
The Triterpenoid CDDO-Methyl Ester Redirects Macrophage Polarization and Reduces Lung Tumor Burden in a Nrf2-Dependent Manner

Abstract
1. Introduction
2. Methods
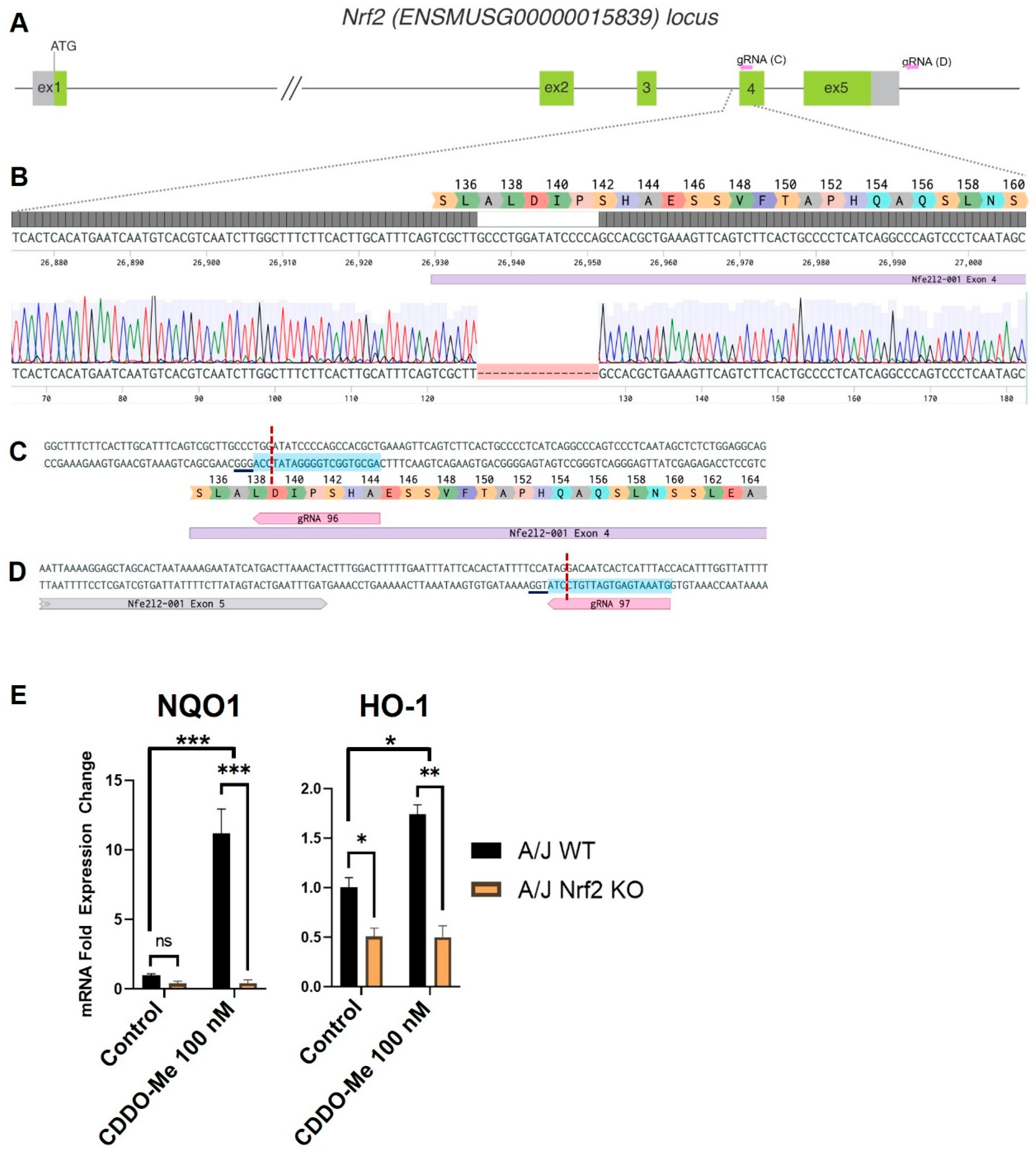
2.1. Generation of Nrf2 KO Mice
2.2. Genotyping of Nrf2 KO Mice
2.3. Cell Culture
2.4. Bone Marrow-Derived Macrophage (BMDM) Isolation
2.5. Quantitative Real-Time PCR
2.6. Treatment of Lung Adenocarcinomas In Vivo
2.7. Flow Cytometry
2.8. ELISAs
2.9. Immunofluorescent Staining
2.10. Western Blotting
2.11. Statistical Analysis
3. Results
3.1. Generation of Constitutive Nrf2 KO A/J Mice
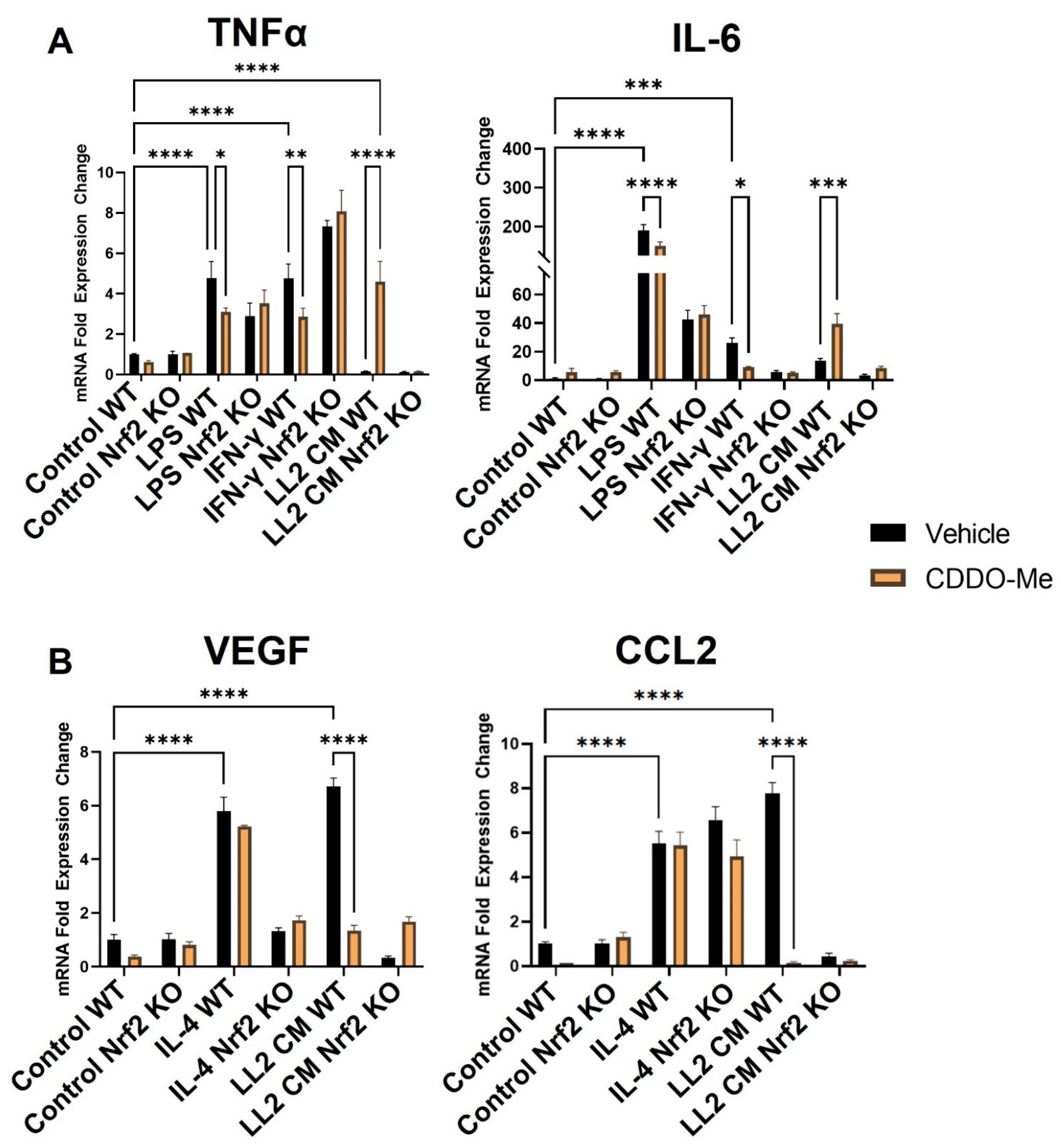
3.2. CDDO-Me Has Anti-Inflammatory Nrf2-Dependent Effects in BMDMs Stimulated with LPS or IFN-γ
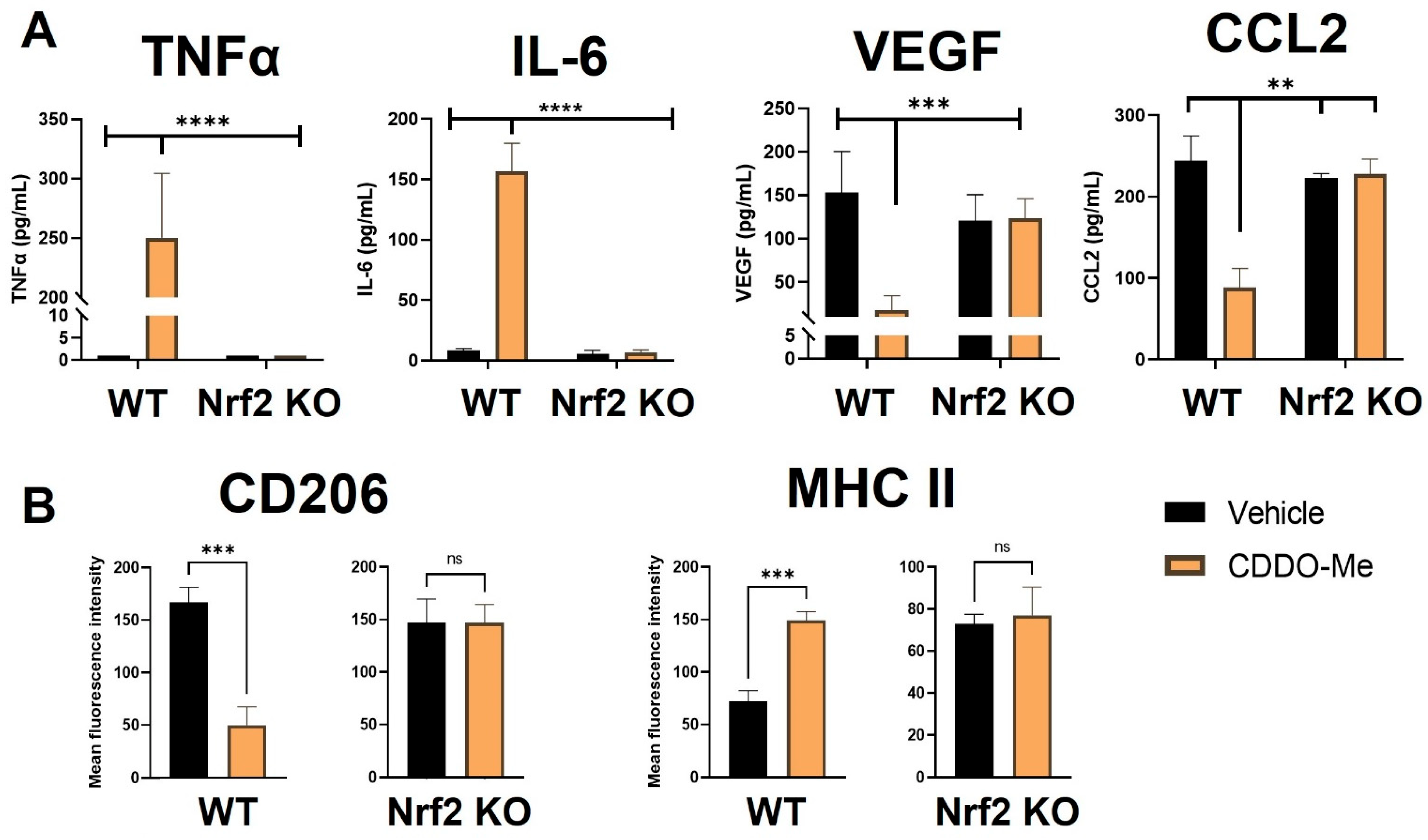
3.3. Conditioned Media from Lung Cancer Cells Reverses the Anti-Inflammatory Effect of CDDO-Me and Promotes a Nrf2-Dependent Anti-Tumor Phenotypic Profile in Tumor-Educated BMDMs
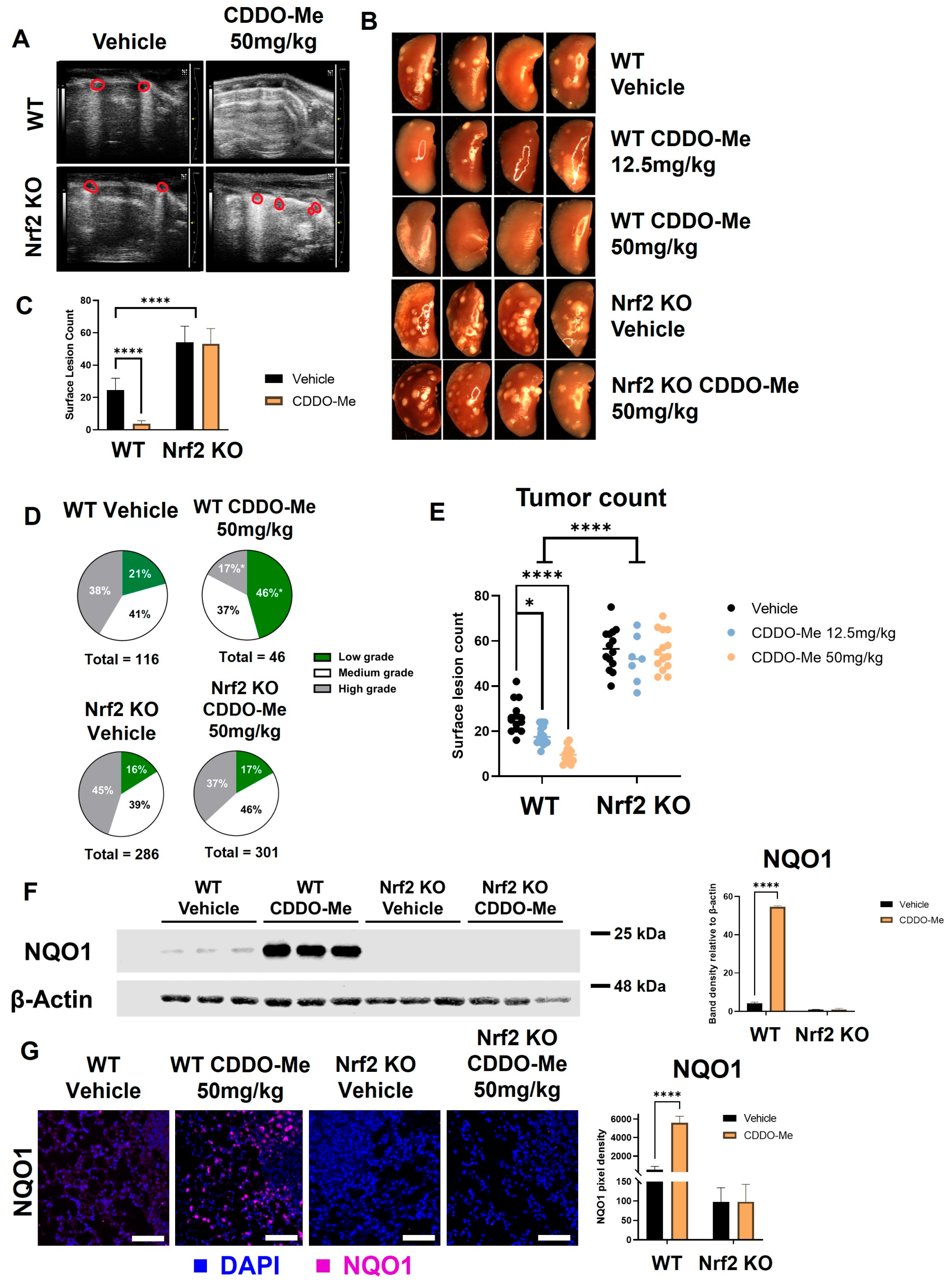
3.4. Nrf2 KO Increases Lung Tumor Burden and CDDO-Me Decreases Lung Tumor Burden in A/J Mice in a Nrf2-Dependent Manner
3.5. Sex-Dependent Differences in Lung Carcinogenesis in A/J Mice
3.6. CDDO-Me Activates the Nrf2 Pathway In Vivo
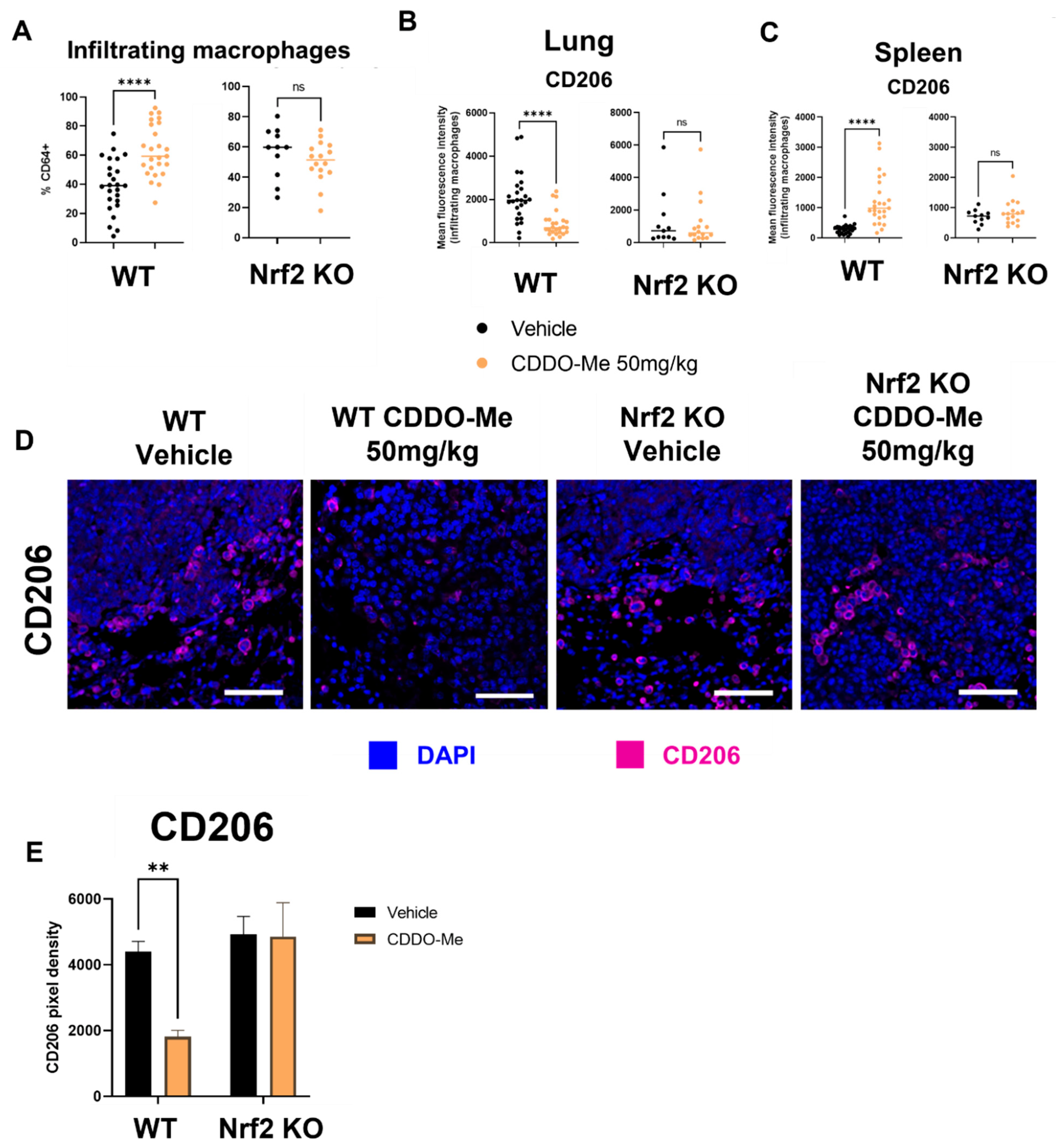
3.7. CDDO-Me Increases Infiltration and Modulates Polarization of Lung Macrophages in a Nrf2- and Context-Dependent Manner
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.; Miller, K.; Fuchs, H.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Nooreldeen, R.; Bach, H. Current and Future Development in Lung Cancer Diagnosis. Int. J. Mol. Sci. 2021, 22, 8661. [Google Scholar] [CrossRef]
- Zimmermann, S.; Peters, S.; Owinokoko, T.; Gadgeel, S. Immune Checkpoint Inhibitors in the Management of Lung Cancer. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 682–695. [Google Scholar] [CrossRef]
- Onoi, K.; Chihara, Y.; Uchino, J.; Shimamoto, T.; Morimoto, Y.; Iwasaku, M.; Kaneko, Y.; Yamada, T.; Takayama, K. Immune Checkpoint Inhibitors for Lung Cancer Treatment: A Review. J. Clin. Med. 2020, 9, 1362. [Google Scholar] [CrossRef]
- Sui, H.; Ma, N.; Wang, Y.; Li, H.; Liu, X.; Su, Y.; Yang, J. Anti-PD-1/PD-L1 Therapy for Non-Small-Cell Lung Cancer: Toward Personalized Medicine and Combination Strategies. J. Immunol. Res. 2018, 2018, 6984948. [Google Scholar] [CrossRef]
- Meyers, D.; Bryan, P.; Banerji, S.; Morris, D. Targeting the PD-1/PD-L1 axis for the treatment of non-small-cell lung cancer. Curr. Oncol. 2018, 25, e324–e334. [Google Scholar] [CrossRef]
- Maung, T.; Ergin, H.; Javed, M.; Inga, E.; Khan, S. Immune Checkpoint Inhibitors in Lung Cancer: Role of Biomarkers and Combination Therapies. Cureus 2020, 12, e8095. [Google Scholar] [CrossRef]
- Xu, F.; Wei, Y.; Tang, Z.; Liu, B.; Dong, J. Tumor-associated macrophages in lung cancer: Friend or foe? (Review). Mol. Med. Rep. 2020, 22, 4107–4115. [Google Scholar]
- Martinez, F.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. A J. Virtual Libr. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Sedighzadeh, S.; Khoshbin, A.; Razi, S.; Keshavarz-Fathi, M.; Rezaei, N. A narrative review of tumor-associated macrophages in lung cancer: Regulation of macrophage polarization and therapeutic implications. Transl. Lung Cancer Res. 2021, 10, 1889–1916. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Conway, E.; Pikor, L.; Kung, S.; Hamilton, M.; Lam, S.; Lam, W.; Bennewith, K. Macrophages, Inflammation, and Lung Cancer. Am. J. Respir. Crit. Care Med. 2015, 193, 116–130. [Google Scholar] [CrossRef]
- Sumitomo, R.; Hirai, T.; Fujita, M.; Murakami, H.; Otake, Y.; Huang, C. M2 tumor-associated macrophages promote tumor progression in non-small-cell lung cancer. Exp. Ther. Med. 2019, 18, 4490–4498. [Google Scholar] [CrossRef]
- Jackute, J.; Zemaitis, M.; Pranys, D.; Sitkauskiene, B.; Miliauskas, S.; Sakalauskas, R. The prognostic influence of tumor infiltrating M1 and M2 phenotype macrophages in resected non-small cell lung cancer. Eur. Respir. J. 2016, 48, OA1775. [Google Scholar]
- Duan, Z.; Luo, Y. Targeting macrophages in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 127. [Google Scholar] [CrossRef]
- Poh, A.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef]
- Kumari, N.; Choi, S. Tumor-associated macrophages in cancer: Recent advancements in cancer nanoimmunotherapies. J. Exp. Clin. Cancer Res. 2022, 41, 68. [Google Scholar] [CrossRef]
- Brüne, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxid. Redox Signal. 2013, 19, 595–637. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.; Wells, G.; Hayes, J.; Cousin, S.; Rumsey, W.; Attucks, O.; Franklin, S.; Levonen, A.-L.; Kensler, T.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Kundu, J.; Surh, Y.-J. Nrf2-Keap1 Signaling as a Potential Target for Chemoprevention of Inflammation-Associated Carcinogenesis. Pharm. Res. 2010, 27, 999–1013. [Google Scholar] [CrossRef]
- Sporn, M.; Liby, K. NRF2 and cancer: The good, the bad and the importance of context, Nature reviews. Cancer 2012, 12, 564–571. [Google Scholar]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Toth, R.; Warfel, N. Strange Bedfellows: Nuclear Factor, Erythroid 2-Like 2 (Nrf2) and Hypoxia-Inducible Factor 1 (HIF-1) in Tumor Hypoxia. Antioxidants 2017, 6, 27. [Google Scholar] [CrossRef]
- de la Vega, M.R.; Chapman, E.; Zhang, D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Lignitto, L.; LeBoeuf, S.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.; Pass, H.; Bhutkar, A.; Tsirigos, A.; Ueberheide, B.; et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019, 178, 316–329.e318. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Kobayashi, E.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Hiramoto, K.; Satoh, H.; Suzuki, T.; Moriguchi, T.; Pi, J.; Shimosegawa, T.; Yamamoto, M. Myeloid lineage-specific deletion of antioxidant system enhances tumor metastasis. Cancer Prev. Res. 2014, 7, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Kuga, A.; Suzuki, M.; Panda, H.; Kitamura, H.; Motohashi, H.; Yamamoto, M. Microenvironmental Activation of Nrf2 Restricts the Progression of Nrf2-Activated Malignant Tumors. Cancer Res. 2020, 80, 3331–3344. [Google Scholar] [CrossRef]
- Zhang, D.; Rennhack, J.; Andrechek, E.; Rockwell, C.; Liby, K. Identification of an Unfavorable Immune Signature in Advanced Lung Tumors from Nrf2-Deficient Mice. Antioxid. Redox Signal. 2018, 29, 1535–1552. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-H.; Hsieh, H.-C.; Shih, F.-H.; Wang, P.-W.; Yang, L.-X.; Shieh, D.-B.; Wang, Y.-C. An innovative NRF2 nano-modulator induces lung cancer ferroptosis and elicits an immunostimulatory tumor microenvironment. Theranostics 2021, 11, 7072–7091. [Google Scholar] [CrossRef]
- Panda, H.; Wen, H.; Suzuki, M.; Yamamoto, M. Multifaceted Roles of the KEAP1-NRF2 System in Cancer and Inflammatory Disease Milieu. Antioxidants 2022, 11, 538. [Google Scholar] [CrossRef]
- Liby, K.; Sporn, M. Synthetic oleanane triterpenoids: Multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol. Rev. 2012, 64, 972–1003. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.; Liby, K.; Stephenson, K.; Holtzclaw, W.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar] [CrossRef]
- Liby, K.; Risingsong, R.; Royce, D.; Williams, C.; Ma, T.; Yore, M.; Sporn, M. Triterpenoids CDDO-methyl ester or CDDO-ethyl amide and rexinoids LG100268 or NRX194204 for prevention and treatment of lung cancer in mice. Cancer Prev. Res. 2009, 2, 1050–1058. [Google Scholar] [CrossRef]
- Liby, K.; Royce, D.; Williams, C.; Risingsong, R.; Yore, M.; Honda, T.; Gribble, G.; Dmitrovsky, E.; Sporn, T.; Sporn, M. The Synthetic Triterpenoids CDDO-Methyl Ester and CDDO-Ethyl Amide Prevent Lung Cancer Induced by Vinyl Carbamate in A/J Mice. Cancer Res. 2007, 67, 2414–2419. [Google Scholar] [CrossRef]
- Chin, M.; Bakris, G.; Block, G.; Chertow, G.; Goldsberry, A.; Inker, L.; Heerspink, H.; O’Grady, M.; Pergola, P.; Wanner, C.; et al. Bardoxolone Methyl Improves Kidney Function in Patients with Chronic Kidney Disease Stage 4 and Type 2 Diabetes: Post-Hoc Analyses from Bardoxolone Methyl Evaluation in Patients with Chronic Kidney Disease and Type 2 Diabetes Study. Am. J. Nephrol. 2018, 47, 40–47. [Google Scholar] [CrossRef]
- Meyer, C.; Chin, M.; Feldman, J.; Goldsberry, A.; McConnell, J.; McCullough, P.; O’Grady, M.; Tapson, V.; Torres, F.; Waxman, A.; et al. Bardoxolone Methyl Increased eGFR in Patients with Pulmonary Arterial Hypertension Associated with Connective Tissue Disease (The LARIAT Study). Am. J. Respir. Crit. Care Med. 2018, 197, A7584. [Google Scholar]
- Singh, A.; Daemen, A.; Nickles, D.; Jeon, S.-M.; Foreman, O.; Sudini, K.; Gnad, F.; Lajoie, S.; Gour, N.; Mitzner, W.; et al. NRF2 Activation Promotes Aggressive Lung Cancer and Associates with Poor Clinical Outcomes. Clin. Cancer Res. 2021, 27, 877–888. [Google Scholar] [CrossRef]
- Sánchez-Ortega, M.; Carrera, A.; Garrido, A. Role of NRF2 in Lung Cancer. Cells 2021, 10, 1879. [Google Scholar] [CrossRef]
- To, C.; Ringelberg, C.; Royce, D.; Williams, C.; Risingsong, R.; Sporn, M.; Liby, K. Dimethyl fumarate and the oleanane triterpenoids, CDDO-imidazolide and CDDO-methyl ester, both activate the Nrf2 pathway but have opposite effects in the A/J model of lung carcinogenesis. Carcinogenesis 2015, 36, 769–781. [Google Scholar] [CrossRef]
- Qin, W.; Dion, S.; Kutny, P.; Zhang, Y.; Cheng, A.; Jillette, N.; Malhotra, A.; Geurts, A.; Chen, Y.; Wang, H. Efficient CRISPR/Cas9-Mediated Genome Editing in Mice by Zygote Electroporation of Nuclease. Genetics 2015, 200, 423–430. [Google Scholar] [CrossRef]
- Moerland, J.; Zhang, D.; Reich, L.; Carapellucci, S.; Lockwood, B.; Leal, A.; Krieger-Burke, T.; Aleiwi, B.; Ellsworth, E.; Liby, K. The novel rexinoid MSU-42011 is effective for the treatment of preclinical Kras-driven lung cancer. Sci. Rep. 2020, 10, 22244. [Google Scholar] [CrossRef]
- Yu, Y.; O’Koren, E.G.; Hotten, D.; Kan, M.; Kopin, D.; Nelson, E.; Que, L.; Gunn, M. A Protocol for the Comprehensive Flow Cytometric Analysis of Immune Cells in Normal and Inflamed Murine Non-Lymphoid Tissues. PLoS ONE 2016, 11, e0150606. [Google Scholar] [CrossRef]
- Gurley, K.; Moser, R.; Kemp, C. Induction of Lung Tumors in Mice with Urethane. Cold Spring Harb. Protoc. 2015, 2015, pdb.prot077446. [Google Scholar] [CrossRef]
- Chan, K.; Lu, R.; Chang, J.; Kan, Y. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef]
- Popp, M.; Maquat, L. Leveraging Rules of Nonsense-Mediated mRNA Decay for Genome Engineering and Personalized Medicine. Cell 2016, 165, 1319–1322. [Google Scholar] [CrossRef]
- Banas, K.; Modarai, S.; Rivera-Torres, N.; Yoo, B.-C.; Bialk, P.; Barrett, C.; Batish, M.; Kmiec, E. Exon skipping induced by CRISPR-directed gene editing regulates the response to chemotherapy in non-small cell lung carcinoma cells. Gene Ther. 2022, 29, 357–367. [Google Scholar] [CrossRef]
- Ying, W.; Cheruku, P.; Bazer, F.; Safe, S.; Zhou, B. Investigation of macrophage polarization using bone marrow derived macrophages. J. Vis. Exp. 2013, 76, e50323. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS–) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, Q.; Xu, M.; Wang, L.; Chen, X.; Feng, Y.; Li, Y.; Zhang, X.; Cui, W.; Jia, X. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol. Cancer 2020, 19, 41. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Reich, L.; Moerland, J.; Leal, A.; Zhang, D.; Carapellucci, S.; Lockwood, B.; Jurutka, P.; Marshall, P.; Wagner, C.; Liby, K. The rexinoid V-125 reduces tumor growth in preclinical models of breast and lung cancer. Sci. Rep. 2022, 12, 293. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Benson, P.; Hu, X.; Pal, A.; Xia, H.; Srivastava, S.; Awasthi, S.; Zaren, H.; Orchard, J.; Awasthi, Y. Gender-related differences in susceptibility of A/J mouse to benzo[a]pyrene-induced pulmonary and forestomach tumorigenesis. Cancer Lett. 1998, 128, 197–204. [Google Scholar] [CrossRef]
- Cho, H.-Y.; Kleeberger, S. Association of Nrf2 with airway pathogenesis: Lessons learned from genetic mouse models. Arch. Toxicol. 2015, 89, 1931–1957. [Google Scholar] [CrossRef]
- Leal, A.; Moerland, J.; Zhang, D.; Carapellucci, S.; Lockwood, B.; Krieger-Burke, T.; Aleiwi, B.; Ellsworth, E.; Liby, K. The RXR Agonist MSU42011 Is Effective for the Treatment of Preclinical HER2+ Breast Cancer and Kras-Driven Lung Cancer. Cancers 2021, 13, 5004. [Google Scholar] [CrossRef]
- Misharin, A.; Morales-Nebreda, L.; Mutlu, G.; Budinger, G.; Perlman, H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 2013, 49, 503–510. [Google Scholar] [CrossRef]
- Jaynes, J.M.; Sable, R.; Ronzetti, M.; Bautista, W.; Knotts, Z.; Abisoye-Ogunniyan, A.; Li, D.; Calvo, R.; Dashnyam, M.; Singh, A.; et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci. Transl. Med. 2020, 12, eaax6337. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Liang, Y.; Wang, L. Shaping Polarization Of Tumor-Associated Macrophages In Cancer Immunotherapy. Front. Immunol. 2022, 13, 888713. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, X.; Liu, L.; Wang, J.; Wu, J.; Sun, C. Role of macrophages in tumor progression and therapy (Review). Int. J. Oncol. 2022, 60, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, X.; Sun, Q.; Zhang, W.; Liu, C.; Ma, W.; Sun, C. Natural compounds: A new perspective on targeting polarization and infiltration of tumor-associated macrophages in lung cancer. Biomed. Pharmacother. 2022, 151, 113096. [Google Scholar] [CrossRef]
- Wang, L.; He, C. Nrf2-mediated anti-inflammatory polarization of macrophages as therapeutic targets for osteoarthritis. Front. Immunol. 2022, 13, 4575. [Google Scholar] [CrossRef]
- Ryan, D.G.; Knatko, E.V.; Casey, A.M.; Hukelmann, J.L.; Naidu, S.D.; Brenes, A.J.; Ekkunagul, T.; Baker, C.; Higgins, M.; Tronci, L.; et al. Nrf2 activation reprograms macrophage intermediary metabolism and suppresses the type I interferon response. Iscience 2022, 25, 103827. [Google Scholar] [CrossRef]
- Kikuchi, K.; Iida, M.; Ikeda, N.; Moriyama, S.; Hamada, M.; Takahashi, S.; Kitamura, H.; Watanabe, T.; Hasegawa, Y.; Hase, K.; et al. Macrophages Switch Their Phenotype by Regulating Maf Expression during Different Phases of Inflammation. J. Immunol. 2018, 201, 635. [Google Scholar] [CrossRef]
- Ball, M.; Shipman, E.; Kim, H.; Liby, K.; Pioli, P. CDDO-Me Redirects Activation of Breast Tumor Associated Macrophages. PLoS ONE 2016, 11, e0149600. [Google Scholar] [CrossRef]
- Torres, G.; Yang, H.; Park, C.; Spezza, P.; Khatwani, N.; Bhandari, R.; Liby, K.; Pioli, P. T Cells and CDDO-Me Attenuate Immunosuppressive Activation of Human Melanoma-Conditioned Macrophages. Front. Immunol. 2022, 13, 768753. [Google Scholar] [CrossRef]
- Satoh, H.; Moriguchi, T.; Takai, J.; Ebina, M.; Yamamoto, M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res 2013, 73, 4158–4168. [Google Scholar] [CrossRef]
- Bauer, A.; Cho, H.; Miller-Degraff, L.; Walker, C.; Helms, K.; Fostel, J.; Yamamoto, M.; Kleeberger, S. Targeted deletion of Nrf2 reduces urethane-induced lung tumor development in mice. PLoS ONE 2011, 6, e26590. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, W.; Blankenhaus, B.; Brunn, M.-L.; Meiners, J.; Breloer, M. Elucidating different pattern of immunoregulation in BALB/c and C57BL/6 mice and their F1 progeny. Sci. Rep. 2021, 11, 1536. [Google Scholar] [CrossRef] [PubMed]
- Nikodemova, M.; Watters, J. Outbred ICR/CD1 mice display more severe neuroinflammation mediated by microglial TLR4/CD14 activation than inbred C57Bl/6 mice. Neuroscience 2011, 190, 67–74. [Google Scholar] [CrossRef]
- Cao, L.; Che, X.; Qiu, X.; Li, Z.; Yang, B.; Wang, S.; Hou, K.; Fan, Y.; Qu, X.; Liu, Y. M2 macrophage infiltration into tumor islets leads to poor prognosis in non-small-cell lung cancer. Cancer Manag. Res. 2019, 11, 6125–6138. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Xiao, Z.; Guo, C.; Pu, Q.; Ma, L.; Liu, C.; Lin, F.; Liao, H.; You, Z.; Liu, L. Prognostic impact of tumor-associated macrophage infiltration in non-small cell lung cancer: A systemic review and meta-analysis. Oncotarget 2016, 7, 34217–34228. [Google Scholar] [CrossRef]
- Hwang, I.; Kim, J.; Ylaya, K.; Chung, E.; Kitano, H.; Perry, C.; Hanaoka, J.; Fukuoka, J.; Chung, J.-Y.; Hewitt, S. Tumor-associated macrophage, angiogenesis and lymphangiogenesis markers predict prognosis of non-small cell lung cancer patients. J. Transl. Med. 2020, 18, 443. [Google Scholar] [CrossRef]
- Jackute, J.; Zemaitis, M.; Pranys, D.; Sitkauskiene, B.; Miliauskas, S.; Vaitkiene, S.; Sakalauskas, R. Distribution of M1 and M2 macrophages in tumor islets and stroma in relation to prognosis of non-small cell lung cancer. BMC Immunol. 2018, 19, 3. [Google Scholar] [CrossRef]
- Yuan, A.; Hsiao, Y.-J.; Chen, H.-Y.; Chen, H.-W.; Ho, C.-C.; Chen, Y.-Y.; Liu, Y.-C.; Hong, T.-H.; Yu, S.-L.; Chen, J.J.; et al. Opposite Effects of M1 and M2 Macrophage Subtypes on Lung Cancer Progression. Sci. Rep. 2015, 5, 14273. [Google Scholar] [CrossRef]
- Brayton, C. Chapter 25—Spontaneous Diseases in Commonly Used Mouse Strains. In The Mouse in Biomedical Research (Second Edition); Fox, J., Davisson, M., Quimby, F., Barthold, S., Newcomer, C., Smith, A., Eds.; Academic Press: Burlington, MA, USA, 2007; pp. 623–717. [Google Scholar]
- Lim, J.; Leprivier, G. The impact of oncogenic RAS on redox balance and implications for cancer development. Cell Death Dis. 2019, 10, 955. [Google Scholar] [CrossRef]
- Riis, S.; Murray, J.; O’Connor, R. IGF-1 Signalling Regulates Mitochondria Dynamics and Turnover through a Conserved GSK-3β-Nrf2-BNIP3 Pathway. Cells 2020, 9, 147. [Google Scholar] [CrossRef]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.; Hampton, G.; Wahl, G. c-Myc Can Induce DNA Damage, Increase Reactive Oxygen Species, and Mitigate p53 Function: A Mechanism for Oncogene-Induced Genetic Instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.; Schumacker, P. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Martinez-Outschoorn, U.; Lisanti, M. Mitochondrial oxidative stress drives tumor progression and metastasis: Should we use antioxidants as a key component of cancer treatment and prevention? BMC Med. 2011, 9, 62. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT Control | WT CDDO-Me 50 mg/kg | Nrf2 KO Control | Nrf2 KO CDDO-Me 50 mg/kg | |
|---|---|---|---|---|
| Surface tumors | 618 | 106 | 1280 | 1289 |
| Mice per group | 23 | 24 | 23 | 24 |
| Average # of tumors per mouse (% WT control) | 26.87 ± 1.9 (100%) | 4.42 ± 0.9 (16.4%) **** | 55.65 ± 2.5 (207%) **** #### | 53.71 ± 2.3 (200%) **** #### |
| Tumor number, size, and burden | ||||
| Number of slides per group | 46 | 48 | 46 | 48 |
| Average # of tumors per slide (% WT control) | 2.52 ± 0.3 (100%) | 0.96 ± 0.1 (38%) ** | 6.22 ± 0.4 (247%) **** #### | 6.25 ± 0.5 (248%) **** #### |
| Average tumor size (mm3) per slide (% WT control) | 0.26 ± 0.07 (100%) | 0.03 ± 0.007 (13%) *** | 0.49 ± 0.15 (189%) *** ### | 0.34 ± 0.10 (131%) *** ### |
| Average tumor burden (mm3) per slide (% WT control) | 0.65 ± 0.1 (100%) | 0.03 ± 0.007 (5%) *** | 3.02 ± 0.04 (467%) *** ### | 2.10 ± 0.3 (324%) *** ### |
| Tumor histopathology | ||||
| Total # low grade (% total) | 24 (21%) | 21 (46%) * | 46 (16%) ## | 51 (17%) ## |
| Total # medium grade (% total) | 44 (38%) | 17 (37%) | 111 (39%) | 139 (46%) |
| Total # high grade (% total) | 48 (41%) | 8 (17%) * | 129 (45%) ## | 111 (37%) # |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moerland, J.A.; Leal, A.S.; Lockwood, B.; Demireva, E.Y.; Xie, H.; Krieger-Burke, T.; Liby, K.T. The Triterpenoid CDDO-Methyl Ester Redirects Macrophage Polarization and Reduces Lung Tumor Burden in a Nrf2-Dependent Manner. Antioxidants 2023, 12, 116. https://doi.org/10.3390/antiox12010116
Moerland JA, Leal AS, Lockwood B, Demireva EY, Xie H, Krieger-Burke T, Liby KT. The Triterpenoid CDDO-Methyl Ester Redirects Macrophage Polarization and Reduces Lung Tumor Burden in a Nrf2-Dependent Manner. Antioxidants. 2023; 12(1):116. https://doi.org/10.3390/antiox12010116
Chicago/Turabian StyleMoerland, Jessica A., Ana S. Leal, Beth Lockwood, Elena Y. Demireva, Huirong Xie, Teresa Krieger-Burke, and Karen T. Liby. 2023. "The Triterpenoid CDDO-Methyl Ester Redirects Macrophage Polarization and Reduces Lung Tumor Burden in a Nrf2-Dependent Manner" Antioxidants 12, no. 1: 116. https://doi.org/10.3390/antiox12010116
APA StyleMoerland, J. A., Leal, A. S., Lockwood, B., Demireva, E. Y., Xie, H., Krieger-Burke, T., & Liby, K. T. (2023). The Triterpenoid CDDO-Methyl Ester Redirects Macrophage Polarization and Reduces Lung Tumor Burden in a Nrf2-Dependent Manner. Antioxidants, 12(1), 116. https://doi.org/10.3390/antiox12010116