
Normal and Pathological NRF2 Signalling in the Central Nervous System

 , , , ,
, , , ,  ,
,
Abstract
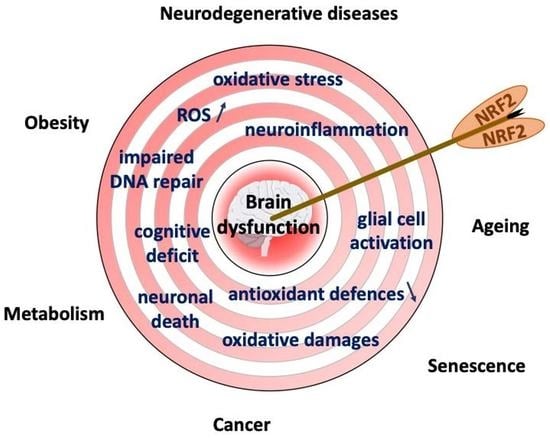
:
1. Beneficial and Harmful Roles of ROS
1.1. Radicals and Other Reactive Species at a Glance
1.2. Significance of ROS
1.3. Redox Homeostasis in Ageing
2. The NRF2-KEAP1 (Kelch-Like ECH-Associated Protein 1)-ARE (Antioxidant Response Element) Signalling Pathway
2.1. Mode of Regulation
2.2. Biological Functions
3. Oxidative Stress, Inflammation, and NRF2 in the Brain
3.1. The Brain, an Ideal Target for Oxidative Attacks
3.2. NRF2, Driver for a Healthy Brain?
3.3. Anti-Inflammatory Contribution of NRF2
4. NRF2 and Brain (Dys)Functions
4.1. NRF2 in Brain Control of Energy Metabolism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | NRF2 Pathway Status | Key Points | References |
|---|---|---|---|
| Fasting (in mice) | downregulation in the brain | reduced FGF21 sensitivity, reduced oxytocin signaling, increased sugar preference | [158] |
| Sugar intake (in mice) | activation in the brain | increased FGF21 sensitivity, activated oxytocin signaling, reduced sugar preference | [158] |
| Diabetes (in db/db diabetic mice) | downregulation in the brain | correlation with blood-brain-barrier permeability | [164,165] |
| downregulation in the brain reversed by FGF21 treament | blood-brain-barrier permeability reversed by FGF21 treament | [165] | |
| Diabetes, obesity (TrspRipKO mice) | downregulation in the brain | reduction in anorectic POMC neurons, loss of leptin sensitivity | [168] |
| downregulation in the brain reversed by Keap1 invalidation | reduction in anorectic POMC neurons, loss of leptin sensitivity reversed by Keap1 invalidation | [168] |
4.2. NRF2 in Brain Ageing
| Characteristics | NRF2 Pathway Status | Key Points | References |
|---|---|---|---|
| Normal ageing | decrease with age in many organs, including the brain | disruption of the antioxidant/oxidant equilibrium | [181,182] |
| Normal ageing | increased activity in liver of long-lived rodents | increased NRF2:ARE binding and antioxidant enzymatic activities | [183] |
| Normal ageing | decrease with age in the brain | decline in antioxidant enzyme activity after 12 months and incraeased superoxide anion in mitochondria | [186,187] |
| Normal ageing | possibly decreased based on blood levels of lipoperoxidation | indication of higher oxidative stress associated with cognitive decline | [188] |
| Normal ageing | possibly decreased based on blood levels of antioxidant enzyme | indication of lower antioxidative response associated with cognitive decline | [189] |
| Normal ageing | not directly implicated | increased oxidative stress associated with increased morbidity | [190] |
| Normal ageing | possibly decreased based on blood levels of antioxidant enzyme | indication of lower antioxidative response associated with cognitive decline | [191] |
| Normal ageing upon NRF2 deletion | inactive due to NRF2 knockout in mouse | reduced midbrain oxidative stress and reduced motor dysfunction in aged mice | [194] |
| Normal ageing upon NRF2 deletion | inactive due to NRF2 knockout in mouse | increased oxidative stress in liver and spleen | [195] |
| Cell culture model of HGPS | decreased due to sequestering of NRF2 by progerin | oxidative stress and aging defects in HGPS depend on NRF2 activity | [198] |
4.3. NRF2 in Age-Related Neurodegenerative Disorders
| Characteristics | NRF2 Pathway Status | Key Points | References |
|---|---|---|---|
| AD mouse model APP/PS1 | genetic ablation of Nfe2l2 | increase in the amyloid level, oxidative and inflammatory markers, autopahgy, gliosis, and cognitive impairments | [120,216,217] |
| AD mouse model APP/PS1 | lentivirus-mediated expression of NRF2 in hippocampi | increase in insoluble Abeta, decrease in astrogliosis, higher HO-1 level, reduction in spatial learning impairments | [218] |
| AD mouse model bigenic APP/TAU | genetic ablation of Nfe2l2 | increase in oxidative and inflammatory markers, cognitive impairments, and lower LTP | [119] |
| AD mouse model APP/PS1 | crossed with GFAP-NRF2, overexpression in astrocytes | reduction in amyloid pathology, gliosis, and cognitive deficits | [219] |
| AD mouse model MAPTP301S | crossed with GFAP-NRF2, overexpression in astrocytes | reduction in Tau pathology, cortical neurodegeneration | [219] |
| AD and AD variant with Lewy bodies | decreased NRF2 levels | hippocampus, cortex | [120,220] |
| PD | preserved or amplified NRF2 levels | substantia nigra neurons | [220,221] |
| α-syn and toxins PD models | decreased NRF2 levels | gliosis, neuronal death, locomotor impairments | [212,223,225,226] |
| α-syn PD mouse model | genetic ablation of Nfe2l2 | higher levels of P-syn, inflammation, microgliosis, and autophagy, loss of TH neurons, increase in cognitive impairments | [126] |
| A53T α-syn PD mouse model | crossed with GFAP-NRF2, overexpression in astrocytes | motor deficits delayed | [227] |
| Amyotrophic Lateral Sclerosis (ALS) | crossed with GFAP-NRF2, overexpression in astrocytes | neurodegeneration delayed | [228] |
| Alexander’s disease | NRF2 overexpression in astrocytes | decrease in GFAP expression and Rosenthal fibres, restoration of body weight | [231] |
4.4. NRF2 in Brain Cancer
| Characteristics | NRF2 Pathway Status | Key Points | References |
|---|---|---|---|
| Glioma cell line | downregulation | increased apoptosis | [237] |
| downregulation | increased autophagy | [238] | |
| overexpression | reversed ERK and PI3K-inhibition-induced inhibition of cell viability | [239] | |
| overexpression | increased proliferation, resistance to ferroptosis, increased oncogenic potential | [240] | |
| downregulation | reduced proliferation | [241] | |
| downregulation | reduced proliferation, reduced mitochondrial oxygen consumption | [242] | |
| downregulation | reduced proliferation | [121] | |
| upregulation | increased proliferation, tumour cell infiltration, and mesenchymal transition | [245] | |
| downregulation | decreased resistance towards chemotherapy | [247] | |
| overexpression | prevention of ROS-induced cell death | [248] | |
| downregulation | enhanced sensitivity towards chemotherapy and irradiation | [251] | |
| overexpression | increased cell survival after chemotherapy | [252] | |
| overexpression | reduced ROS levels, increased increased cell survival | [253] | |
| Human glioma tissue | upregulation (as compared to normal tissue) | potentially implicated in glioma progression | [239] |
| upregulation | worse patient survival | [240] | |
| upregulation (as compared to normal tissue) | potentially implicated in glioma progression | [241] | |
| upregulation | worse patient survival | [241] | |
| upregulation | increased angiogenesis | [242] | |
| upregulation with grade of malignancy | potentially implicated in glioma progression | [243] | |
| cytoplasmic expression | association with worse prognosis | [243] | |
| nuclear expression | association with better prognosis | [243] | |
| overexpression | association with worse prognosis, more tissue necrosis, correlation with high HIF1alpha levels | [244] | |
| overexpression | decreased progression-free survival | [245] | |
| overexpression | worse patient prognosis in anaplastic glioma | [246] | |
| overexpression | upon irradiation and chemotherapy | [251] | |
| overexpression | association with worse prognosis | [252] | |
| Xenograft glioblastomas mouse model | downregulation | reduced tumour growth, reduced proliferation, increased apoptosis, reduced angiogenesis | [241] |
| downregulation | reduced VEGF expression, reduced angiogenesis | [242] | |
| Human lung carcinoma tissue | overexpression | higher risk of brain metastasis | [249] |
| increased mutational rate of NRF2 pathway | higher risk of brain metastasis | [250] |
5. Epigenetic Regulation of NRF2 Signalling
5.1. NRF2-Mediated Transactivation and Chromatin Level Regulation
5.2. Different NRF2 Activity in Brain Cell Types and Approaches for Cell-Type-Specific Targeting
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Di Meo, S.; Napolitano, G.; Venditti, P. Physiological and Pathological Role of ROS: Benefits and Limitations of Antioxidant Treatment. Int. J. Mol. Sci. 2019, 20, 4810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poyton, R.O.; Ball, K.A.; Castello, P.R. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol. Metab. 2009, 20, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Carraro, E.; Schilirò, T.; Biorci, F.; Romanazzi, V.; Degan, R.; Buonocore, D.; Verri, M.; Dossena, M.; Bonetta, S.; Gilli, G. Physical Activity, Lifestyle Factors and Oxidative Stress in Middle Age Healthy Subjects. Int. J. Environ. Res. Public. Health 2018, 15, 1152. [Google Scholar] [CrossRef] [Green Version]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Al-Gubory, K.H. Environmental pollutants and lifestyle factors induce oxidative stress and poor prenatal development. Reprod. Biomed. Online 2014, 29, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Aseervatham, G.S.B.; Sivasudha, T.; Jeyadevi, R.; Arul Ananth, D. Environmental factors and unhealthy lifestyle influence oxidative stress in humans—An overview. Environ. Sci. Pollut. Res. 2013, 20, 4356–4369. [Google Scholar] [CrossRef]
- Knaus, U.G. Oxidants in Physiological Processes. Handb. Exp. Pharmacol. 2021, 264, 27–47. [Google Scholar] [CrossRef]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta Biomembr. 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Jamil, M.; Debbarh, H.; Aboulmaouahib, S.; Aniq Filali, O.; Mounaji, K.; Zarqaoui, M.; Saadani, B.; Louanjli, N.; Cadi, R. Reactive oxygen species in reproduction: Harmful, essential or both? Zygote 2020, 28, 255–269. [Google Scholar] [CrossRef]
- Dennery, P.A. Oxidative stress in development: Nature or nurture? Free Radic. Biol. Med. 2010, 49, 1147–1151. [Google Scholar] [CrossRef]
- Oswald, M.C.W.; Garnham, N.; Sweeney, S.T.; Landgraf, M. Regulation of neuronal development and function by ROS. FEBS Lett. 2018, 592, 679–691. [Google Scholar] [CrossRef]
- Chaudhari, P.; Ye, Z.; Jang, Y.-Y. Roles of reactive oxygen species in the fate of stem cells. Antioxid. Redox Signal. 2014, 20, 1881–1890. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, H.; Iwata, K.; Ogonuki, N.; Inoue, K.; Atsuo, O.; Kanatsu-Shinohara, M.; Morimoto, T.; Yabe-Nishimura, C.; Shinohara, T. ROS are required for mouse spermatogonial stem cell self-renewal. Cell Stem Cell 2013, 12, 774–786. [Google Scholar] [CrossRef] [Green Version]
- Sart, S.; Song, L.; Li, Y. Controlling Redox Status for Stem Cell Survival, Expansion, and Differentiation. Oxid. Med. Cell. Longev. 2015, 2015, 105135. [Google Scholar] [CrossRef] [Green Version]
- Bassoy, E.Y.; Walch, M.; Martinvalet, D. Reactive Oxygen Species: Do They Play a Role in Adaptive Immunity? Front. Immunol. 2021, 12, 755856. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Santos, A.L.; Sinha, S.; Lindner, A.B. The Good, the Bad, and the Ugly of ROS: New Insights on Aging and Aging-Related Diseases from Eukaryotic and Prokaryotic Model Organisms. Oxid. Med. Cell. Longev. 2018, 2018, 1941285. [Google Scholar] [CrossRef]
- Dhoke, N.R.; Geesala, R.; Das, A. Low Oxidative Stress-Mediated Proliferation Via JNK-FOXO3a-Catalase Signaling in Transplanted Adult Stem Cells Promotes Wound Tissue Regeneration. Antioxid. Redox Signal. 2018, 28, 1047–1065. [Google Scholar] [CrossRef] [Green Version]
- Lassègue, B.; Griendling, K.K. Reactive oxygen species in hypertension: An update. Am. J. Hypertens. 2004, 17, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Kröller-Schön, S.; Daiber, A.; Steven, S.; Oelze, M.; Frenis, K.; Kalinovic, S.; Heimann, A.; Schmidt, F.P.; Pinto, A.; Kvandova, M.; et al. Crucial role for Nox2 and sleep deprivation in aircraft noise-induced vascular and cerebral oxidative stress, inflammation, and gene regulation. Eur. Heart J. 2018, 39, 3528–3539. [Google Scholar] [CrossRef]
- Steven, S.; Frenis, K.; Kalinovic, S.; Kvandova, M.; Oelze, M.; Helmstädter, J.; Hahad, O.; Filippou, K.; Kus, K.; Trevisan, C.; et al. Exacerbation of adverse cardiovascular effects of aircraft noise in an animal model of arterial hypertension. Redox Biol. 2020, 34, 101515. [Google Scholar] [CrossRef]
- Kong, H.; Chandel, N.S. Regulation of redox balance in cancer and T cells. J. Biol. Chem. 2018, 293, 7499–7507. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Brennan, M.-L.; Shen, Z.; MacPherson, J.C.; Schmitt, D.; Molenda, C.E.; Hazen, S.L. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J. Biol. Chem. 2002, 277, 46116–46122. [Google Scholar] [CrossRef] [Green Version]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lands, W.E.; Marshall, P.J.; Kulmacz, R.J. Hydroperoxide availability in the regulation of the arachidonate cascade. Adv. Prostaglandin. Thromboxane. Leukot. Res. 1985, 15, 233–235. [Google Scholar] [PubMed]
- Bruegel, M.; Ceglarek, U.; Thiery, J. Eicosanoids: Essential mediators in health and disease/Eicosanoide: Bedeutende Faktoren in der Homöostase und ihre Bedeutung in der Pathogenese multipler Erkrankungen. Lab. Med. 2009, 33, 333–339. [Google Scholar] [CrossRef]
- Yui, K.; Imataka, G.; Nakamura, H.; Ohara, N.; Naito, Y. Eicosanoids Derived From Arachidonic Acid and Their Family Prostaglandins and Cyclooxygenase in Psychiatric Disorders. Curr. Neuropharmacol. 2015, 13, 776–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassoni, D.; Kaur, G.; Weisinger, R.S.; Sinclair, A.J. The role of eicosanoids in the brain. Asia Pac. J. Clin. Nutr. 2008, 17 (Suppl. 1), 220–228. [Google Scholar]
- Wymann, M.P.; Schneiter, R. Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 2008, 9, 162–176. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Flatt, T. A new definition of aging? Front. Genet. 2012, 3, 148. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Sanada, F.; Taniyama, Y.; Muratsu, J.; Otsu, R.; Shimizu, H.; Rakugi, H.; Morishita, R. Source of Chronic Inflammation in Aging. Front. Cardiovasc. Med. 2018, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.-P.-P.; Sulaiman Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.Y.; Kim, D.H.; Lee, E.K.; Chung, K.W.; Chung, S.; Lee, B.; Seo, A.Y.; Chung, J.H.; Jung, Y.S.; Im, E.; et al. Redefining Chronic Inflammation in Aging and Age-Related Diseases: Proposal of the Senoinflammation Concept. Aging Dis. 2019, 10, 367. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Papaconstantinou, J.; Wang, C.Z.; Zhang, M.; Yang, S.; Deford, J.; Bulavin, D.V.; Ansari, N.H. Attenuation of p38α MAPK stress response signaling delays the in vivo aging of skeletal muscle myofibers and progenitor cells. Aging 2015, 7, 718–733. [Google Scholar] [CrossRef]
- Akbari, S.; Amiri, F.T.; Naderi, M.; Shaki, F.; Seyedabadi, M. Sodium arsenite accelerates D-galactose-induced aging in the testis of the rat: Evidence for mitochondrial oxidative damage, NF-kB, JNK, and apoptosis pathways. Toxicology 2022, 470, 153148. [Google Scholar] [CrossRef]
- Ng, A.; Tam, W.W.; Zhang, M.W.; Ho, C.S.; Husain, S.F.; McIntyre, R.S.; Ho, R.C. IL-1β, IL-6, TNF- α and CRP in Elderly Patients with Depression or Alzheimer’s disease: Systematic Review and Meta-Analysis. Sci. Rep. 2018, 8, 12050. [Google Scholar] [CrossRef]
- Chung, H.Y.; Sung, B.; Jung, K.J.; Zou, Y.; Yu, B.P. The molecular inflammatory process in aging. Antioxid. Redox Signal. 2006, 8, 572–581. [Google Scholar] [CrossRef]
- Zhu, Y.; Carvey, P.M.; Ling, Z. Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 2006, 1090, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Maher, P. The effects of stress and aging on glutathione metabolism. Ageing Res. Rev. 2005, 4, 288–314. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Guo, J.; Wei, X.; Niu, C.; Jia, M.; Li, Q.; Meng, D. Bach1: Function, Regulation, and Involvement in Disease. Oxid. Med. Cell. Longev. 2018, 2018, 1347969. [Google Scholar] [CrossRef]
- Davudian, S.; Mansoori, B.; Shajari, N.; Mohammadi, A.; Baradaran, B. BACH1, the master regulator gene: A novel candidate target for cancer therapy. Gene 2016, 588, 30–37. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, S.; Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Bach1 Competes with Nrf2 Leading to Negative Regulation of the Antioxidant Response Element (ARE)-mediated NAD(P)H:Quinone Oxidoreductase 1 Gene Expression and Induction in Response to Antioxidants *. J. Biol. Chem. 2005, 280, 16891–16900. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Chen, S.; Zhao, Q.; Sun, Y.; Nie, H. The Critical Role of Bach2 in Shaping the Balance between CD4+ T Cell Subsets in Immune-Mediated Diseases. Mediators Inflamm. 2019, 2019, 2609737. [Google Scholar] [CrossRef] [Green Version]
- Tsukumo, S.; Unno, M.; Muto, A.; Takeuchi, A.; Kometani, K.; Kurosaki, T.; Igarashi, K.; Saito, T. Bach2 maintains T cells in a naive state by suppressing effector memory-related genes. Proc. Natl. Acad. Sci. USA 2013, 110, 10735–10740. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, H.; Igarashi, K. Expression of the oxidative stress-regulated transcription factor bach2 in differentiating neuronal cells. J. Biochem. 2002, 132, 427–431. [Google Scholar] [CrossRef]
- He, X.; Chen, M.G.; Lin, G.X.; Ma, Q. Arsenic induces NAD(P)H-quinone oxidoreductase I by disrupting the Nrf2 x Keap1 x Cul3 complex and recruiting Nrf2 x Maf to the antioxidant response element enhancer. J. Biol. Chem. 2006, 281, 23620–23631. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Ma, Q. Critical cysteine residues of Kelch-like ECH-associated protein 1 in arsenic sensing and suppression of nuclear factor erythroid 2-related factor 2. J. Pharmacol. Exp. Ther. 2010, 332, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boo, Y.C. Natural Nrf2 Modulators for Skin Protection. Antioxidants 2020, 9, 812. [Google Scholar] [CrossRef]
- Osama, A.; Zhang, J.; Yao, J.; Yao, X.; Fang, J. Nrf2: A dark horse in Alzheimer’s disease treatment. Ageing Res. Rev. 2020, 64, 101206. [Google Scholar] [CrossRef]
- Yang, S.-Y.; Pyo, M.C.; Nam, M.-H.; Lee, K.-W. ERK/Nrf2 pathway activation by caffeic acid in HepG2 cells alleviates its hepatocellular damage caused by t-butylhydroperoxide-induced oxidative stress. BMC Complement. Altern. Med. 2019, 19, 139. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.-R.; Ma, H.; Zou, Z.-Y.; He, K.; Xiao, Y.-B.; Wang, Y.; Feng, M.; Ye, X.-L.; Li, X.-G. Activation of Akt and JNK/Nrf2/NQO1 pathway contributes to the protective effect of coptisine against AAPH-induced oxidative stress. Biomed. Pharmacother. 2017, 85, 313–322. [Google Scholar] [CrossRef]
- Reddy, N.M.; Potteti, H.R.; Vegiraju, S.; Chen, H.-J.; Tamatam, C.M.; Reddy, S.P. PI3K-AKT Signaling via Nrf2 Protects against Hyperoxia-Induced Acute Lung Injury, but Promotes Inflammation Post-Injury Independent of Nrf2 in Mice. PLoS ONE 2015, 10, e0129676. [Google Scholar] [CrossRef] [Green Version]
- Lee, O.-H.; Jain, A.K.; Papusha, V.; Jaiswal, A.K. An auto-regulatory loop between stress sensors INrf2 and Nrf2 controls their cellular abundance. J. Biol. Chem. 2007, 282, 36412–36420. [Google Scholar] [CrossRef] [Green Version]
- Culbreth, M.; Aschner, M. GSK-3β, a double-edged sword in Nrf2 regulation: Implications for neurological dysfunction and disease. F1000Res 2018, 7, 1043. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Zhu, J.; Lei, S.; Dong, Y.; Li, L.; Jiang, B.; Tan, L.; Wu, J.; Yu, S.; et al. GSK-3β downregulates Nrf2 in cultured cortical neurons and in a rat model of cerebral ischemia-reperfusion. Sci. Rep. 2016, 6, 20196. [Google Scholar] [CrossRef] [Green Version]
- Huo, L.; Li, C.-W.; Huang, T.-H.; Lam, Y.C.; Xia, W.; Tu, C.; Chang, W.-C.; Hsu, J.L.; Lee, D.-F.; Nie, L.; et al. Activation of Keap1/Nrf2 signaling pathway by nuclear epidermal growth factor receptor in cancer cells. Am. J. Transl. Res. 2014, 6, 649–663. [Google Scholar]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Xiong, Z.; Huang, C.; Li, J.; Yang, W.; Han, Y.; Paiboonrungruan, C.; Major, M.B.; Chen, K.-N.; Kang, X.; et al. Hyperactivity of the transcription factor Nrf2 causes metabolic reprogramming in mouse esophagus. J. Biol. Chem. 2019, 294, 327–340. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Lin, X.; Meng, D.; Zeng, L.; Zhuang, R.; Huang, S.; Lv, W.; Hu, J. Nrf2 Mediates Metabolic Reprogramming in Non-Small Cell Lung Cancer. Front. Oncol. 2020, 10, 578315. [Google Scholar] [CrossRef]
- Kasai, S.; Mimura, J.; Ozaki, T.; Itoh, K. Emerging Regulatory Role of Nrf2 in Iron, Heme, and Hemoglobin Metabolism in Physiology and Disease. Front. Vet. Sci. 2018, 5, 242. [Google Scholar] [CrossRef] [Green Version]
- Digaleh, H.; Kiaei, M.; Khodagholi, F. Nrf2 and Nrf1 signaling and ER stress crosstalk: Implication for proteasomal degradation and autophagy. Cell. Mol. Life Sci. 2013, 70, 4681–4694. [Google Scholar] [CrossRef]
- Bendavit, G.; Aboulkassim, T.; Hilmi, K.; Shah, S.; Batist, G. Nrf2 Transcription Factor Can Directly Regulate mTOR: Linking Cytoprotective Gene Expression To A Major Metabolic Regulator That Generates Redox Activity. J. Biol. Chem. 2016, 291, 25476–25488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Oh, S.Y.; Lee, D.H.; Lee, Y.S.; Sung, S.H.; Ji, H.W.; Lee, M.J.; Lee, Y.-H.; Rhee, S.G.; Bae, S.H. p62/SQSTM1 is required for the protection against endoplasmic reticulum stress-induced apoptotic cell death. Free Radic. Res. 2016, 50, 1408–1421. [Google Scholar] [CrossRef]
- Bian, S.; Zhao, Y.; Li, F.; Lu, S.; Yang, S.; Liu, M.; Wang, S.; Zhao, D.; Zhang, W.; Wang, J. Knockdown of p62/sequestosome enhances ginsenoside Rh2-induced apoptosis in cervical cancer HeLa cells with no effect on autophagy. Biosci. Biotechnol. Biochem. 2021, 85, 1097–1103. [Google Scholar] [CrossRef]
- Dikovskaya, D.; Dinkova-Kostova, A.T. Measuring Changes in Keap1-Nrf2 Protein Complex Conformation in Individual Cells by FLIM-FRET. Curr. Protoc. Toxicol. 2020, 85, e96. [Google Scholar] [CrossRef]
- Dikovskaya, D.; Appleton, P.L.; Bento-Pereira, C.; Dinkova-Kostova, A.T. Measuring the Interaction of Transcription Factor Nrf2 with Its Negative Regulator Keap1 in Single Live Cells by an Improved FRET/FLIM Analysis. Chem. Res. Toxicol. 2019, 32, 500–512. [Google Scholar] [CrossRef]
- Baird, L.; Swift, S.; Llères, D.; Dinkova-Kostova, A.T. Monitoring Keap1-Nrf2 interactions in single live cells. Biotechnol. Adv. 2014, 32, 1133–1144. [Google Scholar] [CrossRef] [Green Version]
- Heurtaux, T.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Richart, L.; Uriarte Huarte, O.; Schohn, H.; Mittelbronn, M. Apomorphine Reduces A53T α-Synuclein-Induced Microglial Reactivity Through Activation of NRF2 Signalling Pathway. Cell. Mol. Neurobiol. 2021. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, L.; Chen, B.; Fang, Y.; Lin, W.; Zhang, T.; Feng, X.; Tao, X.; Wu, Y.; Fu, X.; et al. Chlorogenic acid exerts neuroprotective effect against hypoxia-ischemia brain injury in neonatal rats by activating Sirt1 to regulate the Nrf2-NF-κB signaling pathway. Cell Commun. Signal. 2022, 20, 84. [Google Scholar] [CrossRef]
- Zhou, L.; Zhou, M.; Tan, H.; Xiao, M. Cypermethrin-induced cortical neurons apoptosis via the Nrf2/ARE signaling pathway. Pestic. Biochem. Physiol. 2020, 165, 104547. [Google Scholar] [CrossRef]
- Zhang, S.; Jin, S.; Zhang, S.; Li, Y.-Y.; Wang, H.; Chen, Y.; Lu, H. Vitexin protects against high glucose-induced endothelial cell apoptosis and oxidative stress via Wnt/β-catenin and Nrf2 signalling pathway. Arch. Physiol. Biochem. 2022, 1–10. [Google Scholar] [CrossRef]
- Mutter, F.E.; Park, B.K.; Copple, I.M. Value of monitoring Nrf2 activity for the detection of chemical and oxidative stress. Biochem. Soc. Trans. 2015, 43, 657–662. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, N.A.; Haskew-Layton, R.E.; Basso, M.; Hushpulian, D.M.; Payappilly, J.B.; Speer, R.E.; Ahn, Y.-H.; Rakhman, I.; Cole, P.A.; Pinto, J.T.; et al. Development of Neh2-luciferase reporter and its application for high throughput screening and real-time monitoring of Nrf2 activators. Chem. Biol. 2011, 18, 752–765. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, G.; Zhu, W.-W.; Zhou, D. Activation of nuclear factor-erythroid 2-related factor 2 (Nrf2) in the basilar artery after subarachnoid hemorrhage in rats. Ann. Clin. Lab. Sci. 2010, 40, 233–239. [Google Scholar]
- Zhong, Q.; Mishra, M.; Kowluru, R.A. Transcription factor Nrf2-mediated antioxidant defense system in the development of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3941–3948. [Google Scholar] [CrossRef]
- Wang, X.J.; Hayes, J.D.; Wolf, C.R. Generation of a Stable Antioxidant Response Element–Driven Reporter Gene Cell Line and Its Use to Show Redox-Dependent Activation of Nrf2 by Cancer Chemotherapeutic Agents. Cancer Res. 2006, 66, 10983–10994. [Google Scholar] [CrossRef] [Green Version]
- Bischoff, L.J.M.; Kuijper, I.A.; Schimming, J.P.; Wolters, L.; Braak, B.; Langenberg, J.P.; Noort, D.; Beltman, J.B.; van de Water, B. A systematic analysis of Nrf2 pathway activation dynamics during repeated xenobiotic exposure. Arch. Toxicol. 2019, 93, 435–451. [Google Scholar] [CrossRef] [Green Version]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, Q.; Wang, Y.; Peng, J.; Shao, L.; Li, X. Irisin protects against sepsis-associated encephalopathy by suppressing ferroptosis via activation of the Nrf2/GPX4 signal axis. Free Radic. Biol. Med. 2022, 187, 171–184. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, P.; Han, K. Hydrogen Attenuated Inflammation Response and Oxidative in Hypoxic Ischemic Encephalopathy via Nrf2 Mediated the Inhibition of NLRP3 and NF-κB. Neuroscience 2022, 485, 23–36. [Google Scholar] [CrossRef]
- Oikawa, D.; Akai, R.; Tokuda, M.; Iwawaki, T. A transgenic mouse model for monitoring oxidative stress. Sci. Rep. 2012, 2, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, Y.; Uchiyama, A.; Sekiguchi, A.; Yamazaki, S.; Fujiwara, C.; Yokoyama, Y.; Ogino, S.; Torii, R.; Hosoi, M.; Akai, R.; et al. Protective effect of dimethyl fumarate for the development of pressure ulcers after cutaneous ischemia-reperfusion injury. Wound Repair Regen. 2020, 28, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox Signaling Mediated by Thioredoxin and Glutathione Systems in the Central Nervous System. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef]
- Lee, K.H.; Cha, M.; Lee, B.H. Crosstalk between Neuron and Glial Cells in Oxidative Injury and Neuroprotection. Int. J. Mol. Sci. 2021, 22, 13315. [Google Scholar] [CrossRef]
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione metabolism in brain: Metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem. 2000, 267, 4912–4916. [Google Scholar] [CrossRef]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef] [Green Version]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Liddell, J.R. Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration? Antioxidants 2017, 6, 65. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Boas, S.M.; Joyce, K.L.; Cowell, R.M. The NRF2-Dependent Transcriptional Regulation of Antioxidant Defense Pathways: Relevance for Cell Type-Specific Vulnerability to Neurodegeneration and Therapeutic Intervention. Antioxidants 2021, 11, 8. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I. Glutathione and Related Molecules in Parkinsonism. Int. J. Mol. Sci. 2021, 22, 8689. [Google Scholar] [CrossRef]
- Burton, N.C.; Kensler, T.W.; Guilarte, T.R. In vivo modulation of the Parkinsonian phenotype by Nrf2. Neurotoxicology 2006, 27, 1094–1100. [Google Scholar] [CrossRef]
- Hubbs, A.F.; Benkovic, S.A.; Miller, D.B.; O’Callaghan, J.P.; Battelli, L.; Schwegler-Berry, D.; Ma, Q. Vacuolar leukoencephalopathy with widespread astrogliosis in mice lacking transcription factor Nrf2. Am. J. Pathol. 2007, 170, 2068–2076. [Google Scholar] [CrossRef] [Green Version]
- Jakel, R.J.; Townsend, J.A.; Kraft, A.D.; Johnson, J.A. Nrf2-mediated protection against 6-hydroxydopamine. Brain Res. 2007, 1144, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Innamorato, N.G.; Jazwa, A.; Rojo, A.I.; García, C.; Fernández-Ruiz, J.; Grochot-Przeczek, A.; Stachurska, A.; Jozkowicz, A.; Dulak, J.; Cuadrado, A. Different susceptibility to the Parkinson’s toxin MPTP in mice lacking the redox master regulator Nrf2 or its target gene heme oxygenase-1. PLoS ONE 2010, 5, e11838. [Google Scholar] [CrossRef] [Green Version]
- Boyanapalli, S.S.S.; Paredes-Gonzalez, X.; Fuentes, F.; Zhang, C.; Guo, Y.; Pung, D.; Saw, C.L.L.; Kong, A.-N.T. Nrf2 knockout attenuates the anti-inflammatory effects of phenethyl isothiocyanate and curcumin. Chem. Res. Toxicol. 2014, 27, 2036–2043. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.A.; Amirahmadi, S.; Ward, C.; Fabry, Z.; Johnson, J.A. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol. Sci. 2010, 114, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Rojo, A.I.; Pajares, M.; García-Yagüe, A.J.; Buendia, I.; Van Leuven, F.; Yamamoto, M.; López, M.G.; Cuadrado, A. Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biol. 2018, 18, 173–180. [Google Scholar] [CrossRef]
- Branca, C.; Ferreira, E.; Nguyen, T.-V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, H.; Sun, Q.; Ji, X.; Zhu, L.; Cong, Z.; Zhou, Y.; Liu, H.; Zhou, M. Nrf2 is required to maintain the self-renewal of glioma stem cells. BMC Cancer 2013, 13, 380. [Google Scholar] [CrossRef] [Green Version]
- Tarantini, S.; Valcarcel-Ares, M.N.; Yabluchanskiy, A.; Tucsek, Z.; Hertelendy, P.; Kiss, T.; Gautam, T.; Zhang, X.A.; Sonntag, W.E.; de Cabo, R.; et al. Nrf2 Deficiency Exacerbates Obesity-Induced Oxidative Stress, Neurovascular Dysfunction, Blood-Brain Barrier Disruption, Neuroinflammation, Amyloidogenic Gene Expression, and Cognitive Decline in Mice, Mimicking the Aging Phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 853–863. [Google Scholar] [CrossRef]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1–Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2017, 9, 41–56. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 175909141989978. [Google Scholar] [CrossRef]
- Anandhan, A.; Nguyen, N.; Syal, A.; Dreher, L.A.; Dodson, M.; Zhang, D.D.; Madhavan, L. NRF2 Loss Accentuates Parkinsonian Pathology and Behavioral Dysfunction in Human α-Synuclein Overexpressing Mice. Aging Dis. 2021, 12, 964–982. [Google Scholar] [CrossRef]
- Bono, S.; Feligioni, M.; Corbo, M. Impaired antioxidant KEAP1-NRF2 system in amyotrophic lateral sclerosis: NRF2 activation as a potential therapeutic strategy. Mol. Neurodegener. 2021, 16, 71. [Google Scholar] [CrossRef]
- Cuadrado, A. Brain-Protective Mechanisms of Transcription Factor NRF2: Toward a Common Strategy for Neurodegenerative Diseases. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 255–277. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-κB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-κB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef] [PubMed]
- Ganesh Yerra, V.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.-S.; Yu, S.; Kong, A.-N. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Pae, H.-O.; Chung, H.-T. Heme oxygenase-1: Its therapeutic roles in inflammatory diseases. Immune Netw. 2009, 9, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Drummond, G.S.; Baum, J.; Greenberg, M.; Lewis, D.; Abraham, N.G. HO-1 overexpression and underexpression: Clinical implications. Arch. Biochem. Biophys. 2019, 673, 108073. [Google Scholar] [CrossRef]
- Subedi, L.; Lee, J.H.; Yumnam, S.; Ji, E.; Kim, S.Y. Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-κB Inhibition and Nrf2/HO-1 Activation. Cells 2019, 8, 194. [Google Scholar] [CrossRef] [Green Version]
- Nitti, M.; Furfaro, A.L.; Mann, G.E. Heme Oxygenase Dependent Bilirubin Generation in Vascular Cells: A Role in Preventing Endothelial Dysfunction in Local Tissue Microenvironment? Front. Physiol. 2020, 11, 23. [Google Scholar] [CrossRef]
- Helou, D.G.; Martin, S.F.; Pallardy, M.; Chollet-Martin, S.; Kerdine-Römer, S. Nrf2 Involvement in Chemical-Induced Skin Innate Immunity. Front. Immunol. 2019, 10, 1004. [Google Scholar] [CrossRef] [Green Version]
- Krajka-Kuźniak, V.; Baer-Dubowska, W. Modulation of Nrf2 and NF-κB Signaling Pathways by Naturally Occurring Compounds in Relation to Cancer Prevention and Therapy. Are Combinations Better Than Single Compounds? Int. J. Mol. Sci. 2021, 22, 8223. [Google Scholar] [CrossRef]
- Yu, M.; Li, H.; Liu, Q.; Liu, F.; Tang, L.; Li, C.; Yuan, Y.; Zhan, Y.; Xu, W.; Li, W.; et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell. Signal. 2011, 23, 883–892. [Google Scholar] [CrossRef]
- Hu, B.; Wei, H.; Song, Y.; Chen, M.; Fan, Z.; Qiu, R.; Zhu, W.; Xu, W.; Wang, F. NF-κB and Keap1 Interaction Represses Nrf2-Mediated Antioxidant Response in Rabbit Hemorrhagic Disease Virus Infection. J. Virol. 2020, 94, e00016-20. [Google Scholar] [CrossRef]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and Their Concerted Modulation in Cancer Pathogenesis and Progression. Cancers 2010, 2, 483–497. [Google Scholar] [CrossRef]
- Guo, X.; Hong, S.; He, H.; Zeng, Y.; Chen, Y.; Mo, X.; Li, J.; Li, L.; Steinmetz, R.; Liu, Q. NFκB promotes oxidative stress-induced necrosis and ischemia/reperfusion injury by inhibiting Nrf2-ARE pathway. Free Radic. Biol. Med. 2020, 159, 125–135. [Google Scholar] [CrossRef]
- Gao, W.; Guo, L.; Yang, Y.; Wang, Y.; Xia, S.; Gong, H.; Zhang, B.-K.; Yan, M. Dissecting the Crosstalk Between Nrf2 and NF-κB Response Pathways in Drug-Induced Toxicity. Front. Cell Dev. Biol. 2021, 9, 809952. [Google Scholar] [CrossRef]
- Liu, G.-H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Zhu, L.; Guan, Q.; Chen, G.; Wang, Q.F.; Yin, H.X.; Hang, C.H.; Shi, J.X.; Wang, H.D. Influence of Nrf2 genotype on pulmonary NF-kappaB activity and inflammatory response after traumatic brain injury. Ann. Clin. Lab. Sci. 2008, 38, 221–227. [Google Scholar]
- Pan, H.; Wang, H.; Wang, X.; Zhu, L.; Mao, L. The absence of Nrf2 enhances NF-κB-dependent inflammation following scratch injury in mouse primary cultured astrocytes. Mediators Inflamm. 2012, 2012, 217580. [Google Scholar] [CrossRef]
- Wang, K.; Zheng, M.; Lester, K.L.; Han, Z. Light-induced Nrf2-/- mice as atrophic age-related macular degeneration model and treatment with nanoceria laden injectable hydrogel. Sci. Rep. 2019, 9, 14573. [Google Scholar] [CrossRef]
- Chang, S.-H.; Lee, J.-S.; Yun, U.J.; Park, K.W. A Role of Stress Sensor Nrf2 in Stimulating Thermogenesis and Energy Expenditure. Biomedicines 2021, 9, 1196. [Google Scholar] [CrossRef]
- Jardim, N.S.; Müller, S.G.; Pase, F.M.; Nogueira, C.W. Nuclear Factor [Erythroid-derived 2]-like 2 and Mitochondrial Transcription Factor A Contribute to Moderate-intensity Swimming Effectiveness against Memory Impairment in Young Mice Induced by Concomitant Exposure to a High-calorie Diet during the Early Life Period. Neuroscience 2021, 452, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.J.; Francis, H.M. The hippocampus and the regulation of human food intake. Psychol. Bull. 2017, 143, 1011–1032. [Google Scholar] [CrossRef]
- Mattson, M.P. Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metab. 2012, 16, 706–722. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, A.R.; Dos Santos, N.B.; Scavone, C.; Munhoz, C.D. Nrf2/ARE Pathway Modulation by Dietary Energy Regulation in Neurological Disorders. Front. Pharmacol. 2019, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Matsui, S.; Sasaki, T.; Kohno, D.; Yaku, K.; Inutsuka, A.; Yokota-Hashimoto, H.; Kikuchi, O.; Suga, T.; Kobayashi, M.; Yamanaka, A.; et al. Neuronal SIRT1 regulates macronutrient-based diet selection through FGF21 and oxytocin signalling in mice. Nat. Commun. 2018, 9, 4604. [Google Scholar] [CrossRef] [Green Version]
- Gälman, C.; Lundåsen, T.; Kharitonenkov, A.; Bina, H.A.; Eriksson, M.; Hafström, I.; Dahlin, M.; Amark, P.; Angelin, B.; Rudling, M. The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARalpha activation in man. Cell Metab. 2008, 8, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Laeger, T.; Henagan, T.M.; Albarado, D.C.; Redman, L.M.; Bray, G.A.; Noland, R.C.; Münzberg, H.; Hutson, S.M.; Gettys, T.W.; Schwartz, M.W.; et al. FGF21 is an endocrine signal of protein restriction. J. Clin. Investig. 2014, 124, 3913–3922. [Google Scholar] [CrossRef] [Green Version]
- Solon-Biet, S.M.; Cogger, V.C.; Pulpitel, T.; Heblinski, M.; Wahl, D.; McMahon, A.C.; Warren, A.; Durrant-Whyte, J.; Walters, K.A.; Krycer, J.R.; et al. Defining the Nutritional and Metabolic Context of FGF21 Using the Geometric Framework. Cell Metab. 2016, 24, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Lundsgaard, A.-M.; Fritzen, A.M.; Sjøberg, K.A.; Myrmel, L.S.; Madsen, L.; Wojtaszewski, J.F.P.; Richter, E.A.; Kiens, B. Circulating FGF21 in humans is potently induced by short term overfeeding of carbohydrates. Mol. Metab. 2017, 6, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Jensen-Cody, S.O.; Flippo, K.H.; Claflin, K.E.; Yavuz, Y.; Sapouckey, S.A.; Walters, G.C.; Usachev, Y.M.; Atasoy, D.; Gillum, M.P.; Potthoff, M.J. FGF21 Signals to Glutamatergic Neurons in the Ventromedial Hypothalamus to Suppress Carbohydrate Intake. Cell Metab. 2020, 32, 273–286.e6. [Google Scholar] [CrossRef] [PubMed]
- Sajja, R.K.; Prasad, S.; Tang, S.; Kaisar, M.A.; Cucullo, L. Blood-brain barrier disruption in diabetic mice is linked to Nrf2 signaling deficits: Role of ABCB10? Neurosci. Lett. 2017, 653, 152–158. [Google Scholar] [CrossRef]
- Yu, Z.; Lin, L.; Jiang, Y.; Chin, I.; Wang, X.; Li, X.; Lo, E.H.; Wang, X. Recombinant FGF21 Protects Against Blood-Brain Barrier Leakage Through Nrf2 Upregulation in Type 2 Diabetes Mice. Mol. Neurobiol. 2019, 56, 2314–2327. [Google Scholar] [CrossRef]
- Furusawa, Y.; Uruno, A.; Yagishita, Y.; Higashi, C.; Yamamoto, M. Nrf2 induces fibroblast growth factor 21 in diabetic mice. Genes Cells 2014, 19, 864–878. [Google Scholar] [CrossRef]
- Sáenz de Urturi, D.; Buqué, X.; Porteiro, B.; Folgueira, C.; Mora, A.; Delgado, T.C.; Prieto-Fernández, E.; Olaizola, P.; Gómez-Santos, B.; Apodaka-Biguri, M.; et al. Methionine adenosyltransferase 1a antisense oligonucleotides activate the liver-brown adipose tissue axis preventing obesity and associated hepatosteatosis. Nat. Commun. 2022, 13, 1096. [Google Scholar] [CrossRef]
- Yagishita, Y.; Uruno, A.; Fukutomi, T.; Saito, R.; Saigusa, D.; Pi, J.; Fukamizu, A.; Sugiyama, F.; Takahashi, S.; Yamamoto, M. Nrf2 Improves Leptin and Insulin Resistance Provoked by Hypothalamic Oxidative Stress. Cell Rep. 2017, 18, 2030–2044. [Google Scholar] [CrossRef] [Green Version]
- Bösl, M.R.; Seldin, M.F.; Nishimura, S.; Taketo, M. Cloning, structural analysis and mapping of the mouse selenocysteine tRNA([Ser]Sec) gene (Trsp). Mol. Gen. Genet. 1995, 248, 247–252. [Google Scholar] [CrossRef]
- Shen, G.; Zhou, L.; Liu, W.; Cui, Y.; Xie, W.; Chen, H.; Yu, W.; Li, W.; Li, H. Di(2-ethylhexyl)phthalate Alters the Synthesis and β-Oxidation of Fatty Acids and Hinders ATP Supply in Mouse Testes via UPLC-Q-Exactive Orbitrap MS-Based Metabonomics Study. J. Agric. Food Chem. 2017, 65, 5056–5063. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Wang, Y.; Zhu, X.; Jiang, M.; Song, E.; Song, Y. New application of the commercial sweetener rebaudioside a as a hepatoprotective candidate: Induction of the Nrf2 signaling pathway. Eur. J. Pharmacol. 2018, 822, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.-F.; Shao, J.; Zhao, S.-Y.; Qiu, Y.-Y.; Teng, L.-P.; Huang, W.; Wang, S.-S.; Cheng, X.-R.; Jiang, Y.-Y. Niga-ichigoside F1 ameliorates high-fat diet-induced hepatic steatosis in male mice by Nrf2 activation. Food Funct. 2018, 9, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; He, J.; Ahmad, H.; Shen, M.; Zhao, Y.; Gan, Z.; Zhang, L.; Zhong, X.; Wang, C.; Wang, T. Dietary Curcumin Supplementation Increases Antioxidant Capacity, Upregulates Nrf2 and Hmox1 Levels in the Liver of Piglet Model with Intrauterine Growth Retardation. Nutrients 2019, 11, 2978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amos, D.; Cook, C.; Santanam, N. Omega 3 rich diet modulates energy metabolism via GPR120-Nrf2 crosstalk in a novel antioxidant mouse model. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 466–488. [Google Scholar] [CrossRef]
- Rajendran, P.; Ammar, R.B.; Al-Saeedi, F.J.; Mohamed, M.E.; ElNaggar, M.A.; Al-Ramadan, S.Y.; Bekhet, G.M.; Soliman, A.M. Kaempferol Inhibits Zearalenone-Induced Oxidative Stress and Apoptosis via the PI3K/Akt-Mediated Nrf2 Signaling Pathway: In Vitro and In Vivo Studies. Int. J. Mol. Sci. 2020, 22, 217. [Google Scholar] [CrossRef]
- Erten, F.; Orhan, C.; Tuzcu, M.; Er, B.; Defo Deeh, P.B.; Sahin, N.; Özercan, I.H.; Juturu, V.; Sahin, K. Salacia chinensis exerts its antidiabetic effect by modulating glucose-regulated proteins and transcription factors in high-fat diet fed-streptozotocin-induced type 2 diabetic rats. J. Food Biochem. 2020, 44, e13513. [Google Scholar] [CrossRef]
- Illesca, P.; Valenzuela, R.; Espinosa, A.; Echeverría, F.; Soto-Alarcon, S.; Campos, C.; Rodriguez, A.; Vargas, R.; Magrone, T.; Videla, L.A. Protective Effects of Eicosapentaenoic Acid Plus Hydroxytyrosol Supplementation Against White Adipose Tissue Abnormalities in Mice Fed a High-Fat Diet. Molecules 2020, 25, 4433. [Google Scholar] [CrossRef]
- Abrescia, P.; Treppiccione, L.; Rossi, M.; Bergamo, P. Modulatory role of dietary polyunsaturated fatty acids in Nrf2-mediated redox homeostasis. Prog. Lipid Res. 2020, 80, 101066. [Google Scholar] [CrossRef]
- Mohamed, A.B.; Rémond, D.; Gual-Grau, A.; Bernalier-Donnadille, A.; Capel, F.; Michalski, M.-C.; Laugerette, F.; Cohade, B.; Hafnaoui, N.; Béchet, D.; et al. A Mix of Dietary Fibres Changes Interorgan Nutrients Exchanges and Muscle-Adipose Energy Handling in Overfed Mini-Pigs. Nutrients 2021, 13, 4202. [Google Scholar] [CrossRef]
- Bruns, D.R.; Drake, J.C.; Biela, L.M.; Peelor, F.F.; Miller, B.F.; Hamilton, K.L. Nrf2 Signaling and the Slowed Aging Phenotype: Evidence from Long-Lived Models. Oxid. Med. Cell. Longev. 2015, 2015, 732596. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Anandhan, A.; Zhang, D.D.; Madhavan, L. An NRF2 Perspective on Stem Cells and Ageing. Front. Aging 2021, 2, 690686. [Google Scholar] [CrossRef]
- Matsumaru, D.; Motohashi, H. The KEAP1-NRF2 System in Healthy Aging and Longevity. Antioxidants 2021, 10, 1929. [Google Scholar] [CrossRef]
- Lewis, K.N.; Wason, E.; Edrey, Y.H.; Kristan, D.M.; Nevo, E.; Buffenstein, R. Regulation of Nrf2 signaling and longevity in naturally long-lived rodents. Proc. Natl. Acad. Sci. USA 2015, 112, 3722–3727. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef] [Green Version]
- Silva-Palacios, A.; Ostolga-Chavarría, M.; Zazueta, C.; Königsberg, M. Nrf2: Molecular and epigenetic regulation during aging. Ageing Res. Rev. 2018, 47, 31–40. [Google Scholar] [CrossRef]
- Tsay, H.J.; Wang, P.; Wang, S.L.; Ku, H.H. Age-associated changes of superoxide dismutase and catalase activities in the rat brain. J. Biomed. Sci. 2000, 7, 466–474. [Google Scholar] [CrossRef]
- Samarghandian, S.; Azimi-Nezhad, M.; Samini, F. Preventive effect of safranal against oxidative damage in aged male rat brain. Exp. Anim. 2015, 64, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Berr, C.; Balansard, B.; Arnaud, J.; Roussel, A.M.; Alpérovitch, A. Cognitive decline is associated with systemic oxidative stress: The EVA study. Etude du Vieillissement Artériel. J. Am. Geriatr. Soc. 2000, 48, 1285–1291. [Google Scholar] [CrossRef]
- Baierle, M.; Nascimento, S.N.; Moro, A.M.; Brucker, N.; Freitas, F.; Gauer, B.; Durgante, J.; Bordignon, S.; Zibetti, M.; Trentini, C.M.; et al. Relationship between inflammation and oxidative stress and cognitive decline in the institutionalized elderly. Oxid. Med. Cell. Longev. 2015, 2015, 804198. [Google Scholar] [CrossRef] [Green Version]
- Schöttker, B.; Saum, K.-U.; Jansen, E.H.J.M.; Holleczek, B.; Brenner, H. Associations of metabolic, inflammatory and oxidative stress markers with total morbidity and multi-morbidity in a large cohort of older German adults. Age Ageing 2016, 45, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Hajjar, I.; Hayek, S.S.; Goldstein, F.C.; Martin, G.; Jones, D.P.; Quyyumi, A. Oxidative stress predicts cognitive decline with aging in healthy adults: An observational study. J. Neuroinflamm. 2018, 15, 17. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [Green Version]
- Hushpulian, D.M.; Ammal Kaidery, N.; Ahuja, M.; Poloznikov, A.A.; Sharma, S.M.; Gazaryan, I.G.; Thomas, B. Challenges and Limitations of Targeting the Keap1-Nrf2 Pathway for Neurotherapeutics: Bach1 De-Repression to the Rescue. Front. Aging Neurosci. 2021, 13, 673205. [Google Scholar] [CrossRef]
- Han, K.; Jin, X.; Guo, X.; Cao, G.; Tian, S.; Song, Y.; Zuo, Y.; Yu, P.; Gao, G.; Chang, Y.-Z. Nrf2 knockout altered brain iron deposition and mitigated age-related motor dysfunction in aging mice. Free Radic. Biol. Med. 2021, 162, 592–602. [Google Scholar] [CrossRef]
- Liu, Z.; Han, K.; Huo, X.; Yan, B.; Gao, M.; Lv, X.; Yu, P.; Gao, G.; Chang, Y.-Z. Nrf2 knockout dysregulates iron metabolism and increases the hemolysis through ROS in aging mice. Life Sci. 2020, 255, 117838. [Google Scholar] [CrossRef]
- Bao, W.-D.; Pang, P.; Zhou, X.-T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.; Xie, D.; Hu, Y.-Z.; et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef] [PubMed]
- Raha, A.A.; Biswas, A.; Henderson, J.; Chakraborty, S.; Holland, A.; Friedland, R.P.; Mukaetova-Ladinska, E.; Zaman, S.; Raha-Chowdhury, R. Interplay of Ferritin Accumulation and Ferroportin Loss in Ageing Brain: Implication for Protein Aggregation in Down Syndrome Dementia, Alzheimer’s, and Parkinson’s Diseases. Int. J. Mol. Sci. 2022, 23, 1060. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Zhang, W.; Wang, L.; Voss, T.C.; Yang, J.; Qu, J.; Liu, G.-H.; Misteli, T. Repression of the Antioxidant NRF2 Pathway in Premature Aging. Cell 2016, 165, 1361–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadowska-Bartosz, I.; Bartosz, G. Effect of antioxidants supplementation on aging and longevity. BioMed Res. Int. 2014, 2014, 404680. [Google Scholar] [CrossRef] [PubMed]
- Conti, V.; Izzo, V.; Corbi, G.; Russomanno, G.; Manzo, V.; De Lise, F.; Di Donato, A.; Filippelli, A. Antioxidant Supplementation in the Treatment of Aging-Associated Diseases. Front. Pharmacol. 2016, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- Grilc, N.K.; Sova, M.; Kristl, J. Drug Delivery Strategies for Curcumin and Other Natural Nrf2 Modulators of Oxidative Stress-Related Diseases. Pharmaceutics 2021, 13, 2137. [Google Scholar] [CrossRef]
- Malavolta, M.; Bracci, M.; Santarelli, L.; Sayeed, M.A.; Pierpaoli, E.; Giacconi, R.; Costarelli, L.; Piacenza, F.; Basso, A.; Cardelli, M.; et al. Inducers of Senescence, Toxic Compounds, and Senolytics: The Multiple Faces of Nrf2-Activating Phytochemicals in Cancer Adjuvant Therapy. Mediators Inflamm. 2018, 2018, 4159013. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef]
- Ngoi, N.Y.L.; Liew, A.Q.X.; Chong, S.J.F.; Davids, M.S.; Clement, M.-V.; Pervaiz, S. The redox-senescence axis and its therapeutic targeting. Redox Biol. 2021, 45, 102032. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. FEBS J. 2022, 197, 111526. [Google Scholar] [CrossRef]
- Nayeri Rad, A.; Shams, G.; Avelar, R.A.; Morowvat, M.H.; Ghasemi, Y. Potential senotherapeutic candidates and their combinations derived from transcriptional connectivity and network measures. Inform. Med. Unlocked 2022, 30, 100920. [Google Scholar] [CrossRef]
- Sikora, E.; Bielak-Zmijewska, A.; Dudkowska, M.; Krzystyniak, A.; Mosieniak, G.; Wesierska, M.; Wlodarczyk, J. Cellular Senescence in Brain Aging. Front. Aging Neurosci. 2021, 13, 646924. [Google Scholar] [CrossRef]
- Martínez-Cué, C.; Rueda, N. Cellular Senescence in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 16. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Evans, S.A.; Fielder, E.; Victorelli, S.; Kruger, P.; Salmonowicz, H.; Weigand, B.M.; Patel, A.D.; Pirtskhalava, T.; Inman, C.L.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, 20, e13296. [Google Scholar] [CrossRef]
- Krzystyniak, A.; Wesierska, M.; Petrazzo, G.; Gadecka, A.; Dudkowska, M.; Bielak-Zmijewska, A.; Mosieniak, G.; Figiel, I.; Wlodarczyk, J.; Sikora, E. Combination of dasatinib and quercetin improves cognitive abilities in aged male Wistar rats, alleviates inflammation and changes hippocampal synaptic plasticity and histone H3 methylation profile. Aging 2022, 14, 572–595. [Google Scholar] [CrossRef]
- Yuan, H.; Xu, Y.; Luo, Y.; Wang, N.-X.; Xiao, J.-H. Role of Nrf2 in cell senescence regulation. Mol. Cell. Biochem. 2021, 476, 247–259. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Profumo, E.; Tucci, P.; Saso, L. A Perspective on Nrf2 Signaling Pathway for Neuroinflammation: A Potential Therapeutic Target in Alzheimer’s and Parkinson’s Diseases. Front. Cell. Neurosci. 2022, 15, 787258. [Google Scholar] [CrossRef]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef] [Green Version]
- Schmidlin, C.J.; Dodson, M.B.; Madhavan, L.; Zhang, D.D. Redox Regulation by NRF2 in Aging and Disease. Free Radic. Biol. Med. 2019, 134, 702–707. [Google Scholar] [CrossRef]
- Joshi, G.; Gan, K.A.; Johnson, D.A.; Johnson, J.A. Increased Alzheimer’s disease-like pathology in the APP/ PS1ΔE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol. Aging 2015, 36, 664–679. [Google Scholar] [CrossRef] [Green Version]
- Ren, P.; Chen, J.; Li, B.; Zhang, M.; Yang, B.; Guo, X.; Chen, Z.; Cheng, H.; Wang, P.; Wang, S.; et al. Nrf2 Ablation Promotes Alzheimer’s Disease-Like Pathology in APP/PS1 Transgenic Mice: The Role of Neuroinflammation and Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 3050971. [Google Scholar] [CrossRef]
- Kanninen, K.; Heikkinen, R.; Malm, T.; Rolova, T.; Kuhmonen, S.; Leinonen, H.; Ylä-Herttuala, S.; Tanila, H.; Levonen, A.-L.; Koistinaho, M.; et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16505–16510. [Google Scholar] [CrossRef] [Green Version]
- Jiwaji, Z.; Tiwari, S.S.; Avilés-Reyes, R.X.; Hooley, M.; Hampton, D.; Torvell, M.; Johnson, D.A.; McQueen, J.; Baxter, P.; Sabari-Sankar, K.; et al. Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to Tau and Aß pathology. Nat. Commun. 2022, 13, 135. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Delaidelli, A.; Richner, M.; Jiang, L.; van der Laan, A.; Bergholdt Jul Christiansen, I.; Ferreira, N.; Nyengaard, J.R.; Vægter, C.B.; Jensen, P.H.; Mackenzie, I.R.; et al. α-Synuclein pathology in Parkinson disease activates homeostatic NRF2 anti-oxidant response. Acta Neuropathol. Commun. 2021, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Barone, M.C.; Sykiotis, G.P.; Bohmann, D. Genetic activation of Nrf2 signaling is sufficient to ameliorate neurodegenerative phenotypes in a Drosophila model of Parkinson’s disease. Dis. Model. Mech. 2011, 4, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Zhao, M.; Wang, B.; Su, Z.; Guo, B.; Qin, L.; Zhang, W.; Zheng, R. The Nrf2-NLRP3-caspase-1 axis mediates the neuroprotective effects of Celastrol in Parkinson’s disease. Redox Biol. 2021, 47, 102134. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Cai, X.; Ding, L.; Liao, J.; Liu, X.; Huang, Y.; Chen, X.; Long, L. Nrf2 Inhibits the Progression of Parkinson’s Disease by Upregulating AABR07032261.5 to Repress Pyroptosis. J. Inflamm. Res. 2022, 15, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Rusnak, M.; Lombroso, P.J.; Sidhu, A. Dopamine promotes striatal neuronal apoptotic death via ERK signaling cascades. Eur. J. Neurosci. 2009, 29, 287–306. [Google Scholar] [CrossRef] [Green Version]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rábano, A.; Kirik, D.; Cuadrado, A. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J. Neurosci. 2012, 32, 17775–17787. [Google Scholar] [CrossRef] [Green Version]
- Vargas, M.R.; Burton, N.C.; Kutzke, J.; Gan, L.; Johnson, D.A.; Schäfer, M.; Werner, S.; Johnson, J.A. Absence of Nrf2 or its selective overexpression in neurons and muscle does not affect survival in ALS-linked mutant hSOD1 mouse models. PLoS ONE 2013, 8, e56625. [Google Scholar] [CrossRef]
- Sigfridsson, E.; Marangoni, M.; Johnson, J.A.; Hardingham, G.E.; Fowler, J.H.; Horsburgh, K. Astrocyte-specific overexpression of Nrf2 protects against optic tract damage and behavioural alterations in a mouse model of cerebral hypoperfusion. Sci. Rep. 2018, 8, 12552. [Google Scholar] [CrossRef] [Green Version]
- Daniels, C.M.L.; Austin, E.V.; Rockney, D.E.; Jacka, E.M.; Hagemann, T.L.; Johnson, D.A.; Johnson, J.A.; Messing, A. Beneficial Effects of Nrf2 Overexpression in a Mouse Model of Alexander Disease. J. Neurosci. 2012, 32, 10507–10515. [Google Scholar] [CrossRef] [Green Version]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53 (Suppl. 3), S26–S36; discussion S36–S38. [Google Scholar] [CrossRef]
- Gan, L.; Johnson, J.A. Oxidative damage and the Nrf2-ARE pathway in neurodegenerative diseases. Biochim. Biophys. Acta 2014, 1842, 1208–1218. [Google Scholar] [CrossRef] [Green Version]
- Vinish, M.; Anand, A.; Prabhakar, S. Altered oxidative stress levels in Indian Parkinson’s disease patients with PARK2 mutations. Acta Biochim. Pol. 2011, 58, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxid. Med. Cell. Longev. 2016, 2016, 8590578. [Google Scholar] [CrossRef]
- Wei, Z.; Li, X.; Li, X.; Liu, Q.; Cheng, Y. Oxidative Stress in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Mol. Neurosci. 2018, 11, 236. [Google Scholar] [CrossRef]
- Pan, H.; Wang, H.; Zhu, L.; Wang, X.; Cong, Z.; Sun, K.; Fan, Y. The involvement of Nrf2-ARE pathway in regulation of apoptosis in human glioblastoma cell U251. Neurol. Res. 2013, 35, 71–78. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, H.-D.; Zhu, L.; Cong, Z.-X.; Li, N.; Ji, X.-J.; Pan, H.; Wang, J.-W.; Li, W.-C. Knockdown of Nrf2 enhances autophagy induced by temozolomide in U251 human glioma cell line. Oncol. Rep. 2013, 29, 394–400. [Google Scholar] [CrossRef] [Green Version]
- Cong, Z.-X.; Wang, H.-D.; Wang, J.-W.; Zhou, Y.; Pan, H.; Zhang, D.-D.; Zhu, L. ERK and PI3K signaling cascades induce Nrf2 activation and regulate cell viability partly through Nrf2 in human glioblastoma cells. Oncol. Rep. 2013, 30, 715–722. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Wirth, A.-K.; Chen, D.; Wruck, C.J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 2017, 6, e371. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.-J.; Chen, S.-H.; Zhu, L.; Pan, H.; Zhou, Y.; Li, W.; You, W.-C.; Gao, C.-C.; Zhu, J.-H.; Jiang, K.; et al. Knockdown of NF-E2-related factor 2 inhibits the proliferation and growth of U251MG human glioma cells in a mouse xenograft model. Oncol. Rep. 2013, 30, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Wang, H.; Zhu, J.; Zhu, L.; Pan, H.; Li, W.; Zhou, Y.; Cong, Z.; Yan, F.; Chen, S. Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Int. J. Cancer 2014, 135, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Haapasalo, J.; Nordfors, K.; Granberg, K.J.; Kivioja, T.; Nykter, M.; Haapasalo, H.; Soini, Y. NRF2, DJ1 and SNRX1 and their prognostic impact in astrocytic gliomas. Histol. Histopathol. 2018, 33, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Wang, H.; Zhu, J.; Tang, Y.; Zhou, Y.; Zhu, L.; Gao, C.; Li, W.; You, W.; Yu, B.; et al. Correlation of Nrf2 and HIF-1α in glioblastoma and their relationships to clinicopathologic features and survival. Neurol. Res. 2013, 35, 1044–1050. [Google Scholar] [CrossRef]
- Pölönen, P.; Jawahar Deen, A.; Leinonen, H.M.; Jyrkkänen, H.-K.; Kuosmanen, S.; Mononen, M.; Jain, A.; Tuomainen, T.; Pasonen-Seppänen, S.; Hartikainen, J.M.; et al. Nrf2 and SQSTM1/p62 jointly contribute to mesenchymal transition and invasion in glioblastoma. Oncogene 2019, 38, 7473–7490. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, M.; Higa, T.; Sonoda, Y.; Murakami, S.; Dodo, M.; Kitamura, H.; Taguchi, K.; Shibata, T.; Watanabe, M.; Suzuki, H.; et al. Activation of the NRF2 pathway and its impact on the prognosis of anaplastic glioma patients. Neuro-Oncology 2015, 17, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Ouyang, L.; He, M.; Luo, M.; Cai, W.; Tu, Y.; Pi, R.; Liu, A. IDH1 R132H mutation regulates glioma chemosensitivity through Nrf2 pathway. Oncotarget 2017, 8, 28865–28879. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.; Dixit, D.; Sharma, V.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Nrf2-driven TERT regulates pentose phosphate pathway in glioblastoma. Cell Death Dis. 2016, 7, e2213. [Google Scholar] [CrossRef] [Green Version]
- Tsakonas, G.; Martín-Bernabé, A.; Rounis, K.; Moreno-Ruiz, P.; Botling, J.; De Petris, L.; Ylipää, A.; Mezheyeuski, A.; Micke, P.; Östman, A.; et al. High Density of NRF2 Expression in Malignant Cells Is Associated with Increased Risk of CNS Metastasis in Early-Stage NSCLC. Cancers 2021, 13, 3151. [Google Scholar] [CrossRef]
- Aljohani, H.M.; Aittaleb, M.; Furgason, J.M.; Amaya, P.; Deeb, A.; Chalmers, J.J.; Bahassi, E.M. Genetic mutations associated with lung cancer metastasis to the brain. Mutagenesis 2018, 33, 137–145. [Google Scholar] [CrossRef]
- Cong, Z.-X.; Wang, H.-D.; Zhou, Y.; Wang, J.-W.; Pan, H.; Zhang, D.-D.; Zhang, L.; Zhu, L. Temozolomide and irradiation combined treatment-induced Nrf2 activation increases chemoradiation sensitivity in human glioblastoma cells. J. Neurooncol. 2014, 116, 41–48. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Reily Rocha, A.; Molina Silva, M.; Rodrigues Gomes, L.; Teatin Latancia, M.; Andrade Tomaz, M.; de Souza, I.; Karolynne Seregni Monteiro, L.; Menck, C.F.M. Revealing Temozolomide Resistance Mechanisms via Genome-Wide CRISPR Libraries. Cells 2020, 9, 2573. [Google Scholar] [CrossRef]
- Escoll, M.; Lastra, D.; Robledinos-Antón, N.; Wandosell, F.; Antón, I.M.; Cuadrado, A. WIP Modulates Oxidative Stress through NRF2/KEAP1 in Glioblastoma Cells. Antioxidants 2020, 9, 773. [Google Scholar] [CrossRef]
- Beltzig, L.; Schwarzenbach, C.; Leukel, P.; Frauenknecht, K.B.M.; Sommer, C.; Tancredi, A.; Hegi, M.E.; Christmann, M.; Kaina, B. Senescence Is the Main Trait Induced by Temozolomide in Glioblastoma Cells. Cancers 2022, 14, 2233. [Google Scholar] [CrossRef]
- Yagishita, Y.; Gatbonton-Schwager, T.N.; McCallum, M.L.; Kensler, T.W. Current Landscape of NRF2 Biomarkers in Clinical Trials. Antioxidants 2020, 9, 716. [Google Scholar] [CrossRef]
- Kornberg, R.D.; Lorch, Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999, 98, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Baker, M. Making sense of chromatin states. Nat. Methods 2011, 8, 717–722. [Google Scholar] [CrossRef]
- Ohnmacht, J.; May, P.; Sinkkonen, L.; Krüger, R. Missing heritability in Parkinson’s disease: The emerging role of non-coding genetic variation. J. Neural Transm. 2020, 127, 729–748. [Google Scholar] [CrossRef] [Green Version]
- Blackwood, E.M.; Kadonaga, J.T. Going the distance: A current view of enhancer action. Science 1998, 281, 60–63. [Google Scholar] [CrossRef] [Green Version]
- Krivega, I.; Dean, A. Enhancer and promoter interactions-long distance calls. Curr. Opin. Genet. Dev. 2012, 22, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Correa, F.; Mallard, C.; Nilsson, M.; Sandberg, M. Activated microglia decrease histone acetylation and Nrf2-inducible anti-oxidant defence in astrocytes: Restoring effects of inhibitors of HDACs, p38 MAPK and GSK3β. Neurobiol. Dis. 2011, 44, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zhu, X.; Kim, Y.; Li, J.; Huang, S.; Saleem, S.; Li, R.; Xu, Y.; Dore, S.; Cao, W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic. Biol. Med. 2012, 52, 928–936. [Google Scholar] [CrossRef]
- Li, Z.; Xu, L.; Tang, N.; Xu, Y.; Ye, X.; Shen, S.; Niu, X.; Lu, S.; Chen, Z. The polycomb group protein EZH2 inhibits lung cancer cell growth by repressing the transcription factor Nrf2. FEBS Lett. 2014, 588, 3000–3007. [Google Scholar] [CrossRef] [Green Version]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.; Alam, J. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J. Biol. Chem. 2001, 276, 20858–20865. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ohta, T.; Maruyama, A.; Hosoya, T.; Nishikawa, K.; Maher, J.M.; Shibahara, S.; Itoh, K.; Yamamoto, M. BRG1 interacts with Nrf2 to selectively mediate HO-1 induction in response to oxidative stress. Mol. Cell. Biol. 2006, 26, 7942–7952. [Google Scholar] [CrossRef] [Green Version]
- Alam, J.; Killeen, E.; Gong, P.; Naquin, R.; Hu, B.; Stewart, D.; Ingelfinger, J.R.; Nath, K.A. Heme activates the heme oxygenase-1 gene in renal epithelial cells by stabilizing Nrf2. Am. J. Physiol. Renal Physiol. 2003, 284, F743–F752. [Google Scholar] [CrossRef] [Green Version]
- Sekine, H.; Okazaki, K.; Ota, N.; Shima, H.; Katoh, Y.; Suzuki, N.; Igarashi, K.; Ito, M.; Motohashi, H.; Yamamoto, M. The Mediator Subunit MED16 Transduces NRF2-Activating Signals into Antioxidant Gene Expression. Mol. Cell. Biol. 2016, 36, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Yu, S.; Chen, J.D.; Kong, A.N. The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains. Oncogene 2013, 32, 514–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, H.; Guan, D.; Liu, X.; Li, J.; Wang, L.; Wu, J.; Zhou, J.; Zhang, W.; Ren, R.; Zhang, W.; et al. SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Res. 2016, 26, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Sugawara, A.; Taniyama, Y.; Uruno, A.; Igarashi, K.; Arima, S.; Ito, S.; Takeuchi, K. Suppression of rat thromboxane synthase gene transcription by peroxisome proliferator-activated receptor gamma in macrophages via an interaction with NRF2. J. Biol. Chem. 2000, 275, 33142–33150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.M.; Rushworth, S.A.; Murray, M.Y.; Bowles, K.M.; MacEwan, D.J. Understanding the role of NRF2-regulated miRNAs in human malignancies. Oncotarget 2013, 4, 1130–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, M.S.; Lee, C.G.; Koo, J.H.; Kim, S.G. miR-125b transcriptionally increased by Nrf2 inhibits AhR repressor, which protects kidney from cisplatin-induced injury. Cell Death Dis. 2013, 4, e899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, M.-K.; Wakabayashi, N.; Itoh, K.; Motohashi, H.; Yamamoto, M.; Kensler, T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003, 278, 8135–8145. [Google Scholar] [CrossRef] [Green Version]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Marstrand, T.T.; Storey, J.D. Identifying and mapping cell-type-specific chromatin programming of gene expression. Proc. Natl. Acad. Sci. USA 2014, 111, E645–E654. [Google Scholar] [CrossRef] [Green Version]
- Winick-Ng, W.; Kukalev, A.; Harabula, I.; Zea-Redondo, L.; Szabó, D.; Meijer, M.; Serebreni, L.; Zhang, Y.; Bianco, S.; Chiariello, A.M.; et al. Cell-type specialization is encoded by specific chromatin topologies. Nature 2021, 599, 684–691. [Google Scholar] [CrossRef]
- Czamara, D.; Eraslan, G.; Page, C.M.; Lahti, J.; Lahti-Pulkkinen, M.; Hämäläinen, E.; Kajantie, E.; Laivuori, H.; Villa, P.M.; Reynolds, R.M.; et al. Integrated analysis of environmental and genetic influences on cord blood DNA methylation in new-borns. Nat. Commun. 2019, 10, 2548. [Google Scholar] [CrossRef] [Green Version]
- Breton, C.V.; Landon, R.; Kahn, L.G.; Enlow, M.B.; Peterson, A.K.; Bastain, T.; Braun, J.; Comstock, S.S.; Duarte, C.S.; Hipwell, A.; et al. Exploring the evidence for epigenetic regulation of environmental influences on child health across generations. Commun. Biol. 2021, 4, 769. [Google Scholar] [CrossRef]
- Wang, Z.; Pan, Q.; Gendron, P.; Zhu, W.; Guo, F.; Cen, S.; Wainberg, M.A.; Liang, C. CRISPR/Cas9-Derived Mutations Both Inhibit HIV-1 Replication and Accelerate Viral Escape. Cell Rep. 2016, 15, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Levings, D.C.; Wang, X.; Kohlhase, D.; Bell, D.A.; Slattery, M. A distinct class of antioxidant response elements is consistently activated in tumors with NRF2 mutations. Redox Biol. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Oyake, T.; Itoh, K.; Motohashi, H.; Hayashi, N.; Hoshino, H.; Nishizawa, M.; Yamamoto, M.; Igarashi, K. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell. Biol. 1996, 16, 6083–6095. [Google Scholar] [CrossRef] [Green Version]
- Itoh-Nakadai, A.; Hikota, R.; Muto, A.; Kometani, K.; Watanabe-Matsui, M.; Sato, Y.; Kobayashi, M.; Nakamura, A.; Miura, Y.; Yano, Y.; et al. The transcription repressors Bach2 and Bach1 promote B cell development by repressing the myeloid program. Nat. Immunol. 2014, 15, 1171–1180. [Google Scholar] [CrossRef]
- Shin, S.; Wakabayashi, N.; Misra, V.; Biswal, S.; Lee, G.H.; Agoston, E.S.; Yamamoto, M.; Kensler, T.W. NRF2 modulates aryl hydrocarbon receptor signaling: Influence on adipogenesis. Mol. Cell. Biol. 2007, 27, 7188–7197. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [Green Version]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef] [Green Version]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030.e16. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.H.; Yu, J.; Ng, R.; Johnson, D.A.; Shen, H.; Honey, C.R.; Johnson, J.A. Preferential expression of antioxidant response element mediated gene expression in astrocytes. J. Neurochem. 2001, 76, 1670–1678. [Google Scholar] [CrossRef]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate Regulation of Glutathione Biosynthesis and Release by Nrf2-Expressing Glia Potently Protects Neurons from Oxidative Stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef]
- Kraft, A.D.; Johnson, D.A.; Johnson, J.A. Nuclear Factor E2-Related Factor 2-Dependent Antioxidant Response Element Activation by tert-Butylhydroquinone and Sulforaphane Occurring Preferentially in Astrocytes Conditions Neurons against Oxidative Insult. J. Neurosci. 2004, 24, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Bell, K.F.S.; Al-Mubarak, B.; Martel, M.-A.; McKay, S.; Wheelan, N.; Hasel, P.; Márkus, N.M.; Baxter, P.; Deighton, R.F.; Serio, A.; et al. Neuronal development is promoted by weakened intrinsic antioxidant defences due to epigenetic repression of Nrf2. Nat. Commun. 2015, 6, 7066. [Google Scholar] [CrossRef] [Green Version]
- Narasimhan, M.; Patel, D.; Vedpathak, D.; Rathinam, M.; Henderson, G.; Mahimainathan, L. Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PLoS ONE 2012, 7, e51111. [Google Scholar] [CrossRef] [Green Version]
- Jing, X.; Shi, H.; Zhang, C.; Ren, M.; Han, M.; Wei, X.; Zhang, X.; Lou, H. Dimethyl fumarate attenuates 6-OHDA-induced neurotoxicity in SH-SY5Y cells and in animal model of Parkinson’s disease by enhancing Nrf2 activity. Neuroscience 2015, 286, 131–140. [Google Scholar] [CrossRef]
- Ahuja, M.; Ammal Kaidery, N.; Yang, L.; Calingasan, N.; Smirnova, N.; Gaisin, A.; Gaisina, I.N.; Gazaryan, I.; Hushpulian, D.M.; Kaddour-Djebbar, I.; et al. Distinct Nrf2 Signaling Mechanisms of Fumaric Acid Esters and Their Role in Neuroprotection against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Experimental Parkinson’s-Like Disease. J. Neurosci. 2016, 36, 6332–6351. [Google Scholar] [CrossRef] [Green Version]
- Campolo, M.; Casili, G.; Biundo, F.; Crupi, R.; Cordaro, M.; Cuzzocrea, S.; Esposito, E. The Neuroprotective Effect of Dimethyl Fumarate in an MPTP-Mouse Model of Parkinson’s Disease: Involvement of Reactive Oxygen Species/Nuclear Factor-κB/Nuclear Transcription Factor Related to NF-E2. Antioxid. Redox Signal. 2017, 27, 453–471. [Google Scholar] [CrossRef] [Green Version]
- Campolo, M.; Casili, G.; Lanza, M.; Filippone, A.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Multiple mechanisms of dimethyl fumarate in amyloid β-induced neurotoxicity in human neuronal cells. J. Cell. Mol. Med. 2018, 22, 1081–1094. [Google Scholar] [CrossRef]
- Rajput, M.S.; Nirmal, N.P.; Rathore, D.; Dahima, R. Dimethyl Fumarate Mitigates Tauopathy in Aβ-Induced Neuroblastoma SH-SY5Y Cells. Neurochem. Res. 2020, 45, 2641–2652. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Suo, X.; Xia, X.; Yu, C.; Dou, Y. Dimethyl Fumarate is a Potential Therapeutic Option for Alzheimer’s Disease. J. Alzheimers Dis. 2022, 85, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.-Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, K.; Anzawa, H.; Liu, Z.; Ota, N.; Kitamura, H.; Onodera, Y.; Alam, M.M.; Matsumaru, D.; Suzuki, T.; Katsuoka, F.; et al. Enhancer remodeling promotes tumor-initiating activity in NRF2-activated non-small cell lung cancers. Nat. Commun. 2020, 11, 5911. [Google Scholar] [CrossRef]
- Preissl, S.; Fang, R.; Huang, H.; Zhao, Y.; Raviram, R.; Gorkin, D.U.; Zhang, Y.; Sos, B.C.; Afzal, V.; Dickel, D.E.; et al. Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat. Neurosci. 2018, 21, 432–439. [Google Scholar] [CrossRef] [Green Version]
- Mich, J.K.; Graybuck, L.T.; Hess, E.E.; Mahoney, J.T.; Kojima, Y.; Ding, Y.; Somasundaram, S.; Miller, J.A.; Kalmbach, B.E.; Radaelli, C.; et al. Functional enhancer elements drive subclass-selective expression from mouse to primate neocortex. Cell Rep. 2021, 34, 108754. [Google Scholar] [CrossRef]
- Gui, Y.; Grzyb, K.; Thomas, M.H.; Ohnmacht, J.; Garcia, P.; Buttini, M.; Skupin, A.; Sauter, T.; Sinkkonen, L. Single-nuclei chromatin profiling of ventral midbrain reveals cell identity transcription factors and cell-type-specific gene regulatory variation. Epigenetics Chromatin 2021, 14, 43. [Google Scholar] [CrossRef]
- Ahuja, M.; Ammal Kaidery, N.; Attucks, O.C.; McDade, E.; Hushpulian, D.M.; Gaisin, A.; Gaisina, I.; Ahn, Y.H.; Nikulin, S.; Poloznikov, A.; et al. Bach1 derepression is neuroprotective in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2111643118. [Google Scholar] [CrossRef]
- Wang, X.J.; Hayes, J.D.; Henderson, C.J.; Wolf, C.R. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 19589–19594. [Google Scholar] [CrossRef] [Green Version]
- Armand, E.J.; Li, J.; Xie, F.; Luo, C.; Mukamel, E.A. Single-Cell Sequencing of Brain Cell Transcriptomes and Epigenomes. Neuron 2021, 109, 11–26. [Google Scholar] [CrossRef]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [Green Version]
- Schaub, M.A.; Boyle, A.P.; Kundaje, A.; Batzoglou, S.; Snyder, M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012, 22, 1748–1759. [Google Scholar] [CrossRef] [Green Version]
- Deplancke, B.; Alpern, D.; Gardeux, V. The Genetics of Transcription Factor DNA Binding Variation. Cell 2016, 166, 538–554. [Google Scholar] [CrossRef] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Tsai, W.-C.; Hueng, D.-Y.; Lin, C.-R.; Yang, T.C.K.; Gao, H.-W. Nrf2 Expressions Correlate with WHO Grades in Gliomas and Meningiomas. Int. J. Mol. Sci. 2016, 17, 722. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
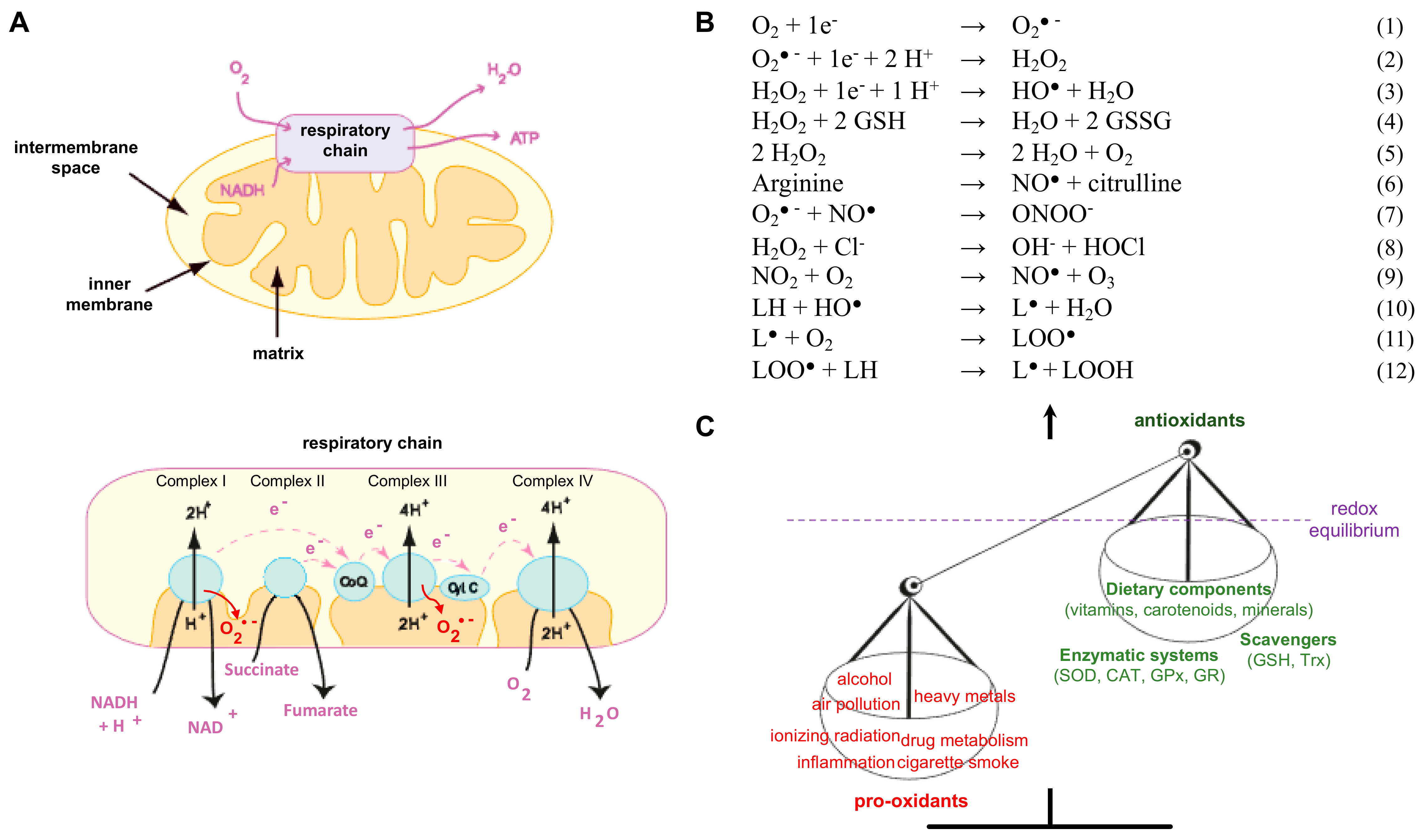
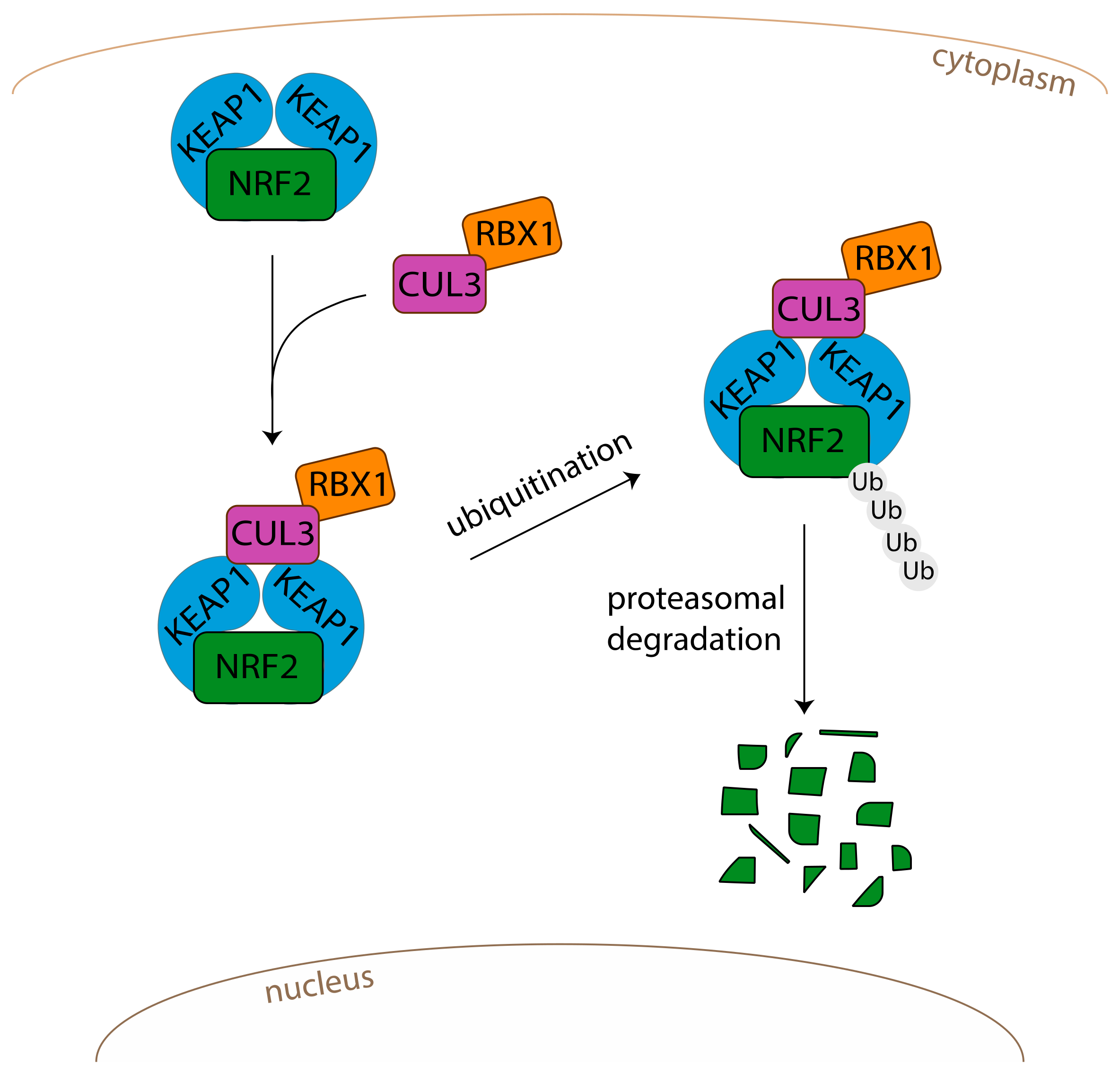
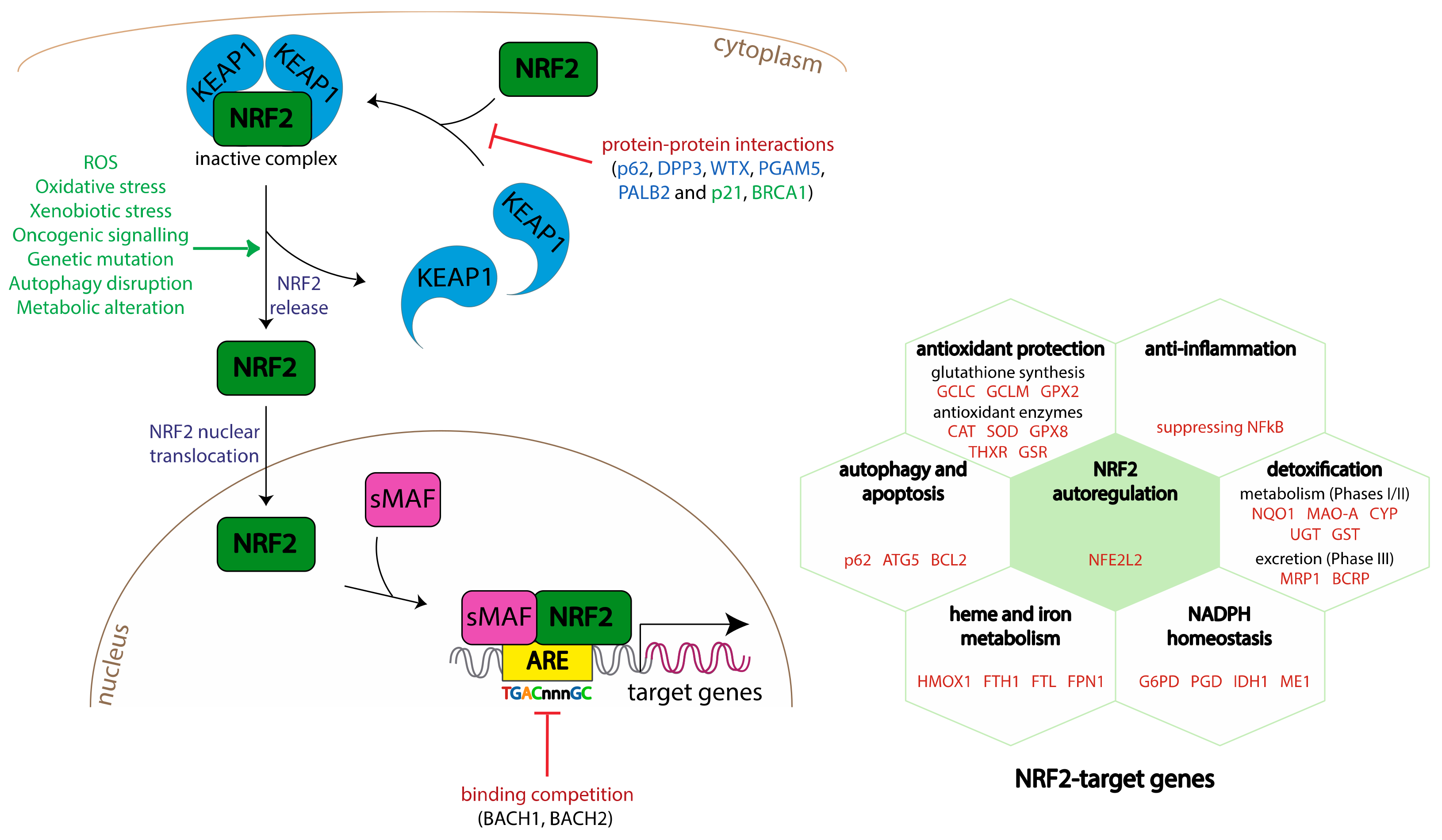

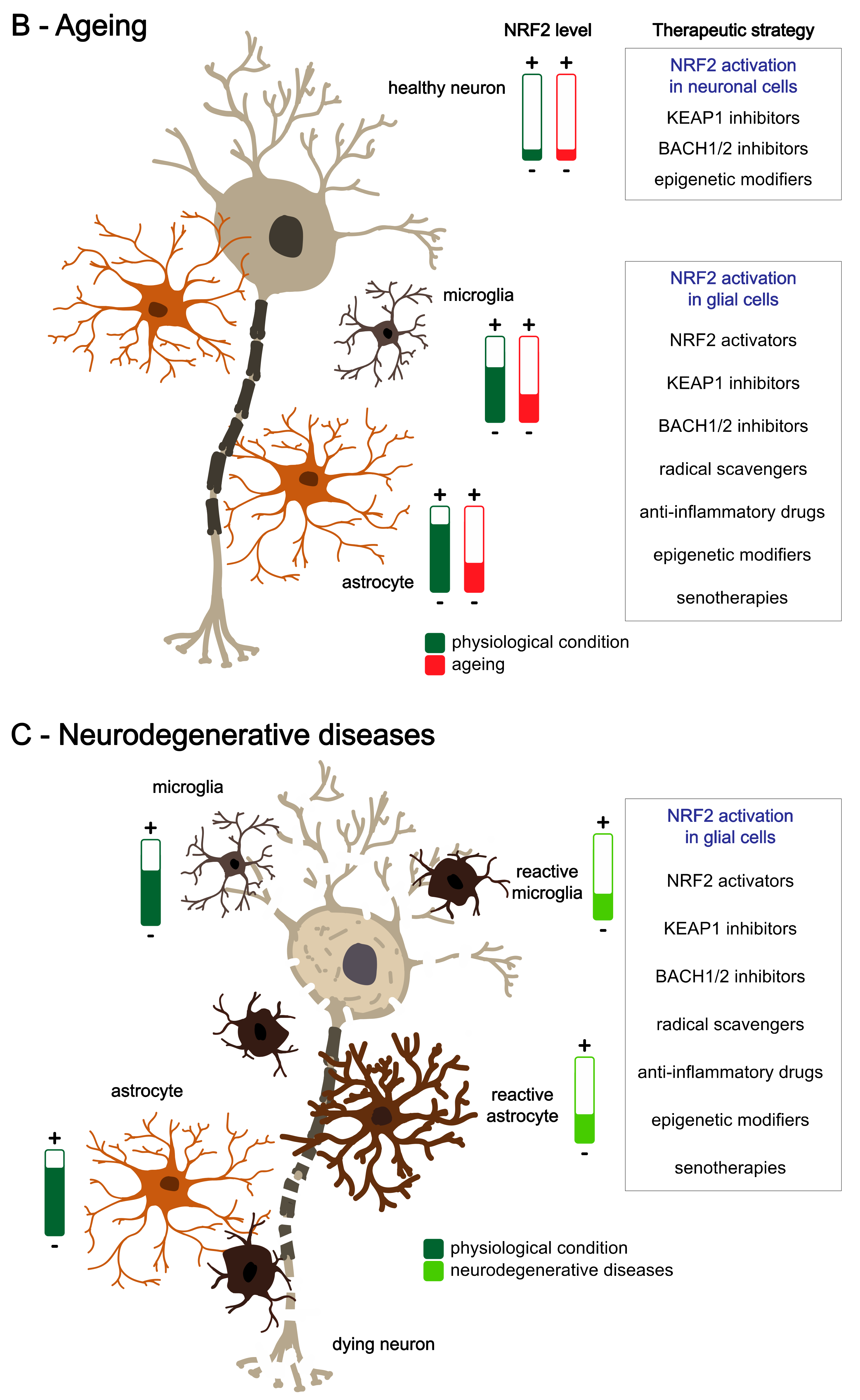
| Activities | Description | Methods | Principle | Observations | Refs |
|---|---|---|---|---|---|
| Interaction KEAP1-NRF2 | The complex NRF2-KEAP1 is sequestered in the cytosol before being rapidly ubiquitinated and degradated by the proteasome. | Fluorescence Lifetime Imaging–Förster Resonance Energy Transfer (FLIM-FRET) approach in cells expressing fluorescently tagged NRF2 and KEAP1 | evaluation of conformation changes in KEAP1-NRF2 complex in cells/establishment of NRF2 activation kinetics | Conformational changes (spatial distribution) can result in functional inactivation of the complex and consequential induction of an NRF2-mediated stress response. | [84,85,86] |
| NRF2 translocation | Free NRF2 will enter in the nucleus and activate the transcription of NRF2 target genes. | Western blots | evaluation of the NRF2 localisation (cytoplasmic versus nuclear levels) | increase in NRF2 level in the nucleus | [87,88,89] |
| immunofluorescence (cells, tissues) | NRF2 staining in the nucleus | increase in NRF2 level in the nucleus | [87,90] | ||
| transcriptomic analyses | analyses of NRF2 target genes expression | increase in gene expression | [53,87,91] | ||
| NRF2 binding to AREs | Free NRF2 enters in the nucleus and binds to specific sequences (Antioxidant Response Elements, ARE). This binding will activate the transcription of genes that have ARE sequences on their promoter. | ARE luciferase reporter kit | Cells are transfected by an ARE luciferase reporter vector constitutively expressing Renilla luciferase vector. | Amount of light generated after treatments is positively correlated with the ARE luciferase reporter activity as a consequence of NRF2 binding to ARE sequence. | [92] |
| Electrophoretic-Mobility Shift Assay (EMSA) | Nuclear extracts are incubated with an NRF2 probe (oligonucleotide containing the ARE consensus sequence). Protein/DNA complexes are separated on a nondenaturing polyacrylamide gel and transferred to a membrane. Detection is performed using Streptavidin-HRP Conjugate and a chemiluminescent substrate. | The shifted bands corresponding to the protein/DNA complexes (interaction of NRF2 with its ARE-probe) are visualized. | [93] | ||
| DNA-binding ELISA for activated NRF2 transcription factor | NRF2 transcription factor (from nuclear extracts) binds to DNA sequence (containing the NRF2 consensus binding site) immobilized in the well. Incubation with primary and secondary antibodies specifically quantifies the amount of activated transcription factor. | This method is 100-fold more sensitive than EMSAs. Colorimetric readout enables easy, quantitative analysis by spectrophotometry at 450 nm. | [94] | ||
| NRF2/ARE luciferase reporter stable cell lines | After treatments, the NRF2 transcription factor will enter in the nucleus and bind to its response element. As a consequence, luciferase is expressed, and light will be generated in an enzymatic assay (addition of luciferin). | Amount of light generated is positively correlated with the level of NRF2 activation. | [95] | ||
| high-content imaging of cell lines expressing fluorescent-tagged NRF2 (fluorescent protein reporter cell lines, expressing GFP tagged Nfe2l2) | High-throughput live confocal imaging is used to measure the temporal dynamics of the NRF2 pathway after treatments. | Fluorescent protein signal intensity is correlated to NRF2 response. | [96] | ||
| GSH production | NRF2 regulates GSH biosynthesizing enzymes (GCLM, GCLC) and plays a key role in the regulation of cellular GSH homeostasis | GSH quantification (GSH assay kit) | GSH is oxidized by the sulfhydryl reagent 5,5’-dithio-bis(2-nitrobenzoic acid). The formed derivative is measurable at 412 nm. The glutathione disulphide (GSSG) formed can be recycled to GSH by glutathione reductase in the presence of NADPH. | Increase in the GSH levels may confirm NRF2 recruitment. Glutathione quantification can be performed in any biological fluids, tissues, and cell extracts. | [97] |
| Nfe2l2manipulation | activation/inhibition of the NRF2 signalling pathway and inactive-luciferase-tagged Nfe2l2, can be set up in transgenic mouse models | transgenic mouse models | Transgenic NRF2−/− mice (NRF2 pathway is downregulated) or KEAP1−/− mice (NRF2 pathway is upregulated) can be useful in order to verify the implication of the NRF2 pathway during pathologies or ageing. | Transgenic animal models are commercially available. | [98,99] |
| OKD48 transgenic mice | The OKD48 construct has a 3xARE promoter, human Nfe2l2, and Flag-tagged luciferase. Upon stress, OKD48 is transcriptionally induced by the 3xARE element, and luciferase activity (luminescence) is observed only in cells experiencing oxidative stress. | OKD48-luc model is a highly specific and sensitive system for screening NRF2 activity | [100,101] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heurtaux, T.; Bouvier, D.S.; Benani, A.; Helgueta Romero, S.; Frauenknecht, K.B.M.; Mittelbronn, M.; Sinkkonen, L. Normal and Pathological NRF2 Signalling in the Central Nervous System. Antioxidants 2022, 11, 1426. https://doi.org/10.3390/antiox11081426
Heurtaux T, Bouvier DS, Benani A, Helgueta Romero S, Frauenknecht KBM, Mittelbronn M, Sinkkonen L. Normal and Pathological NRF2 Signalling in the Central Nervous System. Antioxidants. 2022; 11(8):1426. https://doi.org/10.3390/antiox11081426
Chicago/Turabian StyleHeurtaux, Tony, David S. Bouvier, Alexandre Benani, Sergio Helgueta Romero, Katrin B. M. Frauenknecht, Michel Mittelbronn, and Lasse Sinkkonen. 2022. "Normal and Pathological NRF2 Signalling in the Central Nervous System" Antioxidants 11, no. 8: 1426. https://doi.org/10.3390/antiox11081426
APA StyleHeurtaux, T., Bouvier, D. S., Benani, A., Helgueta Romero, S., Frauenknecht, K. B. M., Mittelbronn, M., & Sinkkonen, L. (2022). Normal and Pathological NRF2 Signalling in the Central Nervous System. Antioxidants, 11(8), 1426. https://doi.org/10.3390/antiox11081426