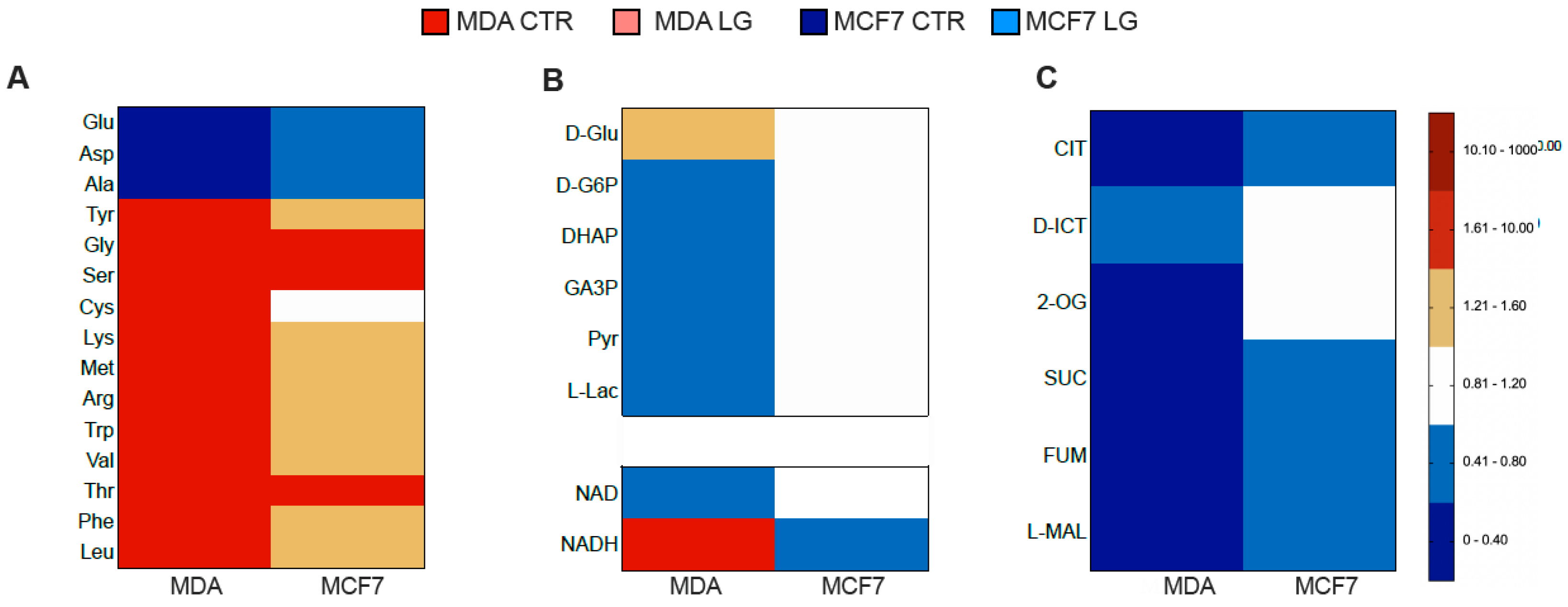
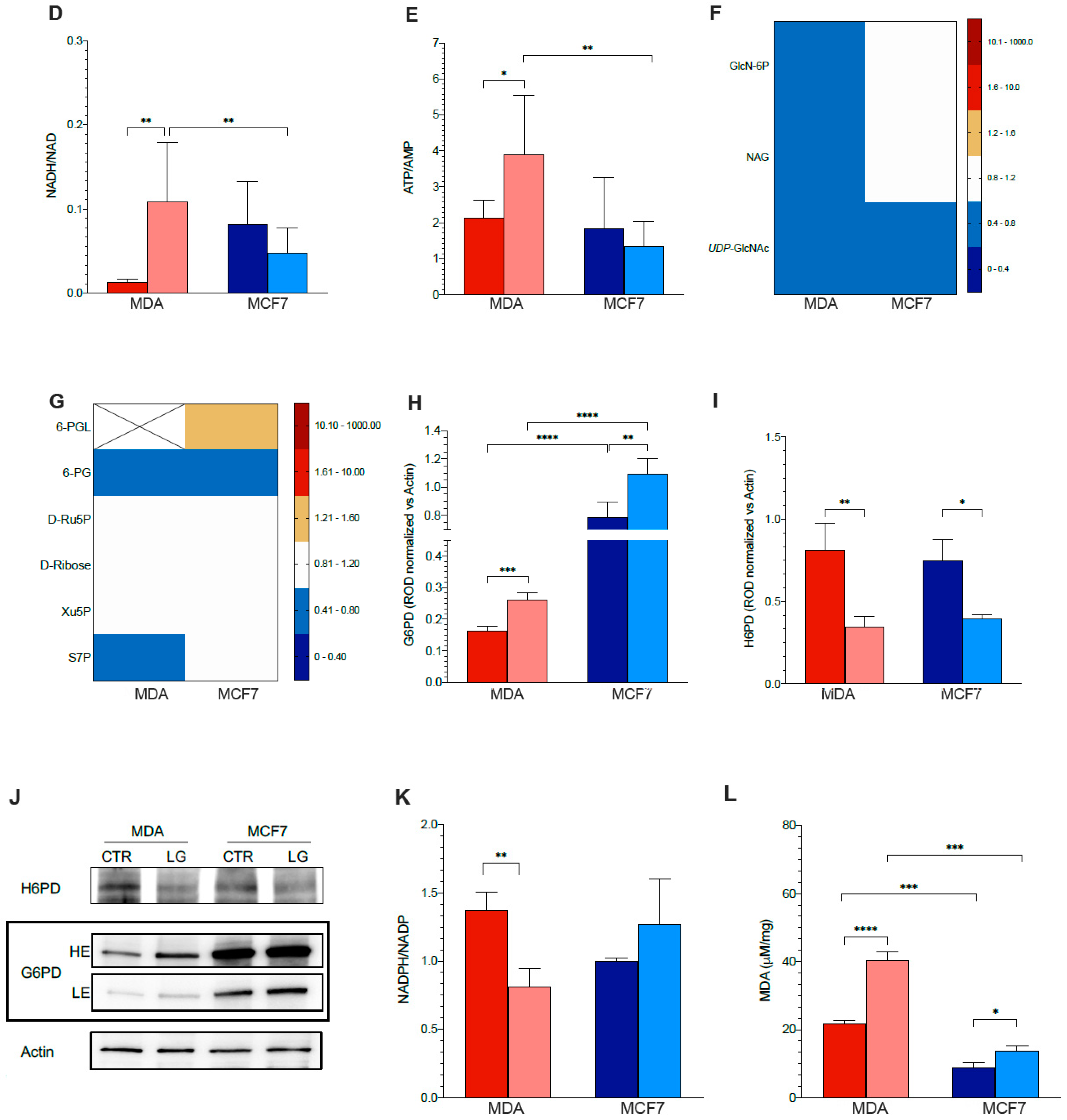
3.4. Permissive Role for Glutamine in NADH Oxidation and NADP Reduction in Cancer Cell
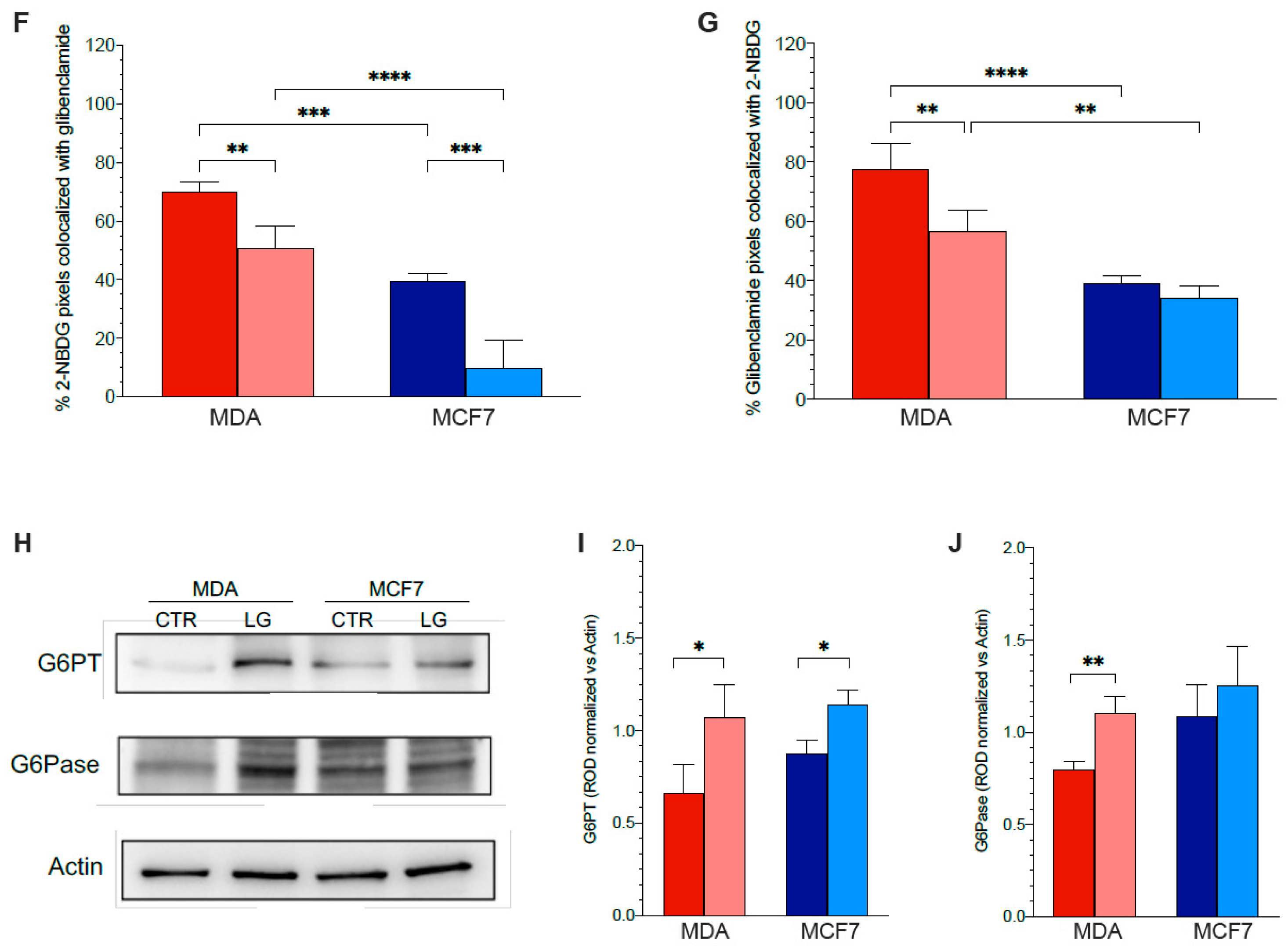
3.5. Reticular PPP as a Selective Target of Glutamine Shortage
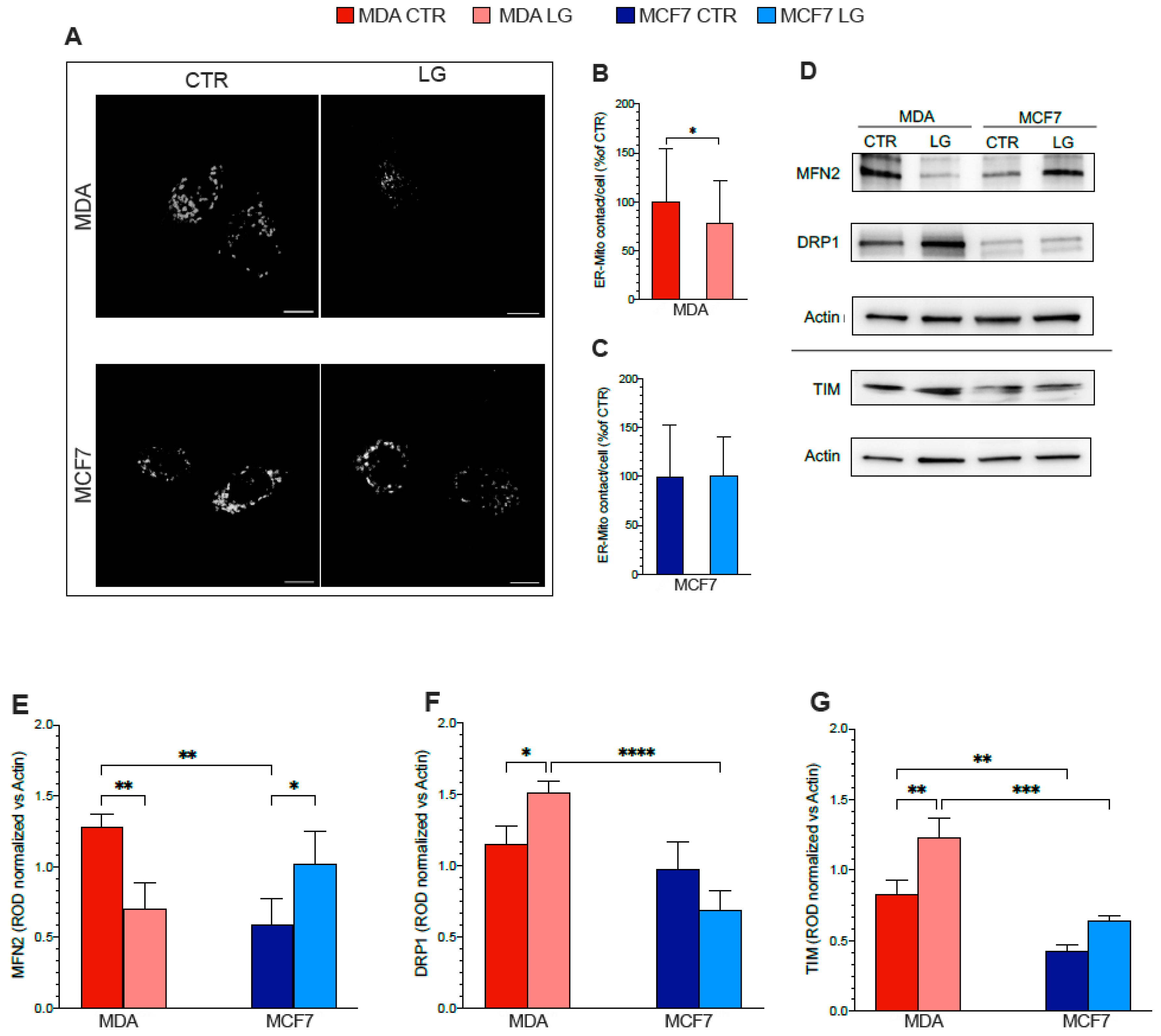
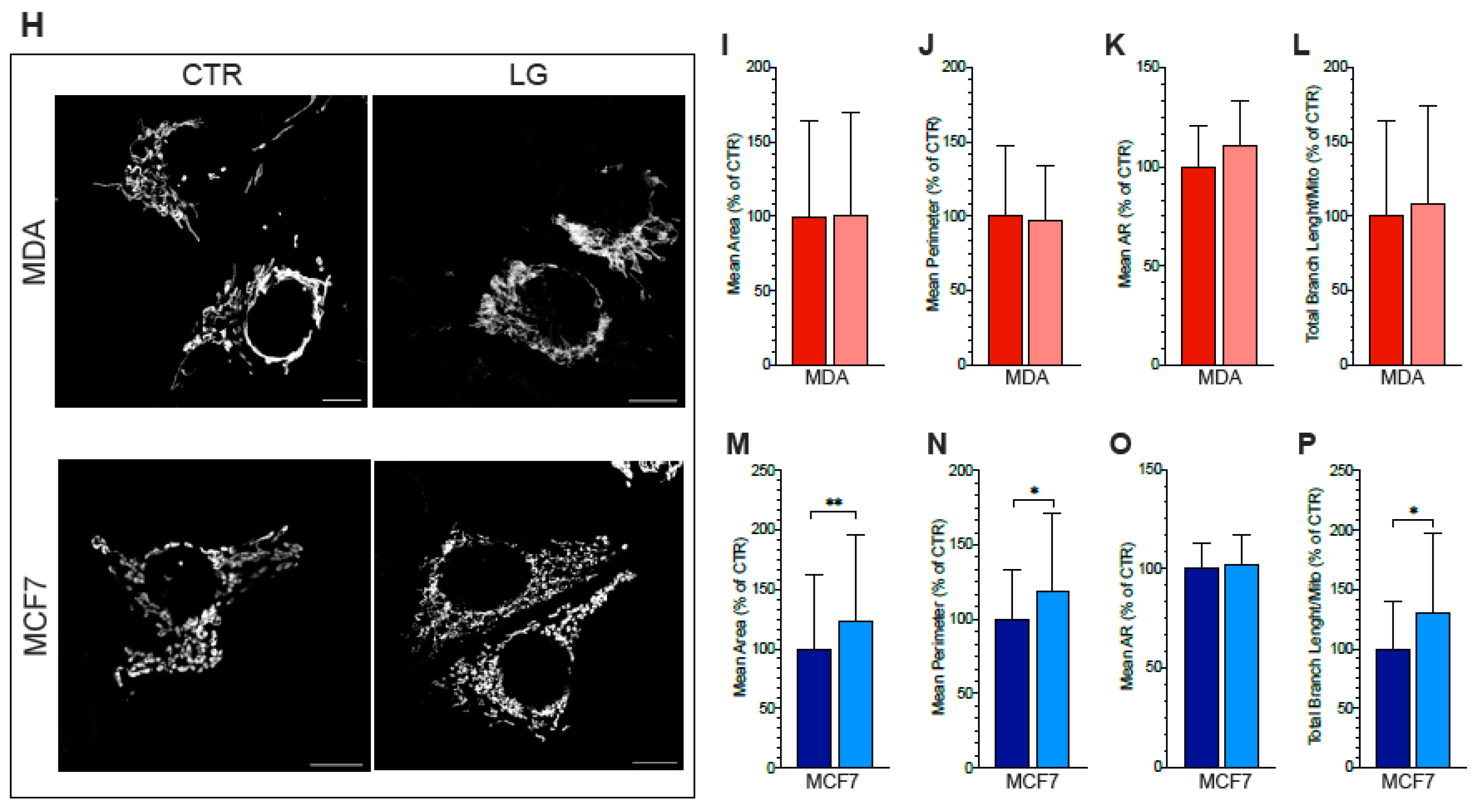
3.6. ER–Mitochondria Connection is Dependent upon ER-PPP Activity
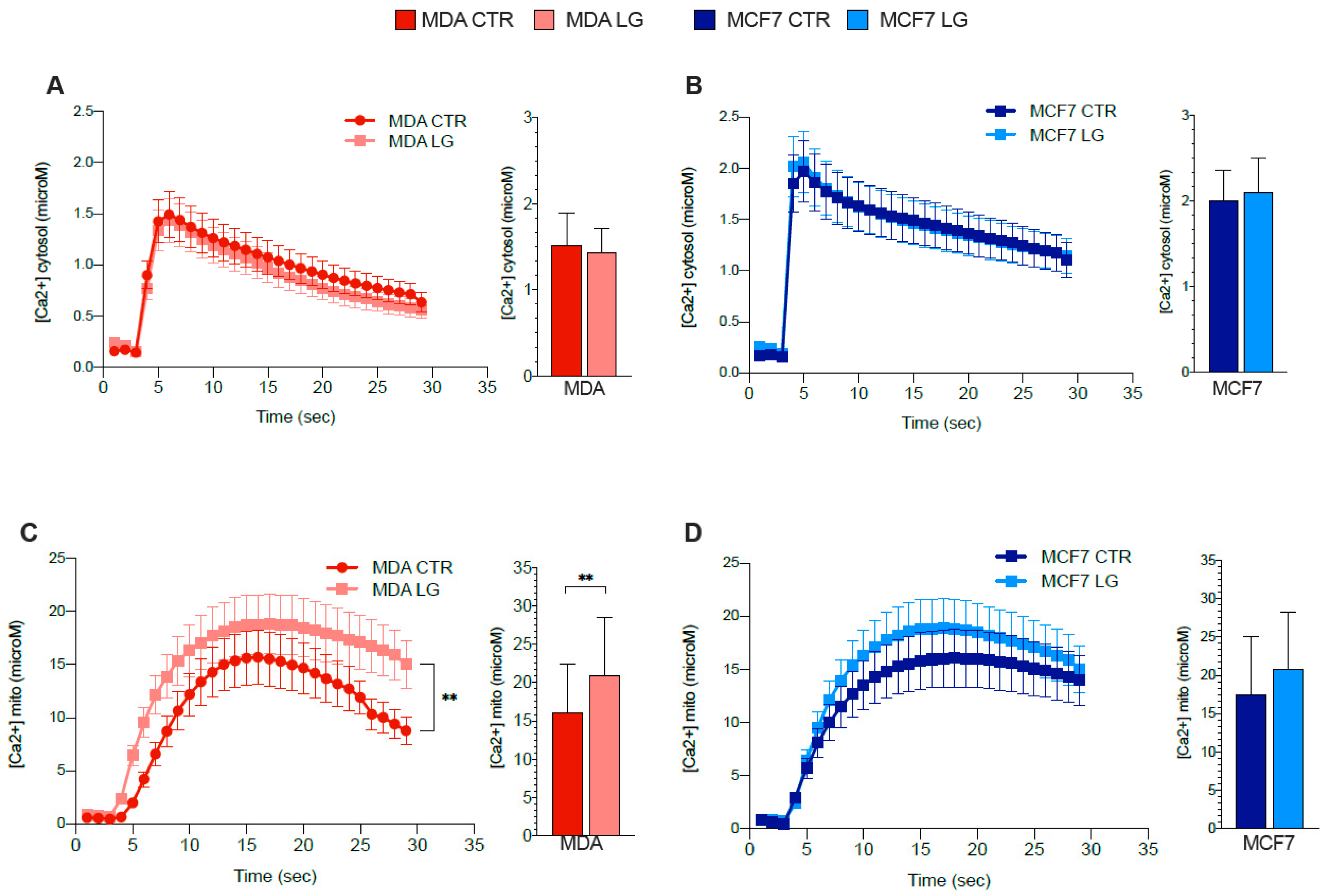
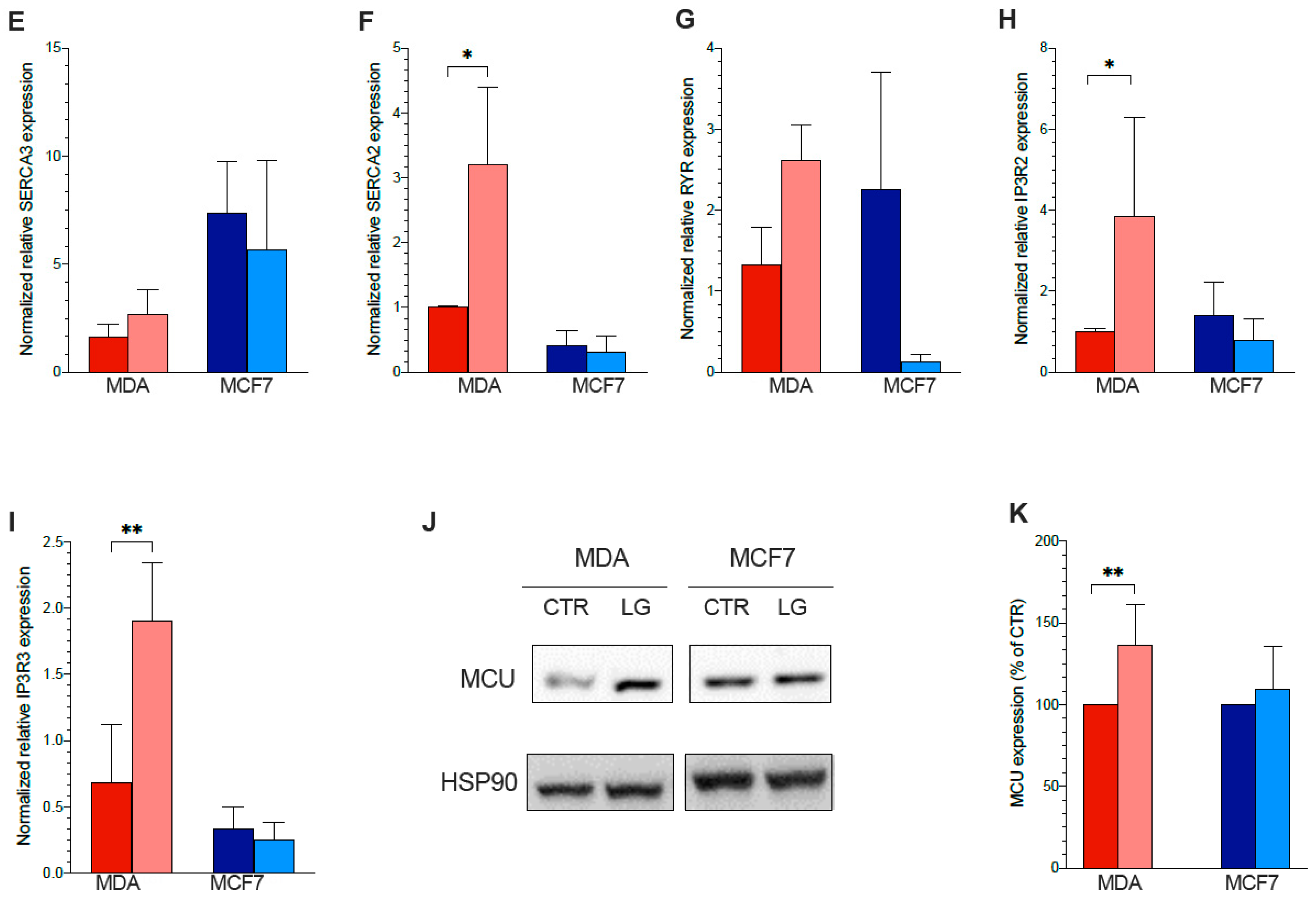
3.7. ER–Mitochondrial Ca2+ Exchanges
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warburg, O. Über den stoffwechsel der carcinomzelle. Naturwissenschaften 1924, 12, 1131–1137. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. Warburg report on the metabolism of tumors. J. Chem. Educ. 1930, 7, 179. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Icard, P.; Poulain, L.; Lincet, H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim. Biophys. Acta 2012, 1825, 111–116. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A., 3rd; Peters, E.C.; Driggers, E.M.; Hsieh-Wilson, L.C. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 2012, 337, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Polat, I.H.; Tarrado-Castellarnau, M.; Benito, A.; Hernandez-Carro, C.; Centelles, J.; Marin, S.; Cascante, M. Glutamine Modulates Expression and Function of Glucose 6-Phosphate Dehydrogenase via NRF2 in Colon Cancer Cells. Antioxidants 2021, 10, 1349. [Google Scholar] [CrossRef] [PubMed]
- Senesi, S.; Csala, M.; Marcolongo, P.; Fulceri, R.; Mandl, J.; Banhegyi, G.; Benedetti, A. Hexose-6-phosphate dehydrogenase in the endoplasmic reticulum. Biol. Chem. 2010, 391, 1–8. [Google Scholar] [CrossRef]
- Marcolongo, P.; Senesi, S.; Giunti, R.; Csala, M.; Fulceri, R.; Bánhegyi, G.; Benedetti, A. Expression of hexose-6-phosphate dehydrogenase in rat tissues. J. Steroid. Biochem. Mol. Biol. 2011, 126, 57–64. [Google Scholar] [CrossRef]
- Tsachaki, M.; Mladenovic, N.; Štambergová, H.; Birk, J.; Odermatt, A. Hexose-6-phosphate dehydrogenase controls cancer cell proliferation and migration through pleiotropic effects on the unfolded-protein response, calcium homeostasis, and redox balance. FASEB J. 2018, 32, 2690–2705. [Google Scholar] [CrossRef]
- Marini, C.; Cossu, V.; Kumar, M.; Milanese, M.; Cortese, K.; Bruno, S.; Bellese, G.; Carta, S.; Zerbo, R.A.; Torazza, C.; et al. The Role of Endoplasmic Reticulum in the Differential Endurance against Redox Stress in Cortical and Spinal Astrocytes from the Newborn SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Antioxidants 2021, 10, 1392. [Google Scholar] [CrossRef]
- Marini, C.; Cossu, V.; Bonifacino, T.; Bauckneht, M.; Torazza, C.; Bruno, S.; Castellani, P.; Ravera, S.; Milanese, M.; Venturi, C.; et al. Mechanisms underlying the predictive power of high skeletal muscle uptake of FDG in amyotrophic lateral sclerosis. EJNMMI Res. 2020, 10, 76. [Google Scholar] [CrossRef]
- Marini, C.; Cossu, V.; Bauckneht, M.; Carta, S.; Lanfranchi, F.; D’Amico, F.; Ravera, S.; Orengo, A.M.; Ghiggi, C.; Ballerini, F.; et al. Mitochondrial Generated Redox Stress Differently Affects the Endoplasmic Reticulum of Circulating Lymphocytes and Monocytes in Treatment-Naïve Hodgkin’s Lymphoma. Antioxidants 2022, 11, 762. [Google Scholar] [CrossRef]
- Kruger, N.J. The Bradford Method for Protein Quantitation. Methods Mol. Biol. 1994, 32, 9–15. [Google Scholar] [CrossRef]
- Cossu, V.; Marini, C.; Piccioli, P.; Rocchi, A.; Bruno, S.; Orengo, A.M.; Emionite, L.; Bauckneht, M.; Grillo, F.; Capitanio, S.; et al. Obligatory role of endoplasmic reticulum in brain FDG uptake. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1184–1196. [Google Scholar] [CrossRef] [PubMed]
- Bauckneht, M.; Cossu, V.; Castellani, P.; Piccioli, P.; Orengo, A.M.; Emionite, L.; Di Giulio, F.; Donegani, M.I.; Miceli, A.; Raffa, S.; et al. FDG uptake tracks the oxidative damage in diabetic skeletal muscle: An experimental study. Mol. Metab. 2020, 31, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Buschiazzo, A.; Cossu, V.; Bauckneht, M.; Orengo, A.M.; Piccioli, P.; Emionite, L.; Bianchi, G.; Grillo, F.; Rocchi, A.; Di Giulio, F.; et al. Effect of starvation on brain glucose metabolism and 18F-2-fluoro-2-deoxyglucose uptake: An experimental in-vivo and ex-vivo study. EJNMMI Res. 2018, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, L.; Reivic, H.M.; Kennedy, C.; Rosiers, M.H.D.; Patlak, C.S.; Pettigrew, K.D.; Sakurada, O.; Shinohara, M. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: Theory, procedure, and normal values in the conscious and anesthetized albino rat. J. Neurochem. 1977, 28, 897–916. [Google Scholar] [CrossRef] [PubMed]
- Marini, C.; Ravera, S.; Buschiazzo, A.; Bianchi, G.; Orengo, A.M.; Bruno, S.; Bottoni, G.; Emionite, L.; Pastorino, F.; Monteverde, E.; et al. Discovery of a novel glucose metabolism in cancer: The role of endoplasmic reticulum beyond glycolysis and pentose phosphate shunt. Sci. Rep. 2016, 6, 25092. [Google Scholar] [CrossRef] [PubMed]
- Cieri, D.; Vicario, M.; Giacomello, M.; Vallese, F.; Filadi, R.; Wagner, T.; Pozzan, T.; Pizzo, P.; Scorrano, L.; Brini, M.; et al. SPLICS: A split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ. 2018, 25, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Vallese, F.; Catoni, C.; Cieri, D.; Barazzuol, L.; Ramirez, O.; Calore, V.; Bonora, M.; Giamogante, F.; Pinton, P.; Brini, M.; et al. An expanded palette of improved SPLICS reporters detects multiple organelle contacts in vitro and in vivo. Nat. Commun. 2020, 11, 6069. [Google Scholar] [CrossRef]
- Chaudhry, A.; Shi, R.; Luciani, D.S. A pipeline for multidimensional confocal analysis of mitochondrial morphology, function, and dynamics in pancreatic β-cells. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E87–E101. [Google Scholar] [CrossRef]
- Simon, P. Q-Gene: Processing quantitative real-time RT-PCR data. Bioinformatics 2003, 19, 1439–1440. [Google Scholar] [CrossRef]
- Gwangwa, M.V.; Joubert, A.M.; Visagie, M.H. Effects of glutamine deprivation on oxidative stress and cell survival in breast cell lines. Biol. Res. 2019, 52, 15. [Google Scholar] [CrossRef]
- Dilshara, M.G.; Jeong, J.W.; Prasad Tharanga Jayasooriya, R.G.; Neelaka Molagoda, I.M.; Lee, S.; Park, S.R.; Choi, Y.H.; Kim, G.Y. Glutamine deprivation sensitizes human breast cancer MDA-MB-231 cells to TRIAL-mediated apoptosis. Biochem. Biophys. Res. Commun. 2017, 485, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Reda, A.; Refaat, A.; Abd-Rabou, A.A.; Mahmoud, A.M.; Adel, M.; Sabert, S.; Ali, S.S. Role of mitochondria in rescuing glycolytically inhibited subpopulation of triple negative but not hormone-responsive breast cancer cells. Sci. Rep. 2019, 9, 13748. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Cossu, V.; Bonanomi, M.; Bauckneht, M.; Ravera, S.; Righi, N.; Miceli, A.; Morbelli, S.; Orengo, A.M.; Piccioli, P.; Bruno, S.; et al. Two high-rate pentose-phosphate pathways in cancer cells. Sci. Rep. 2020, 10, 22111. [Google Scholar] [CrossRef]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 71–87.e7. [Google Scholar] [CrossRef]
- Caracó, C.; Aloj, L.; Chen, L.Y.; Chou, J.Y.; Eckelman, W.C. Cellular release of [18F]2-fluoro-2-deoxyglucose as a function of the glucose-6-phosphatase enzyme system. J. Biol. Chem. 2000, 275, 18489–18494. [Google Scholar] [CrossRef]
- Reivich, M.; Alavi, A.; Wolf, A.; Fowler, J.; Russell, J.; Arnett, C.; MacGregor, R.R.; Shiue, C.J.; Atkins, H.; Anand, A.; et al. Glucose metabolic rate kinetic model parameter determination in humans: The lumped constants and rate constants for [18F]fluorodeoxyglucose and [llC]deoxyglucose. J. Cereb. Blood Flow Metab. 1985, 5, 179–192. [Google Scholar] [CrossRef]
- Suda, S.; Shinohara, M.; Miyaoka, M.; Lucignani, G.; Kennedy, C.; Sokoloff, L. The lumped constant of the deoxyglucose method in hypoglycemia: Effects of moderate hypoglycemia on local cerebral glucose utilization in the rat. J. Cereb. Blood Flow Metab. 1990, 10, 499–509. [Google Scholar] [CrossRef]
- Bøtker, H.E.; Goodwin, G.W.; Holden, J.E.; Doenst, T.; Gjedde, A.; Taegtmeyer, H. Myocardial glucose uptake measured with fluorodeoxyglucose: A proposed method to account for variable lumped constants. J. Nucl. Med. 1999, 40, 1186–1196. [Google Scholar]
- Graham, M.M.; Muzi, M.; Spence, A.M.; O’Sullivan, F.; Lewellen, T.K.; Link, J.M.; Krohn, K.A. The FDG lumped constant in normal human brain. J. Nucl. Med. 2002, 43, 1157–1166. [Google Scholar]
- Cossu, V.; Bauckneht, M.; Bruno, S.; Orengo, A.M.; Emionite, L.; Balza, E.; Castellani, P.; Piccioli, P.; Miceli, A.; Raffa, S.; et al. The Elusive Link Between Cancer FDG Uptake and Glycolytic Flux Explains the Preserved Diagnostic Accuracy of PET/CT in Diabetes. Transl. Oncol. 2020, 13, 100752. [Google Scholar] [CrossRef] [PubMed]
- Bustos, G.; Ahumada-Castro, U.; Silva-Pavez, E.; Puebla, A.; Lovy, A.; Cesar Cardenas, J. The ER-mitochondria Ca2+ signaling in cancer progression: Fueling the monster. Int. Rev. Cell Mol. Biol. 2021, 363, 49–121. [Google Scholar] [CrossRef] [PubMed]
- Varga, K.; Hollósi, A.; Pászty, K.; Hegedűs, L.; Szakács, G.; Tímár, J.; Papp, B.; Enyedi, Á.; Padányi, R. Expression of calcium pumps is differentially regulated by histone deacetylase inhibitors and estrogen receptor alpha in breast cancer cells. BMC Cancer 2018, 18, 1029. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of Pyruvate Kinase M2 by Reactive Oxygen Species Contributes to Cellular Antioxidant Responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Suo, C.; Li, S.; Zhang, H.; Gao, P. Metabolic reprogramming for cancer cells and their microenvironment: Beyond the Warburg Effect. BBA 2018, 1870, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Z.; Hoshino, A.; Zheng, H.D.; Morley, M.; Arany, Z.; Rabinowitz, J.D. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat. Metab. 2019, 1, 404–415. [Google Scholar] [CrossRef]
- Botman, D.; Tigchelaar, W.; Van Noorden, C.J.F. Determination of glutamate dehydrogenase activity and its kinetics in mouse tissues using metabolic mapping (quantitative enzyme histochemistry). J. Histochem. Cytochem. 2014, 62, 802–812. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Bachelard, H.S. Specificity and kinetic properties of monosaccharide uptake into guinea pig cerebral cortex in vitro. J. Neurochem. 1971, 18, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Kung, H.-N.; Marks, J.R.; Chi, J.-T. Glutamine Synthetase Is a Genetic Determinant of Cell Type–Specific Glutamine Independence in Breast Epithelia. PLoS Genet. 2011, 7, e1002229. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, X.; Zhao, F. MCU-Dependent mROS Generation Regulates Cell Metabolism and Cell Death Modulated by the AMPK/PGC-1α/SIRT3 Signaling Pathway. Front. Med. 2021, 8, 674986. [Google Scholar] [CrossRef] [PubMed]
- Schömel, N.; Geisslinger, G.; Wegner, M.S. Influence of glycosphingolipids on cancer cell energy metabolism. Prog. Lipid Res. 2020, 79, 101050. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marini, C.; Cossu, V.; Carta, S.; Greotti, E.; Gaglio, D.; Bertola, N.; Chiesa, S.; Bruno, S.; Vitale, F.; Bonanomi, M.; et al. Fundamental Role of Pentose Phosphate Pathway within the Endoplasmic Reticulum in Glutamine Addiction of Triple-Negative Breast Cancer Cells. Antioxidants 2023, 12, 43. https://doi.org/10.3390/antiox12010043
Marini C, Cossu V, Carta S, Greotti E, Gaglio D, Bertola N, Chiesa S, Bruno S, Vitale F, Bonanomi M, et al. Fundamental Role of Pentose Phosphate Pathway within the Endoplasmic Reticulum in Glutamine Addiction of Triple-Negative Breast Cancer Cells. Antioxidants. 2023; 12(1):43. https://doi.org/10.3390/antiox12010043
Chicago/Turabian StyleMarini, Cecilia, Vanessa Cossu, Sonia Carta, Elisa Greotti, Daniela Gaglio, Nadia Bertola, Sabrina Chiesa, Silvia Bruno, Francesca Vitale, Marcella Bonanomi, and et al. 2023. "Fundamental Role of Pentose Phosphate Pathway within the Endoplasmic Reticulum in Glutamine Addiction of Triple-Negative Breast Cancer Cells" Antioxidants 12, no. 1: 43. https://doi.org/10.3390/antiox12010043
APA StyleMarini, C., Cossu, V., Carta, S., Greotti, E., Gaglio, D., Bertola, N., Chiesa, S., Bruno, S., Vitale, F., Bonanomi, M., Porro, D., Riondato, M., Orengo, A. M., Bauckneht, M., Morbelli, S., Ravera, S., & Sambuceti, G. (2023). Fundamental Role of Pentose Phosphate Pathway within the Endoplasmic Reticulum in Glutamine Addiction of Triple-Negative Breast Cancer Cells. Antioxidants, 12(1), 43. https://doi.org/10.3390/antiox12010043