
Nature-Inspired Hybrids (NIH) Improve Proteostasis by Activating Nrf2-Mediated Protective Pathways in Retinal Pigment Epithelial Cells

 ,
,  , , ,
, , ,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
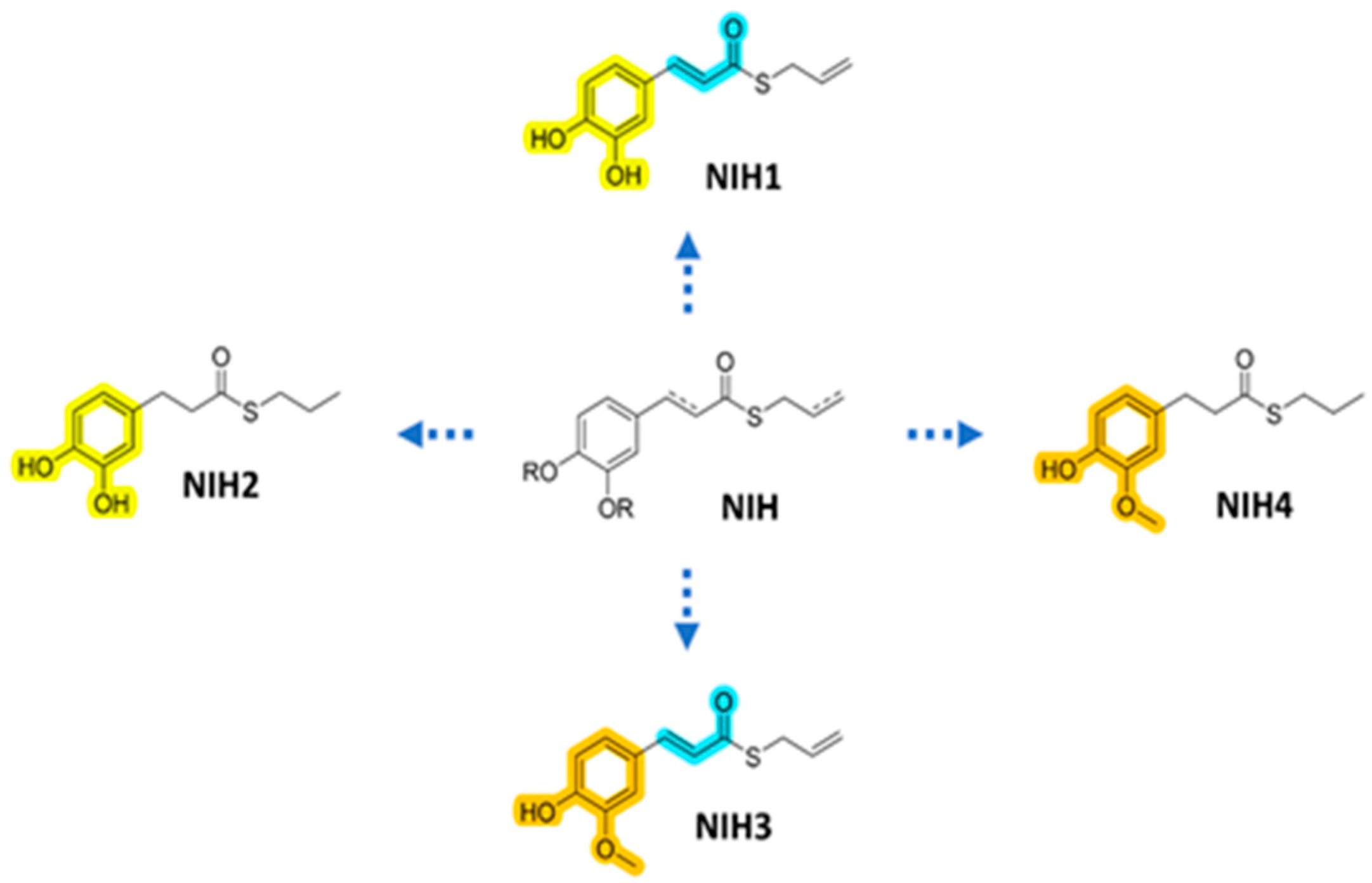
2.1. NIH Syntheses and Characterization
2.2. Cell Culture and Treatments
2.3. Protein Extraction and Western Blotting
2.4. RNA Extraction, Retrotranscription and Real-Time Quantitative PCR
2.5. Cell Viability Assay
2.6. ELISA
2.7. Statistical Analyses
3. Results
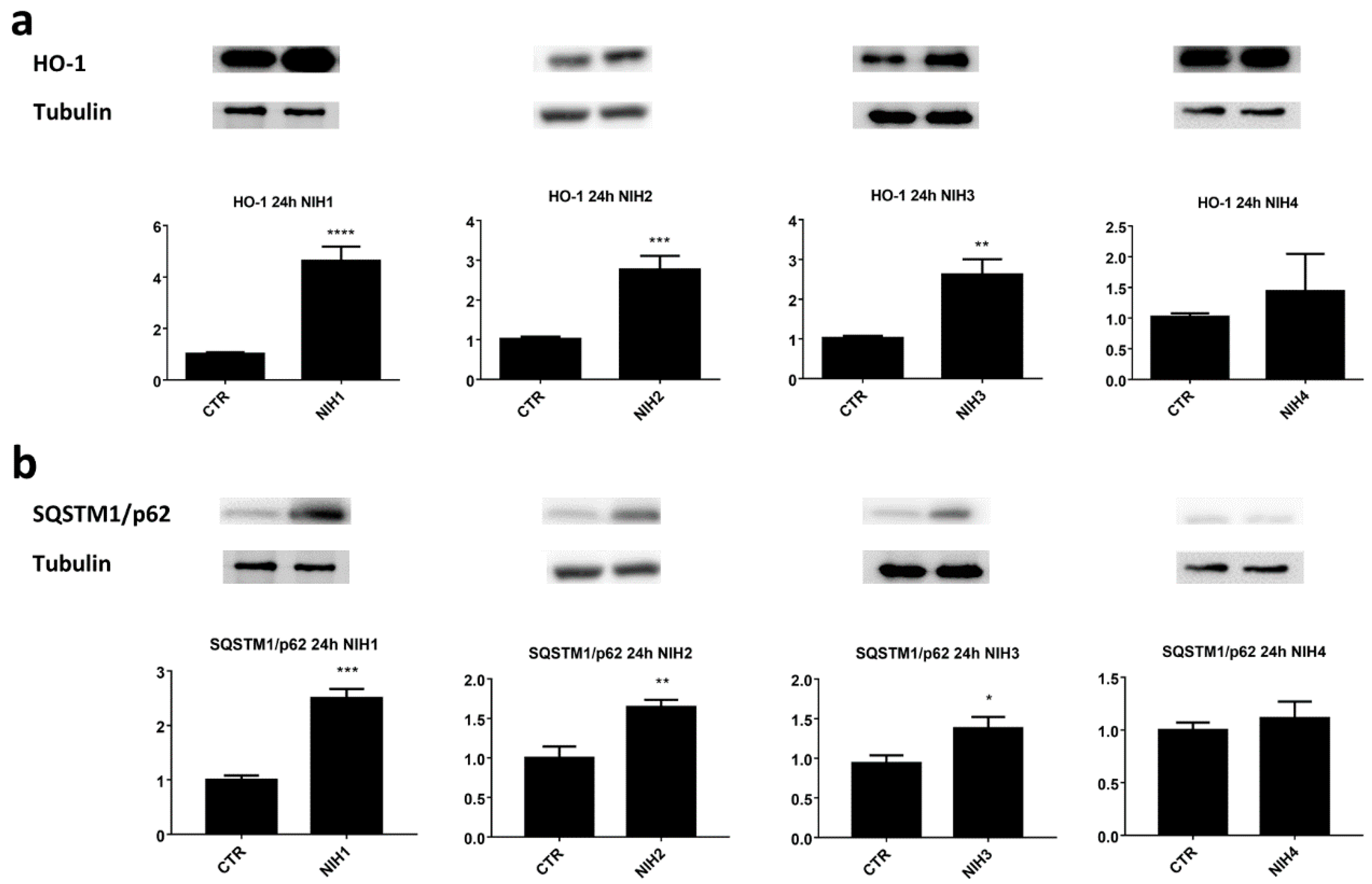
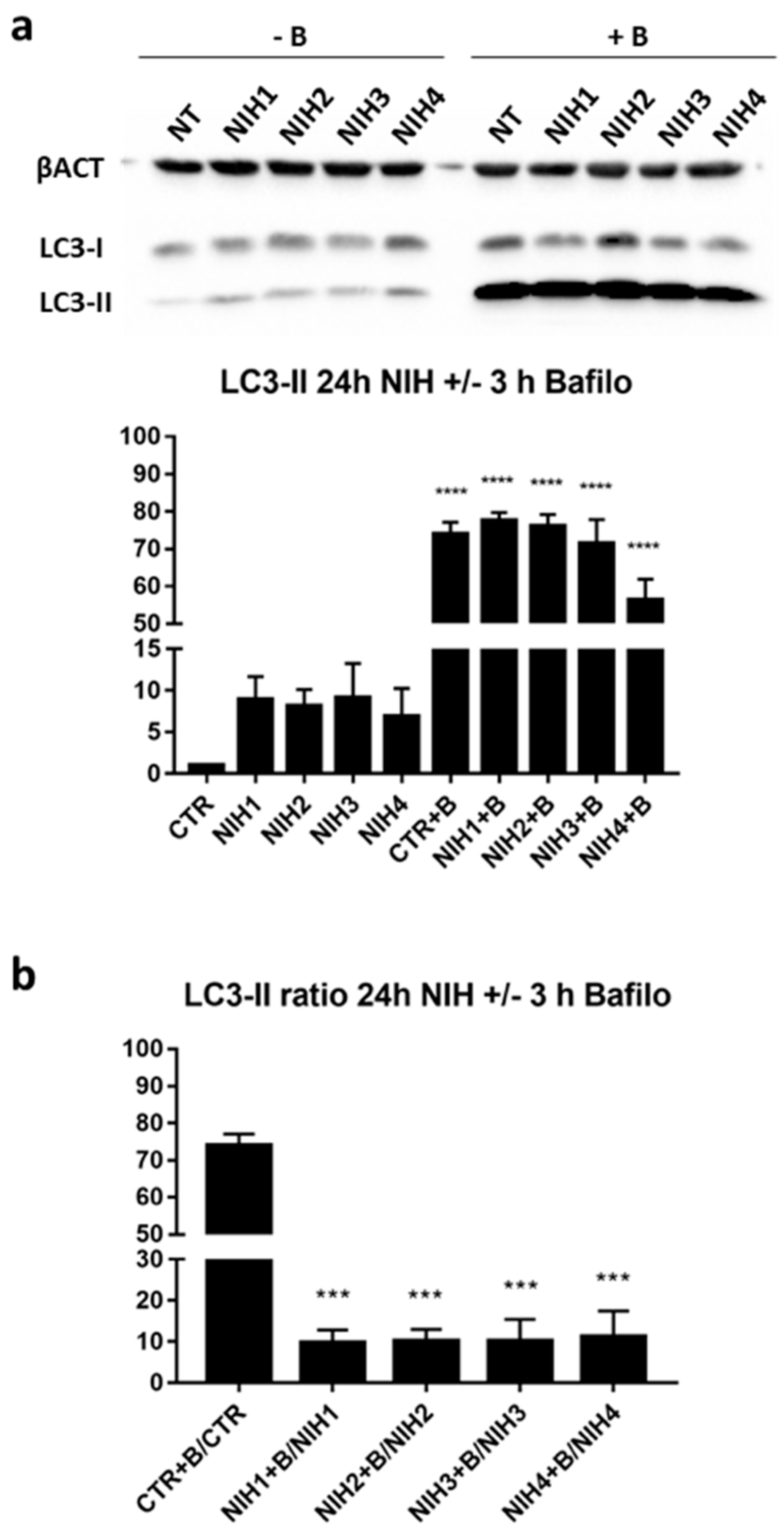
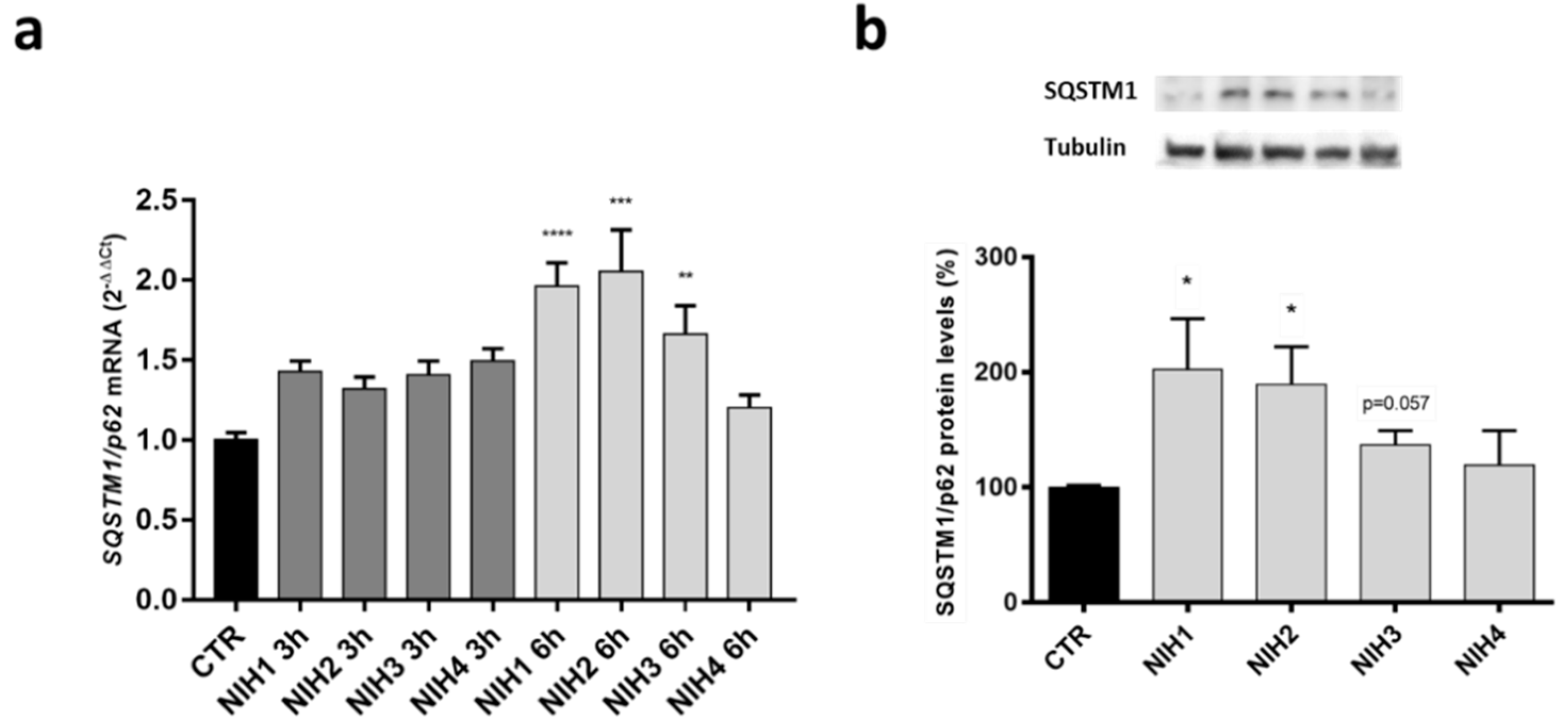
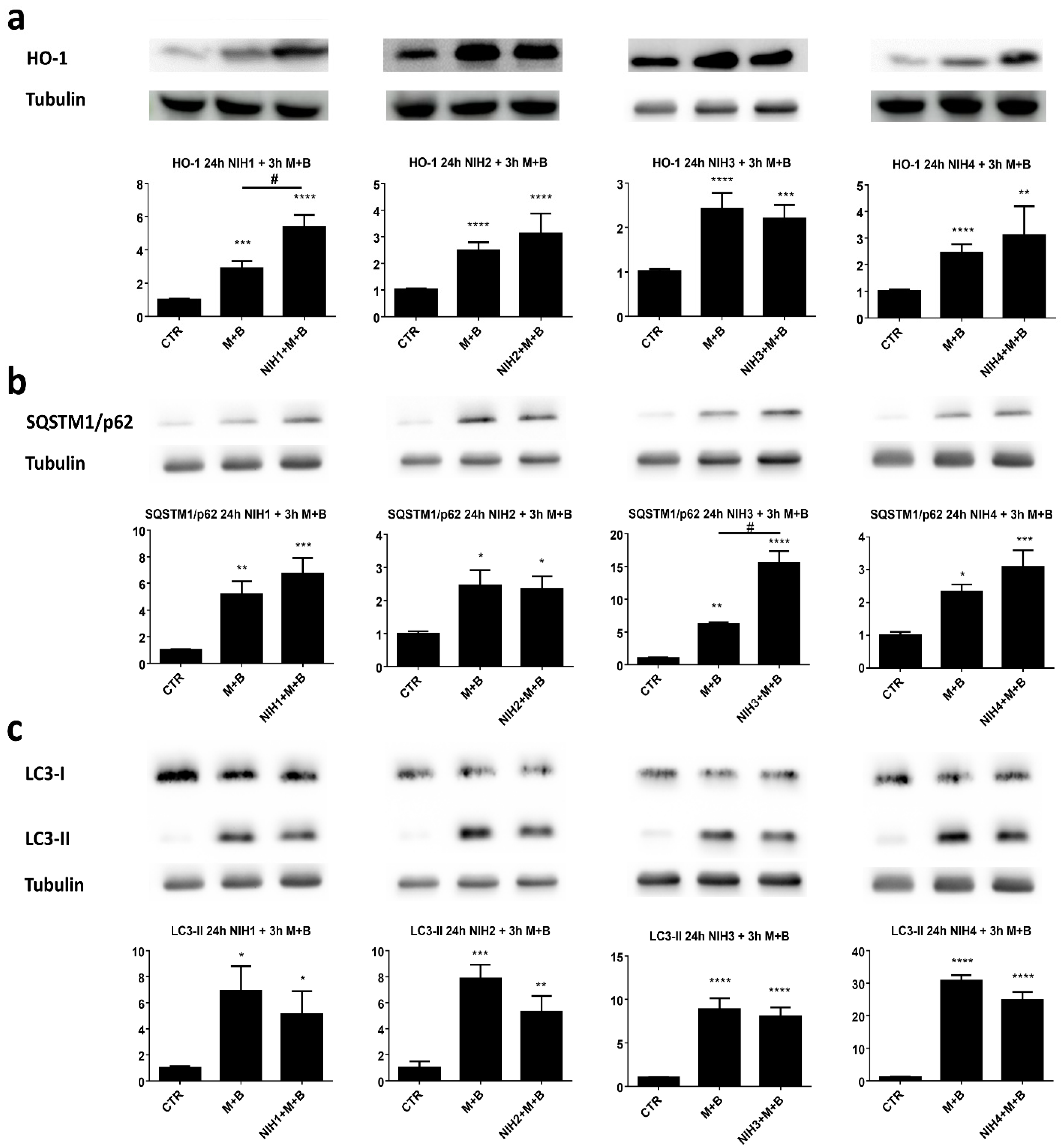
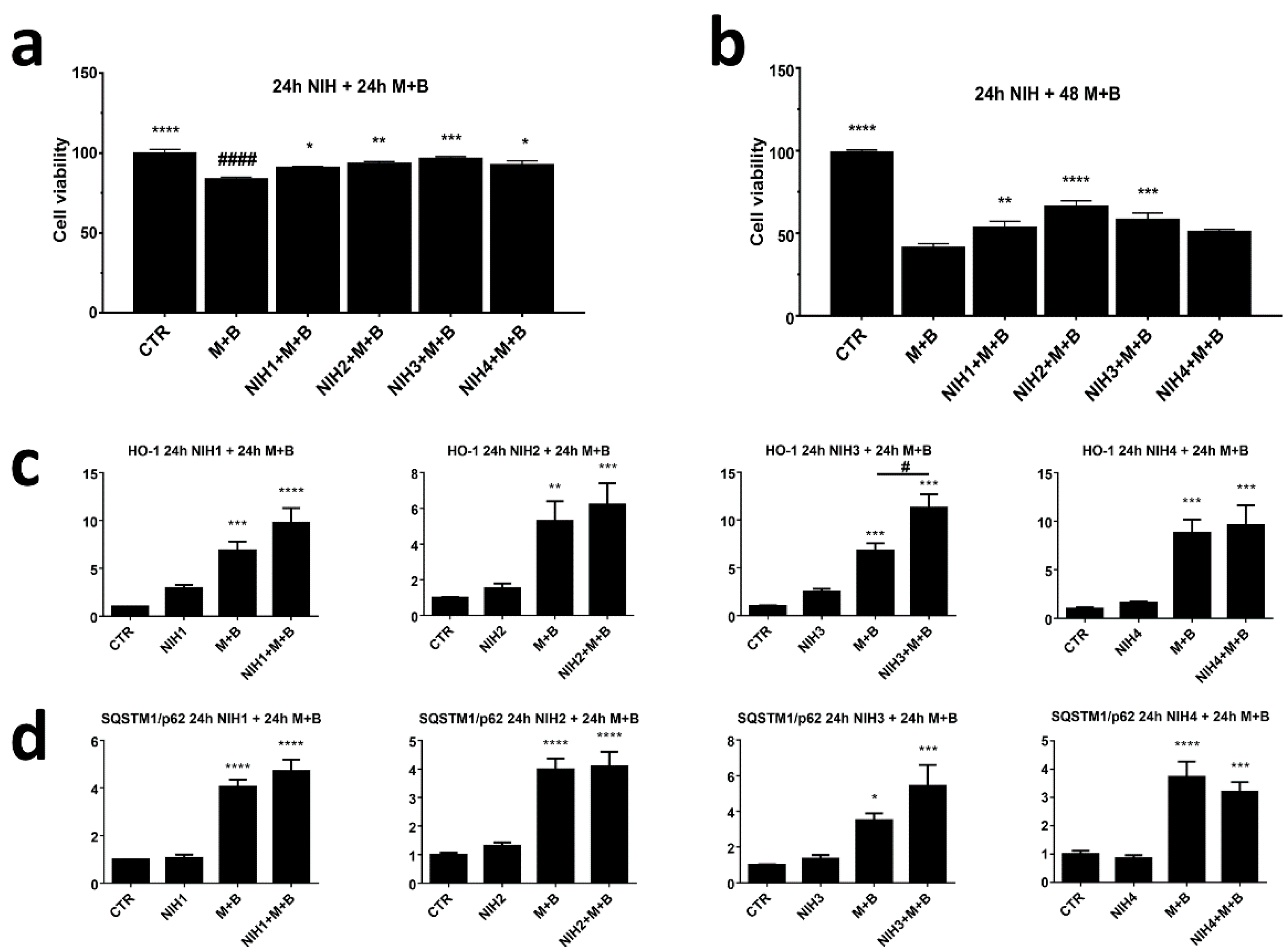
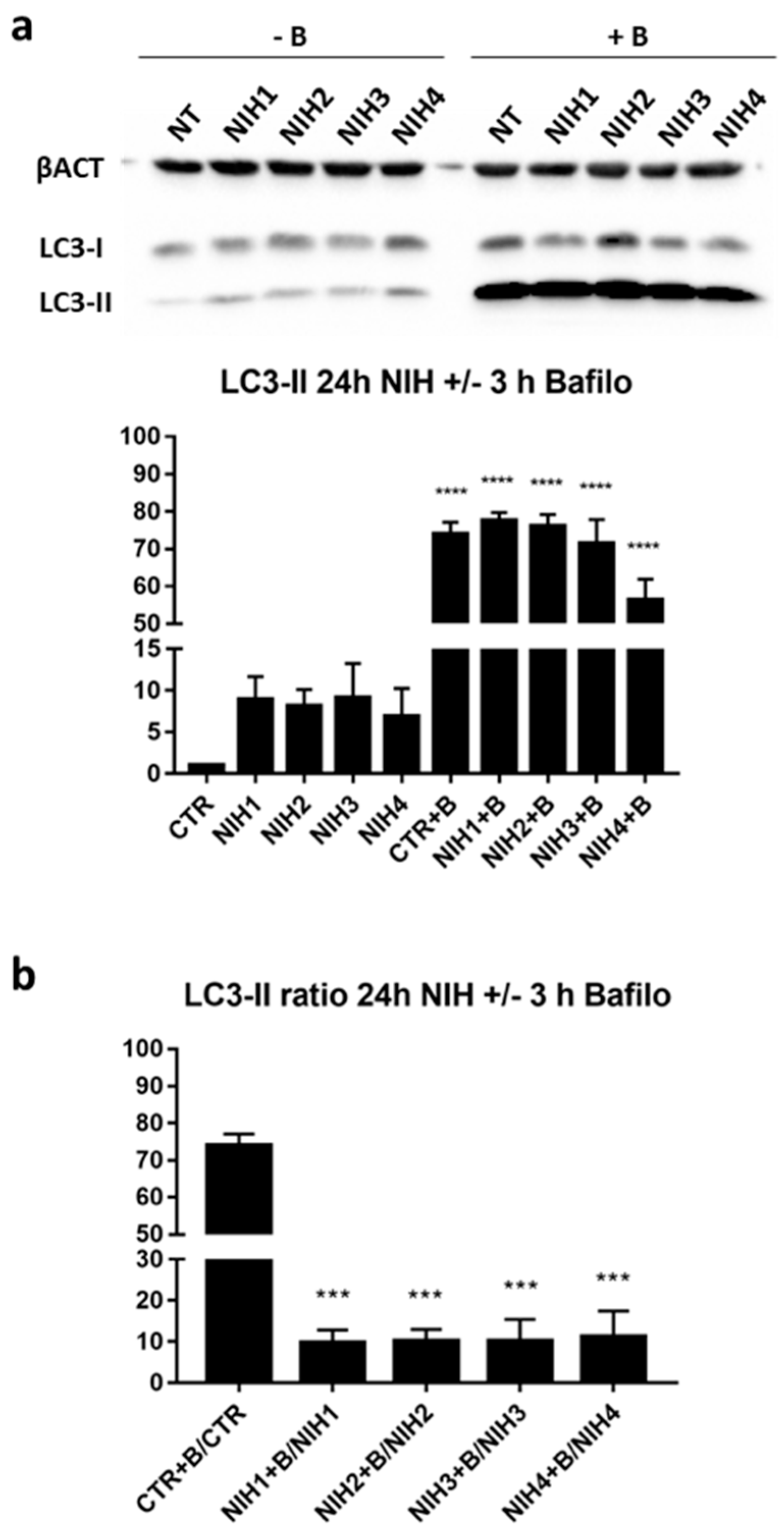
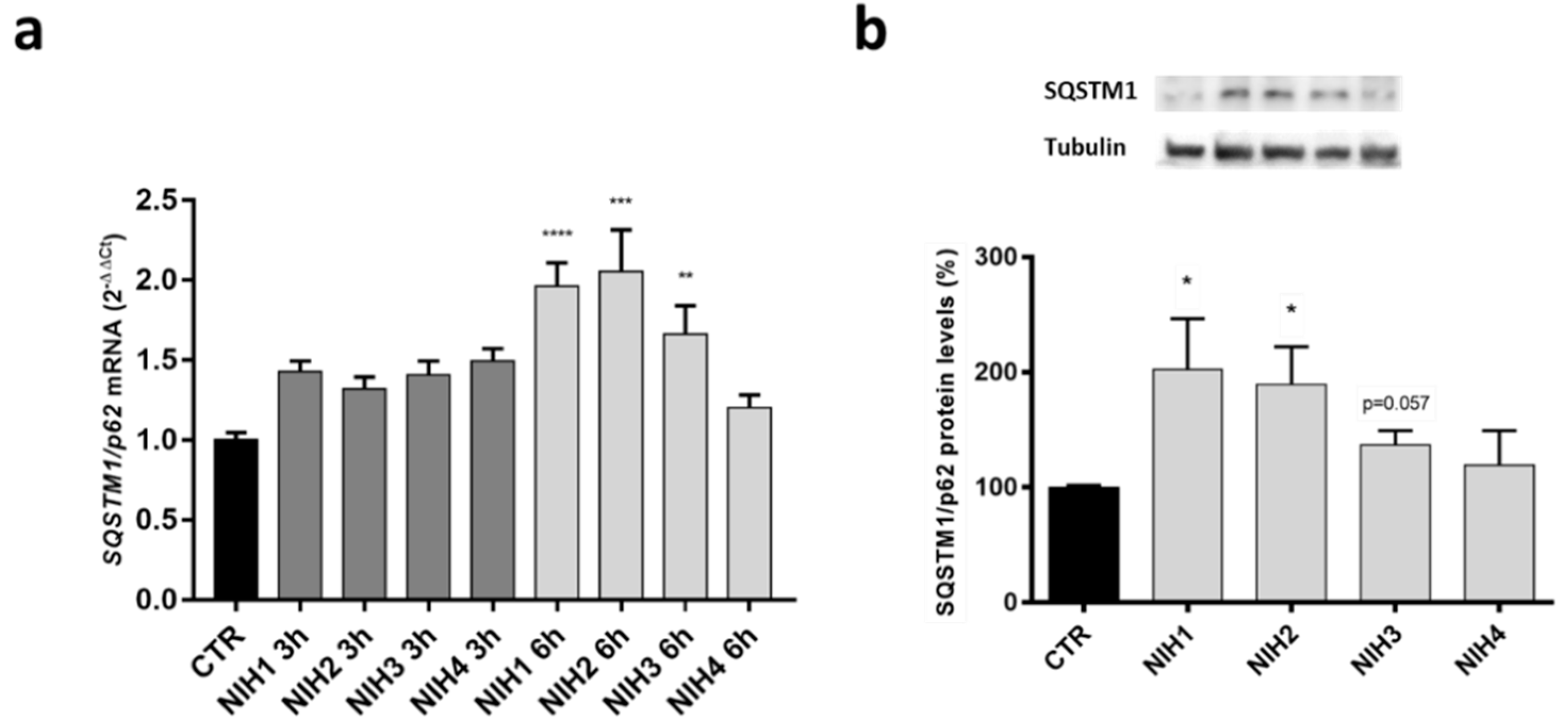
3.1. The Nrf2 Activators NIH1–3 Upregulate HO-1 and SQSTM1/p62 Protein Levels, Leading to a Decrease in Basal Autophagy Flux
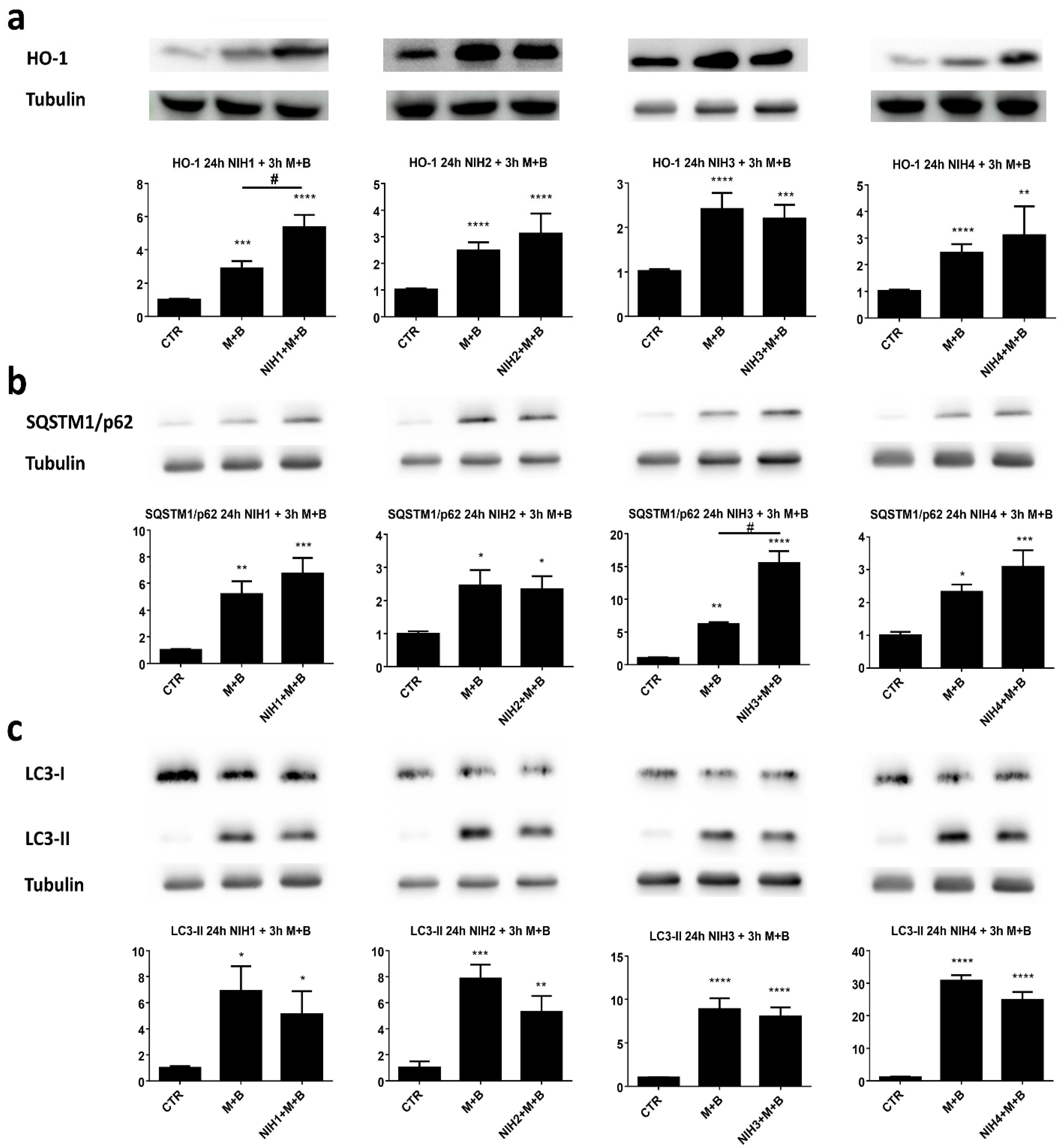
3.2. NIH Compounds Protect Cellular Viability of ARPE-19 Cell upon Prolonged Proteasome and Autophagy Dysfunction
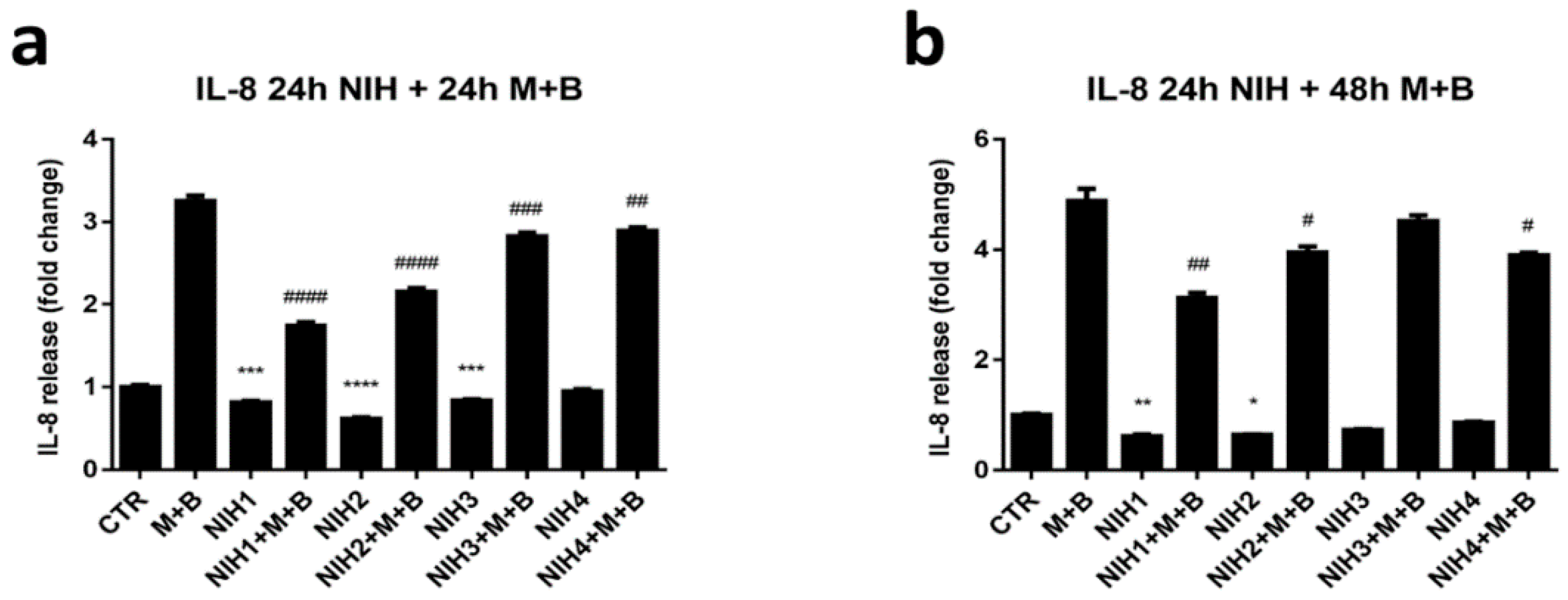
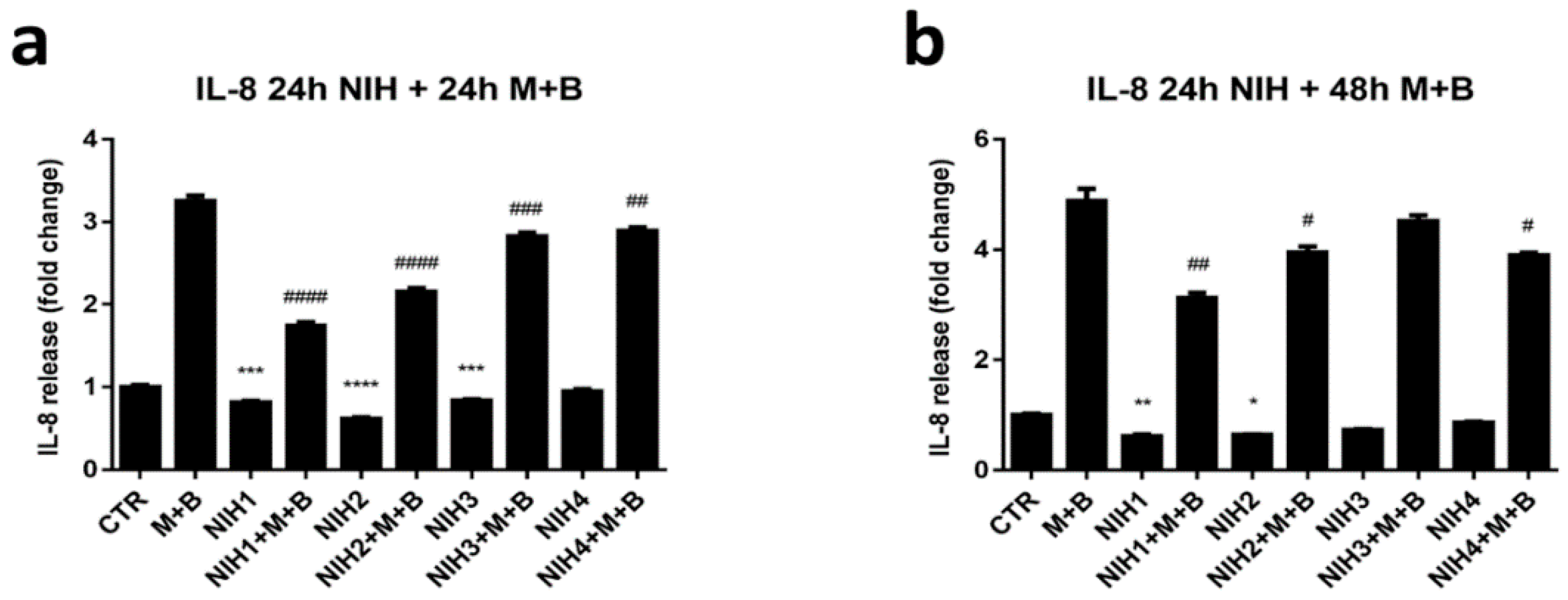
3.3. NIH Compounds Decrease IL-8 Secretion in Oxidative-Stress-Prone Environment Created by Dysfunctional Protein Clearance
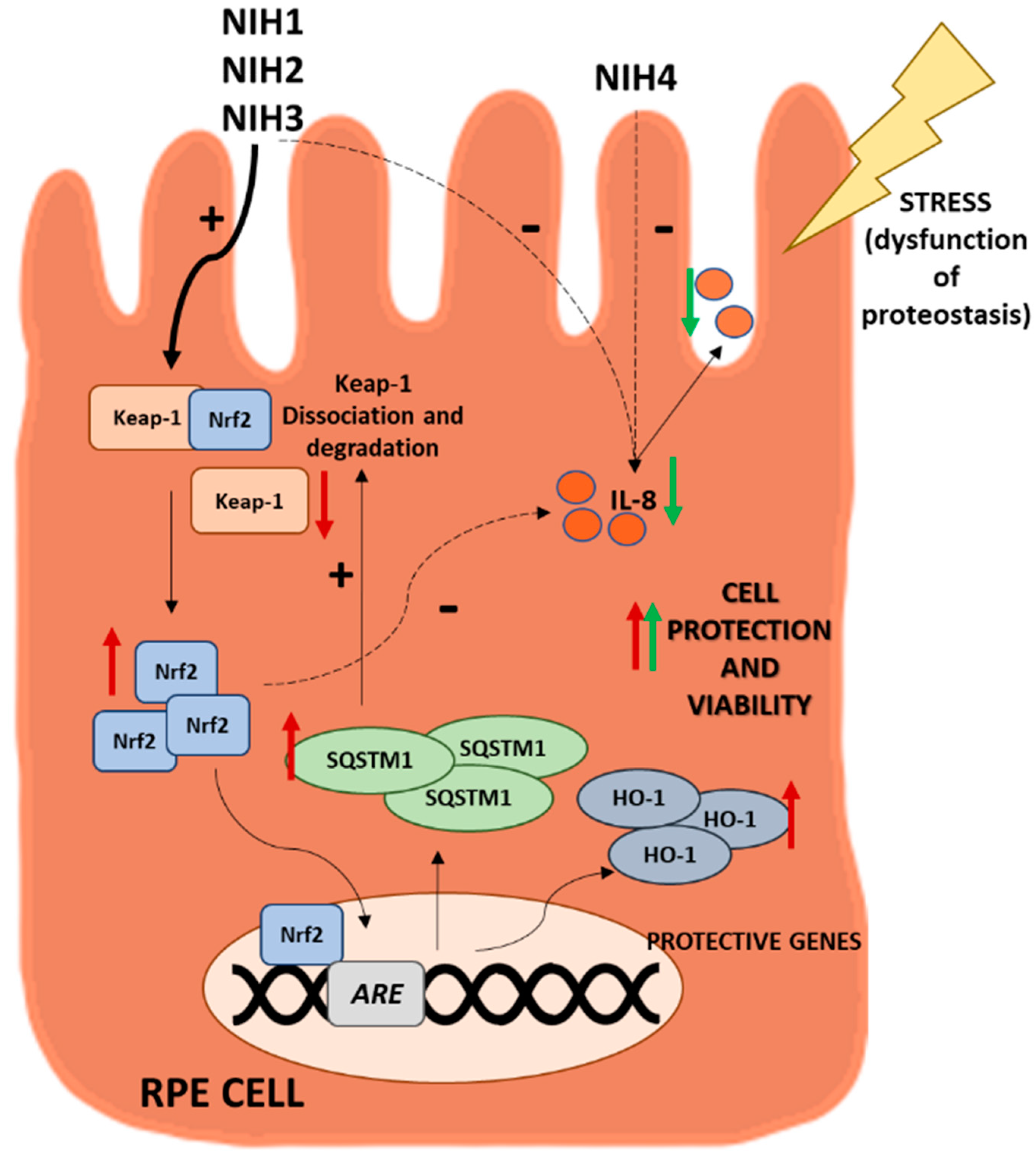
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaarniranta, K.; Blasiak, J.; Liton, P.; Boulton, M.; Klionsky, D.J.; Sinha, D. Autophagy in age-related macular degeneration. Autophagy 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Jayesh, S.S.; Junnuthula, V.; Kolimi, P.; Dyawanapelly, S. Novel and investigational therapies for wet and dry age-related macular degeneration. Drug Discov. Today. 2022, 27, 2322–2332. [Google Scholar] [CrossRef] [PubMed]
- Ferris, F.L.; Fine, S.L.; Hyman, L. Age-related macular degeneration and blindness due to neovascular maculopathy. Arch. Ophthalmol. 1984, 102, 1640–1642. [Google Scholar] [CrossRef]
- Ammar, M.J.; Hsu, J.; Chiang, A.; Ho, A.C.; Regillo, C.D. Age-related macular degeneration threapy: A review. Curr. Opin. Ophthalmol. 2020, 31, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Amadio, M.; Govoni, S.; Pascale, A. Targeting VEGF in eye neovascularization: What’s new? A comprehensive review on current therapies and oligonucleotide-based interventions under development. Pharmacol. Res. 2016, 103, 253–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molday, R.S.; Moritz, O.L. Photoreceptros at a glance. J. Cell Sci. 2015, 128, 4039–4045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volland, S.; Esteve-Rudd, J.; Hoo, J.; Yee, C.; Williams, D.S. A comparison of some organizational characteristics of the mouse central retina and the human macula. PLoS ONE 2015, 10, e0125631. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.; Temple, S. Retinal pigment epithelial cell proliferation. Exp. Biol. Med. 2015, 240, 1079–1086. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, M.; Serrao, S.; Lombardo, G. Challenges in age-related macular degeneration: From risk factors to novel diagnostics and prevention strategies. Front. Med. 2022, 9, 887104. [Google Scholar] [CrossRef]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: An insight into cullin-ring ubiquitin ligases. Antioxid. Redox Signal. 2010, 13, 1699–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef] [PubMed]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of Nrf2 and PGC-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Synowiec, E.; Sliwinski, T.; Danisz, K.; Blasiak, J.; Sklodowska, A.; Romaniuk, D.; Watala, C.; Szaflik, J.; Szaflik, J.P. Association between polymorphism of the NQO1, NOS3 and NFE2L2 genes and AMD. Front. Biosci. 2013, 18, 80–90. [Google Scholar]
- Zhao, Z.; Chen, Y.; Wang, J.; Sternberg, P.; Freeman, M.L.; Grossniklaus, H.E.; Cai, J. Age-related retinopathy in NRF2-deficient mice. PLoS ONE 2011, 6, e19456. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Ding, Z.; Du, K.; Ye, X.; Cheng, S. Reactive oxygen species as a link between antioxidant pathways and autophagy. Oxidative Med. Cell. Longev. 2021, 2021, 5583215. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Johansson, I.; Monsen, V.T.; Pettersen, K.; Mildenberger, J.; Misund, K.; Kaarniranta, K.; Schonberg, S.; Bjorkoy, G. The marine n3 PUFA DHA evokes cytoprotection against oxidative stress and protein misfolding by inducing autophagy and NFE2L2 in human retinal pigment epithelial cells. Autophagy 2015, 11, 1636–1651. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Kirkin, V.; Dikic, I.; Johansen, T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009, 8, 1986–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajares, M.; Jimenez-Moreno, N.; Garcia-Yague, A.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rabano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [Green Version]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction ant their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.T.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef]
- Viiri, J.; Amadio, M.; Marchesi, N.; Hyttinen, J.M.; Kivinen, N.; Sironen, R.; Rilla, K.; Akhtar, S.; Provenzani, A.; D’Agostino, V.G.; et al. Autophagy activation clears ELAVL1/HuR-mediated accumulation of SQSTM1/p62 during proteasomal inhibition in human retinal pigment epithelial cells. PLoS ONE 2013, 8, e69563. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; Leon, R.; Lopez, M.G.; Oliva, B.; et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: A systems medicine approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [Green Version]
- Syed, M.U.A.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar]
- Cascella, R.; Ragazzo, M.; Strafella, C.; Missiroli, F.; Borgiani, P.; Angelucci, F.; Marsella, L.T.; Cusumano, A.; Novelli, G.; Ricci, F.; et al. Age-related macular degeneration: Insights into inflammatory genes. J. Ophthalmol. 2014, 2014, 582842. [Google Scholar] [CrossRef]
- Zhou, A.; Jiang, Z.; Lu, H.; Xu, Z.; Tong, R.; Shi, J.; Jia, G. Recent advances of natural polyphenols activators for Keap1-Nrf2 signaling pathway. Chem. Biodivers. 2019, 16, e1900400. [Google Scholar] [CrossRef] [PubMed]
- Gersch, M.; Kreuzer, J.; Sieber, S.A. Electrophilic natural products and their biological targets. Nat. Prod. Rep. 2012, 29, 659–682. [Google Scholar] [CrossRef] [PubMed]
- Basagni, F.; Lanni, C.; Minarini, A.; Rosini, M. Lights and shadows of electrophile signaling: Focus on the Nrf2-Keap1 pathway. Future Med. Chem. 2019, 11, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Serafini, M.M.; Catanzaro, M.; Fagiani, F.; Simoni, E.; Caporaso, R.; Dacrema, M.; Romanoni, I.; Govoni, S.; Racchi, M.; Daglia, M.; et al. Modulation of Keap1/Nrf2/ARE signaling pathway by curcuma- and garlic-derived hybrids. Front. Pharmacol. 2020, 10, 1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Nuccio, C.; Bernardo, A.; Troiano, C.; Brignone, M.A.; Falchi, M.; Greco, A.; Rosini, M.; Basagni, F.; Lanni, C.; Serafini, M.M.; et al. NRF2 and PPAR-γ pathways in oligodendrocyte progenitors: Focus on ROS protection, mitochondrial biogenesis and promotion of cell differentiation. Int. J. Mol. Sci. 2020, 21, 7216. [Google Scholar] [CrossRef]
- Simoni, E.; Serafini, M.M.; Caporaso, R.; Marchetti, C.; Racchi, M.; Minarini, A.; Bartolini, M.; Lanni, C.; Rosini, M. Targeting the Nrf2/amyloid-beta liaison in Alzheimer’s disease: A rational approach. ACS Chem. Neurosci. 2017, 7, 1618–1627. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, M.; Lanni, C.; Basagni, F.; Rosini, M.; Govoni, S.; Amadio, M. Eye-light on age-related macular degeneration: Targeting Nrf2-pathway as a novel therapeutic strategy for retinal pigment epithelium. Front. Pharmacol. 2020, 11, 844. [Google Scholar] [CrossRef] [PubMed]
- Rossino, M.G.; Amato, R.; Amadio, M.; Rosini, M.; Basagni, F.; Cammalleri, M.; Dal Monte, M.; Casini, G. A nature-inspired Nrf2 activator protects retinal explants from oxidative stress and neurodegeneration. Antioxidants 2021, 10, 1296. [Google Scholar] [CrossRef]
- Simoni, E.; Serafini, M.M.; Bartolini, M.; Caporaso, R.; Pinto, A.; Necchi, D.; Fiori, J.; Andrisano, V.; Minarini, A.; Lanni, C.; et al. Nature-inspired multifunctional ligands: Focusing on amyloid-based molecular mechanisms of Alzheimer’s disease. ChemMedChem 2016, 11, 1309–1317. [Google Scholar] [CrossRef]
- Marchesi, N.; Thongon, N.; Pascale, A.; Provenzani, A.; Koskela, A.; Korhonen, E.; Smedowski, A.; Govoni, S.; Kauppinen, A.; Kaarniranta, K.; et al. Autophagy stimulus promotes early HuR protein activation and p62/SQSTM1 protein synthesis in ARPE-19 cells by triggering Erk1/2, p38MAPK, and JNK kinase pathways. Oxidative Med. Cell. Longev. 2018, 2018, 4956080. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.J.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.C.; Grosso, R.A.; Fader, C.M. Hallmarks of aging: An autophagic perspective. Front. Endocrinol. 2018, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Robledinos-Anton, N.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Activators and inhibitors of NRF2: A review of their potential for clinical development. Oxidative Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Johansen, T. Aggrephagy: Selective disposal of protein aggregates by macrophagy. Int. J. Cell Biol. 2012, 2012, 736905. [Google Scholar] [CrossRef] [Green Version]
- Silva-Islas, C.A.; Maldonado, P.A. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.B.; Rahu, N. Oxidative stress and inflammation: What polyphenols can do for us? Oxidative Med. Cell. Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef] [Green Version]
- Fagiani, F.; Catanzaro, M.; Buoso, E.; Basagni, F.; Di Marino, D.; Raniolo, S.; Amadio, M.; Frost, E.H.; Corsini, E.; Racchi, M.; et al. Targeting cytokine release through the differential modulation of Nrf2 and NF-κB pathways by electrophilic/non-electrophilic compounds. Front. Pharmacol. 2020, 11, 1256. [Google Scholar] [CrossRef]
- Saviranta, N.M.M.; Veeroos, L.; Granlund, L.J.; Hassinen, V.H.; Kaarniranta, K.; Karjalainen, R.O. Plant flavonol quercetin and isoflavone biochanin A differentially induce protection against oxidative stress and inflammation in ARPE-19 cells. Food Res. Int. 2011, 44, 109–113. [Google Scholar] [CrossRef]
- Sharifi-Rad, J.; El Rauess, Y.; Rizk, A.A.; Sadaka, C.; Zgheib, R.; Zam, W.; Sestito, S.; Rapposelli, S.; Neffe-Skocinska, K.; Zielinska, D.; et al. Turmeric and its major compound curcumin on health: Bioactive effects and safety profiles for food, pharmaceutical, biotechnological and medicinal applications. Front. Pharmacol. 2020, 11, 01021. [Google Scholar] [CrossRef]
- Park, J.-Y.; Sohn, H.-Y.; Koh, Y.O.; Jo, C. Curcumin activates Nrf2 through PKCδ-mediated p62 phosphorylation at Ser351. Sci. Rep. 2021, 11, 8430. [Google Scholar] [CrossRef]
- Shin, J.W.; Chun, K.-Y.; Kim, D.-H.; Kim, S.-J.; Kim, S.H.; Cho, N.-C.; Na, H.-K.; Surh, Y.-J. Curcumin induces stabilization of Nrf2 protein through Keap1 cysteine modification. Biochem. Pharmacol. 2020, 173, 113820. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggrawal, B.B.; Gupta, S.C.; Sung, B. Curcumin: An orally bioactive blocker of TNF and other pro-inflammatory biomarkers. Br. J. Pharmacol. 2013, 169, 1672–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koskela, A.; Manai, F.; Basagni, F.; Liukkonen, M.; Rosini, M.; Govoni, S.; Monte, M.D.; Smedowski, A.; Kaarniranta, K.; Amadio, M. Nature-Inspired Hybrids (NIH) Improve Proteostasis by Activating Nrf2-Mediated Protective Pathways in Retinal Pigment Epithelial Cells. Antioxidants 2022, 11, 1385. https://doi.org/10.3390/antiox11071385
Koskela A, Manai F, Basagni F, Liukkonen M, Rosini M, Govoni S, Monte MD, Smedowski A, Kaarniranta K, Amadio M. Nature-Inspired Hybrids (NIH) Improve Proteostasis by Activating Nrf2-Mediated Protective Pathways in Retinal Pigment Epithelial Cells. Antioxidants. 2022; 11(7):1385. https://doi.org/10.3390/antiox11071385
Chicago/Turabian StyleKoskela, Ali, Federico Manai, Filippo Basagni, Mikko Liukkonen, Michela Rosini, Stefano Govoni, Massimo Dal Monte, Adrian Smedowski, Kai Kaarniranta, and Marialaura Amadio. 2022. "Nature-Inspired Hybrids (NIH) Improve Proteostasis by Activating Nrf2-Mediated Protective Pathways in Retinal Pigment Epithelial Cells" Antioxidants 11, no. 7: 1385. https://doi.org/10.3390/antiox11071385
APA StyleKoskela, A., Manai, F., Basagni, F., Liukkonen, M., Rosini, M., Govoni, S., Monte, M. D., Smedowski, A., Kaarniranta, K., & Amadio, M. (2022). Nature-Inspired Hybrids (NIH) Improve Proteostasis by Activating Nrf2-Mediated Protective Pathways in Retinal Pigment Epithelial Cells. Antioxidants, 11(7), 1385. https://doi.org/10.3390/antiox11071385