
Role of Oxidative Stress in the Pathogenesis of Atherothrombotic Diseases

 , and
, and
Abstract
:
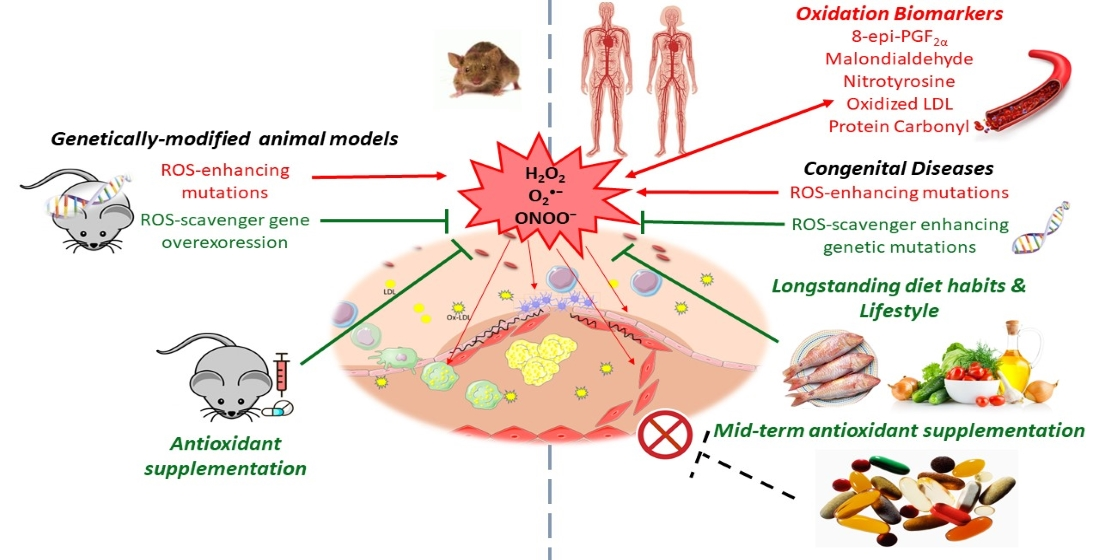
1. Introduction
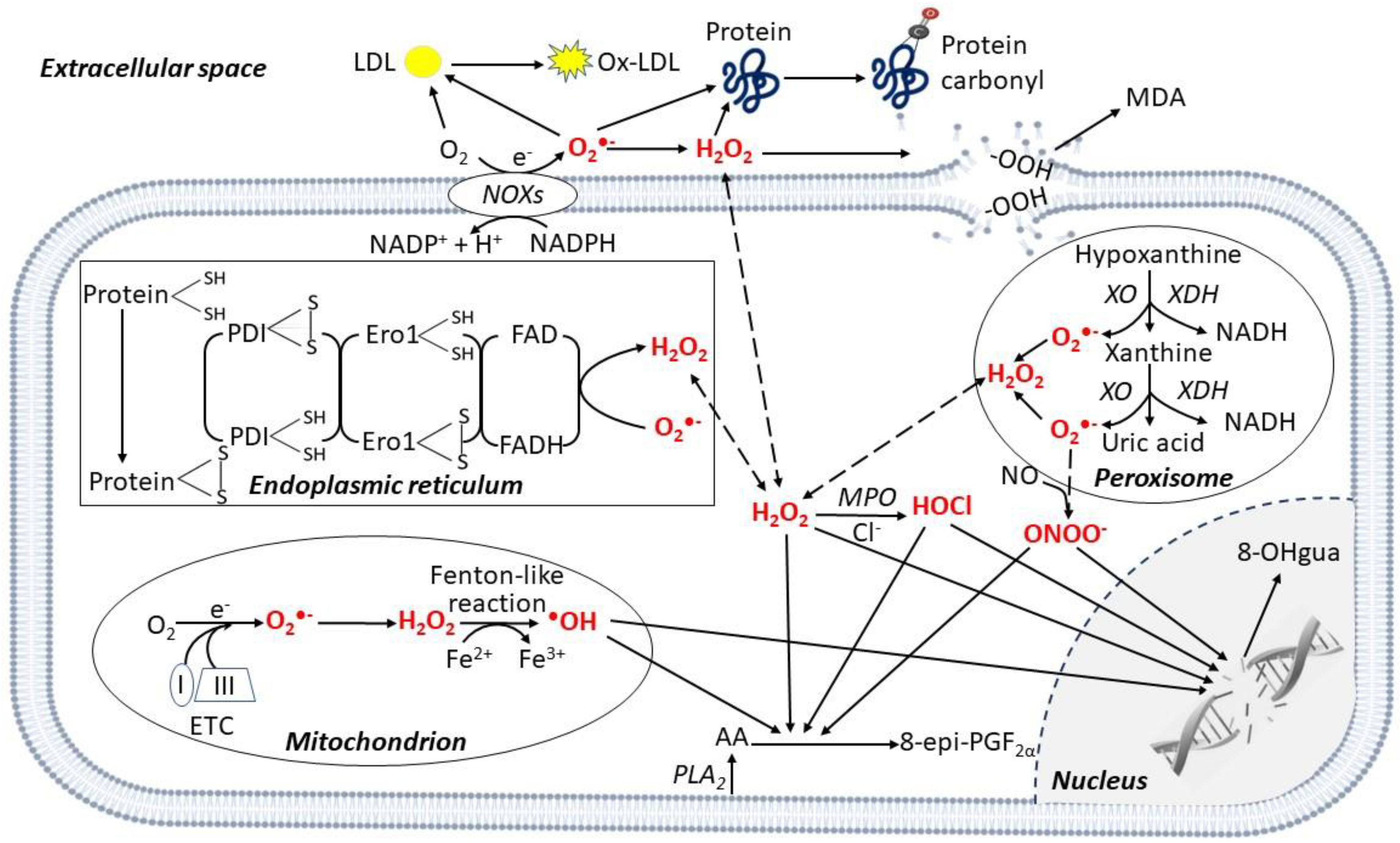
2. ROS Generation
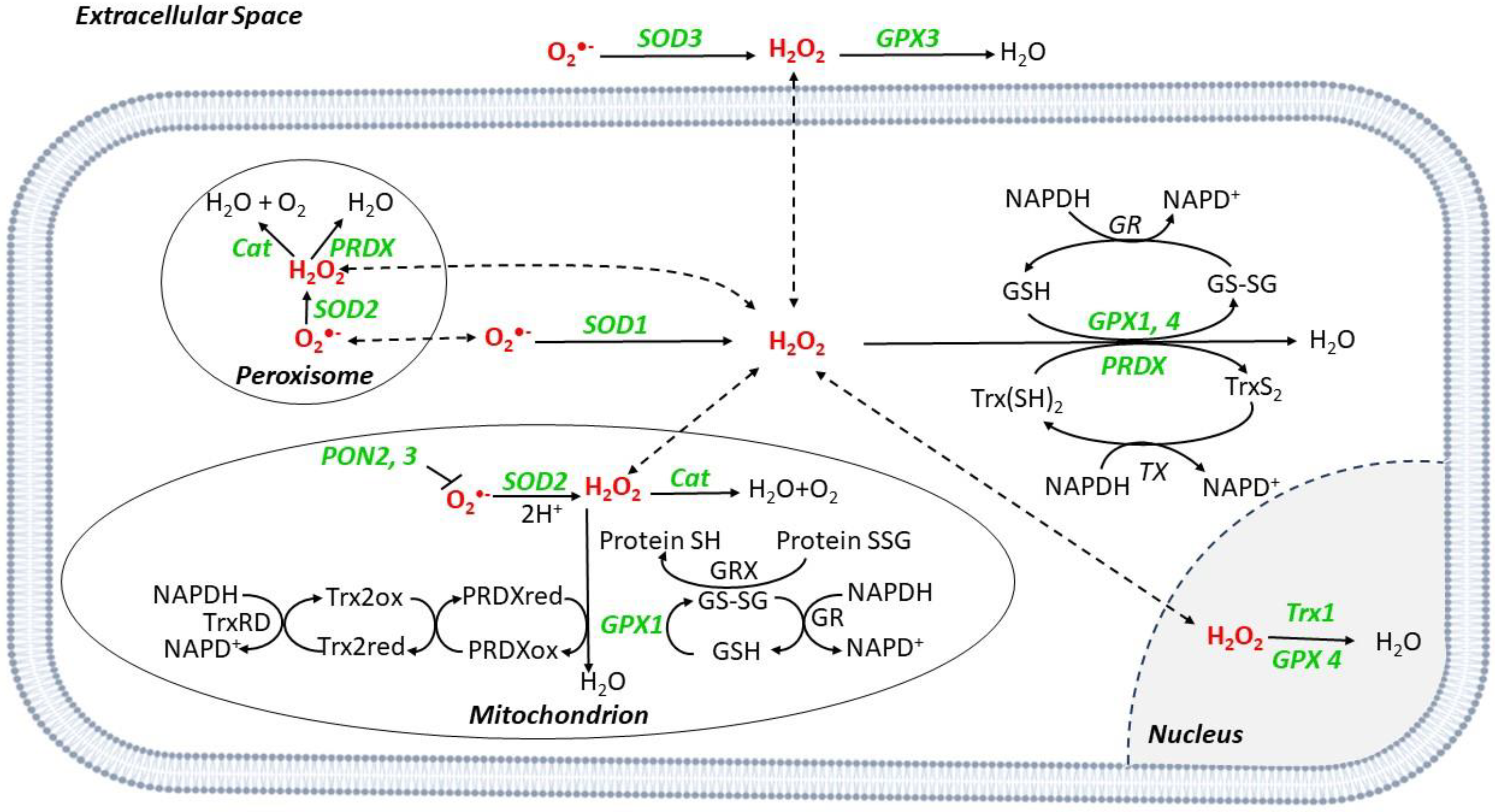
3. Scavenger Systems
4. Human Circulating Biomarkers of Oxidative Stress
5. Pharmacological Interventions
5.1. Antioxidant Compounds
5.2. Studies in Animals
5.3. Intervention Studies in Humans

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study (Year) | Study Population | Design and Study Duration | CV Functional Surrogates or Oxidative Stress Biomarkers | Results |
|---|---|---|---|---|
| Ashor et al. (2014) [221] | Adults with T1DM and T2DM, hypertension, heart failure and healthy subjects (n = 1129) | Meta-analysis of 44 RCT on vitamin C (<500 mg/od to >2 g/od) on endothelial function. Treatment duration: 1 day to 8 weeks | Endothelial function evaluated as FMD, plethysmography, pulse wave analysis and forearm blood flow | Standardized mean difference for endothelial function: 0.50, 95% CI = 0.34–0.66; p < 0.001 |
| Montero et al. (2014) [222] | T2DM (n = 296) | Meta-analysis of 10 trials: Vitamin E or Vitamin C (n = 148) vs. placebo (n = 148) Treatment duration: 2–52 weeks | Endothelial function, evaluated as FMD or PORH or plethysmography | Standardized mean difference for endothelial function: 0.35, 95% CI = −0.17–0.88; p = 0.18 |
| Derosa G et al. (2016) [223] | T2DM (n = 105) | Randomized study: alpha lipoic acid (ALA) 600 mg/od (n = 54) Vs. placebo (n = 51) Follow-up: 3 months | Serum SOD, erythrocyte GPX, plasma MDA | SOD comparison of within-group variations: ALA 16.7 U/mL vs. placebo 1.9 U/mL; p < 0.05 GPX comparison of within-group variations: ALA 22.4 EE/U vs. placebo 0.7 EE/U; p < 0.05 MDA comparison of within-group variations: ALA −8.9 nmol/mL vs. placebo −3.1 nmol/mL; p < 0.05 |
| Imamura et al. (2017) [224] | T2DM (n = 50) | Randomied study: Resveratrol 100 mg/od (n = 25) vs. placebo (n = 25) Treatment duration: 12 weeks | Arterial stiffness assessed by cardio-ankle vascular index | Within-group difference in cardio-ankle vascular index: resveratrol −0.4 ± 0.7 vs. placebo 0.1 ± 0.5; p < 0.01 |
| Mansournia et al. (2018) [225] | T2DM (n = 1053) | Meta-analysis of 33 studies: vitamin D vs. placebo Follow-up: 6 weeks–12 months | Serum CRP, eNOS, MDA | CRP-weighted mean difference between vitamin D vs. placebo: −0.27, 95% CI = −0.35–0.20; p < 0.001 NO-weighted mean difference between between vitamin D vs. placebo: 4.33, 95% CI = 0.96–7.70; p < 0.001 MDA-weighted mean difference between between vitamin D and placebo: –0.43, 95% CI = −0.62–0.25; p < 0.001 |
| Sattarinezhad et al. (2018) [226] | T2DM and nephropathy (n = 60) | Randomized study: Resveratrol 500 mg/od (n = 30) vs. placebo (n = 30) Follow-up: 90 days | Serum markers of NO, mSOD and MDA | NO markers’ comparison of within-group variation: resveratrol 4.4 ± 5.61 μmol/l vs. placebo −0.5 ± 5.0 μmol/L; p < 0.01 SOD comparison of within-group variation: resveratrol 4.8 ± 5.3 U/L vs. placebo −4.2 ± 9.3 U/L; p < 0.01 MDA comparison of within-group variations: resveratrol −0.4 ± 0.9 nmol/mL vs. placebo 0.9 ± 1.3 nmol/mL; p < 0.01 |
| Seyyedebrahimi et al. (2018) [227] | T2DM (n = 60) | Randomized study: Resveratrol 800 mg/od (n = 30) vs. placebo (n = 30) Follow-up: 2 months | Ferric-reducing ability in plasma (FRAP) | Percentage of FRAP change: resveratrol 44.41 ± 138.52% vs. placebo 15.30 ± 88.72%; p = 0.002 |
| Hoseini et al. (2019) [228] | T2DM (n = 46) | Randomized study: Resveratrol 500 mg/od (n = 23) vs. placebo (n = 23) Follow-up: 4 weeks | Plasma MDA and ferric-reducing ability (FRAP) | Difference between resveratrol and placebo MDA: −0.21 μmol/L, 95% CI = −0.41–0.005; p = 0.04 FRAP: 58.88 mmol/L, 95% CI = 17.33–100.44; p = 0.006 |
| Mendoza-Nùñez et al. (2019) [229] | Adults aged 60–74 years with T2DM (n = 135) | ALA 600 mg/od (n = 50) vs. placebo (n = 50) Follow-up: 6 months | Erythrocyte SOD/GPx, plasma 8-epi-PGF2α | Comparison of within-group variations SOD/GPx: ALA −0.004 vs. placebo −0.005 vs. control 0.005; p < 0.05 Comparison of within-group variations 8-epi-PGF2α: ALA −43 vs. placebo −29 vs. control 13; p < 0.05 |
| Raygan et al. (2019) [220] | T2DM with BMI ≥ 25 g/m2 and coronary heart disease, with 2- and 3- vessels (n = 60) | Randomized study: Melatonin 10 mg/od (n = 30) vs. placebo (n = 30) Follow-up:12 weeks | Plasma GSH, NO and MDA | Within-group change of GSH Melatonin +64.7 ± 105.7 mmol/L Placebo −11.1 ± 137.6 mmol/L; p = 0.02 Comparison of within-group variations NO melatonin +0.9 ± 4.7 mmol/L vs. placebo −3.3 ± 9.6 mmol/L; p = 0.03 Comparison of within-group variations MDA melatonin −0.2 ± 0.3 mmol/L vs. placebo +0.1 ± 0.5 mmol/L; p = 0.007 |
| Dalan et al. (2020) [230] | T2DM (n = 166) | Randomized study: Vitamin E 400 UI/od (n = 84) vs. placebo (n = 82) Follow-up: 24 weeks | Endothelial function assessed as peripheral arterial tonometry- reactive hyperaemia index (EndoPAT-RHI) | Difference of EndoPAT-RHI Vitamin E vs. placebo −0.02, 95% CI −0.10–0.06; p = 0.690 |
| Study (Year) | Study Population | Design and Study Duration | Primary Endpoints | Results |
|---|---|---|---|---|
| De Lorgeril et al. (1994) [233] | Adults aged < 70 yrs with a MI within 6 months (n = 605) | Randomized study: Mediterranean alpha-linolenic acid-rich diet (n = 302) versus Usual diet (n = 303) Mean follow-up: 27 months | Non-fatal acute MI and CV death | Primary Endpoint Mediterranean diet n = 8 Usual diet n = 33 RR 0.27, 95% CI 0.12–0.59, p = 0.001 |
| Yusuf et al. (2000) [234] | High CV Risk for previous CV events or T2DM+1 CV risk factor (n = 9541) | Randomized study: Vitamin E 400 UI/od (n = 4761) vs. placebo (n = 4780) Mean follow-up: 4.5 years | MI, stroke, or CV death | Primary endpoint: Vitamin E n = 772 (16.2%) Placebo n = 739 (15.5%) RR: 1.05, 95% CI 0.95–1.16; p = 0.33 |
| Knoops et al. (2004) [235] | Healthy elderly from 2 European cohorts (FINE n = 726 and SENECA n = 1613) | Pooled analysis on the effect of Mediterranean diet, quitting smoking and engaging physical activity on mortality Mean follow-up: 10 years | All-cause mortality, Death from CAD, CV death | All-cause mortality Mediterranean diet HR: 0.77, 95% CI 0.68–0.88 Death from CAD Mediterranean diet HR: 0.61, 95% CI 0.43–0.88 CV Death Mediterranean Diet HR: 0.71, 95% CI 0.58–0.88 |
| Whelthon et al. (2004) [236] | Adults with and without CV disease (n = 228,864) | Metanalysis of 19 observational studies (14 cohort studies and 5 case-control studies) comparing regular fish consumption (mean intake 36 g/od or 2.2 servings/week) vs. little/no fish consumption Mean follow-up of cohort studies: 15 years | Fatal and Total CAD | Fatal CAD Regular Fish consumption RR: 0.83, 95% CI 0.76 to 0.90; p < 0.005 Total CAD Regular Fish Consumption RR: 0.86, 95% CI 0.81–0.92; p < 0.005 |
| Lee et al. (2005) [232] | Healthy women aged ≥ 45 (n = 39,876) | Randomized study: Vitamin E 600 UI/eod (n = 19,937) vs. placebo (n = 19,939) Mean follow-up: 10.1 years | Nonfatal MI, nonfatal stroke, or CV death | Primary endpoint: Vitamin E n = 482 (2.4%) Placebo n = 517 (2.5%) RR: 0.93, 95% CI 0.82–1.05; p = 0.26 |
| Cook et al. (2007) [237] | Female aged ≥ 40 with previous CV event or with ≥3 CV risk factors (hypertension, high cholesterol, DM, history of MI, BMI ≥30 kg/m2, current cigarette smoking) (n = 8171) | Randomized study, 2X2 Factorial design: Vitamin E 600 UI/eod (n = 4087), Vitamin C 500 mg/od (n = 4083) vs. placebo (n = 4084) Mean follow-up: 9.4 years | MI, stroke, CABG or PTCA, CV death | Primary endpoint: Vitamin E n = 708 (17.3%) Placebo n = 742 (18.1%) RR: 0.94, 95% CI 0.85–1.04; p = 0.23 Vitamine C n = 731 (17.9%), Placebo n = 719 (17.5%), RR: 1.02, 95% CI 0.92–1.13; p = 0.71 |
| Sesso et al. (2008) [238] | Male aged ≥ 50 years, including 5.1% with prevalent CV disease, as MI and stroke (n = 14,641) | Randomized study, 2 × 2 factorial Design: Vitamin E 400 UI/eod (n = 7329) + Vitamin C 500 mg/od (n = 7315) vs. placebo (n = 7312 vs. Vitamin E or n = 7326 vs. Vitamin C) alone Mean follow-up: 8.0 years | Non-fatal MI, non-fatal stroke, CV death | Primary endpoint: Vitamin E n = 620, 1.09 events per 1000 person–years Placebo n = 625, 1.09 events per 1000 person–year HR: 1.01, 95% CI 0.90–1.13; p = 0.86 Vitamin C n = 619, 1.08 events per 1000 person–years Placebo n = 626, 1.09 events per 1000 person–years HR: 0.99, 95% CI 0.89–1.11; p = 0.91 |
| Myung et al. (2013) [239] | Adults with and without CV disease (n = 294,478) | Metanalysis of 50 RCT evaluating the effect of several compounds (Vitamins Q10 coenzyme, calcium, n3-fatty acids) Follow-up: 6 months–12 years | CV death, MI, stroke, angina, sudden cardiac death | Primary endpoint All compounds RR 1.00, 95% CI 0.98–1.02 Vitamin B6 RR 0.92, 95% CI 0.85–0.99 |
| Bowman et al. (2018) [240] | T2DM without ASCVD (n = 15,480) | Randomized study: n-3 fatty acid 1 g/od (n = 7740) vs. placebo (n = 7740) Mean follow-up: 7.4 years | Non-fatal MI or stroke, TIA, vascular death | Primary endpoint n-3 fatty acid group n = 689 (8.9%) Placebo n = 712 (9.2%) RR: 0.97, 95% CI 0.87–1.08; p = 0.55 |
| Estruch et al. (2018) [241] | Subjects at high CV risk (T2DM or ≥3 CV risk factors, as smoking, hypertension, elevated LDL cholesterol, low HDL cholesterol, overweight or obesity, or a family history of premature CHD) (n = 7447) | Randomized study: mediterranean diet with extra-virgin olive oil integration (n = 2543) vs. mediterranean diet with mixed nuts integration (n = 2454) vs. dietary fat reduction advice as control (n = 2450) Median follow-up: 4.8 years | MI, stroke, CV death | Primary endpoint Mediterranean diet with extra-virgin olive oil n = 98 (3.8%) Incidence rate 8.1 per 1000 person–years HR vs. control: 0.69, 95% CI 0.53–0.92; p < 0.05 Mediterranean diet with nuts n = 83 (3.4%) Incidence rate 8.0 per 1000 person–years HR vs. control: 0.72, 95% CI 0.53–0.94; p < 0.05 Control group n = 109 (4.4%) Incidence 11.2 per 1000 person–years |
| Manson et al. (2019) [242] | Men aged ≥50 years and women aged ≥ 55 years without CV disease (n = 25,871) | Randomized study: Vitamin D 2000 UI/od + n-3 fatty acid 1 g/od (n = 12,927) vs. placebo (n = 12,944) Median follow-up: 5.3 years | MI, stroke, CV death | Primary endpoint Vitamin D + n-3 fatty acid group n = 96 (0.03%) Placebo group n = 409 (0.03%) HR: 0.97, 95% CI 0.85–1.12; p = 0.69 |
| Khan et al. (2021) [243] | Adults with and without CV disease (n = 149,051) | Metanalysis of 38 RCTs evaluating the effect of EPA alone (4 RCTs) or of EPA+DHA (34 RCTs) vs. placebo or low-dose fatty acid supplementation. Mean follow-up: 2.0 years | CV death, non-fatal MI, CHD | CV death Overall RR 0.93, lower limit 0.88-upper limit 0.98; p = 0.01 EPA RR 0.82, lower limit 0.68, upper limit 0.99; p = 0.04 EPA+DHA RR 0.94, lower limit 0.89, upper limit 0.99; p = 0.02 Non-fatal MI Overall RR 0.87, lower limit 0.81, upper limit 0.93; p < 0.01 EPA RR 0.72, lower limit 0.62, upper limit 0.84; p < 0.01 EPA+DHA RR 0.92, lower limit 0.85, upper limit 1.00; p = 0.05 CHD Overall RR 0.91, lower limit 0.87, upper limit 0.96; p < 0.01 EPA RR 0.73, lower limit 0.62, upper limit 0.85; p < 0.01 EPA+DHA RR 0.94, lower limit 0.89, upper limit 0.99; p = 0.01 |
| Mohan et al. (2021) [244] | Adults with and without CV event (PURE n = 147,645 ONTARGET/TRASCEND n = 31,491 ORIGIN n = 12,422) | Pooled analysis of individual participant data from a cohort study and 3 RCTs (ONTARGET, TRASCEND, ORIGIN) comparing high fish intake (≥175 g/weekly) vs. little/no fish intake (<50 g/monthly) Median follow-up: PURE: 9.1 years; ONTARGET/TRASCEND: 4.5 years; ORIGIN 6.2 years | MI, stroke, congestive heart failure, or sudden death, all-cause mortality | Primary Endpoints PURE Subjects without prior CV event >175 g/weekly fish HR: 0.94, 95% CI 0.88–1.01 Subjects with prior CV event >175 g/weekly fish HR: 0.89, 95% CI 0.74–1.06 ONTARGET/TRASCEND Subjects with prior CV event >175 g/weekly fish HR: 0.88, 95% CI 0.80–0.97; p < 0.05 ORIGIN Subjects without prior CV event >175 g/weekly fish HR: 0.94, 95% CI 0.88–1.04 Subjects with prior CV event >175 g/weekly fish HR: 0.86, 95% CI 0.80–0.92; p < 0.05 |
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ma, Y.; Liang, X.; Li, C.; Li, R.; Tong, X.; Zhang, R.; Shan, X.; Yang, J.; Ma, X.; Lu, W.; et al. 5-HT(2A) Receptor and 5-HT Degradation Play a Crucial Role in Atherosclerosis by Modulating Macrophage Foam Cell Formation, Vascular Endothelial Cell Inflammation, and Hepatic Steatosis. J. Atheroscler. Thromb. 2022, 29, 322–336. [Google Scholar] [CrossRef]
- Brown, D.I.; Griendling, K.K. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ. Res. 2015, 116, 531–549. [Google Scholar] [CrossRef] [Green Version]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxid. Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef]
- Andreadou, I.; Efentakis, P.; Frenis, K.; Daiber, A.; Schulz, R. Thiol-based redox-active proteins as cardioprotective therapeutic agents in cardiovascular diseases. Basic Res. Cardiol. 2021, 116, 44. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Nayor, M.; Brown, K.J.; Vasan, R.S. The Molecular Basis of Predicting Atherosclerotic Cardiovascular Disease Risk. Circ. Res. 2021, 128, 287–303. [Google Scholar] [CrossRef]
- Niu, X.-L.; Madamanchi, N.R.; Vendrov, A.E.; Tchivilev, I.; Rojas, M.; Madamanchi, C.; Brandes, R.P.; Krause, K.-H.; Humphries, J.; Smith, A.; et al. Nox activator 1: A potential target for modulation of vascular reactive oxygen species in atherosclerotic arteries. Circulation 2010, 121, 549–559. [Google Scholar] [CrossRef] [Green Version]
- Judkins, C.P.; Diep, H.; Broughton, B.R.S.; Mast, A.E.; Hooker, E.U.; Miller, A.A.; Selemidis, S.; Dusting, G.J.; Sobey, C.G.; Drummond, G.R. Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE−/− mice. Am. J. Physiol.-Heart Circ. Physiol. 2010, 298, H24–H32. [Google Scholar] [CrossRef] [Green Version]
- Sheehan, A.L.; Carrell, S.; Johnson, B.; Stanic, B.; Banfi, B.; Miller, F.J., Jr. Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis 2011, 216, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef] [Green Version]
- Quesada, I.M.; Lucero, A.; Amaya, C.; Meijles, D.N.; Cifuentes, M.E.; Pagano, P.J.; Castro, C. Selective inactivation of NADPH oxidase 2 causes regression of vascularization and the size and stability of atherosclerotic plaques. Atherosclerosis 2015, 242, 469–475. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Keaney, J.F., Jr.; Schulz, E.; Levison, B.; Shan, L.; Sakuma, M.; Zhang, X.; Shi, C.; Hazen, S.L.; Simon, D.I. Decreased neointimal formation in Nox2-deficient mice reveals a direct role for NADPH oxidase in the response to arterial injury. Proc. Natl. Acad. Sci. USA 2004, 101, 13014–13019. [Google Scholar] [CrossRef] [Green Version]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef]
- Douglas, G.; Bendall, J.K.; Crabtree, M.J.; Tatham, A.L.; Carter, E.E.; Hale, A.B.; Channon, K.M. Endothelial-specific Nox2 overexpression increases vascular superoxide and macrophage recruitment in ApoE−/− mice. Cardiovasc. Res. 2012, 94, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Barry-Lane, P.A.; Patterson, C.; van der Merwe, M.; Hu, Z.; Holland, S.M.; Yeh, E.T.; Runge, M.S. p47phox is required for atherosclerotic lesion progression in ApoE−/− mice. J. Clin. Investig. 2001, 108, 1513–1522. [Google Scholar] [CrossRef]
- Vara, D.; Mailer, R.K.; Tarafdar, A.; Wolska, N.; Heestermans, M.; Konrath, S.; Spaeth, M.; Renné, T.; Schröder, K.; Pula, G. NADPH Oxidases Are Required for Full Platelet Activation In Vitro and Thrombosis In Vivo but Dispensable for Plasma Coagulation and Hemostasis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 683–697. [Google Scholar] [CrossRef]
- Xu, S.; Chamseddine, A.H.; Carrell, S.; Miller, F.J. Nox4 NADPH oxidase contributes to smooth muscle cell phenotypes associated with unstable atherosclerotic plaques. Redox Biol. 2014, 2, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Ohara, Y.; Peterson, T.E.; Harrison, D.G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Investig. 1993, 91, 2546–2551. [Google Scholar] [CrossRef] [Green Version]
- Schröder, K.; Vecchione, C.; Jung, O.; Schreiber, J.G.; Shiri-Sverdlov, R.; van Gorp, P.J.; Busse, R.; Brandes, R.P. Xanthine oxidase inhibitor tungsten prevents the development of atherosclerosis in ApoE knockout mice fed a Western-type diet. Free Radic. Biol. Med. 2006, 41, 1353–1360. [Google Scholar] [CrossRef]
- Nomura, J.; Busso, N.; Ives, A.; Matsui, C.; Tsujimoto, S.; Shirakura, T.; Tamura, M.; Kobayashi, T.; So, A.; Yamanaka, Y. Xanthine oxidase inhibition by febuxostat attenuates experimental atherosclerosis in mice. Sci. Rep. 2014, 4, 4554. [Google Scholar] [CrossRef] [Green Version]
- Tiyerili, V.; Camara, B.; Becher, M.U.; Schrickel, J.W.; Lütjohann, D.; Mollenhauer, M.; Baldus, S.; Nickenig, G.; Andrié, R.P. Neutrophil-derived myeloperoxidase promotes atherogenesis and neointima formation in mice. Int. J. Cardiol. 2016, 204, 29–36. [Google Scholar] [CrossRef]
- Brennan, M.-L.; Anderson, M.M.; Shih, D.M.; Qu, X.-D.; Wang, X.; Mehta, A.C.; Lim, L.L.; Shi, W.; Hazen, S.L.; Jacob, J.S. Increased atherosclerosis in myeloperoxidase-deficient mice. J. Clin. Investig. 2001, 107, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Song, R.; Dasgupta, C.; Mulder, C.; Zhang, L. MicroRNA-210 Controls Mitochondrial Metabolism and Protects Heart Function in Myocardial Infarction. Circulation 2022, 145, 1140–1153. [Google Scholar] [CrossRef]
- Guzik, T.J.; Sadowski, J.; Kapelak, B.; Jopek, A.; Rudzinski, P.; Pillai, R.; Korbut, R.; Channon, K.M. Systemic regulation of vascular NAD(P)H oxidase activity and nox isoform expression in human arteries and veins. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1614–1620. [Google Scholar] [CrossRef] [Green Version]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybylowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Sibley, C.T.; Estwick, T.; Zavodni, A.; Huang, C.Y.; Kwan, A.C.; Soule, B.P.; Long Priel, D.A.; Remaley, A.T.; Rudman Spergel, A.K.; Turkbey, E.B.; et al. Assessment of atherosclerosis in chronic granulomatous disease. Circulation 2014, 130, 2031–2039. [Google Scholar] [CrossRef] [Green Version]
- Pignatelli, P.; Sanguigni, V.; Lenti, L.; Ferro, D.; Finocchi, A.; Rossi, P.; Violi, F. gp91phox-Dependent Expression of Platelet CD40 Ligand. Circulation 2004, 110, 1326–1329. [Google Scholar] [CrossRef] [Green Version]
- Manea, A.; Manea, S.-A.; Gan, A.M.; Constantin, A.; Fenyo, I.M.; Raicu, M.; Muresian, H.; Simionescu, M. Human monocytes and macrophages express NADPH oxidase 5; a potential source of reactive oxygen species in atherosclerosis. Biochem. Biophys. Res. Commun. 2015, 461, 172–179. [Google Scholar] [CrossRef]
- Sugiyama, S.; Okada, Y.; Sukhova, G.K.; Virmani, R.; Heinecke, J.W.; Libby, P. Macrophage Myeloperoxidase Regulation by Granulocyte Macrophage Colony-Stimulating Factor in Human Atherosclerosis and Implications in Acute Coronary Syndromes. Am. J. Pathol. 2001, 158, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Investig. 1994, 94, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Lassègue, B.; Clempus, R.E. Vascular NAD(P)H oxidases: Specific features, expression, and regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R277–R297. [Google Scholar] [CrossRef] [Green Version]
- Gimenez, M.; Schickling, B.M.; Lopes, L.R.; Miller, F.J., Jr. Nox1 in cardiovascular diseases: Regulation and pathophysiology. Clin. Sci. 2016, 130, 151–165. [Google Scholar] [CrossRef]
- Zafari, A.M.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Shah, A.; Harrison, D.G.; Taylor, W.R.; Griendling, K.K. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension 1998, 32, 488–495. [Google Scholar] [CrossRef] [Green Version]
- Lassègue, B.; Griendling, K.K. NADPH oxidases: Functions and pathologies in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [Green Version]
- Johar, S.; Cave, A.C.; Narayanapanicker, A.; Grieve, D.J.; Shah, A.M. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. 2006, 20, 1546–1548. [Google Scholar] [CrossRef] [Green Version]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential Roles of the NADPH-Oxidase 1 and 2 in Platelet Activation and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 846–854. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell. Neurosci. 2017, 10, 301. [Google Scholar] [CrossRef] [Green Version]
- Pignatelli, P.; Carnevale, R.; Di Santo, S.; Bartimoccia, S.; Sanguigni, V.; Lenti, L.; Finocchi, A.; Mendolicchio, L.; Soresina, A.R.; Plebani, A.; et al. Inherited human gp91phox deficiency is associated with impaired isoprostane formation and platelet dysfunction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Chen, X.; Tabet, F.; Yao, G.; He, G.; Quinn, M.T.; Pagano, P.J.; Schiffrin, E.L. Expression of a Functionally Active gp91phox-Containing Neutrophil-Type NAD(P)H Oxidase in Smooth Muscle Cells From Human Resistance Arteries. Circ. Res. 2002, 90, 1205–1213. [Google Scholar] [CrossRef] [Green Version]
- Ago, T.; Kitazono, T.; Ooboshi, H.; Iyama, T.; Han, Y.H.; Takada, J.; Wakisaka, M.; Ibayashi, S.; Utsumi, H.; Iida, M. Nox4 as the Major Catalytic Component of an Endothelial NAD(P)H Oxidase. Circulation 2004, 109, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Lener, B.; Kozieł, R.; Pircher, H.; Hütter, E.; Greussing, R.; Herndler-Brandstetter, D.; Hermann, M.; Unterluggauer, H.; Jansen-Dürr, P. The NADPH oxidase Nox4 restricts the replicative lifespan of human endothelial cells. Biochem. J. 2009, 423, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.F.; Qiao, M.; Schröder, K.; Zhao, Q.; Asmis, R. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ. Res. 2010, 106, 1489–1497. [Google Scholar] [CrossRef] [Green Version]
- Asmis, R.; Begley, J.G. Oxidized LDL promotes peroxide-mediated mitochondrial dysfunction and cell death in human macrophages: A caspase-3-independent pathway. Circ. Res. 2003, 92, e20–e29. [Google Scholar] [CrossRef] [Green Version]
- Sorescu, D.; Weiss, D.; Lassègue, B.; Clempus, R.E.; Szöcs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide Production and Expression of Nox Family Proteins in Human Atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [Green Version]
- Ndrepepa, G. Myeloperoxidase—A bridge linking inflammation and oxidative stress with cardiovascular disease. Clin. Chim. Acta 2019, 493, 36–51. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell. 2012, 48, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial Dysfunction in Atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tabas, I. Emerging roles of mitochondria ROS in atherosclerotic lesions: Causation or association? J. Atheroscler. Thromb. 2014, 21, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Vendrov, K.C.; Simmons, B.P.; Schuck, R.N.; Stouffer, G.A.; Lee, C.R. Urinary 11-dehydro-thromboxane B2 levels are associated with vascular inflammation and prognosis in atherosclerotic cardiovascular disease. Prostaglandins Other Lipid Mediat. 2018, 134, 24–31. [Google Scholar] [CrossRef]
- Han, A.-P.; Yu, C.; Lu, L.; Fujiwara, Y.; Browne, C.; Chin, G.; Fleming, M.; Leboulch, P.; Orkin, S.H.; Chen, J.-J. Heme-regulated eIF2α kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001, 20, 6909–6918. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Wang, X.; Li, C.; Li, Q.; An, Y.A.; Luo, X.; Deng, Y.; Gillette, T.G.; Scherer, P.E.; Wang, Z.V. Integrated Stress Response Couples Mitochondrial Protein Translation With Oxidative Stress Control. Circulation 2021, 144, 1500–1515. [Google Scholar] [CrossRef]
- He, M.; Lu, Y.; Xu, S.; Mao, L.; Zhang, L.; Duan, W.; Liu, C.; Pi, H.; Zhang, Y.; Zhong, M.; et al. MiRNA-210 modulates a nickel-induced cellular energy metabolism shift by repressing the iron–sulfur cluster assembly proteins ISCU1/2 in Neuro-2a cells. Cell Death Dis. 2014, 5, e1090. [Google Scholar] [CrossRef]
- Yu, E.P.K.; Reinhold, J.; Yu, H.; Starks, L.; Uryga, A.K.; Foote, K.; Finigan, A.; Figg, N.; Pung, Y.-F.; Logan, A.; et al. Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2322–2332. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Roberts, L.J.; Shi, M.J.; Zhou, L.C.; Ballard, B.R.; Richardson, A.; Guo, Z.M. Retardation of atherosclerosis by overexpression of catalase or both Cu/Zn-superoxide dismutase and catalase in mice lacking apolipoprotein E. Circ. Res. 2004, 95, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhou, L.; Wang, Z.; Roberts, L.J., 2nd; Lin, X.; Zhao, Y.; Guo, Z. Overexpression of antioxidant enzymes in ApoE-deficient mice suppresses benzo(a)pyrene-accelerated atherosclerosis. Atherosclerosis 2009, 207, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Parastatidis, I.; Weiss, D.; Joseph, G.; Taylor, W.R. Overexpression of catalase in vascular smooth muscle cells prevents the formation of abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2389–2396. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, G.Z.; Rabinovitch, P.S.; Tabas, I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-κB-mediated inflammation in macrophages. Circ. Res. 2014, 114, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, W.; Wang, N.; Tall, A.R.; Tabas, I. Mitochondrial Oxidative Stress Promotes Atherosclerosis and Neutrophil Extracellular Traps in Aged Mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e99–e107. [Google Scholar] [CrossRef] [Green Version]
- Guo, X. Overexpression of Peroxiredoxin 4 Attenuates Atherosclerosis in Apolipoprotein E Knockout Mice. Antioxid. Redox Signal. 2012, 17, 1362–1375. [Google Scholar] [CrossRef] [Green Version]
- Kisucka, J.; Chauhan, A.K.; Patten, I.S.; Yesilaltay, A.; Neumann, C.; Van Etten, R.A.; Krieger, M.; Wagner, D.D. Peroxiredoxin1 prevents excessive endothelial activation and early atherosclerosis. Circ. Res. 2008, 103, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Park, J.G.; Yoo, J.Y.; Jeong, S.J.; Choi, J.H.; Lee, M.R.; Lee, M.N.; Hwa Lee, J.; Kim, H.C.; Jo, H.; Yu, D.Y.; et al. Peroxiredoxin 2 deficiency exacerbates atherosclerosis in apolipoprotein E-deficient mice. Circ. Res. 2011, 109, 739–749. [Google Scholar] [CrossRef]
- Kader, T.; Porteous, C.M.; Jones, G.T.; Dickerhof, N.; Narayana, V.K.; Tull, D.; Taraknath, S.; McCormick, S.P.A. Ribose-cysteine protects against the development of atherosclerosis in apoE-deficient mice. PLoS ONE 2020, 15, e0228415. [Google Scholar] [CrossRef]
- Forgione, M.A.; Cap, A.; Liao, R.; Moldovan, N.I.; Eberhardt, R.T.; Lim, C.C.; Jones, J.; Goldschmidt-Clermont, P.J.; Loscalzo, J. Heterozygous Cellular Glutathione Peroxidase Deficiency in the Mouse. Circulation 2002, 106, 1154–1158. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Torzewski, M.; Degreif, A.; Rossmann, H.; Canisius, A.; Lackner, K.J. Impact of Glutathione Peroxidase-1 Deficiency on Macrophage Foam Cell Formation and Proliferation: Implications for Atherogenesis. PLoS ONE 2013, 8, e72063. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.; Stefanovic, N.; Pete, J.; Calkin, A.C.; Giunti, S.; Thallas-Bonke, V.; Jandeleit-Dahm, K.A.; Allen, T.J.; Kola, I.; Cooper, M.E.; et al. Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein E-deficient mice. Circulation 2007, 115, 2178–2187. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Luo, Y.; Zhang, W.; He, Y.; Dai, S.; Zhang, R.; Huang, Y.; Bernatchez, P.; Giordano, F.J.; Shadel, G.; et al. Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. Am. J. Pathol. 2007, 170, 1108–1120. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Zhou, H.J.; Zhang, H.; Huang, Y.; Hinojosa-Kirschenbaum, F.; Fan, P.; Yao, L.; Belardinelli, L.; Tellides, G.; Giordano, F.J.; et al. Thioredoxin-2 Inhibits Mitochondrial Reactive Oxygen Species Generation and Apoptosis Stress Kinase-1 Activity to Maintain Cardiac Function. Circulation 2015, 131, 1082–1097. [Google Scholar] [CrossRef] [Green Version]
- Kameritsch, P.; Singer, M.; Nuernbergk, C.; Rios, N.; Reyes, A.M.; Schmidt, K.; Kirsch, J.; Schneider, H.; Müller, S.; Pogoda, K.; et al. The mitochondrial thioredoxin reductase system (TrxR2) in vascular endothelium controls peroxynitrite levels and tissue integrity. Proc. Natl. Acad. Sci. USA 2021, 118, e1921828118. [Google Scholar] [CrossRef]
- Dayal, S.; Gu, S.X.; Hutchins, R.D.; Wilson, K.M.; Wang, Y.; Fu, X.; Lentz, S.R. Deficiency of Superoxide Dismutase Impairs Protein C Activation and Enhances Susceptibility to Experimental Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1798–1804. [Google Scholar] [CrossRef] [Green Version]
- Vendrov, A.E.; Stevenson, M.D.; Alahari, S.; Pan, H.; Wickline, S.A.; Madamanchi, N.R.; Runge, M.S. Attenuated Superoxide Dismutase 2 Activity Induces Atherosclerotic Plaque Instability During Aging in Hyperlipidemic Mice. J. Am. Heart Assoc. 2017, 6, e006775. [Google Scholar] [CrossRef] [Green Version]
- Ng, D.S.; Chu, T.; Esposito, B.; Hui, P.; Connelly, P.W.; Gross, P.L. Paraoxonase-1 deficiency in mice predisposes to vascular inflammation, oxidative stress, and thrombogenicity in the absence of hyperlipidemia. Cardiovasc. Pathol. 2008, 17, 226–232. [Google Scholar] [CrossRef]
- Tward, A.; Xia, Y.-R.; Wang, X.-P.; Shi, Y.-S.; Park, C.; Castellani, L.W.; Lusis, A.J.; Shih, D.M. Decreased Atherosclerotic Lesion Formation in Human Serum Paraoxonase Transgenic Mice. Circulation 2002, 106, 484–490. [Google Scholar] [CrossRef] [Green Version]
- Mackness, B.; Quarck, R.; Verreth, W.; Mackness, M.; Holvoet, P. Human paraoxonase-1 overexpression inhibits atherosclerosis in a mouse model of metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1545–1550. [Google Scholar] [CrossRef] [Green Version]
- Devarajan, A.; Bourquard, N.; Hama, S.; Navab, M.; Grijalva, V.R.; Morvardi, S.; Clarke, C.F.; Vergnes, L.; Reue, K.; Teiber, J.F.; et al. Paraoxonase 2 deficiency alters mitochondrial function and exacerbates the development of atherosclerosis. Antioxid. Redox Signal. 2011, 14, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.J.; Hama, S.Y.; Bourquard, N.; Navab, M.; Reddy, S.T. Adenovirus mediated expression of human paraoxonase 2 protects against the development of atherosclerosis in apolipoprotein E-deficient mice. Mol. Genet. Metab. 2006, 89, 368–373. [Google Scholar] [CrossRef]
- Góth, L. A new type of inherited catalase deficiencies: Its characterization and comparison to the Japanese and Swiss type of acatalasemia. Blood Cells Mol. Dis. 2001, 27, 512–517. [Google Scholar] [CrossRef]
- Góth, L.; Eaton, J.W. Hereditary catalase deficiencies and increased risk of diabetes. Lancet 2000, 356, 1820–1821. [Google Scholar] [CrossRef]
- Sharpe, M.A.; Ollosson, R.; Stewart, V.C.; Clark, J.B. Oxidation of nitric oxide by oxomanganese-salen complexes: A new mechanism for cellular protection by superoxide dismutase/catalase mimetics. Biochem. J. 2002, 366, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Shuvalova, Y.; Kaminnyi, A.I.; Meshkov, A.; Shirokov, R.; Samko, A. Association between polymorphisms of eNOS and GPx-1 genes, activity of free-radical processes and in-stent restenosis. Mol. Cell. Biochem. 2012, 370, 241–249. [Google Scholar] [CrossRef]
- Wagner, A.H.; Kautz, O.; Fricke, K.; Zerr-Fouineau, M.; Demicheva, E.; Güldenzoph, B.; Bermejo, J.L.; Korff, T.; Hecker, M. Upregulation of Glutathione Peroxidase Offsets Stretch-Induced Proatherogenic Gene Expression in Human Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1894–1901. [Google Scholar] [CrossRef]
- Garin, M.C.; James, R.W.; Dussoix, P.; Blanché, H.; Passa, P.; Froguel, P.; Ruiz, J. Paraoxonase polymorphism Met-Leu54 is associated with modified serum concentrations of the enzyme. A possible link between the paraoxonase gene and increased risk of cardiovascular disease in diabetes. J. Clin. Investig. 1997, 99, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Morita, H.; Kurihara, H.; Sugiyama, T.; Kato, N.; Ebihara, A.; Hamada, C.; Kurihara, Y.; Shindo, T.; Oh-hashi, Y.; et al. Evidence for association between paraoxonase gene polymorphisms and atherosclerotic diseases. Atherosclerosis 2000, 149, 435–442. [Google Scholar] [CrossRef]
- Fortunato, G.; Rubba, P.; Panico, S.; Trono, D.; Tinto, N.; Mazzaccara, C.; De Michele, M.; Iannuzzi, A.; Vitale, D.F.; Salvatore, F.; et al. A paraoxonase gene polymorphism, PON 1 (55), as an independent risk factor for increased carotid intima-media thickness in middle-aged women. Atherosclerosis 2003, 167, 141–148. [Google Scholar] [CrossRef]
- Okuda, M.; Inoue, N.; Azumi, H.; Seno, T.; Sumi, Y.; Hirata, K.; Kawashima, S.; Hayashi, Y.; Itoh, H.; Yodoi, J.; et al. Expression of glutaredoxin in human coronary arteries: Its potential role in antioxidant protection against atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1483–1487. [Google Scholar] [CrossRef] [Green Version]
- Urbonavicius, S.; Lindholt, J.S.; Vorum, H.; Urbonaviciene, G.; Henneberg, E.W.; Honoré, B. Proteomic identification of differentially expressed proteins in aortic wall of patients with ruptured and nonruptured abdominal aortic aneurysms. J. Vasc. Surg. 2009, 49, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Juul, K.; Tybjærg-Hansen, A.; Marklund, S.; Heegaard, N.H.H.; Steffensen, R.; Sillesen, H.; Jensen, G.; Nordestgaard, B.G. Genetically Reduced Antioxidative Protection and Increased Ischemic Heart Disease Risk. Circulation 2004, 109, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Mohammedi, K.; Bellili-Muñoz, N.; Marklund, S.L.; Driss, F.; Le Nagard, H.; Patente, T.A.; Fumeron, F.; Roussel, R.; Hadjadj, S.; Marre, M.; et al. Plasma extracellular superoxide dismutase concentration, allelic variations in the SOD3 gene and risk of myocardial infarction and all-cause mortality in people with type 1 and type 2 diabetes. Cardiovasc. Diabetol. 2015, 14, 845. [Google Scholar] [CrossRef] [Green Version]
- del Río, L.A.; Sandalio, L.M.; Palma, J.M.; Bueno, P.; Corpas, F.J. Metabolism of oxygen radicals in peroxisomes and cellular implications. Free Radic. Biol. Med. 1992, 13, 557–580. [Google Scholar] [CrossRef]
- Lin, S.J.; Shyue, S.K.; Liu, P.L.; Chen, Y.H.; Ku, H.H.; Chen, J.W.; Tam, K.B.; Chen, Y.L. Adenovirus-mediated overexpression of catalase attenuates oxLDL-induced apoptosis in human aortic endothelial cells via AP-1 and C-Jun N-terminal kinase/extracellular signal-regulated kinase mitogen-activated protein kinase pathways. J. Mol. Cell. Cardiol. 2004, 36, 129–139. [Google Scholar] [CrossRef]
- Bellinger, F.P.; Raman, A.V.; Reeves, M.A.; Berry, M.J. Regulation and function of selenoproteins in human disease. Biochem. J. 2009, 422, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.R. The glutathione peroxidases. Cell. Mol. Life Sci. 2000, 57, 1825–1835. [Google Scholar] [CrossRef]
- Weiss, N.; Zhang, Y.Y.; Heydrick, S.; Bierl, C.; Loscalzo, J. Overexpression of cellular glutathione peroxidase rescues homocyst(e)ine-induced endothelial dysfunction. Proc. Natl. Acad. Sci. USA 2001, 98, 12503–12508. [Google Scholar] [CrossRef] [Green Version]
- Blankenberg, S.; Rupprecht, H.J.; Bickel, C.; Torzewski, M.; Hafner, G.; Tiret, L.; Smieja, M.; Cambien, F.; Meyer, J.; Lackner, K.J. Glutathione Peroxidase 1 Activity and Cardiovascular Events in Patients with Coronary Artery Disease. N. Engl. J. Med. 2003, 349, 1605–1613. [Google Scholar] [CrossRef] [Green Version]
- Loeper, J.; Goy, J.; Rozensztajn, L.; Bedu, O.; Moisson, P. Lipid peroxidation and protective enzymes during myocardial infarction. Clin. Chim. Acta 1991, 196, 119–125. [Google Scholar] [CrossRef]
- Espinola-Klein, C.; Rupprecht, H.J.; Bickel, C.; Schnabel, R.; Genth-Zotz, S.; Torzewski, M.; Lackner, K.; Munzel, T.; Blankenberg, S.; Investigators, A. Glutathione peroxidase-1 activity, atherosclerotic burden, and cardiovascular prognosis. Am. J. Cardiol. 2007, 99, 808–812. [Google Scholar] [CrossRef]
- Rubattu, S.; Forte, M.; Raffa, S. Circulating Leukocytes and Oxidative Stress in Cardiovascular Diseases: A State of the Art. Oxid. Med. Cell. Longev. 2019, 2019, 2650429. [Google Scholar] [CrossRef]
- Mackness, B.; Davies, G.K.; Turkie, W.; Lee, E.; Roberts, D.H.; Hill, E.; Roberts, C.; Durrington, P.N.; Mackness, M.I. Paraoxonase Status in Coronary Heart Disease. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1451–1457. [Google Scholar] [CrossRef] [Green Version]
- Mackness, B.; Durrington, P.; McElduff, P.; Yarnell, J.; Azam, N.; Watt, M.; Mackness, M. Low Paraoxonase Activity Predicts Coronary Events in the Caerphilly Prospective Study. Circulation 2003, 107, 2775–2779. [Google Scholar] [CrossRef] [Green Version]
- Fortunato, G.; Taranto, M.D.D.; Bracale, U.M.; Guercio, L.D.; Carbone, F.; Mazzaccara, C.; Morgante, A.; D’Armiento, F.P.; D’Armiento, M.; Porcellini, M.; et al. Decreased Paraoxonase-2 Expression in Human Carotids During the Progression of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 594–600. [Google Scholar] [CrossRef] [Green Version]
- Turanov, A.A.; Kehr, S.; Marino, S.M.; Yoo, M.-H.; Carlson, B.A.; Hatfield, D.L.; Gladyshev, V.N. Mammalian thioredoxin reductase 1: Roles in redox homoeostasis and characterization of cellular targets. Biochem. J. 2010, 430, 285–293. [Google Scholar] [CrossRef]
- Sun, Q.A.; Kirnarsky, L.; Sherman, S.; Gladyshev, V.N. Selenoprotein oxidoreductase with specificity for thioredoxin and glutathione systems. Proc. Natl. Acad. Sci. USA 2001, 98, 3673–3678. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Faraci, F.M.; Didion, S.P. Vascular protection: Superoxide dismutase isoforms in the vessel wall. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1367–1373. [Google Scholar] [CrossRef] [Green Version]
- Tainer, J.A.; Getzoff, E.D.; Richardson, J.S.; Richardson, D.C. Structure and mechanism of copper, zinc superoxide dismutase. Nature 1983, 306, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.K.; Bose, S.K.; McCord, J.M. The toxicity of high-dose superoxide dismutase suggests that superoxide can both initiate and terminate lipid peroxidation in the reperfused heart. Free Radic. Biol. Med. 1994, 16, 195–200. [Google Scholar] [CrossRef]
- Chen, Y.; Hou, M.; Li, Y.; Traverse, J.H.; Zhang, P.; Salvemini, D.; Fukai, T.; Bache, R.J. Increased superoxide production causes coronary endothelial dysfunction and depressed oxygen consumption in the failing heart. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H133–H141. [Google Scholar] [CrossRef] [PubMed]
- Sudhahar, V.; Okur, M.N.; Bagi, Z.; O’Bryan, J.P.; Hay, N.; Makino, A.; Patel, V.S.; Phillips, S.A.; Stepp, D.; Ushio-Fukai, M.; et al. Akt2 (Protein Kinase B Beta) Stabilizes ATP7A, a Copper Transporter for Extracellular Superoxide Dismutase, in Vascular Smooth Muscle. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 529–541. [Google Scholar] [CrossRef] [Green Version]
- Barajas, B.; Che, N.; Yin, F.; Rowshanrad, A.; Orozco, L.D.; Gong, K.W.; Wang, X.; Castellani, L.W.; Reue, K.; Lusis, A.J.; et al. NF-E2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Chew, P.; Yuen, D.Y.; Stefanovic, N.; Pete, J.; Coughlan, M.T.; Jandeleit-Dahm, K.A.; Thomas, M.C.; Rosenfeldt, F.; Cooper, M.E.; de Haan, J.B. Antiatherosclerotic and renoprotective effects of ebselen in the diabetic apolipoprotein E/GPx1-double knockout mouse. Diabetes 2010, 59, 3198–3207. [Google Scholar] [CrossRef] [Green Version]
- Habib, A.; Petrucci, G.; Rocca, B. Pathophysiology of Thrombosis in Peripheral Artery Disease. Curr. Vasc. Pharmacol. 2020, 18, 204–214. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G.; Rocca, B.; Patrono, C. The key contribution of platelet and vascular arachidonic acid metabolism to the pathophysiology of atherothrombosis. Cardiovasc. Res. 2021, 117, 2001–2015. [Google Scholar] [CrossRef]
- Patrono, C.; FitzGerald, G.A. Isoprostanes: Potential Markers of Oxidant Stress in Atherothrombotic Disease. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2309–2315. [Google Scholar] [CrossRef]
- Audoly, L.P.; Rocca, B.; Fabre, J.E.; Koller, B.H.; Thomas, D.; Loeb, A.L.; Coffman, T.M.; FitzGerald, G.A. Cardiovascular responses to the isoprostanes iPF(2alpha)-III and iPE(2)-III are mediated via the thromboxane A(2) receptor in vivo. Circulation 2000, 101, 2833–2840. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.-S.; Ramamurthy, S.K.; Lin, X.; Le Breton, G.C. Cell signalling through thromboxane A2 receptors. Cell. Signal. 2004, 16, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C.; Rocca, B. Measurement of Thromboxane Biosynthesis in Health and Disease. Front Pharmacol 2019, 10, 1244. [Google Scholar] [CrossRef]
- Petrucci, G.; Zaccardi, F.; Giaretta, A.; Cavalca, V.; Capristo, E.; Cardillo, C.; Pitocco, D.; Porro, B.; Schinzari, F.; Toffolo, G.; et al. Obesity is associated with impaired responsiveness to once-daily low-dose aspirin and in vivo platelet activation. J. Thromb. Haemost. 2019, 17, 885–895. [Google Scholar] [CrossRef]
- Davi, G.; Guagnano, M.T.; Ciabattoni, G.; Basili, S.; Falco, A.; Marinopiccoli, M.; Nutini, M.; Sensi, S.; Patrono, C. Platelet activation in obese women: Role of inflammation and oxidant stress. JAMA 2002, 288, 2008–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davi, G.; Alessandrini, P.; Mezzetti, A.; Minotti, G.; Bucciarelli, T.; Costantini, F.; Cipollone, F.; Bon, G.B.; Ciabattoni, G.; Patrono, C. In vivo formation of 8-Epi-prostaglandin F2 alpha is increased in hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3230–3235. [Google Scholar] [CrossRef]
- Zaccardi, F.; Rizzi, A.; Petrucci, G.; Ciaffardini, F.; Tanese, L.; Pagliaccia, F.; Cavalca, V.; Ciminello, A.; Habib, A.; Squellerio, I.; et al. In Vivo Platelet Activation and Aspirin Responsiveness in Type 1 Diabetes. Diabetes 2016, 65, 503–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davì, G.; Chiarelli, F.; Santilli, F.; Pomilio, M.; Vigneri, S.; Falco, A.; Basili, S.; Ciabattoni, G.; Patrono, C. Enhanced Lipid Peroxidation and Platelet Activation in the Early Phase of Type 1 Diabetes Mellitus. Circulation 2003, 107, 3199–3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santilli, F.; Zaccardi, F.; Liani, R.; Petrucci, G.; Simeone, P.; Pitocco, D.; Tripaldi, R.; Rizzi, A.; Formoso, G.; Pontecorvi, A.; et al. In vivo thromboxane-dependent platelet activation is persistently enhanced in subjects with impaired glucose tolerance. Diabetes/Metab. Res. Rev. 2020, 36, e3232. [Google Scholar] [CrossRef]
- Pascale, S.; Petrucci, G.; Dragani, A.; Habib, A.; Zaccardi, F.; Pagliaccia, F.; Pocaterra, D.; Ragazzoni, E.; Rolandi, G.; Rocca, B.; et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood 2012, 119, 3595–3603. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Jung, S.; Kim, S.Y.; Lee, S.-H.; Lee, J.H. Prehypertension-Associated Elevation in Circulating Lysophosphatidlycholines, Lp-PLA2 Activity, and Oxidative Stress. PLoS ONE 2014, 9, e96735. [Google Scholar] [CrossRef] [Green Version]
- Keaney, J.F.; Larson, M.G.; Vasan, R.S.; Wilson, P.W.F.; Lipinska, I.; Corey, D.; Massaro, J.M.; Sutherland, P.; Vita, J.A.; Benjamin, E.J. Obesity and Systemic Oxidative Stress. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 434–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwedhelm, E.; Bartling, A.; Lenzen, H.; Tsikas, D.; Maas, R.; Brümmer, J.; Gutzki, F.M.; Berger, J.; Frölich, J.C.; Böger, R.H. Urinary 8-iso-prostaglandin F2alpha as a risk marker in patients with coronary heart disease: A matched case-control study. Circulation 2004, 109, 843–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roest, M.; Voorbij, H.A.M.; Van der Schouw, Y.T.; Peeters, P.H.M.; Teerlink, T.; Scheffer, P.G. High levels of urinary F2-isoprostanes predict cardiovascular mortality in postmenopausal women. J. Clin. Lipidol. 2008, 2, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Noberasco, G.; Odetti, P.; Boeri, D.; Maiello, M.; Adezati, L. Malondialdehyde (MDA) level in diabetic subjects. Relationship with blood glucose and glycosylated hemoglobin. Biomed. Pharmacother. 1991, 45, 193–196. [Google Scholar] [CrossRef]
- Cavalca, V.; Cighetti, G.; Bamonti, F.; Loaldi, A.; Bortone, L.; Novembrino, C.; De Franceschi, M.; Belardinelli, R.; Guazzi, M.D. Oxidative Stress and Homocysteine in Coronary Artery Disease. Clin. Chem. 2001, 47, 887–892. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.F.; Jacob, R.F.; Jeffers, B.; Ghadanfar, M.M.; Preston, G.M.; Buch, J.; Mason, R.P. Serum levels of thiobarbituric acid reactive substances predict cardiovascular events in patients with stable coronary artery disease: A longitudinal analysis of the PREVENT study. J. Am. Coll. Cardiol. 2004, 44, 1996–2002. [Google Scholar] [CrossRef] [Green Version]
- Tanriverdi, H.; Evrengul, H.; Kuru, O.; Tanriverdi, S.; Seleci, D.; Enli, Y.; Kaftan, H.A.; Kilic, M. Cigarette Smoking Induced Oxidative Stress may Impair Endothelial Function and Coronary Blood Flow in Angiographically Normal Coronary Arteries. Circ. J. 2006, 70, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Kotur-Stevuljevic, J.; Memon, L.; Stefanovic, A.; Spasic, S.; Spasojevic-Kalimanovska, V.; Bogavac-Stanojevic, N.; Kalimanovska-Ostric, D.; Jelić-Ivanovic, Z.; Zunic, G. Correlation of oxidative stress parameters and inflammatory markers in coronary artery disease patients. Clin. Biochem. 2007, 40, 181–187. [Google Scholar] [CrossRef]
- Kubihal, C.; Naik, H. Effect of smoking on vitamin C and MDA: A cross sectional comparative study. Int. J. Res. Med. Sci. 2019, 7, 746–749. [Google Scholar] [CrossRef]
- Ehara, S.; Ueda, M.; Naruko, T.; Haze, K.; Itoh, A.; Otsuka, M.; Komatsu, R.; Matsuo, T.; Itabe, H.; Takano, T.; et al. Elevated levels of oxidized low density lipoprotein show a positive relationship with the severity of acute coronary syndromes. Circulation 2001, 103, 1955–1960. [Google Scholar] [CrossRef]
- Shimada, K.; Mokuno, H.; Matsunaga, E.; Miyazaki, T.; Sumiyoshi, K.; Miyauchi, K.; Daida, H. Circulating oxidized low-density lipoprotein is an independent predictor for cardiac event in patients with coronary artery disease. Atherosclerosis 2004, 174, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Kiechl, S.; Willeit, J.; Mayr, M.; Miller, E.R.; Kronenberg, F.; Xu, Q.; Bergmark, C.; Weger, S.; Oberhollenzer, F.; et al. Oxidized Phospholipids Predict the Presence and Progression of Carotid and Femoral Atherosclerosis and Symptomatic Cardiovascular Disease. J. Am. Coll. Cardiol. 2006, 47, 2219–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-C.; Tang, Y.; Chen, Y.; Huang, X.-H.; Zhang, M.; Chen, J.; Sun, Y.-G.; Li, Y.-G. Oxidized Low-Density Lipoprotein and C-Reactive Protein Have Combined Utility for Better Predicting Prognosis After Acute Coronary Syndrome. Cell Biochem. Biophys. 2014, 68, 379–385. [Google Scholar] [CrossRef]
- Gao, S.; Zhao, D.; Wang, M.; Zhao, F.; Han, X.; Qi, Y.; Liu, J. Association Between Circulating Oxidized LDL and Atherosclerotic Cardiovascular Disease: A Meta-analysis of Observational Studies. Can. J. Cardiol. 2017, 33, 1624–1632. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Mercuri, F.; Quagliaro, L.; Assaloni, R.; Motz, E.; Tonutti, L.; Taboga, C. Detection of nitrotyrosine in the diabetic plasma: Evidence of oxidative stress. Diabetologia 2001, 44, 834–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shishehbor, M.H.; Aviles, R.J.; Brennan, M.-L.; Fu, X.; Goormastic, M.; Pearce, G.L.; Gokce, N.; Keaney, J.; John, F.; Penn, M.S.; et al. Association of Nitrotyrosine Levels With Cardiovascular Disease and Modulation by Statin Therapy. JAMA 2003, 289, 1675–1680. [Google Scholar] [CrossRef] [Green Version]
- Kilhovd, B.K.; Berg, T.J.; Birkeland, K.I.; Thorsby, P.; Hanssen, K.F. Serum levels of advanced glycation end products are increased in patients with type 2 diabetes and coronary heart disease. Diabetes Care 1999, 22, 1543–1548. [Google Scholar] [CrossRef]
- De Cristofaro, R.; Rocca, B.; Vitacolonna, E.; Falco, A.; Marchesani, P.; Ciabattoni, G.; Landolfi, R.; Patrono, C.; Davì, G. Lipid and protein oxidation contribute to a prothrombotic state in patients with type 2 diabetes mellitus. J. Thromb. Haemost. 2003, 1, 250–256. [Google Scholar] [CrossRef]
- Mutlu-Türkoglu, Ü.; Akalýn, Z.; Ýlhan, E.; Yýlmaz, E.; Bilge, A.; Niþancý, Y.; Uysal, M. Increased plasma malondialdehyde and protein carbonyl levels and lymphocyte DNA damage in patients with angiographically defined coronary artery disease. Clin. Biochem. 2005, 38, 1059–1065. [Google Scholar] [CrossRef]
- Semba, R.D.; Ferrucci, L.; Sun, K.; Beck, J.; Dalal, M.; Varadhan, R.; Walston, J.; Guralnik, J.M.; Fried, L.P. Advanced glycation end products and their circulating receptors predict cardiovascular disease mortality in older community-dwelling women. Aging Clin. Exp. Res. 2009, 21, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Pirinccioglu, A.G.; Gökalp, D.; Pirinccioglu, M.; Kizil, G.; Kizil, M. Malondialdehyde (MDA) and protein carbonyl (PCO) levels as biomarkers of oxidative stress in subjects with familial hypercholesterolemia. Clin. Biochem. 2010, 43, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Vegi, P.; Kutty, A. Protein carbonyl content as a stable Oxidative stress marker in Type II Diabetes. Int. J. Biol. Med. Res. 2012, 3, 2362–2365. [Google Scholar]
- van Eupen, M.G.A.; Schram, M.T.; Colhoun, H.M.; Scheijen, J.L.J.M.; Stehouwer, C.D.A.; Schalkwijk, C.G. Plasma levels of advanced glycation endproducts are associated with type 1 diabetes and coronary artery calcification. Cardiovasc. Diabetol. 2013, 12, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNair, E.; Qureshi, M.; Prasad, K.; Pearce, C. Atherosclerosis and the Hypercholesterolemic AGE-RAGE Axis. Int. J. Angiol. 2016, 25, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Kopytek, M.; Ząbczyk, M.; Mazur, P.; Undas, A.; Natorska, J. Accumulation of advanced glycation end products (AGEs) is associated with the severity of aortic stenosis in patients with concomitant type 2 diabetes. Cardiovasc. Diabetol. 2020, 19, 92. [Google Scholar] [CrossRef]
- Sharifi-Zahabi, E.; Sharafabad, F.H.; Abdollahzad, H.; Malekahmadi, M.; Rad, N.B. Circulating Advanced Glycation End Products and Their Soluble Receptors in Relation to All-Cause and Cardiovascular Mortality: A Systematic Review and Meta-analysis of Prospective Observational Studies. Adv. Nutr. 2021, 12, 2157–2171. [Google Scholar] [CrossRef]
- Gianazza, E.; Brioschi, M.; Fernandez, A.M.; Banfi, C. Lipoxidation in cardiovascular diseases. Redox Biol. 2019, 23, 101119. [Google Scholar] [CrossRef]
- Uchida, K. Role of reactive aldehyde in cardiovascular diseases. Free. Radic. Biol. Med. 2000, 28, 1685–1696. [Google Scholar] [CrossRef]
- Nair, V.; Cooper, C.S.; Vietti, D.E.; Turner, G.A. The chemistry of lipid peroxidation metabolites: Crosslinking reactions of malondialdehyde. Lipids 1986, 21, 6–10. [Google Scholar] [CrossRef]
- Tsikas, D.; Suchy, M.-T.; Niemann, J.; Tossios, P.; Schneider, Y.; Rothmann, S.; Gutzki, F.-M.; Frölich, J.C.; Stichtenoth, D.O. Glutathione promotes prostaglandin H synthase (cyclooxygenase)-dependent formation of malondialdehyde and 15(S)-8-iso-prostaglandin F2α. FEBS Lett. 2012, 586, 3723–3730. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, E.A.; Myasoedova, V.A.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Small Dense Low-Density Lipoprotein as Biomarker for Atherosclerotic Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 1273042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 2021, 11, 613780. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the Pathogenesis of Atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, G.; Song, X.; Wang, L.; Li, Z.; Wu, H.; Dong, H. Golgi apparatus fragmentation participates in oxidized low-density lipoprotein-induced endothelial cell injury. J. Cell. Biochem. 2019, 120, 18862–18870. [Google Scholar] [CrossRef]
- Wraith, K.S.; Magwenzi, S.; Aburima, A.; Wen, Y.; Leake, D.; Naseem, K.M. Oxidized low-density lipoproteins induce rapid platelet activation and shape change through tyrosine kinase and Rho kinase–signaling pathways. Blood 2013, 122, 580–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsikas, D.; Mitschke, A.; Gutzki, F.M. Measurement of 3-nitro-tyrosine in human plasma and urine by gas chromatography-tandem mass spectrometry. Methods Mol. Biol. 2012, 828, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Schopfer, F.J.; Baker, P.R.S.; Freeman, B.A. NO-dependent protein nitration: A cell signaling event or an oxidative inflammatory response? Trends Biochem. Sci. 2003, 28, 646–654. [Google Scholar] [CrossRef]
- Suzuki, Y.J.; Carini, M.; Butterfield, D.A. Protein carbonylation. Antioxid. Redox Signal. 2010, 12, 323–325. [Google Scholar] [CrossRef]
- Levine, R.L. Carbonyl modified proteins in cellular regulation, aging, and disease. Free Radic. Biol. Med. 2002, 32, 790–796. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Levine, R.L. Protein oxidation. Ann. N. Y. Acad. Sci. 2000, 899, 191–208. [Google Scholar] [CrossRef]
- Dean, R.T.; Fu, S.; Stocker, R.; Davies, M.J. Biochemistry and pathology of radical-mediated protein oxidation. Biochem. J. 1997, 324, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.X.; Requena, J.R.; Jenkins, A.J.; Lyons, T.J.; Baynes, J.W.; Thorpe, S.R. The advanced glycation end product, Nepsilon-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J. Biol. Chem. 1996, 271, 9982–9986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.-J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cao, Q.; Gao, W.; Li, B.; Xia, Z.; Zhao, B. Melatonin protects against focal cerebral ischemia-reperfusion injury in diabetic mice by ameliorating mitochondrial impairments: Involvement of the Akt-SIRT3-SOD2 signaling pathway. Aging 2021, 13, 16105–16123. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Liu, X.; Ahmad, N. Resveratrol, in its natural combination in whole grape, for health promotion and disease management. Ann. N. Y. Acad. Sci. 2015, 1348, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Daiber, A.; Förstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1633–1646. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Labinskyy, N.; Mukhopadhyay, P.; Pinto, J.T.; Bagi, Z.; Ballabh, P.; Zhang, C.; Pacher, P.; Csiszar, A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1876–H1881. [Google Scholar] [CrossRef] [Green Version]
- Niki, E. Lipid oxidation that is, and is not, inhibited by vitamin E: Consideration about physiological functions of vitamin E. Free Radic. Biol. Med. 2021, 176, 1–15. [Google Scholar] [CrossRef]
- Jiang, Q. Natural forms of vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Christen, S.; Shigenaga, M.K.; Ames, B.N. γ-Tocopherol, the major form of vitamin E in the US diet, deserves more attention. Am. J. Clin. Nutr. 2001, 74, 714–722. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Meza, C.A.; Clarke, H.; Kim, J.S.; Hickner, R.C. Vitamin D and Endothelial Function. Nutrients 2020, 12, 575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinari, C.; Rizzi, M.; Squarzanti, D.F.; Pittarella, P.; Vacca, G.; Renò, F. 1α,25-Dihydroxycholecalciferol (Vitamin D3) induces NO-dependent endothelial cell proliferation and migration in a three-dimensional matrix. Cell. Physiol. Biochem. 2013, 31, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Spoelstra-de Man, A.M.E.; Elbers, P.W.G.; Oudemans-Van Straaten, H.M. Vitamin C: Should we supplement? Curr. Opin. Crit. Care 2018, 24, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Levine, M. Vitamin C: The known and the unknown and Goldilocks. Oral Dis. 2016, 22, 463–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buettner, G.R.; Jurkiewicz, B.A. Ascorbate free radical as a marker of oxidative stress: An EPR study. Free Radic. Biol. Med. 1993, 14, 49–55. [Google Scholar] [CrossRef]
- Oudemans-van Straaten, H.M.; Spoelstra-de Man, A.M.; de Waard, M.C. Vitamin C revisited. Crit. Care 2014, 18, 460. [Google Scholar] [CrossRef] [Green Version]
- Endo, N.; Nishiyama, K.; Otsuka, A.; Kanouchi, H.; Taga, M.; Oka, T. Antioxidant activity of vitamin B6 delays homocysteine-induced atherosclerosis in rats. Br. J. Nutr. 2006, 95, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Dalto, D.B.; Matte, J.J. Pyridoxine (Vitamin B6) and the Glutathione Peroxidase System; a Link between One-Carbon Metabolism and Antioxidation. Nutrients 2017, 9, 189. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, C.S.; Reed, D.J. Glutathione redox cycle-driven recovery of reduced glutathione after oxidation by tertiary-butyl hydroperoxide in preimplantation mouse embryos. Arch. Biochem. Biophys. 1995, 321, 6–12. [Google Scholar] [CrossRef]
- Packer, L.; Kraemer, K.; Rimbach, G. Molecular aspects of lipoic acid in the prevention of diabetes complications. Nutrition 2001, 17, 888–895. [Google Scholar] [CrossRef]
- Peteliuk, V.; Rybchuk, L.; Bayliak, M.; Storey, K.B.; Lushchak, O. Natural sweetener Stevia rebaudiana: Functionalities, health benefits and potential risks. EXCLI J 2021, 20, 1412–1430. [Google Scholar] [CrossRef] [PubMed]
- Prata, C.; Zambonin, L.; Rizzo, B.; Maraldi, T.; Angeloni, C.; Vieceli Dalla Sega, F.; Fiorentini, D.; Hrelia, S. Glycosides from Stevia rebaudiana Bertoni Possess Insulin-Mimetic and Antioxidant Activities in Rat Cardiac Fibroblasts. Oxid. Med. Cell. Longev. 2017, 2017, 3724545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punia, R.; Raina, K.; Agarwal, R.; Singh, R.P. Acacetin enhances the therapeutic efficacy of doxorubicin in non-small-cell lung carcinoma cells. PLoS ONE 2017, 12, e0182870. [Google Scholar] [CrossRef]
- Wu, Y.; Song, F.; Li, Y.; Li, J.; Cui, Y.; Hong, Y.; Han, W.; Wu, W.; Lakhani, I.; Li, G.; et al. Acacetin exerts antioxidant potential against atherosclerosis through Nrf2 pathway in apoE−/− Mice. J. Cell. Mol. Med. 2021, 25, 521–534. [Google Scholar] [CrossRef]
- Liu, H.; Yang, L.; Wu, H.J.; Chen, K.H.; Lin, F.; Li, G.; Sun, H.Y.; Xiao, G.S.; Wang, Y.; Li, G.R. Water-soluble acacetin prodrug confers significant cardioprotection against ischemia/reperfusion injury. Sci. Rep. 2016, 6, 36435. [Google Scholar] [CrossRef]
- Oppedisano, F.; Macrì, R.; Gliozzi, M.; Musolino, V.; Carresi, C.; Maiuolo, J.; Bosco, F.; Nucera, S.; Caterina Zito, M.; Guarnieri, L.; et al. The Anti-Inflammatory and Antioxidant Properties of n-3 PUFAs: Their Role in Cardiovascular Protection. Biomedicines 2020, 8, 306. [Google Scholar] [CrossRef]
- Li, R.; Jia, Z.; Zhu, H. Regulation of Nrf2 Signaling. React. Oxyg. Species 2019, 8, 312–322. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, C.; Dong, Y.; Wang, S.; Song, P.; Viollet, B.; Zou, M.H. Activation of the AMP-activated protein kinase by eicosapentaenoic acid (EPA, 20:5 n-3) improves endothelial function in vivo. PLoS ONE 2012, 7, e35508. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Jayachandran, M.; Bai, W.; Xu, B. A critical review on the health benefits of fish consumption and its bioactive constituents. Food Chem. 2022, 369, 130874. [Google Scholar] [CrossRef]
- Chi, C.F.; Hu, F.Y.; Wang, B.; Li, Z.R.; Luo, H.Y. Influence of Amino Acid Compositions and Peptide Profiles on Antioxidant Capacities of Two Protein Hydrolysates from Skipjack Tuna (Katsuwonus pelamis) Dark Muscle. Mar. Drugs 2015, 13, 2580–2601. [Google Scholar] [CrossRef]
- Nadtochiy, S.M.; Redman, E.K. Mediterranean diet and cardioprotection: The role of nitrite, polyunsaturated fatty acids, and polyphenols. Nutrition 2011, 27, 733–744. [Google Scholar] [CrossRef] [Green Version]
- Lou-Bonafonte, J.M.; Gabás-Rivera, C.; Navarro, M.A.; Osada, J. PON1 and Mediterranean Diet. Nutrients 2015, 7, 4068–4092. [Google Scholar] [CrossRef] [Green Version]
- Ren, B.C.; Zhang, W.; Zhang, W.; Ma, J.X.; Pei, F.; Li, B.Y. Melatonin attenuates aortic oxidative stress injury and apoptosis in STZ-diabetes rats by Notch1/Hes1 pathway. J. Steroid. Biochem. Mol. Biol. 2021, 212, 105948. [Google Scholar] [CrossRef]
- Perneby, C.; Granström, E.; Beck, O.; Fitzgerald, D.; Harhen, B.; Hjemdahl, P. Optimization of an Enzyme Immunoassay for 11-Dehydro-Thromboxane B2 in Urine: Comparison with GC-MS. Thromb. Res. 1999, 96, 427–436. [Google Scholar] [CrossRef]
- Vural, H.; Sabuncu, T.; Arslan, S.O.; Aksoy, N. Melatonin inhibits lipid peroxidation and stimulates the antioxidant status of diabetic rats. J. Pineal Res. 2001, 31, 193–198. [Google Scholar] [CrossRef]
- Guo, R.; Liu, B.; Wang, K.; Zhou, S.; Li, W.; Xu, Y. Resveratrol ameliorates diabetic vascular inflammation and macrophage infiltration in db/db mice by inhibiting the NF-κB pathway. Diabetes Vasc. Dis. Res. 2014, 11, 92–102. [Google Scholar] [CrossRef]
- Ullevig, S.L.; Zhao, Q.; Zamora, D.; Asmis, R. Ursolic acid protects diabetic mice against monocyte dysfunction and accelerated atherosclerosis. Atherosclerosis 2011, 219, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Vasamsetti, S.B.; Karnewar, S.; Gopoju, R.; Gollavilli, P.N.; Narra, S.R.; Kumar, J.M.; Kotamraju, S. Resveratrol attenuates monocyte-to-macrophage differentiation and associated inflammation via modulation of intracellular GSH homeostasis: Relevance in atherosclerosis. Free Radic. Biol. Med. 2016, 96, 392–405. [Google Scholar] [CrossRef]
- Huang, J.P.; Hsu, S.C.; Li, D.E.; Chen, K.H.; Kuo, C.Y.; Hung, L.M. Resveratrol Mitigates High-Fat Diet-Induced Vascular Dysfunction by Activating the Akt/eNOS/NO and Sirt1/ER Pathway. J. Cardiovasc. Pharmacol. 2018, 72, 231–241. [Google Scholar] [CrossRef]
- Otero, P.; Bonet, B.; Herrera, E.; Rabano, A. Development of atherosclerosis in the diabetic BALB/c mice: Prevention with Vitamin E administration. Atherosclerosis 2005, 182, 259–265. [Google Scholar] [CrossRef]
- Yi, X.; Maeda, N. alpha-Lipoic acid prevents the increase in atherosclerosis induced by diabetes in apolipoprotein E-deficient mice fed high-fat/low-cholesterol diet. Diabetes 2006, 55, 2238–2244. [Google Scholar] [CrossRef]
- Geeraert, B.; Crombé, F.; Hulsmans, M.; Benhabilès, N.; Geuns, J.M.; Holvoet, P. Stevioside inhibits atherosclerosis by improving insulin signaling and antioxidant defense in obese insulin-resistant mice. Int. J. Obes. 2010, 34, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Han, W.-M.; Chen, X.-C.; Li, G.-R.; Wang, Y. Acacetin protects against high glucose-induced endothelial cells injury by preserving mitochondrial function via activating Sirt1/Sirt3/AMPK signals. Front. Pharmacol. 2020, 11, 2179. [Google Scholar] [CrossRef]
- Wang, H.-H.; Hung, T.-M.; Wei, J.; Chiang, A.-N. Fish oil increases antioxidant enzyme activities in macrophages and reduces atherosclerotic lesions in apoE-knockout mice. Cardiovasc. Res. 2004, 61, 169–176. [Google Scholar] [CrossRef]
- Casós, K.; Sáiz, M.P.; Ruiz-Sanz, J.I.; Mitjavila, M.T. Atherosclerosis prevention by a fish oil-rich diet in apoE−/− mice is associated with a reduction of endothelial adhesion molecules. Atherosclerosis 2008, 201, 306–317. [Google Scholar] [CrossRef]
- Casós, K.; Zaragozá, M.C.; Zarkovic, N.; Zarkovic, K.; Andrisic, L.; Portero-Otín, M.; Cacabelos, D.; Mitjavila, M.T. A fish oil-rich diet reduces vascular oxidative stress in apoE–/– mice. Free Radic. Res. 2010, 44, 821–829. [Google Scholar] [CrossRef]
- Matsumoto, M.; Sata, M.; Fukuda, D.; Tanaka, K.; Soma, M.; Hirata, Y.; Nagai, R. Orally administered eicosapentaenoic acid reduces and stabilizes atherosclerotic lesions in ApoE-deficient mice. Atherosclerosis 2008, 197, 524–533. [Google Scholar] [CrossRef]
- Mercer, J.R.; Yu, E.; Figg, N.; Cheng, K.K.; Prime, T.A.; Griffin, J.L.; Masoodi, M.; Vidal-Puig, A.; Murphy, M.P.; Bennett, M.R. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/−/ApoE−/− mice. Free Radic. Biol. Med. 2012, 52, 841–849. [Google Scholar] [CrossRef]
- Raygan, F.; Ostadmohammadi, V.; Bahmani, F.; Reiter, R.J.; Asemi, Z. Melatonin administration lowers biomarkers of oxidative stress and cardio-metabolic risk in type 2 diabetic patients with coronary heart disease: A randomized, double-blind, placebo-controlled trial. Clin. Nutr. 2019, 38, 191–196. [Google Scholar] [CrossRef]
- Ashor, A.W.; Lara, J.; Mathers, J.C.; Siervo, M. Effect of vitamin C on endothelial function in health and disease: A systematic review and meta-analysis of randomised controlled trials. Atherosclerosis 2014, 235, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Montero, D.; Walther, G.; Stehouwer, C.D.; Houben, A.J.; Beckman, J.A.; Vinet, A. Effect of antioxidant vitamin supplementation on endothelial function in type 2 diabetes mellitus: A systematic review and meta-analysis of randomized controlled trials. Obes. Rev. 2014, 15, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; D’Angelo, A.; Romano, D.; Maffioli, P. A Clinical Trial about a Food Supplement Containing α-Lipoic Acid on Oxidative Stress Markers in Type 2 Diabetic Patients. Int. J. Mol. Sci. 2016, 17, 1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamura, H.; Yamaguchi, T.; Nagayama, D.; Saiki, A.; Shirai, K.; Tatsuno, I. Resveratrol Ameliorates Arterial Stiffness Assessed by Cardio-Ankle Vascular Index in Patients With Type 2 Diabetes Mellitus. Int. Heart J. 2017, 58, 577–583. [Google Scholar] [CrossRef] [Green Version]
- Mansournia, M.A.; Ostadmohammadi, V.; Doosti-Irani, A.; Ghayour-Mobarhan, M.; Ferns, G.; Akbari, H.; Ghaderi, A.; Talari, H.R.; Asemi, Z. The Effects of Vitamin D Supplementation on Biomarkers of Inflammation and Oxidative Stress in Diabetic Patients: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Horm. Metab. Res. 2018, 50, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Sattarinezhad, A.; Roozbeh, J.; Shirazi Yeganeh, B.; Omrani, G.R.; Shams, M. Resveratrol reduces albuminuria in diabetic nephropathy: A randomized double-blind placebo-controlled clinical trial. Diabetes Metab. 2019, 45, 53–59. [Google Scholar] [CrossRef]
- Seyyedebrahimi, S.; Khodabandehloo, H.; Nasli Esfahani, E.; Meshkani, R. The effects of resveratrol on markers of oxidative stress in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled clinical trial. Acta Diabetol. 2018, 55, 341–353. [Google Scholar] [CrossRef]
- Hoseini, A.; Namazi, G.; Farrokhian, A.; Reiner, Ž.; Aghadavod, E.; Bahmani, F.; Asemi, Z. The effects of resveratrol on metabolic status in patients with type 2 diabetes mellitus and coronary heart disease. Food Funct. 2019, 10, 6042–6051. [Google Scholar] [CrossRef]
- Mendoza-Núñez, V.M.; García-Martínez, B.I.; Rosado-Pérez, J.; Santiago-Osorio, E.; Pedraza-Chaverri, J.; Hernández-Abad, V.J. The Effect of 600 mg Alpha-lipoic Acid Supplementation on Oxidative Stress, Inflammation, and RAGE in Older Adults with Type 2 Diabetes Mellitus. Oxid. Med. Cell. Longev. 2019, 2019, 3276958. [Google Scholar] [CrossRef] [Green Version]
- Dalan, R.; Goh, L.L.; Lim, C.J.; Seneviratna, A.; Liew, H.; Seow, C.J.; Xia, L.; Chew, D.E.K.; Leow, M.K.S.; Boehm, B.O. Impact of Vitamin E supplementation on vascular function in haptoglobin genotype stratified diabetes patients (EVAS Trial): A randomised controlled trial. Nutr. Diabetes 2020, 10, 13. [Google Scholar] [CrossRef]
- Ishida, K.; Morimoto, S.; Horiuchi, S.; Kimura, M.; Ishikawa, T.; Kimura, S.; Yamashita, K.; Takano, N.; Seki, Y.; Bokuda, K.; et al. Comparison of the usefulness of the cardio-ankle vascular index and augmentation index as an index of arteriosclerosis in patients with essential hypertension. Hypertens Res. 2022, 45, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.M.; Cook, N.R.; Gaziano, J.M.; Gordon, D.; Ridker, P.M.; Manson, J.E.; Hennekens, C.H.; Buring, J.E. Vitamin E in the primary prevention of cardiovascular disease and cancer: The Women’s Health Study: A randomized controlled trial. JAMA 2005, 294, 56–65. [Google Scholar] [CrossRef] [PubMed]
- de Lorgeril, M.; Renaud, S.; Mamelle, N.; Salen, P.; Martin, J.L.; Monjaud, I.; Guidollet, J.; Touboul, P.; Delaye, J. Mediterranean alpha-linolenic acid-rich diet in secondary prevention of coronary heart disease. Lancet 1994, 343, 1454–1459. [Google Scholar] [CrossRef]
- Yusuf. Vitamin E Supplementation and Cardiovascular Events in High-Risk Patients. N. Engl. J. Med. 2000, 342, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.T.; de Groot, L.C.; Kromhout, D.; Perrin, A.E.; Moreiras-Varela, O.; Menotti, A.; van Staveren, W.A. Mediterranean diet, lifestyle factors, and 10-year mortality in elderly European men and women: The HALE project. JAMA 2004, 292, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Whelton, S.P.; He, J.; Whelton, P.K.; Muntner, P. Meta-Analysis of observational studies on fish intake and coronary heart disease. Am. J. Cardiol. 2004, 93, 1119–1123. [Google Scholar] [CrossRef]
- Cook, N.R.; Albert, C.M.; Gaziano, J.M.; Zaharris, E.; MacFadyen, J.; Danielson, E.; Buring, J.E.; Manson, J.E. A randomized factorial trial of vitamins C and E and beta carotene in the secondary prevention of cardiovascular events in women: Results from the Women’s Antioxidant Cardiovascular Study. Arch. Intern. Med. 2007, 167, 1610–1618. [Google Scholar] [CrossRef] [Green Version]
- Sesso, H.D.; Buring, J.E.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Glynn, R.J.; Gaziano, J.M. Vitamins E and C in the prevention of cardiovascular disease in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2008, 300, 2123–2133. [Google Scholar] [CrossRef] [Green Version]
- Myung, S.K.; Ju, W.; Cho, B.; Oh, S.W.; Park, S.M.; Koo, B.K.; Park, B.J. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: Systematic review and meta-analysis of randomised controlled trials. BMJ 2013, 346, f10. [Google Scholar] [CrossRef] [Green Version]
- Bowman, L.; Mafham, M.; Wallendszus, K.; Stevens, W.; Buck, G.; Barton, J.; Murphy, K.; Aung, T.; Haynes, R.; Cox, J.; et al. Effects of n-3 Fatty Acid Supplements in Diabetes Mellitus. N. Engl. J. Med. 2018, 379, 1540–1550. [Google Scholar] [CrossRef]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N. Engl. J. Med. 2018, 378, e34. [Google Scholar] [CrossRef]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N. Engl. J. Med. 2019, 380, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Lone, A.N.; Khan, M.S.; Virani, S.S.; Blumenthal, R.S.; Nasir, K.; Miller, M.; Michos, E.D.; Ballantyne, C.M.; Boden, W.E.; et al. Effect of omega-3 fatty acids on cardiovascular outcomes: A systematic review and meta-analysis. EClinicalMedicine 2021, 38, 100997. [Google Scholar] [CrossRef] [PubMed]
- Mohan, D.; Mente, A.; Dehghan, M.; Rangarajan, S.; O’Donnell, M.; Hu, W.; Dagenais, G.; Wielgosz, A.; Lear, S.; Wei, L.; et al. Associations of Fish Consumption With Risk of Cardiovascular Disease and Mortality Among Individuals With or Without Vascular Disease From 58 Countries. JAMA Intern. Med. 2021, 181, 631–649. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA 2007, 297, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Aranda, N.; Valls, R.M.; Romeu, M.; Sánchez-Martos, V.; Albaladejo, R.; Fernández-Castillejo, S.; Nogués, R.; Catalán, Ú.; Pedret, A.; Espinel, A.; et al. Consumption of seafood and its estimated heavy metals are associated with lipid profile and oxidative lipid damage on healthy adults from a Spanish Mediterranean area: A cross-sectional study. Environ. Res. 2017, 156, 644–651. [Google Scholar] [CrossRef]
- Hungate, B.A.; Van GROENIGEN, K.J.; Six, J.; Jastrow, J.D.; Luo, Y.; De GRAAFF, M.A.; van Kessel, C.; Osenberg, C.W. Assessing the effect of elevated carbon dioxide on soil carbon: A comparison of four meta-analyses. Glob. Chang. Biol. 2009, 15, 2020–2034. [Google Scholar] [CrossRef]
- Jayedi, A.; Shab-Bidar, S. Fish Consumption and the Risk of Chronic Disease: An Umbrella Review of Meta-Analyses of Prospective Cohort Studies. Adv. Nutr. 2020, 11, 1123–1133. [Google Scholar] [CrossRef]
- Lee, S.R.; An, E.J.; Kim, J.; Bae, Y.S. Function of NADPH Oxidases in Diabetic Nephropathy and Development of Nox Inhibitors. Biomol. Ther. 2020, 28, 25–33. [Google Scholar] [CrossRef]
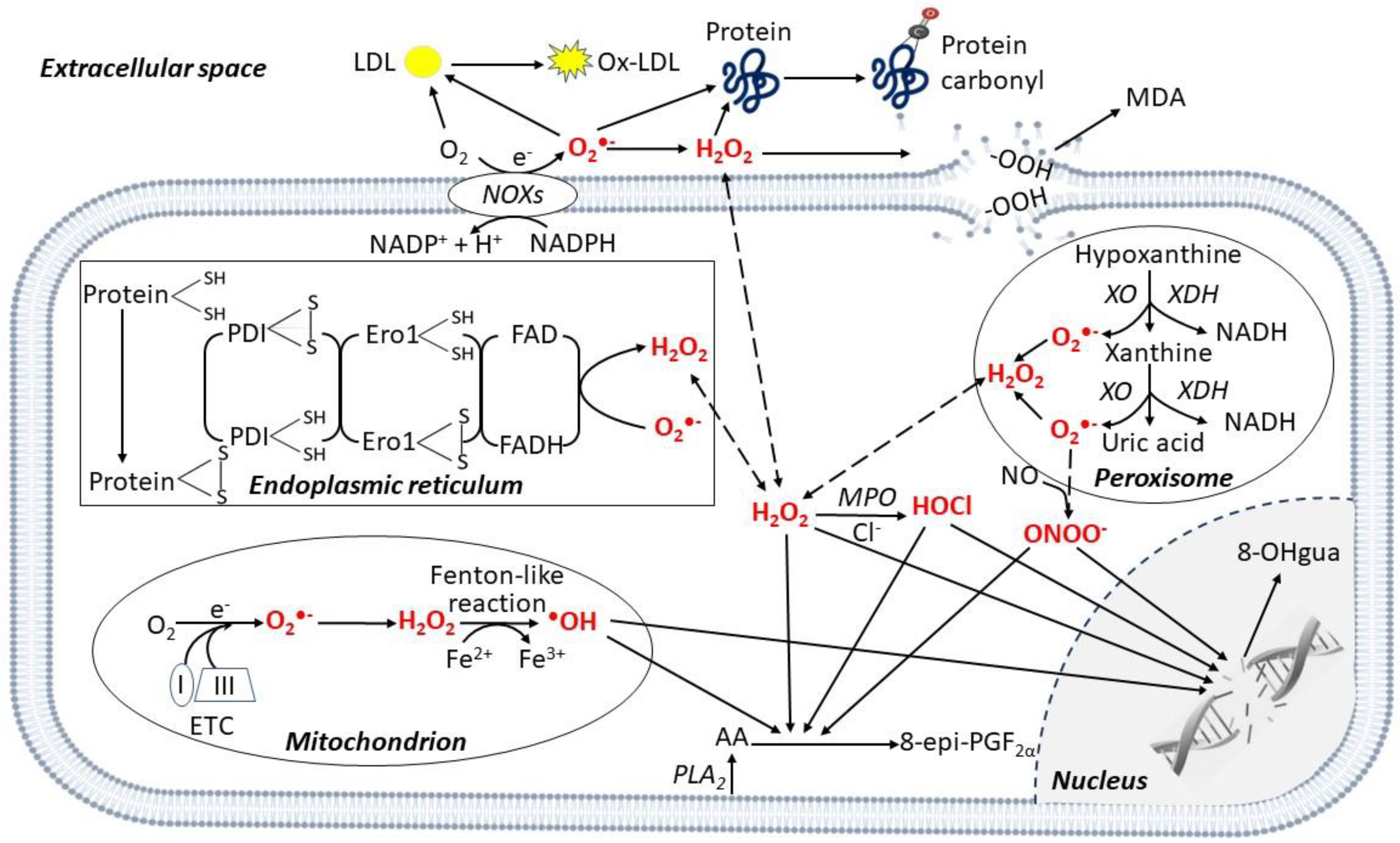
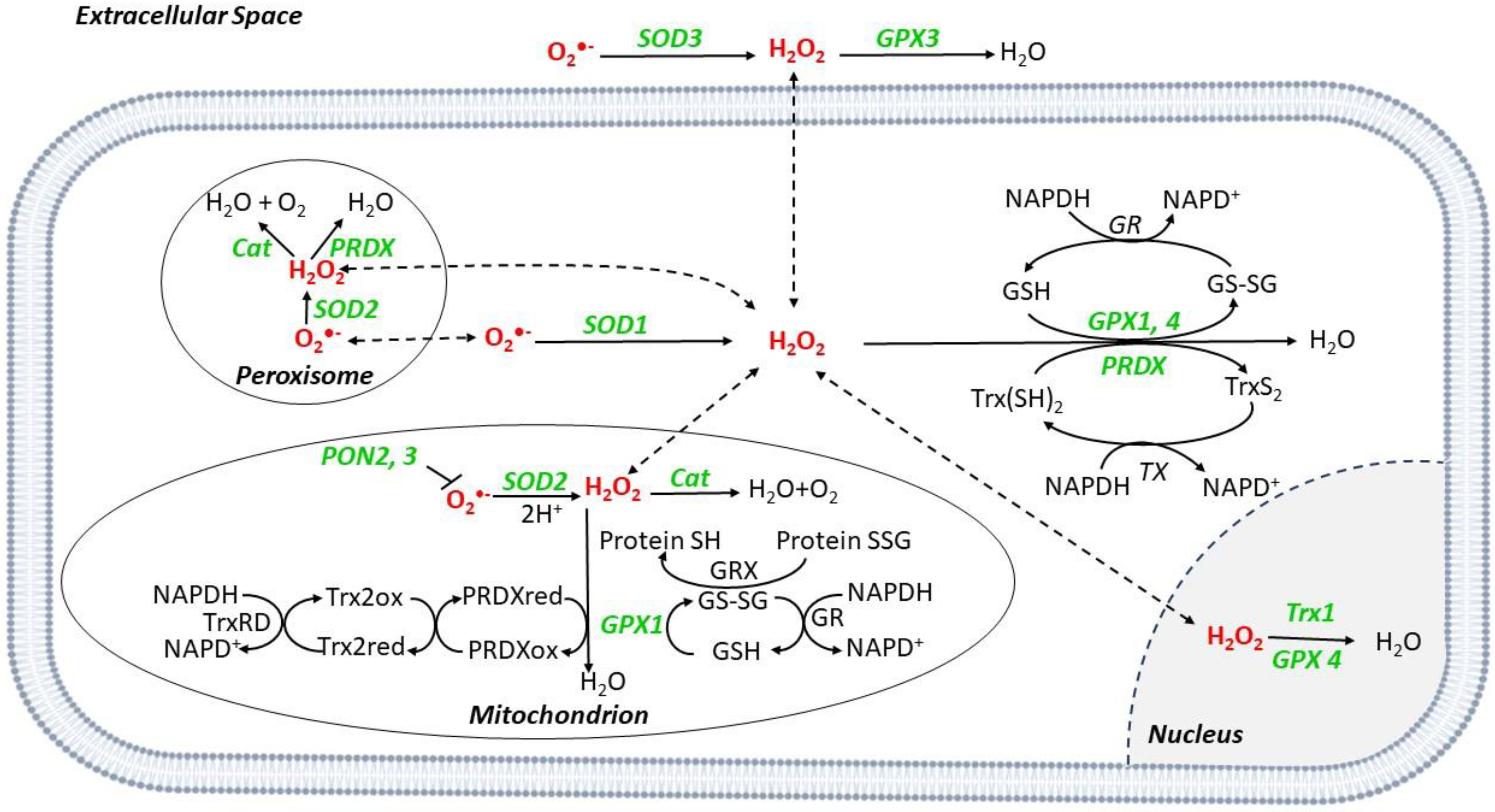
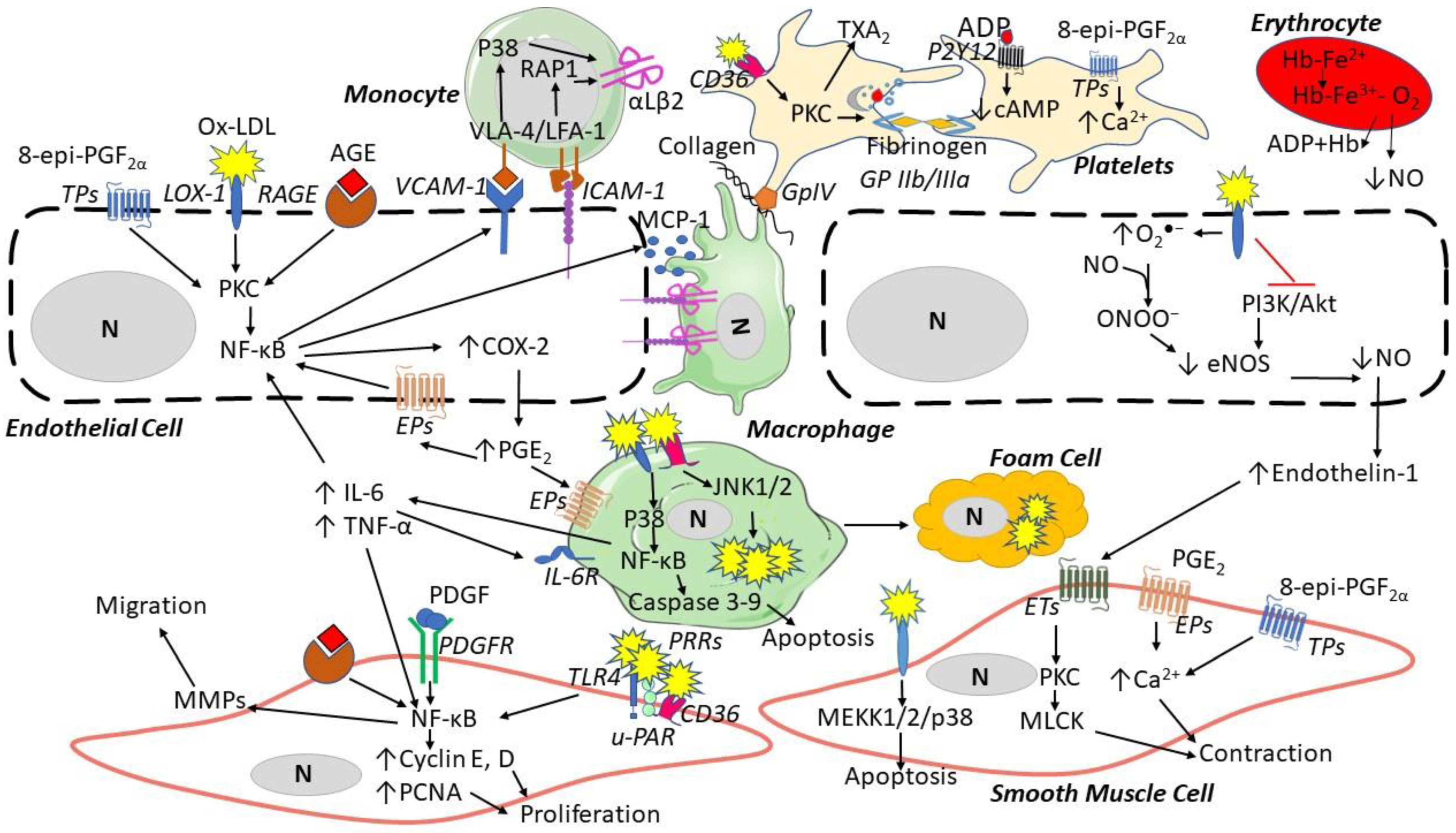
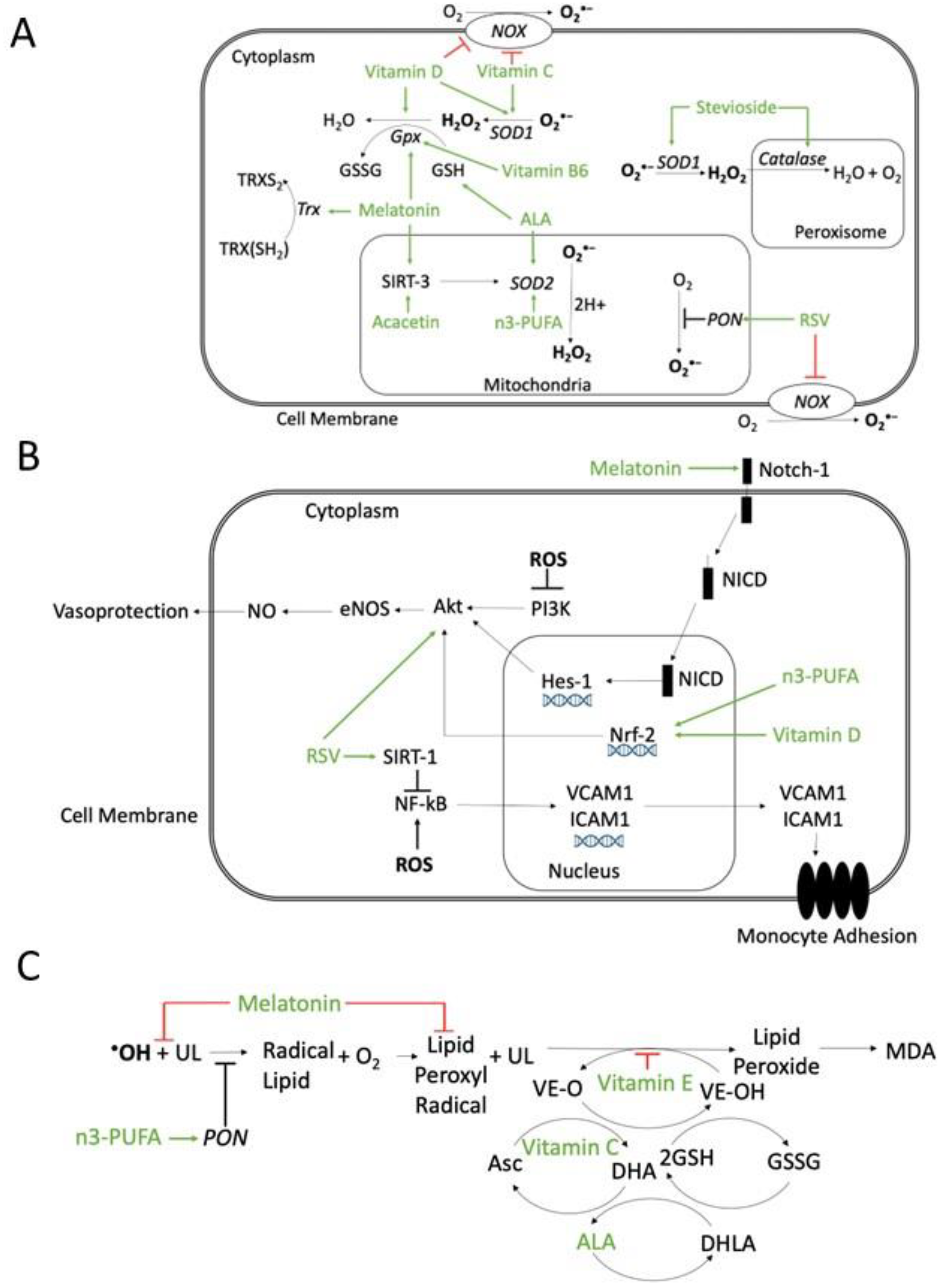
| Genetic Background and/or Experimental Setting | Phenotype |
|---|---|
| Animal Models | |
| ApoE−/− | ↑ NOXA-1, NOX2, and O2•− in the aortic atherosclerotic lesions, as assessed by DHE and L-012 vs. WT mice [10,11] |
| ApoE−/−/NOX1−/− | ↓ O2•− levels in the aorta, as assessed by L-012, macrophage infiltration and MDA in atherosclerotic lesions vs. ApoE−/− [12,13] |
| ApoE−/− on HFD and a NOX2 inhibitor | ↓ O2•− levels assessed by DHE and atherosclerotic lesion areas vs. ApoE−/− [14] |
| NOX2−/− with vascular wire-injury | ↓ O2•− from platelets and in the aorta (by DCF and DHE, respectively), ↓ macrophage infiltration, cellular proliferation, and platelet adhesion on injured aortas vs. WT [15,16] |
| ApoE−/−/NOX2−/− | ↓ O2•− as assessed by L-012, macrophage infiltration and number of lesions in the aorta ↑ NO in the aorta vs. ApoE−/− [11] |
| ApoE−/−/EC NOX2+/+ | ↑ O2•− levels, as assessed by L-012 and DHE, VCAM-1, and macrophage infiltration into early aortic lesions vs. ApoE−/− [17] |
| ApoE−/−/p47phox−/− | ↓ O2•− levels, as assessed by DHE, macrophage infiltration, and atherosclerotic lesion burden vs. ApoE−/− [18] |
| ApoE−/−/gp91phox−/− | ↓ O2•− levels, as assessed by DHE and atherosclerosis ↑ NO in the aorta vs. ApoE−/− [11] |
| NOX1−/−/NOX2−/−/NOX4−/− | ↓ O2•− from platelets, as assessed by EPR, platelet adhesion and aggregation in vitro vs. WT platelets [19] |
| ApoE−/−/LDLr−/− | ↑ NOX4 and O2•− in the aortic lesions vs. WT [20] |
| Rabbits on HFD with or withour XO inhibitor | ↓ O2•− levels in the aorta, assessed by L-012 ↑ endothelium-dependent relaxation in response to acetylcholine vs. HFD animals [21] |
| ApoE−/− on a XO inhibitor | ↓ O2•− as assessed by DHE, chemokine CK, IL-1α, IL-1β, and MCP-1 expression, and atherosclerotic lesions vs. ApoE−/− [22,23] |
| ApoE−/−/MPO−/− bone marrow | ↓ O2•− as assessed by DHE and atherosclerotic lesions, ↑ NO in the aorta vs. ApoE−/− [24] |
| LDLr−/− transplanted with MPO−/− bone marrow | ↑ Macrophage infiltration and atherosclerotic lesion area vs. LDLr−/−/MPO WT [25] |
| MicroRNA-210−/− | ↑ Mitochondrial ROS after I/R vs. WT [26] |
| Human studies | |
| NOX mRNA expression | ↑ NOX2 and NOX4 in coronary arteries from CAD patients vs. non-CAD [27,28] |
| Congenital NOX2 deficiency | ↓ Atherosclerosis, ox-LDL, and 8-epi-PGF2α vs. controls [29] ↓ O2•− as assessed by L-012 and 8-epi-PGF2α from platelets, ↑ NO upon collagen stimulation vs. controls [30] |
| Immunohistochemistry of NOX5 in carotid plaques | ↑ NOX5 vs. non-atherosclerotic sections [31] |
| Immunohistochemistry of MPO in arteries from transplanted hearts | ↑ MPO in the fibrous cap and lipid core vs. other lesion’s parts and normal arteries [32,33] |
| Genetic Background and/or Experimental Setting | Phenotype |
|---|---|
| Animal models | |
| ApoE−/−/Cat+/+ | ↓ Plasma, aortic 8-epi-PGF2α, size and progression of atherosclerotic lesions [60] VCAM-1, ICAM-1, BaP-induced monocyte adhesion to ECs vs. ApoE−/− [61] |
| Cat+/+ in SMCs | ↓ MMP1, TNFα, apoptosis in aortas vs. WT [62] |
| LDLr−/−/mCat+/+ | ↓ MCP-1, Phosphorylation of RelA (NF-κB), macrophage infiltration into the atherosclerotic lesions, [63] ↓ Neutrophil extracellular traps and myeloid-cell accumulation in the atherosclerotic lesions vs. LDLr−/− [64] |
| ApoE−/−/PRDX4+/+ | ↓ Ox-LDL levels in the plaques, CD3+ T cells, collagen in the fibrous caps, intimal lesions of the atherosclerotic aortic valves vs. ApoE−/− [65] |
| PRDX1−/− | ↑ Endothelial and soluble P-selectin, Von Willebrand factor vs. WT [66] |
| ApoE−/−/PRDX1−/− | ↑ Macrophage infiltration into the atherosclerotic lesions and atherosclerotic lesion size vs. ApoE−/− [66] |
| ApoE−/−/PRDX2−/− | ↑ Activation of p65, c-Jun, JNKs, p38 MPK, VCAM-1, ICAM-1, MCP-1, TNFα in the plaques vs. ApoE−/− [67] |
| ApoE−/−/GPX1+/+ | ↓ Aortic 8-epi-PGF2α, number and size of atherosclerotic lesions vs. ApoE−/− [68] |
| GPX1+/- | ↑ Plasma 8-epi-PGF2α, perivascular matrix deposition vs. WT [69] |
| ApoE−/−/GPX1−/− | ↑ Ox-LDL, macrophages infiltration, foam cells formation and proliferation, atherosclerotic lesions size, [70] VCAM-1, VEGF-1, p-63 activation, macrophages in aortas vs. ApoE−/− [71] |
| Trx2+/+ | ↑ Total antioxidants and NO, ↓ Plasma 8-epi-PGF2α in the atherosclerotic lesions vs. WT [72] |
| Trx2−/− | ↑ ONOO−, arterial hypertrophy, vascular stiffness, apoptosis, fibrosis, ↓ NO vs. WT [73,74] |
| ApoE−/−/SOD1+/+ | ↓ 8-epi-PGF2α in the plasma and aortas, size of atherosclerotic lesions vs. ApoE−/− [60] |
| SOD1−/− | ↑ O2•− in the aorta assessed by LCD and susceptibility to experimental thrombosis vs. WT [75] |
| ApoE−/−/SOD2+/- | ↑ 8-OHgua VCAM-1, Calpain-2, Caspase-3, MMP-2 in intimal VSMC, T-cell content and ↓ Collagen in the plaque vs. ApoE−/− [76] |
| PON1−/− | ↑ O2•− in the aorta as assessed by LCD, VCAM-1, ICAM-1, P-selectin vs. WT [77] |
| ApoE−/−/PON1+/+ | ↓ Ox-LDL and atherosclerotic lesion size vs. ApoE−/− [78] |
| LDL−/−/adenovirus-mediated PON1 gene transfer | ↓ Ox-LDL levels in plasma and plaques vs. LDL−/− [79] |
| ApoE−/−/PON2−/− | ↑ O2•− levels in the supernatants of aorta lysates, as assessed by DHE and atherosclerotic lesion size vs. ApoE−/− [80] |
| ApoE−/− injected with adenovirus PON2 (AdPON2) | ↓ Ox-LDL and serum lipid hydroperoxides vs. ApoE−/− [81] |
| Human studies | |
| Congenital Cat deficiency | ↓ Cat levels, ↑ H2O2, atherosclerosis, and DM vs. subjects without acatalasemia [82,83,84] |
| 599C/T allele of the GPX1 gene | ↓ GPX activity, ox-LDL and ↑ MDA and risk of restenosis vs. non-carriers 599C/T allele [85] |
| Upregulation GPX1 in ECs in vitro | ↓ CD40 protein, MCP-1 and VCAM-1 [86] |
| M/L54 PON1 polymorphisms | ↓ Serum PON1 activity and ↑ CHD in carriers M/L54 PON1 DM patients vs. non-carrier DM patients [87] |
| M/L55 and Q/R 192 PON1 polymorphism | ↓ Serum PON1 activity and ↑ CAD, carotid thickening and plaques in M/L54 and Q/R 192 PON1 carriers vs. non-carriers [88,89] |
| Immunofluorescence in carotid lesions | ↓ PON2 expression in atherosclerotic lesions vs. healthy tissues [81] |
| Immunohistochemistry in coronary arteries | ↑ Trx expression in VSMCs and macrophages of atherosclerotic vs. healthy coronary arteries [90] |
| Proteomics in aortic aneurysm tissues | ↑ PRDX2 expression in patients with ruptured vs. non-ruptured aneurysms [91] |
| SOD3 R213G polymorphism | ↓ SOD3 activity and ↑ ischemic heart diseases [92] |
| T-allele of rs2284659 variant of SOD3 promoter | ↑ SOD3 plasma levels and ↓ Circulating 8-epi-PGF2α, oxidation protein products, MI, in rs2284659 carriers DM patients vs. non-carrier DM patients [93] |
| Study (Year) | Study Population | Design of the Study | Main Results |
|---|---|---|---|
| 8-epi-PGF2α | |||
| Davi et al. (1997) [125] | Hypercholesterolemic patients (n = 40) vs. matched controls (n = 40) | Cross-sectional study | 8-epi-PGF2α: 473 ± 305 vs. 205 ± 95 pg/mg creatinine; p = 0.0001 in hypercholesterolemic patients vs. controls 8-epi-PGF2α correlated with 11-dehydro-TXB2 in hypercholesterolemic patients, rho = 0.512; p = 0.0001 |
| Davi et al. (2002) [124] | Healthy obese women (n = 44) vs. non obese matched controls (n = 24) | Cross-sectional study | 8-epi-PGF2α: 523 (293–685) vs. 187 (140–225) pg/mg creatinine; p < 0.001 in obese women vs. controls 8-epi-PGF2α correlated with 11-dehydro-TXB2 in obese women, rho = 0.61; p < 0.001 |
| Keaney et al. (2003) [131] | Adult subjects (n = 2828) | Cohort study | 8-epi-PGF2α: 240 ± 145 vs. 148 ± 100 ng/mmol creatinine; p < 0.0001 in smokers vs. non-smokers 8-epi-PGF2α: 181 ± 128 vs. 157 ± 108 ng/mmol creatinine; p < 0.0001 in DM vs. non-DM subjects 8-epi-PGF2α independently significantly correlated with smoking, BMI, and history of CVD. |
| Schwedhelm et al. (2004) [132] | CAD patients (n = 93) vs. matched controls (n = 93) | Case-control study | 8-epi-PGF2α: 139 (93–231) vs. 77 (61–101) pmol/mmol creatinine; p < 0.001 in CAD vs. controls 8-epi-PGF2α correlated with 2,3-dinor-5,6-dihydro-8-iso-PGF2α, and CRP in CAD patients, rho = 0.225, p < 0.01, and rho = 0.321, p < 0.001, respectively 8-epi-PGF2α correlated with DM, hypertension, smoking, hyperlipidemia, and BMI for all subjects; p < 0.001 for trend |
| Roest et al. (2008) [133] | Postmenopausal women (n = 12,239) including women who died of CHD (n = 141) and stroke (n = 109) vs. controls (n = 142) | Nested prospective case-cohort study Follow-up: 18 years | 8-epi-PGF2α: 0.31 (0.23–0.46) vs. 0.23 (0.18–0.31) ng/mg creatinine; in smokers (n = 128) vs. non-smokers (n = 264), p < 0.001 CVD mortality risk higher for the highest of 8-iso PGF2α vs. the lowest quartile, OR: 1.8 (95% CI; 1.1–3.1; p = 0.02) |
| Pascale et al. (2012) [129] | Patients with ET (n = 38) | Cross-sectional study. | 8-epi-PGF2α correlated with 11-dehydro-TXB2, rho = 0.55, p = 0.008 |
| Zaccardi et al. (2016) [126] | T1DM patients (n = 51) vs. matched healthy controls (n = 63) | Cross-sectional study | 8-epi-PGF2α: 796 ± 218 vs. 468 ± 235 pg/mg creatinine; p < 0.001 in T1DM patients vs. controls 8-epi-PGF2α correlated with 11-dehydro-TXB2 in T1DM patients, rho = 0.75; p < 0.001 |
| Petrucci et al. (2019) [123] | Healthy obese subjects (n = 19) vs. matched controls (n = 19) | Cross-sectional study | 8-epi-PGF2α: 826 (129–549) vs. 555 (425–693) pg/mg creatinine; p = 0.03 in obese subjects vs. controls 8-epi-PGF2α correlated with 11-dehydro-TXB2 in obese subjects, rho = 0.55; p = 0.02 |
| Santilli et al. (2020) [128] | Subjects with IGT (n = 48), T2DM patients since <1 year (n = 60), and T2DM patients since >1 year (n = 58) | Cross-sectional study | 8-epi-PGF2α: 594 (411–876) vs. 618 (402–1060) vs. 466 (371–716) pg/mg creatinine; p = 0.0138 in IGT subjects vs. new DM vs. established DM 8-epi-PGF2α correlated with 11-dehydro-TXB2 in IGT and DM |
| MDA | |||
| Noberasco et al. (1991) [134] | DM patients (n = 67) vs. matched healthy controls (n = 40) | Cross-sectional study | MDA: 3.69 ± 0.28 vs. 1.92 ± 0.13 nmol/mL; z = 4.48, α < 0.01 in DM patients vs. controls MDA is correlated with glycosylated hemoglobin in DM patients (rho = 0.29, α < 0.05) |
| Cavalca et al. (2001) [135] | CAD patients (n = 40) vs. matched healthy controls (n = 70) | Cross-sectional study | Total MDA: 2.6 (3.8–1.7) vs. 1.3 (2.2–0.9) µmol/L; p < 0.00001 in CAD patients vs. controls Free MDA: 0.5 (1.3–0.2) vs. 0.3 (0.7–0.05) µmol/L; p < 0.03 in unstable vs. stable angina group |
| Walter et al. (2004) [136] | CAD patients (n = 643) | Prospective cohort study Follow-up: 2 years | CAD patients in the highest vs. lowest quartile of MDA: MI (n = 51) RR: 2.94 (95% CI 1.75–4.94; p < 0.0001) Angina (n = 149) RR: 2.58 (95% CI 1.98–3.37; p < 0.0001) CABG/PTCA (n = 139) RR: 2.14 (95% CI 1.61–2.84; p < 0.0001) |
| Tanriverdi et al. (2006) [137] | Smokers (n = 36) vs. matched non-smokers controls (n = 51) | Cross-sectional study | MDA: 1.91 ± 1.3 vs. 1.18 ± 0.9 nmol/mL; p = 0.003 in smokers vs. controls SOD: 4267.7 ± 2842.8 vs. 2812 ± 665.4 U/gHb; p = 0.008 in smokers vs. controls GSH: 7.1 ± 1.8 vs. 8.5 ± 3.6 μmol/gHb; p = 0.019 in smokers vs. controls |
| Kotur-Stevuljevic et al. (2007) [138] | CAD (n = 141) vs. non-CAD controls (n = 47) | Cross-sectional study | MDA: 3.22 (1.336–7.762) vs. 2.66 (1.021–6.902) μmol/L; p < 0.001 in CAD patients vs. controls MDA in CAD patients independently correlated with fibrinogen and CRP: β = 0.262; p < 0.01and β = 0.331; p < 0.001, respectively |
| Kubihal et al. (2019) [139] | Healthy smokers (n = 75) vs. matched non-smokers controls (n = 25) | Cross-sectional study | MDA: 5.15 ± 0.39 vs. 4.11 ± 0.55 nmol/mL; p < 0.0001 in smokers vs. controls Vitamin C: 10.35 ± 1.44 vs. 13.9 ± 1.45 mg/L; p < 0.0001 in smokers vs. controls |
| Ox-LDL | |||
| Ehara et al. (2001) [140] | Patients with acute MI (n = 45) vs. matched healthy controls (n = 46) | Cross-sectional study | Ox-LDL: 1.95 ± 1.42 vs. 0.58 ± 0.23 ng/5µg LDL; p < 0.0001 in patients with MI vs. controls |
| Shimada et al. (2004) [141] | CAD patients (n = 238) with (n = 162) vs. without cardiac events controls (n = 76) | Prospective cohort study Follow-up: over 4 years | Ox-LDL: 20.3 (17.5–30) vs. 17.6 (13.2–24.7) U/mL; p = 0.002 in patients with events vs. controls Cardiac event risk in patients in the highest vs. lowest quartile of ox-LDL, HR: 3.15 (95% CI 1.47–6.76; p = 0.003) |
| Tsimikas et al. (2006) [142] | Men and women aged 40-80 years (n = 826) | Prospective study Follow-up: 5 years | Ox-LDL circulating levels associated with the incidence and progression of carotid atherosclerosis, β = 0.17; p = 0.001, OR: 1.44 (95% CI 1.06–1.96; p = 0.02) and femoral atherosclerosis, β = 0.16; p = 0.003, RR: 1.34 (95% CI 1.05–1.71; p = 0.018) |
| Zhang et al. (2014) [143] | ACS patients (n = 425) | Prospective cohort study Median follow-up: 30 months | Ox-LDL: 283.22 ± 38.93 vs. 198.62 ± 56.42 mmol/L; p < 0.01 in event vs. event free patients hsCRP: 20.75 ± 5.37 vs. 14.22 ± 4.18 mg/L; p < 0.01 in patients with or without events Ox-LDL and hsCRP correlated rho = 0.67, p < 0.01 |
| Gao et al. (2017) [144] | Adults with vs. without CVD (n = 8644) | Meta-analysis of 12 observational studies | Summary effect size of increased circulating ox-LDL was 1.79 (95% CI 1.56–2.05) for ASCVD. There was no statistical heterogeneity observed across studies (Q = 15.22; p = 0.230; I2 = 21.2%) |
| Nitrotyrosine | |||
| Ceriello et al. (2001) [145] | T2DM patients (n = 40) vs. matched healthy controls (n = 35) | Cross-sectional study | Nitrotyrosine: 0.251 ± 0.141 µmol/L vs. <10 nmol/L in T2DM patients vs. healthy controls Nitrotyrosine correlated with plasma glucose concentration in T2DM patients, rho = 0.38; p < 0.02 |
| Shishehbor et al. (2003) [146] | Patients with CAD (n = 100) PAD (n = 36) vs. non-CAD controls (n = 108) | Cross-sectional study | Nitrotyrosine: 9.1 (4.8–13.8) vs. 5.2 (2.2–8.4) μmol/mol tyrosine; p < 0.001 in CAD patients vs. controls; 9.6 vs. 5.7 μmol/mol tyrosine; p = 0.001 in CAD patients with DM vs. non-DM patients. CAD risk in the upper vs. lower quartile in CAD patients without PAD, OR: 4.4 (95% CI 1.8–10.6; p < 0.001) CAD in the upper vs. lower quartile in CAD patients with PAD, OR: 26.3 (95% CI 2.9–238; p < 0.001) Atherosclerosis prevalence: 46% vs. 3%; p < 0.001 in CAD plus PAD patients in the highest quartile of nitrotyrosine vs. lowest quartile |
| Protein carbonyl | |||
| Kilhovd et al. (1999) [147] | T2DM patients (n = 53, vs. matched non-DM subjects (n = 34) | Cross-sectional study | AGEs: 7.4 (4.4–10.9) vs. 4.2 (1.6–6.4) U/mL; p < 0.0001 in T2DM patients vs. controls; 8.1 [6,4,5,6,7,8,9,10,9] vs. 7.1 (3.5–9.8) U/mL, p = 0.03 in T2DM with CHD vs. without CHD AGEs associated with CHD in T2DM patients, OR: 2.4 (95% CI 1.2–4.8; p = 0.008) |
| De Cristofaro et al. (2003) [148] | T2DM patients (n = 72) vs. matched healthy controls (n = 72) | Cross-sectional study | Protein carbonyls: 6.1 ± 1.4 vs. 4.6 ± 1 × 10−6 w/w; p < 0.05 in T2DM patients vs. controls Protein carbonyls correlated with 8-epi-PGF2α in T2DM patients, rho = 0.242; p = 0.039 |
| Mutlu-Türkoglu et al. (2005) [149] | CAD patients (n = 30) vs. matched healthy controls (n = 30) | Cross-sectional study | Protein carbonyls: 1.1 ± 0.05 vs. 0.9 ± 0.02 nmol/mg protein, p < 0.01 in CAD patients vs. controls |
| Semba et al. (2009) [150] | Dwelling women, aged ≥65 years (n = 559) | Prospective study Follow-up: 4.5 years | CVD mortality in dwelling women (n = 54), CVD in subjects in the highest quartile of AGEs: HR 2.29 (95% CI, 1.21–4.34; p = 0.01) |
| Pirinccioglu et al. (2010) [151] | Hypercholesteraemic patients (n = 25) vs. matched healthy controls (n = 25) | Cross-sectional study | Protein carbonyls: 2.12 ± 0.26 vs. 1.52 ± 0.28 nmol/mg protein; p < 0.001 in hypercholesteraemic patients vs. controls Protein carbonyls are correlated with MDA and IMT in hypercholesterolemic patients, rho = 0.77; p < 0.001, and rho = 0.82; p < 0.001, respectively |
| Vegi et.al (2012) [152] | T2DM patients (n = 60) vs. matched healthy controls (n = 60) | Cross-sectional study | Protein carbonyls: 1.68 ± 0.47 vs. 0.7 ± 0.34 nmol/L; p < 0.001 in T2DM patients vs. controls |
| Van Eupen et al. (2013) ([153] | T1DM patients (n = 165) vs. matched non-DM controls (n = 169) | Cross-sectional study | Plasma levels in protein- bound Nε-(carboxymethyl) lysine: 105 (102–107) vs. 93 (90–95) nmol/mmol LYS; p < 0.001 in T1DM patients vs. controls Plasma levels in protein-bound Pentosidine: 0.69 (0.65-0.73) vs. 0.51 (0.48-0.54) nmol/mmol LYS; p < 0.001 in T1DM patients vs. controls Plasma levels in protein-bound Pentosidine: 0.81 [0.70–0.93] vs. 0.67 (0.63–0.71) nmol/mmol LYS; p = 0.028 in T1DM patients with moderate to high CAC vs. low CAC score |
| McNair et al. (2016) [154] | Hypercholesterolemic ACS patients (n = 55) vs. matched normocholesterolemic ACS controls (n = 45) | Cross-sectional study | AGEs: 1213 ± 68.6 vs. 642 ± 22 ng/mL, p = 0.001 in hypercholesterolemic patients vs. controls AGE/sRAGE ratio: 1.71 ± 0.16 vs. 0.49 ± 0.02; p < 0.001 in hypercholesterolemic patients vs. controls AGEs are correlated with total cholesterol, LDL-C, and triglycerides, rho = 0.664, rho = 0.66, and rho = 0.741; p < 0.001, respectively |
| Kopytek et al. (2020) [155] | T2DM patients with atherosclerosis (n = 50) vs. matched non-DM with atherosclerosis controls (n = 76) | Cross-sectional study | AGEs: 9.55 (8.56–10.92) vs. 0.73 (0.68–0.77) ng/mL; p < 0.0001 in T2DM patients with atherosclerosis vs. non-DM with atherosclerosis Valvular AGEs in all DM patients are associated with AVA rho = 0.68; p < 0.0001 |
| Sharifi-Zahabi et al. (2021) [156] | Adults with and without DM and CVD (n = 3718) | Systematic review and meta-analysis of Prospective Observational Studies | AGEs associated with increased risk of the following: all-cause mortality (pooled effect measure: 1.05; 95% CI: 1.01, 1.09; p = 0.018), and CVD mortality (pooled effect measure: 1.08; 95% CI: 1.01, 1.14; p = 0.015) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrucci, G.; Rizzi, A.; Hatem, D.; Tosti, G.; Rocca, B.; Pitocco, D. Role of Oxidative Stress in the Pathogenesis of Atherothrombotic Diseases. Antioxidants 2022, 11, 1408. https://doi.org/10.3390/antiox11071408
Petrucci G, Rizzi A, Hatem D, Tosti G, Rocca B, Pitocco D. Role of Oxidative Stress in the Pathogenesis of Atherothrombotic Diseases. Antioxidants. 2022; 11(7):1408. https://doi.org/10.3390/antiox11071408
Chicago/Turabian StylePetrucci, Giovanna, Alessandro Rizzi, Duaa Hatem, Giulia Tosti, Bianca Rocca, and Dario Pitocco. 2022. "Role of Oxidative Stress in the Pathogenesis of Atherothrombotic Diseases" Antioxidants 11, no. 7: 1408. https://doi.org/10.3390/antiox11071408
APA StylePetrucci, G., Rizzi, A., Hatem, D., Tosti, G., Rocca, B., & Pitocco, D. (2022). Role of Oxidative Stress in the Pathogenesis of Atherothrombotic Diseases. Antioxidants, 11(7), 1408. https://doi.org/10.3390/antiox11071408