
Alcohol-Induced Oxidative Stress and the Role of Antioxidants in Alcohol Use Disorder: A Systematic Review


Abstract
1. Introduction
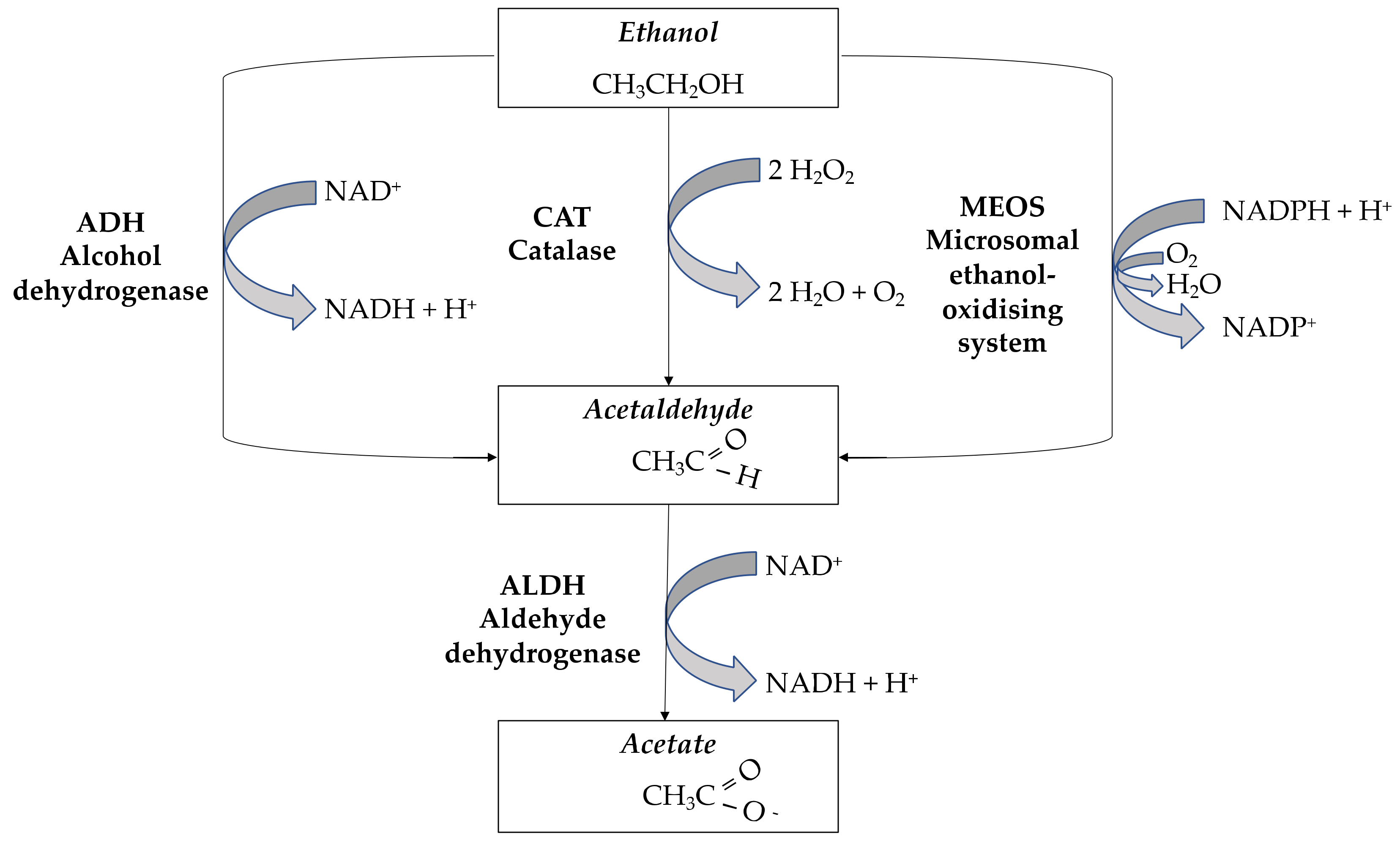
1.1. Ethanol Metabolism
1.2. Alcohol and Oxidative Stress
1.3. Comorbid Mental Disorders and AUD
2. Methods
3. Results and Discussion
3.1. AUD and Oxidative Stress in Preclinical and Clinical Studies
3.2. Genetics of Alcohol-Related Disorders
3.3. Candidate Gene Studies on AUD and Oxidative Stress
3.4. GWAS on AUD and Oxidative Stress
3.5. Oxidative Stress, AUD, and DNA Damage
3.6. Oxidative Stress and the Pathophysiology of AUD
3.7. Oxidative Stress, Immune System, and Neurodegeneration in AUD
3.8. Oxidative Stress, AUD, and Potential Therapeutic Target and Agents
3.9. Registered Clinical Trials on Potential Therapeutic Targets and Agents in AUD-Related Oxidative Stress
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schuckit, M.A. Alcohol-use disorders. Lancet 2009, 373, 492–501. [Google Scholar] [CrossRef]
- Peacock, A.; Leung, J.; Larney, S.; Colledge, S.; Hickman, M.; Rehm, J.; Giovino, G.A.; West, R.; Hall, W.; Griffiths, P.; et al. Global statistics on alcohol, tobacco and illicit drug use: 2017 status report. Addiction 2018, 113, 1905–1926. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.; Cichon, S.; Treutlein, J.; Ridinger, M.; Mattheisen, M.; Hoffmann, P.; Herms, S.; Wodarz, N.; Soyka, M.; Zill, P.; et al. Genome-wide significant association between alcohol dependence and a variant in the ADH gene cluster. Addict. Biol. 2012, 17, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Hasin, D.S.; Stinson, F.S.; Ogburn, E.; Grant, B.F. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: Results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch. Gen. Psychiatry 2007, 64, 830–842. [Google Scholar] [CrossRef]
- Rehm, J. The Risks Associated With Alcohol Use and Alcoholism. Alcohol Res. Heal. 2011, 34, 135. [Google Scholar]
- Griswold, M.G.; Fullman, N.; Hawley, C.; Arian, N.; Zimsen, S.R.M.; Tymeson, H.D.; Venkateswaran, V.; Tapp, A.D.; Forouzanfar, M.H.; Salama, J.S.; et al. Alcohol use and burden for 195 countries and territories, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2018, 392, 1015–1035. [Google Scholar] [CrossRef]
- Walters, R.K.; Polimanti, R.; Johnson, E.C.; McClintick, J.N.; Adams, M.J.; Adkins, A.E.; Aliev, F.; Bacanu, S.A.; Batzler, A.; Bertelsen, S.; et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat. Neurosci. 2018, 21, 1656–1669. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013; ISBN 0-89042-555-8. [Google Scholar]
- Kranzler, H.R.; Zhou, H.; Kember, R.L.; Vickers Smith, R.; Justice, A.C.; Damrauer, S.; Tsao, P.S.; Klarin, D.; Baras, A.; Reid, J.; et al. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat. Commun. 2019, 10, 1499. [Google Scholar] [CrossRef]
- Saunders, J.B.; Aasland, O.G.; Babor, T.F.; de la Fuente, J.R.; Grant, M. Development of the Alcohol Use Disorders Identification Test (AUDIT): WHO Collaborative Project on Early Detection of Persons with Harmful Alcohol Consumption—II. Addiction 1993, 88, 791–804. [Google Scholar] [CrossRef]
- Mansoori, A.A.; Jain, S.K. ADH1B, ALDH2, GSTM1 and GSTT1 Gene Polymorphic Frequencies among Alcoholics and Controls in the Arcadian Population of Central India. Asian Pac. J. Cancer Prev. 2018, 19, 725–731. [Google Scholar] [CrossRef]
- Edenberg, H.J. The Genetics of Alcohol Metabolism: Role of Alcohol Dehydrogenase and Aldehyde Dehydrogenase Variants. Alcohol Res. Health 2007, 30, 5. [Google Scholar]
- Tawa, E.A.; Hall, S.D.; Lohoff, F.W. Overview of the Genetics of Alcohol Use Disorder. Alcohol Alcohol 2016, 51, 507–514. [Google Scholar] [CrossRef]
- Das, S.K.; Vasudevan, D.M. Alcohol-induced oxidative stress. Life Sci. 2007, 81, 177–187. [Google Scholar] [CrossRef]
- Zakhari, S. Overview: How Is Alcohol Metabolized by the Body? Alcohol Res. Health 2006, 29, 245. [Google Scholar]
- Gemma, S.; Vichi, S.; Testai, E. Individual susceptibility and alcohol effects: Biochemical and genetic aspects. Ann. Ist. Super. Sanita 2006, 42, 8–16. [Google Scholar]
- Steiner, J.L.; Lang, C.H. Etiology of alcoholic cardiomyopathy: Mitochondria, oxidative stress and apoptosis. Int. J. Biochem. Cell Biol. 2017, 89, 125–135. [Google Scholar] [CrossRef]
- Peana, A.T.; Sánchez-Catalán, M.J.; Hipólito, L.; Rosas, M.; Porru, S.; Bennardini, F.; Romualdi, P.; Caputi, F.F.; Candeletti, S.; Polache, A.; et al. Mystic Acetaldehyde: The Never-Ending Story on Alcoholism. Front. Behav. Neurosci. 2017, 11, 81. [Google Scholar] [CrossRef]
- El-Mas, M.M.; Abdel-Rahman, A.A. Role of Alcohol Oxidative Metabolism in Its Cardiovascular and Autonomic Effects. Adv. Exp. Med. Biol. 2019, 1193, 1–33. [Google Scholar] [CrossRef]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Oliveira de Araújo Melo, C.; Cidália Vieira, T.; Duarte Gigonzac, M.A.; Soares Fortes, J.; Moreira Duarte, S.S.; da Cruz, A.D.; Silva, D.d.M.E. Evaluation of polymorphisms in repair and detoxification genes in alcohol drinkers and non-drinkers using capillary electrophoresis. Electrophoresis 2020, 41, 254–258. [Google Scholar] [CrossRef]
- Huang, M.C.; Chen, C.H.; Peng, F.C.; Tang, S.H.; Chen, C.C. Alterations in oxidative stress status during early alcohol withdrawal in alcoholic patients. J. Formos. Med. Assoc. 2009, 108, 560–569. [Google Scholar] [CrossRef]
- Hovatta, I.; Juhila, J.; Donner, J. Oxidative stress in anxiety and comorbid disorders. Neurosci. Res. 2010, 68, 261–275. [Google Scholar] [CrossRef]
- Kimura, M.; Yokoyama, A.; Higuchi, S. Aldehyde dehydrogenase-2 as a therapeutic target. Expert Opin. Ther. Targets 2019, 23, 955–966. [Google Scholar] [CrossRef]
- Chen, C.H.; Ferreira, J.C.B.; Gross, E.R.; Mochly-Rosen, D. Targeting aldehyde dehydrogenase 2: New therapeutic opportunities. Physiol. Rev. 2014, 94, 1–34. [Google Scholar] [CrossRef]
- Haorah, J.; Ramirez, S.H.; Floreani, N.; Gorantla, S.; Morsey, B.; Persidsky, Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic. Biol. Med. 2008, 45, 1542–1550. [Google Scholar] [CrossRef]
- Mansoori, A.A.; Jain, S.K. Molecular Links between Alcohol and Tobacco Induced DNA Damage, Gene Polymorphisms and Patho-physiological Consequences: A Systematic Review of Hepatic Carcinogenesis. Asian Pac. J. Cancer Prev. 2015, 16, 4803–4812. [Google Scholar] [CrossRef]
- Hu, X.; Oroszi, G.; Chun, J.; Smith, T.L.; Goldman, D.; Schuckit, M.A. An expanded evaluation of the relationship of four alleles to the level of response to alcohol and the alcoholism risk. Alcohol. Clin. Exp. Res. 2005, 29, 8–16. [Google Scholar] [CrossRef]
- Yan, T.; Zhao, Y. Acetaldehyde induces phosphorylation of dynamin-related protein 1 and mitochondrial dysfunction via elevating intracellular ROS and Ca2+ levels. Redox Biol. 2020, 28, 101381. [Google Scholar] [CrossRef]
- Kamal, H.; Tan, G.C.; Ibrahim, S.F.; Shaikh, M.F.; Mohamed, I.N.; Mohamed, R.M.P.; Hamid, A.A.; Ugusman, A.; Kumar, J. Alcohol Use Disorder, Neurodegeneration, Alzheimer’s and Parkinson’s Disease: Interplay between Oxidative Stress, Neuroimmune Response and Excitotoxicity. Front. Cell. Neurosci. 2020, 14, 282. [Google Scholar] [CrossRef]
- Kim, S.R.; Jeong, H.Y.; Yang, S.; Choi, S.P.; Seo, M.Y.; Yun, Y.K.; Choi, Y.; Baik, S.H.; Park, J.S.; Gwon, A.R.; et al. Effects of chronic alcohol consumption on expression levels of APP and Aβ-producing enzymes. BMB Rep. 2011, 44, 135–139. [Google Scholar] [CrossRef]
- Agar, E.; Bosnak, M.; Amanvermez, R.; Demir, S.; Ayyildiz, M.; Celik, C. The effect of ethanol on lipid peroxidation and glutathione level in the brain stem of rat. Neuroreport 1999, 10, 1799–1801. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Muneer, P.M.; Alikunju, S.; Mishra, V.; Schuetz, H.; Szlachetka, A.M.; Burnham, E.L.; Haorah, J. Activation of NLRP3 inflammasome by cholesterol crystals in alcohol consumption induces atherosclerotic lesions. Brain. Behav. Immun. 2017, 62, 291–305. [Google Scholar] [CrossRef] [PubMed]
- González-Reimers, E.; Santolaria-Fernández, F.; Martín-González, M.C.; Fernández-Rodríguez, C.M.; Quintero-Platt, G. Alcoholism: A systemic proinflammatory condition. World J. Gastroenterol. 2014, 20, 14660–14671. [Google Scholar] [CrossRef] [PubMed]
- Blanco, A.M.; Vallés, S.L.; Pascual, M.; Guerri, C. Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes. J. Immunol. 2005, 175, 6893–6899. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Carniglia, A.; Keyes, K.M.; Hasin, D.S.; Cerdá, M. Psychiatric comorbidities in alcohol use disorder. Lancet. Psychiatry 2019, 6, 1068–1080. [Google Scholar] [CrossRef]
- Sánchez-Peña, J.F.; Álvarez-Cotoli, P.; Rodríguez-Solano, J.J. Psychiatric disorders associated with alcoholism: 2 year follow-up of treatment. Actas Esp. Psiquiatr. 2012, 40, 129–135. [Google Scholar]
- Tobore, T.O. On the Neurobiological Role of Oxidative Stress in Alcohol-Induced Impulsive, Aggressive and Suicidal Behavior. Subst. Use Misuse 2019, 54, 2290–2303. [Google Scholar] [CrossRef]
- Kathryn Mchugh, R.; Weiss, R.D. Alcohol Use Disorder and Depressive Disorders. Alcohol Res. 2019, 40, e1–e8. [Google Scholar] [CrossRef]
- Archibald, L.; Brunette, M.F.; Wallin, D.J.; Green, A.I. Alcohol Use Disorder and Schizophrenia or Schizoaffective Disorder. Alcohol Res. 2019, 40, e1–e9. [Google Scholar] [CrossRef]
- Helle, A.C.; Watts, A.L.; Trull, T.J.; Sher, K.J. Alcohol Use Disorder and Antisocial and Borderline Personality Disorders. Alcohol Res. 2019, 40, e1–e16. [Google Scholar] [CrossRef]
- Anker, J.J.; Kushner, M.G. Co-Occurring Alcohol Use Disorder and Anxiety: Bridging Psychiatric, Psychological, and Neurobiological Perspectives. Alcohol Res. 2019, 40, e1–e12. [Google Scholar] [CrossRef]
- Sullivan, E.V.; Pfefferbaum, A. Brain-behavior relations and effects of aging and common comorbidities in alcohol use disorder: A review. Neuropsychology 2019, 33, 760–780. [Google Scholar] [CrossRef]
- Edwards, A.C.; Ohlsson, H.; Sundquist, J.; Sundquist, K.; Kendler, K.S. Alcohol Use Disorder and Risk of Suicide in a Swedish Population-Based Cohort. Am. J. Psychiatry 2020, 177, 627–634. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Pemberton, P.W.; Smith, A.; Warnes, T.W. Non-invasive monitoring of oxidant stress in alcoholic liver disease. Scand. J. Gastroenterol. 2005, 40, 1102–1108. [Google Scholar] [CrossRef]
- Xu, H.; Li, H.; Liu, D.; Wen, W.; Xu, M.; Frank, J.A.; Chen, J.; Zhu, H.; Grahame, N.J.; Luo, J. Chronic Voluntary Alcohol Drinking Causes Anxiety-like Behavior, Thiamine Deficiency, and Brain Damage of Female Crossed High Alcohol Preferring Mice. Front. Pharmacol. 2021, 12, 614396. [Google Scholar] [CrossRef]
- Sommavilla, M.; Sánchez-Villarejo, M.V.; Almansa, I.; Sánchez-Vallejo, V.; Barcia, J.M.; Romero, F.J.; Miranda, M. The effects of acute ethanol exposure and ageing on rat brain glutathione metabolism. Free Radic. Res. 2012, 46, 1076–1081. [Google Scholar] [CrossRef]
- Uys, J.D.; Mulholland, P.J.; Townsend, D.M. Glutathione and redox signaling in substance abuse. Biomed. Pharmacother. 2014, 68, 799–807. [Google Scholar] [CrossRef]
- Dries, S.S.; Seibert, B.S.; Bastiani, M.F.; Linden, R.; Perassolo, M.S. Evaluation of oxidative stress biomarkers and liver and renal functional parameters in patients during treatment a mental health unit to treat alcohol dependence. Drug Chem. Toxicol. 2022, 45, 861–867. [Google Scholar] [CrossRef]
- Plotnikov, E.; Korotkova, E.; Voronova, O.; Sazhina, N.; Petrova, E.; Artamonov, A.; Chernyavskaya, L.; Dorozhko, E. Comparative investigation of antioxidant activity of human serum blood by amperometric, voltammetric and chemiluminescent methods. Arch. Med. Sci. 2016, 12, 1071–1076. [Google Scholar] [CrossRef]
- Thome, J.; Foley, P.; Gsell, W.; Davids, E.; Wodarz, N.; Wiesbeck, G.A.; Böning, J.; Riederer, P. Increased concentrations of manganese superoxide dismutase in serum of alcohol-dependent patients. Alcohol Alcohol 1997, 32, 65–69. [Google Scholar] [CrossRef]
- Peng, F.C.; Tang, S.H.; Huang, M.C.; Chen, C.C.; Kuo, T.L.; Yin, S.J. Oxidative status in patients with alcohol dependence: A clinical study in Taiwan. J. Toxicol. Environ. Health A 2005, 68, 1497–1509. [Google Scholar] [CrossRef]
- Huang, M.C.; Chen, C.C.; Peng, F.C.; Tang, S.H.; Chen, C.H. The correlation between early alcohol withdrawal severity and oxidative stress in patients with alcohol dependence. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Kapaki, E.; Liappas, I.; Lyras, L.; Paraskevas, G.P.; Mamali, I.; Theotoka, I.; Bourboulis, N.; Liosis, I.; Petropoulou, O.; Soldatos, K. Oxidative Damage to Plasma Proteins in Patients with Chronic Alcohol Dependence: The Effect of Smoking. Vivo 2007, 21, 523–528. [Google Scholar] [PubMed]
- Budzyński, J.; Ziółkowski, M.; Kłopocka, M.; Czarnecki, D. Oxidoreductive homeostasis in alcohol-dependent male patients and the risk of alcohol drinking relapse in a 6-month follow-up. Alcohol 2016, 50, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Yamaki, N.; Matsushita, S.; Hara, S.; Yokoyama, A.; Hishimoto, A.; Higuchi, S. Telomere shortening in alcohol dependence: Roles of alcohol and acetaldehyde. J. Psychiatr. Res. 2019, 109, 27–32. [Google Scholar] [CrossRef]
- Chen, C.H.; Pan, C.H.; Chen, C.C.; Huang, M.C. Increased oxidative DNA damage in patients with alcohol dependence and its correlation with alcohol withdrawal severity. Alcohol. Clin. Exp. Res. 2011, 35, 338–344. [Google Scholar] [CrossRef]
- Huang, M.C.; Chen, C.C.; Pan, C.H.; Chen, C.H. Comparison of oxidative DNA damage between alcohol-dependent patients with and without delirium tremens. Alcohol. Clin. Exp. Res. 2014, 38, 2523–2528. [Google Scholar] [CrossRef]
- Wu, S.Y.; Chen, C.Y.; Huang, T.L.; Tsai, M.C. Brain-derived neurotrophic factor and glutathione peroxidase as state biomarkers in alcohol use disorder patients undergoing detoxification. Medicine 2020, 99, e19938. [Google Scholar] [CrossRef]
- Kang, J.I.; Hwang, S.S.; Choi, J.R.; Lee, S.T.; Kim, J.; Hwang, I.S.; Kim, H.W.; Kim, C.H.; Kim, S.J. Telomere length in alcohol dependence: A role for impulsive choice and childhood maltreatment. Psychoneuroendocrinology 2017, 83, 72–78. [Google Scholar] [CrossRef]
- Chen, C.H.; Walker, J.; Momenan, R.; Rawlings, R.; Heilig, M.; Hommer, D.W. Relationship between liver function and brain shrinkage in patients with alcohol dependence. Alcohol. Clin. Exp. Res. 2012, 36, 625–632. [Google Scholar] [CrossRef]
- Flatscher-Bader, T.; Van Der Brug, M.; Hwang, J.W.; Gochee, P.A.; Matsumoto, I.; Niwa, S.I.; Wilce, P.A. Alcohol-responsive genes in the frontal cortex and nucleus accumbens of human alcoholics. J. Neurochem. 2005, 93, 359–370. [Google Scholar] [CrossRef]
- Matthews, B.A.; Kish, S.J.; Xu, X.; Boileau, I.; Rusjan, P.M.; Wilson, A.A.; Digiacomo, D.; Houle, S.; Meyer, J.H. Greater monoamine oxidase a binding in alcohol dependence. Biol. Psychiatry 2014, 75, 756–764. [Google Scholar] [CrossRef]
- Bota, A.B.; Simmons, J.G.; DiBattista, A.; Wilson, K. Carnitine in Alcohol Use Disorders: A Scoping Review. Alcohol. Clin. Exp. Res. 2021, 45, 666–674. [Google Scholar] [CrossRef]
- De Ternay, J.; Naassila, M.; Nourredine, M.; Louvet, A.; Bailly, F.; Sescousse, G.; Maurage, P.; Cottencin, O.; Carrieri, P.M.; Rolland, B. Therapeutic Prospects of Cannabidiol for Alcohol Use Disorder and Alcohol-Related Damages on the Liver and the Brain. Front. Pharmacol. 2019, 10, 627. [Google Scholar] [CrossRef]
- Pervin, Z.; Stephen, J.M. Effect of alcohol on the central nervous system to develop neurological disorder: Pathophysiological and lifestyle modulation can be potential therapeutic options for alcohol-induced neurotoxication. AIMS Neurosci. 2021, 8, 390–413. [Google Scholar] [CrossRef]
- Crews, F.T.; Sarkar, D.K.; Qin, L.; Zou, J.; Boyadjieva, N.; Vetreno, R.P. Neuroimmune Function and the Consequences of Alcohol Exposure. Alcohol Res. 2015, 37, 331. [Google Scholar]
- Read, E.; Zhu, J.; Yang, G. Disrupted H 2 S Signaling by Cigarette Smoking and Alcohol Drinking: Evidence from Cellular, Animal, and Clinical Studies. Antioxidants 2021, 10, 49. [Google Scholar] [CrossRef]
- Crews, F.T.; Vetreno, R.P. Neuroimmune basis of alcoholic brain damage. Int. Rev. Neurobiol. 2014, 118, 315–357. [Google Scholar] [CrossRef]
- Chastain, L.G.; Sarkar, D.K. Role of microglia in regulation of ethanol neurotoxic action. Int. Rev. Neurobiol. 2014, 118, 81–103. [Google Scholar] [CrossRef]
- Manzo-Avalos, S.; Saavedra-Molina, A. Cellular and mitochondrial effects of alcohol consumption. Int. J. Environ. Res. Public Health 2010, 7, 4281–4304. [Google Scholar] [CrossRef]
- Hurley, T.D.; Edenberg, H.J. Genes Encoding Enzymes Involved in Ethanol Metabolism. Alcohol Res. 2012, 34, 339. [Google Scholar]
- Shen, Y.C.; Fan, J.H.; Edenberg, H.J.; Li, T.K.; Cui, Y.H.; Wang, Y.F.; Tian, C.H.; Zhou, C.F.; Zhou, R.L.; Wang, J.; et al. Polymorphism of ADH and ALDH Genes among Four Ethnic Groups in China and Effects upon the Risk for Alcoholism. Alcohol. Clin. Exp. Res. 1997, 21, 1272–1277. [Google Scholar] [CrossRef]
- Muramatsu, T.; Zu-Cheng, W.; Yi-Ru, F.; Kou-Bao, H.; Heqin, Y.; Yamada, K.; Higuchi, S.; Harada, S.; Kono, H. Alcohol and aldehyde dehydrogenase genotypes and drinking behavior of Chinese living in Shanghai. Hum. Genet. 1995, 96, 151–154. [Google Scholar] [CrossRef]
- Higuchi, S.; Matsushita, S.; Muramatsu, T.; Murayama, M.; Hayashida, M. Alcohol and aldehyde dehydrogenase genotypes and drinking behavior in Japanese. Alcohol. Clin. Exp. Res. 1996, 20, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, S.; Matsushita, S.; Masaki, T.; Yokoyama, A.; Kimura, M.; Go, S.; Mochizuki, H. Influence of genetic variations of ethanol-metabolizing enzymes on phenotypes of alcohol-related disorders. Ann. N. Y. Acad. Sci. 2004, 1025, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.C.; López-Granero, C.; Ferrer, B.; Tinkov, A.A.; Skalny, A.V.; Paoliello, M.M.B.; Aschner, M. BXD Recombinant Inbred Mice as a Model to Study Neurotoxicity. Biomolecules 2021, 11, 1762. [Google Scholar] [CrossRef] [PubMed]
- Metten, P.; Phillips, T.J.; Crabbe, J.C.; Tarantino, L.M.; McClearn, G.E.; Plomin, R.; Erwin, V.G.; Belknap, J.K. High genetic susceptibility to ethanol withdrawal predicts low ethanol consumption. Mamm. Genome 1998, 9, 983–990. [Google Scholar] [CrossRef]
- Demarest, K.; Koyner, J.; Mccaughran, J.; Cipp, L.; Hitzemann, R. Further characterization and high-resolution mapping of quantitative trait loci for ethanol-induced locomotor activity. Behav. Genet. 2001, 31, 79–91. [Google Scholar] [CrossRef]
- Xu, Y.; Demarest, K.; Hitzemann, R.; Sikela, J.M. Gene coding variant in Cas1 between the C57bl/6J and DBA/2J inbred mouse strains: Linkage to a QTL for ethanol-induced locomotor activation. Alcohol. Clin. Exp. Res. 2002, 26, 1–7. [Google Scholar] [CrossRef]
- Sun, Y.; Chang, S.; Wang, F.; Sun, H.; Ni, Z.; Yue, W.; Zhou, H.; Gelernter, J.; Malison, R.T.; Kalayasiri, R.; et al. Genome-wide association study of alcohol dependence in male Han Chinese and cross-ethnic polygenic risk score comparison. Transl. Psychiatry 2019, 9, 249. [Google Scholar] [CrossRef]
- Verhulst, B.; Neale, M.C.; Kendler, K.S. The heritability of alcohol use disorders: A meta-analysis of twin and adoption studies. Psychol. Med. 2015, 45, 1061. [Google Scholar] [CrossRef]
- Gelernter, J.; Kranzler, H.R.; Sherva, R.; Almasy, L.; Koesterer, R.; Smith, A.H.; Anton, R.; Preuss, U.W.; Ridinger, M.; Rujescu, D.; et al. Genome-wide association study of alcohol dependence:significant findings in African- and European-Americans including novel risk loci. Mol. Psychiatry 2014, 19, 41–49. [Google Scholar] [CrossRef]
- Jorgenson, E.; Thai, K.K.; Hoffmann, T.J.; Sakoda, L.C.; Kvale, M.N.; Banda, Y.; Schaefer, C.; Risch, N.; Mertens, J.; Weisner, C.; et al. Genetic contributors to variation in alcohol consumption vary by race/ethnicity in a large multi-ethnic genome-wide association study. Mol. Psychiatry 2017, 22, 1359–1367. [Google Scholar] [CrossRef]
- Koechling, U.M.; Amit, Z.; Negrete, J.C. Family history of alcoholism and the mediation of alcohol intake by catalase: Further evidence for catalase as a marker of the propensity to ingest alcohol. Alcohol. Clin. Exp. Res. 1995, 19, 1096–1104. [Google Scholar] [CrossRef]
- Forsberg, L.; Lyrenäs, L.; De Faire, U.; Morgenstern, R. A common functional C-T substitution polymorphism in the promoter region of the human catalase gene influences transcription factor binding, reporter gene transcription and is correlated to blood catalase levels. Free Radic. Biol. Med. 2001, 30, 500–505. [Google Scholar] [CrossRef]
- Plemenitas, A.; Kastelic, M.; Porcelli, S.; Serretti, A.; Rus Makovec, M.; Kores Plesnicar, B.; Dolžan, V. Genetic variability in CYP2E1 and catalase gene among currently and formerly alcohol-dependent male subjects. Alcohol Alcohol 2015, 50, 140–145. [Google Scholar] [CrossRef]
- Covolo, L.; Gelatti, U.; Talamini, R.; Garte, S.; Trevisi, P.; Franceschi, S.; Franceschini, M.; Barbone, F.; Tagger, A.; Ribero, M.L.; et al. Alcohol dehydrogenase 3, glutathione S-transferase M1 and T1 polymorphisms, alcohol consumption and hepatocellular carcinoma (Italy). Cancer Causes Control 2005, 16, 831–838. [Google Scholar] [CrossRef]
- MacGregor, S.; Lind, P.A.; Bucholz, K.K.; Hansell, N.K.; Madden, P.A.F.; Richter, M.M.; Montgomery, G.W.; Martin, N.G.; Heath, A.C.; Whitfield, J.B. Associations of ADH and ALDH2 gene variation with self report alcohol reactions, consumption and dependence: An integrated analysis. Hum. Mol. Genet. 2009, 18, 580–593. [Google Scholar] [CrossRef]
- Bierut, L.J.; Goate, A.M.; Breslau, N.; Johnson, E.O.; Bertelsen, S.; Fox, L.; Agrawal, A.; Bucholz, K.K.; Grucza, R.; Hesselbrock, V.; et al. ADH1B is associated with alcohol dependence and alcohol consumption in populations of European and African ancestry. Mol. Psychiatry 2012, 17, 445–450. [Google Scholar] [CrossRef]
- Tsuchihashi-Makaya, M.; Serizawa, M.; Yanai, K.; Katsuya, T.; Takeuchi, F.; Fujioka, A.; Yamori, Y.; Ogihara, T.; Kato, N. Gene-environmental interaction regarding alcohol-metabolizing enzymes in the Japanese general population. Hypertens. Res. 2009, 32, 207–213. [Google Scholar] [CrossRef]
- Sherva, R.; Rice, J.P.; Neuman, R.J.; Rochberg, N.; Saccone, N.L.; Bierut, L.J. Associations and interactions between SNPs in the alcohol metabolizing genes and alcoholism phenotypes in European Americans. Alcohol. Clin. Exp. Res. 2009, 33, 848–857. [Google Scholar] [CrossRef]
- Carr, L.G.; Foroud, T.; Stewart, T.; Castelluccio, P.; Edenberg, H.J.; Li, T.K. Influence of ADH1B polymorphism on alcohol use and its subjective effects in a Jewish population. Am. J. Med. Genet. 2002, 112, 138–143. [Google Scholar] [CrossRef]
- Biernacka, J.M.; Geske, J.R.; Schneekloth, T.D.; Frye, M.A.; Cunningham, J.M.; Choi, D.S.; Tapp, C.L.; Lewis, B.R.; Drews, M.S.; Pietrzak, T.L.; et al. Replication of genome wide association studies of alcohol dependence: Support for association with variation in ADH1C. PLoS ONE 2013, 8, e58798. [Google Scholar] [CrossRef]
- Birley, A.J.; James, M.R.; Dickson, P.A.; Montgomery, G.W.; Heath, A.C.; Martin, N.G.; Whitfield, J.B. ADH single nucleotide polymorphism associations with alcohol metabolism in vivo. Hum. Mol. Genet. 2009, 18, 1533–1542. [Google Scholar] [CrossRef]
- Nishiyori, A.; Shibata, A.; Ogimoto, I.; Uchimura, N.; Egami, H.; Nakamura, J.; Sakata, R.; Fukuda, K. Alcohol drinking frequency is more directly associated with alcohol use disorder than alcohol metabolizing enzymes among male Japanese. Psychiatry Clin. Neurosci. 2005, 59, 38–44. [Google Scholar] [CrossRef]
- Wall, T.L.; Carr, L.G.; Ehlers, C.L. Protective association of genetic variation in alcohol dehydrogenase with alcohol dependence in Native American Mission Indians. Am. J. Psychiatry 2003, 160, 41–46. [Google Scholar] [CrossRef]
- Chambers, G.K.; Marshall, S.J.; Robinson, G.M.; Maguire, S.; Newton-Howes, J.; Chong, N.L. The genetics of alcoholism in Polynesians: Alcohol and aldehyde dehydrogenase genotypes in young men. Alcohol. Clin. Exp. Res. 2002, 26, 949–955. [Google Scholar] [CrossRef]
- Edenberg, H.J.; Xuei, X.; Chen, H.J.; Tian, H.; Wetherill, L.F.; Dick, D.M.; Almasy, L.; Bierut, L.; Bucholz, K.K.; Goate, A.; et al. Association of alcohol dehydrogenase genes with alcohol dependence: A comprehensive analysis. Hum. Mol. Genet. 2006, 15, 1539–1549. [Google Scholar] [CrossRef]
- Teixeira, T.M.; da Silva, H.D.; Goveia, R.M.; Ribolla, P.E.M.; Alonso, D.P.; Alves, A.A.; Melo e Silva, D.; Collevatti, R.G.; Bicudo, L.A.; Bérgamo, N.A.; et al. First description and evaluation of SNPs in the ADH and ALDH genes in a population of alcoholics in Central-West Brazil. Alcohol 2017, 65, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Lind, P.A.; Eriksson, C.J.P.; Wilhelmsen, K.C. The role of aldehyde dehydrogenase-1 (ALDH1A1) polymorphisms in harmful alcohol consumption in a Finnish population. Hum. Genomics 2008, 3, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Montane-Jaime, K.; Shafe, S.; Joseph, R.; Crooks, H.; Carr, L.G.; Ehlers, C.L. Association of ALDH1 promoter polymorphisms with alcohol-related phenotypes in Trinidad and Tobago. J. Stud. Alcohol Drugs 2007, 68, 192–196. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shin, S.; Stewart, R.; Ferri, C.P.; Kim, J.M.; Shin, I.S.; Kim, S.W.; Yang, S.J.; Yoon, J.S. An investigation of associations between alcohol use disorder and polymorphisms on ALDH2, BDNF, 5-HTTLPR, and MTHFR genes in older Korean men. Int. J. Geriatr. Psychiatry 2010, 25, 441–448. [Google Scholar] [CrossRef]
- Lin, C.L.; Chien, R.N.; Chen, L.W.; Huang, T.S.; Shyu, Y.C.; Yeh, C.T.; Liang, K.H. The Aldehyde Dehydrogenase ALDH2*2 Allele, Associated with Alcohol Drinking Behavior, Dates Back to Prehistoric Times. Biomolecules 2021, 11, 1376. [Google Scholar] [CrossRef]
- Chen, Y.C.; Lu, R.B.; Peng, G.S.; Wang, M.F.; Wang, H.K.; Ko, H.C.; Chang, Y.C.; Lu, J.J.; Li, T.K.; Yin, S.J. Alcohol Metabolism and Cardiovascular Response in an Alcoholic Patient Homozygous for the ALDH2*2 Variant Gene Allele. Alcohol. Clin. Exp. Res. 1999, 23, 1853–1860. [Google Scholar] [CrossRef]
- Li, D.; Zhao, H.; Gelernter, J. Strong protective effect of the aldehyde dehydrogenase gene (ALDH2) 504lys (*2) allele against alcoholism and alcohol-induced medical diseases in Asians. Hum. Genet. 2012, 131, 725–737. [Google Scholar] [CrossRef]
- Zaso, M.J.; Goodhines, P.A.; Wall, T.L.; Park, A. Meta-Analysis on Associations of Alcohol Metabolism Genes With Alcohol Use Disorder in East Asians. Alcohol Alcohol 2019, 54, 216–224. [Google Scholar] [CrossRef]
- Luczak, S.E.; Glatt, S.J.; Wall, T.J. Meta-analyses of ALDH2 and ADH1B with alcohol dependence in Asians. Psychol. Bull. 2006, 132, 607–621. [Google Scholar] [CrossRef]
- Li, D.; Zhao, H.; Gelernter, J. Strong association of the alcohol dehydrogenase 1B gene (ADH1B) with alcohol dependence and alcohol-induced medical diseases. Biol. Psychiatry 2011, 70, 504–512. [Google Scholar] [CrossRef]
- Castaldelli-Maia, J.M.; Malbergier, A.; de Oliveira, A.B.P.; Amaral, R.A.; Negrão, A.B.; Gonçalves, P.D.; Ventriglio, A.; de Berardis, D.; De Antonio, J.; Firigato, I.; et al. Exploring the Role of Alcohol Metabolizing Genotypes in a 12-Week Clinical Trial of Naltrexone for Alcohol Use Disorder. Biomolecules 2021, 11, 1495. [Google Scholar] [CrossRef]
- Zhao, R.; Zhang, R.; Li, W.; Liao, Y.; Tang, J.; Miao, Q.; Hao, W. Genome-wide DNA methylation patterns in discordant sib pairs with alcohol dependence. Asia-Pac. Psychiatry 2013, 5, 39–50. [Google Scholar] [CrossRef]
- Zhang, R.; Miao, Q.; Wang, C.; Zhao, R.; Li, W.; Haile, C.N.; Hao, W.; Zhang, X.Y. Genome-wide DNA methylation analysis in alcohol dependence. Addict. Biol. 2013, 18, 392–403. [Google Scholar] [CrossRef]
- Quillen, E.E.; Chen, X.D.; Almasy, L.; Yang, F.; He, H.; Li, X.; Wang, X.Y.; Liu, T.Q.; Hao, W.; Deng, H.W.; et al. ALDH2 is associated to alcohol dependence and is the major genetic determinant of “daily maximum drinks” in a GWAS study of an isolated rural Chinese sample. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014, 165B, 103–110. [Google Scholar] [CrossRef]
- Treutlein, J.; Cichon, S.; Ridinger, M.; Wodarz, N.; Soyka, M.; Zill, P.; Maier, W.; Moessner, R.; Gaebel, W.; Dahmen, N.; et al. Genome-wide association study of alcohol dependence. Arch. Gen. Psychiatry 2009, 66, 773–784. [Google Scholar] [CrossRef]
- Clarke, T.K.; Adams, M.J.; Davies, G.; Howard, D.M.; Hall, L.S.; Padmanabhan, S.; Murray, A.D.; Smith, B.H.; Campbell, A.; Hayward, C.; et al. Genome-wide association study of alcohol consumption and genetic overlap with other health-related traits in UK Biobank (N = 112 117). Mol. Psychiatry 2017, 22, 1376–1384. [Google Scholar] [CrossRef]
- Sanchez-Roige, S.; Fontanillas, P.; Elson, S.L.; Gray, J.C.; de Wit, H.; Davis, L.K.; MacKillop, J.; Palmer, A.A. Genome-wide association study of alcohol use disorder identification test (AUDIT) scores in 20 328 research participants of European ancestry. Addict. Biol. 2019, 24, 121–131. [Google Scholar] [CrossRef]
- Li, W.; Thygesen, J.H.; O’Brien, N.L.; Heydtmann, M.; Smith, I.; Degenhardt, F.; Nöthen, M.M.; Morgan, M.Y.; Bass, N.J.; McQuillin, A. The influence of regression models on genome-wide association studies of alcohol dependence: A comparison of binary and quantitative analyses. Psychiatr. Genet. 2021, 31, 13–20. [Google Scholar] [CrossRef]
- Lai, D.; Wetherill, L.; Bertelsen, S.; Carey, C.E.; Kamarajan, C.; Kapoor, M.; Meyers, J.L.; Anokhin, A.P.; Bennett, D.A.; Bucholz, K.K.; et al. Genome-wide association studies of alcohol dependence, DSM-IV criterion count and individual criteria. Genes. Brain. Behav. 2019, 18, e12579. [Google Scholar] [CrossRef]
- Xu, K.; Kranzler, H.R.; Sherva, R.; Sartor, C.E.; Almasy, L.; Koesterer, R.; Zhao, H.; Farrer, L.A.; Gelernter, J. Genomewide Association Study for Maximum Number of Alcoholic Drinks in European Americans and African Americans. Alcohol. Clin. Exp. Res. 2015, 39, 1137–1147. [Google Scholar] [CrossRef]
- Gelernter, J.; Zhou, H.; Nuñez, Y.Z.; Mutirangura, A.; Malison, R.T.; Kalayasiri, R. Genomewide Association Study of Alcohol Dependence and Related Traits in a Thai Population. Alcohol. Clin. Exp. Res. 2018, 42, 861–868. [Google Scholar] [CrossRef]
- Park, B.L.; Kim, J.W.; Cheong, H.S.; Kim, L.H.; Lee, B.C.; Seo, C.H.; Kang, T.C.; Nam, Y.W.; Kim, G.B.; Shin, H.D.; et al. Extended genetic effects of ADH cluster genes on the risk of alcohol dependence: From GWAS to replication. Hum. Genet. 2013, 132, 657–668. [Google Scholar] [CrossRef]
- Takeuchi, F.; Isono, M.; Nabika, T.; Katsuya, T.; Sugiyama, T.; Yamaguchi, S.; Kobayashi, S.; Ogihara, T.; Yamori, Y.; Fujioka, A.; et al. Confirmation of ALDH2 as a Major locus of drinking behavior and of its variants regulating multiple metabolic phenotypes in a Japanese population. Circ. J. 2011, 75, 911–918. [Google Scholar] [CrossRef]
- Cadet, J.; Delatour, T.; Douki, T.; Gasparutto, D.; Pouget, J.P.; Ravanat, J.L.; Sauvaigo, S. Hydroxyl radicals and DNA base damage. Mutat. Res. 1999, 424, 9–21. [Google Scholar] [CrossRef]
- Duncan, J.W.; Johnson, S.; Zhang, X.; Zheng, B.; Luo, J.; Ou, X.M.; Stockmeier, C.A.; Wang, J.M. Up-Regulation of PKR Signaling Pathway by Ethanol Displays an Age of Onset-Dependent Relationship. Alcohol. Clin. Exp. Res. 2016, 40, 2320–2328. [Google Scholar] [CrossRef][Green Version]
- Agudelo, M.; Gandhi, N.; Saiyed, Z.; Pichili, V.; Thangavel, S.; Khatavkar, P.; Yndart-Arias, A.; Nair, M. Effects of alcohol on histone deacetylase 2 (HDAC2) and the neuroprotective role of trichostatin A (TSA). Alcohol. Clin. Exp. Res. 2011, 35, 1550–1556. [Google Scholar] [CrossRef]
- Muneer, A.; Minhas, F.A. Telomere Biology in Mood Disorders: An Updated, Comprehensive Review of the Literature. Clin. Psychopharmacol. Neurosci. 2019, 17, 343–363. [Google Scholar] [CrossRef]
- Pavanello, S.; Hoxha, M.; Dioni, L.; Bertazzi, P.A.; Snenghi, R.; Nalesso, A.; Ferrara, S.D.; Montisci, M.; Baccarelli, A. Shortened telomeres in individuals with abuse in alcohol consumption. Int. J. Cancer 2011, 129, 983–992. [Google Scholar] [CrossRef]
- El-Terras, A.; Soliman, M.M.; Alkhedaide, A.; Attia, H.F.; Alharthy, A.; Banaja, A.E. Carbonated soft drinks induce oxidative stress and alter the expression of certain genes in the brains of Wistar rats. Mol. Med. Rep. 2016, 13, 3147–3154. [Google Scholar] [CrossRef][Green Version]
- Matthews, B.A.; Tong, J.; Attwells, S.; Xu, X.; Le, A.; Kish, S.J.; Meyer, J.H. Elevated monoamine oxidase A activity and protein levels in rodent brain during acute withdrawal after chronic intermittent ethanol vapor exposure. Drug Alcohol Depend. 2018, 185, 398–405. [Google Scholar] [CrossRef]
- Duncan, J.W.; Zhang, X.; Wang, N.; Johnson, S.; Harris, S.; Udemgba, C.; Ou, X.M.; Youdim, M.B.; Stockmeier, C.A.; Wang, J.M. Binge ethanol exposure increases the Krüppel-like factor 11-monoamine oxidase (MAO) pathway in rats: Examining the use of MAO inhibitors to prevent ethanol-induced brain injury. Neuropharmacology 2016, 105, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Crews, F.T. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J. Neuroinflammation 2012, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liu, Y.; Hong, J.S.; Crews, F.T. NADPH oxidase and aging drive microglial activation, oxidative stress, and dopaminergic neurodegeneration following systemic LPS administration. Glia 2013, 61, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.K.; Thompson, J.; Chen, L.; Dagda, M.; Dertien, J.; Dossou, K.S.S.; Moaddel, R.; Bergeson, S.E.; Kruman, I.I. Differential sensitivity of prefrontal cortex and hippocampus to alcohol-induced toxicity. PLoS ONE 2014, 9, e106945. [Google Scholar] [CrossRef] [PubMed]
- Boyadjieva, N.I.; Sarkar, D.K. Cyclic adenosine monophosphate and brain-derived neurotrophic factor decreased oxidative stress and apoptosis in developing hypothalamic neuronal cells: Role of microglia. Alcohol. Clin. Exp. Res. 2013, 37, 1370–1379. [Google Scholar] [CrossRef]
- Rendón-Ramírez, A.; Cortés-Couto, M.; Martínez-Rizo, A.B.; Muñiz-Hernández, S.; Velázquez-Fernández, J.B. Oxidative damage in young alcohol drinkers: A preliminary study. Alcohol 2013, 47, 501–504. [Google Scholar] [CrossRef]
- Morley, K.C.; Lagopoulos, J.; Logge, W.; Chitty, K.; Baillie, A.; Haber, P.S. Neurometabolite Levels in Alcohol Use Disorder Patients During Baclofen Treatment and Prediction of Relapse to Heavy Drinking. Front. Psychiatry 2018, 9, 412. [Google Scholar] [CrossRef]
- Perfilova, V.N.; Ostrovskii, O.V.; Verovskii, V.E.; Popova, T.A.; Lebedeva, S.A.; Dib, H. Effect of Citrocard on functional activity of cardiomyocyte mitochondria during chronic alcohol intoxication. Bull. Exp. Biol. Med. 2007, 143, 341–343. [Google Scholar] [CrossRef]
- Ledesma, J.C.; Baliño, P.; Aragon, C.M.G. Reduction in central H2O2 levels prevents voluntary ethanol intake in mice: A role for the brain catalase-H2O2 system in alcohol binge drinking. Alcohol. Clin. Exp. Res. 2014, 38, 60–67. [Google Scholar] [CrossRef]
- Peana, A.T.; Muggironi, G.; Fois, G.; Diana, M. Alpha-lipoic acid reduces ethanol self-administration in rats. Alcohol. Clin. Exp. Res. 2013, 37, 1816–1822. [Google Scholar] [CrossRef]
- Ferreira Seiva, F.R.; Amauchi, J.F.; Ribeiro Rocha, K.K.; Souza, G.A.; Ebaid, G.X.; Burneiko, R.M.; Novelli, E.L.B. Effects of N-acetylcysteine on alcohol abstinence and alcohol-induced adverse effects in rats. Alcohol 2009, 43, 127–135. [Google Scholar] [CrossRef]
- Walter, N.A.R.; Denmark, D.A.L.; Kozell, L.B.; Buck, K.J. A Systems Approach Implicates a Brain Mitochondrial Oxidative Homeostasis Co-expression Network in Genetic Vulnerability to Alcohol Withdrawal. Front. Genet. 2017, 7, 218. [Google Scholar] [CrossRef]
- Guillot, A.; Ren, T.; Jourdan, T.; Pawlosky, R.J.; Han, E.; Kim, S.J.; Zhang, L.; Koob, G.F.; Gao, B. Targeting liver aldehyde dehydrogenase-2 prevents heavy but not moderate alcohol drinking. Proc. Natl. Acad. Sci. USA 2019, 116, 25974–25981. [Google Scholar] [CrossRef]
- Chen, C.H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Hurley, T.D.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef]
- Rivera-Meza, M.; Vásquez, D.; Quintanilla, M.E.; Lagos, D.; Rojas, B.; Herrera-Marschitz, M.; Israel, Y. Activation of mitochondrial aldehyde dehydrogenase (ALDH2) by ALDA-1 reduces both the acquisition and maintenance of ethanol intake in rats: A dual mechanism? Neuropharmacology 2019, 146, 175–183. [Google Scholar] [CrossRef]
- Maheshwari, R.K.; Singh, A.K.; Gaddipati, J.; Srimal, R.C. Multiple biological activities of curcumin: A short review. Life Sci. 2006, 78, 2081–2087. [Google Scholar] [CrossRef]
- Cannon, C.P.; Braunwald, E.; McCabe, C.H.; Rader, D.J.; Rouleau, J.L.; Belder, R.; Joyal, S.V.; Hill, K.A.; Pfeffer, M.A.; Skene, A.M. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N. Engl. J. Med. 2004, 350, 1495–1504. [Google Scholar] [CrossRef]
- Petrella, C.; Carito, V.; Carere, C.; Ferraguti, G.; Ciafrè, S.; Natella, F.; Bello, C.; Greco, A.; Ralli, M.; Mancinelli, R.; et al. Oxidative stress inhibition by resveratrol in alcohol-dependent mice. Nutrition 2020, 79–80, 110783. [Google Scholar] [CrossRef]
- Hwang, C.L.; Bian, J.T.; Thur, L.A.; Peters, T.A.; Piano, M.R.; Phillips, S.A. Tetrahydrobiopterin Restores Microvascular Dysfunction in Young Adult Binge Drinkers. Alcohol. Clin. Exp. Res. 2020, 44, 407–414. [Google Scholar] [CrossRef]
- Manzardo, A.M.; Penick, E.C.; Knop, J.; Nickel, E.J.; Hall, S.; Jensen, P.; Miller, C.C.; Gabrielli, W.F. Neonatal vitamin K might reduce vulnerability to alcohol dependence in Danish men. J. Stud. Alcohol 2005, 66, 586–592. [Google Scholar] [CrossRef]
- Carito, V.; Ceccanti, M.; Cestari, V.; Natella, F.; Bello, C.; Coccurello, R.; Mancinelli, R.; Fiore, M. Olive polyphenol effects in a mouse model of chronic ethanol addiction. Nutrition 2017, 33, 65–69. [Google Scholar] [CrossRef]
- Da Costa e Silva, L.D.; Pereira, P.; Regner, G.G.; Boaretto, F.B.M.; Hoffmann, C.; Pflüger, P.; da Silva, L.L.; Steffens, L.R.; Morás, A.M.; Moura, D.J.; et al. DNA damage and oxidative stress induced by seizures are decreased by anticonvulsant and neuroprotective effects of lobeline, a candidate to treat alcoholism. Metab. Brain Dis. 2018, 33, 53–61. [Google Scholar] [CrossRef]
- Mohebbi, E.; Molavi, M.; Mohammadzadeh, M.; Hosseinzadeh, H.; Amin, B. Clavulanic acid improves ethanol withdrawal symptoms in rats. Iran. J. Basic Med. Sci. 2020, 23, 730–736. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Topic of the Study | Aim of the Study | Number of Patients Included | Significant Findings, Safety, Disease Response, and Disease Control | Type of Study | Reference |
|---|---|---|---|---|---|
| Evaluation of oxidative stress biomarkers, liver, and renal function parameters in patients during AD treatment | To compare oxidative stress and renal and hepatic function parameters upon admission and discharge from the hospital | 28 | Chlorpromazine showed influence over hepatic function markers and oxidative stress parameters (i.e., CAT and GPX); carbamazepine influenced hepatic function and ferric reducing antioxidant power; SOD levels were lower, and GPX and ferric reducing antioxidant power presented higher levels at discharge | Prospective cohort study | [51] |
| Influence of heavy drinking on the onset of age-related diseases by measuring telomere length | To measure telomere length of Japanese patients with AD and search for an association between telomere length and genetic variants of ADH1B and ALDH2 | 255 | Telomere length was almost 50% shorter in AD patients relative to the controls. There were no associations between ADH1B and ALDH2 genotypes and telomere length | Cohort study | [58] |
| The correlation between early alcohol withdrawal severity and oxidative stress in AD patients | To explore the correlation between alcohol withdrawal severity and two oxidative stress markers: MDA and SOD | 95 | Compared to the controls, serum MDA levels were significantly elevated, and SOD activity was significantly lowered in alcoholic patients; clinical withdrawal severity was significantly positively correlated with serum MDA levels | Cohort study | [55] |
| Increased oxidative DNA damage in AD patients and its correlation with alcohol withdrawal severity | To compare serum 8-OHdG levels between patients with AD and healthy controls and to investigate the correlation between this marker and the severity of alcohol withdrawal syndrome | 142 | The oxidative DNA damage persisted after 1 week of detoxification. The alcohol withdrawal syndrome severity was correlated with the increase in oxidative stress | Prospective cohort study | [59] |
| Comparison of oxidative DNA damage between AD patients with and without delirium tremens | To investigate levels of 8-hydroxy-2’-deoxyguanosine (8-OhdG) as a marker of oxidative DNA damage in AD patients | 74 | AD patients with delirium tremens had higher serum 8-OhdG levels than those without delirium tremens, suggesting that higher oxidative stress carries a greater risk of the occurrence of delirium tremens | Prospective cohort study | [60] |
| BDNF and GPX as state biomarkers in AUD patients undergoing detoxification | To investigate the serum levels of BDNF and oxidative stress markers in AUD patients during alcohol detoxification | 34 | Serum levels of oxidative stress markers were significantly higher in the AUD group than in control group, while BDNF levels were lower; after alcohol detoxification treatment, the GPX levels in the AUD group dropped, and the BDNF levels rose | Cohort study | [61] |
| Telomere length in AD and its role in impulsive choice and childhood maltreatment | To examine whether delayed discounting and childhood trauma are related to leukocyte telomere length in AD patients, who are considered to have a higher impulsive choice and shorter telomere length | 253 | Patients with AD and high childhood trauma showed a significant relationship between delay discounting and leukocyte telomere length, while those with low trauma showed no association between them | Prospective study | [62] |
| Alterations in oxidative stress status during early alcohol withdrawal in alcoholic patients | To investigate serial alterations in various oxidative stress markers during early detoxification in alcoholic patients | 140 | Marked oxidative stress in alcoholic patients without severe liver disease was observed; the attenuation of a raised MDA level and lowering of CAT activity appeared after one week of detoxification; alcoholic patients did not scavenge free radicals as readily as controls | Prospective cohort study | [22] |
| Oxidative damage to plasma proteins in patients with chronic AD and the effect of smoking | To examine the oxidative status of plasma proteins as markers of oxidative stress in subjects with chronic AD with smoking as a cofounding factor | 132 | Systemic oxidative stress in chronic AD was attributed mainly to alcohol consumption, while smoking may act synergistically | Prospective cohort study | [56] |
| Relationship between liver function and brain shrinkage in AD patients | To assess the correlations between liver function and brain volume measurements in AD patients | 235 | The results showed that higher liver function levels correlated with brain volume shrinkage in AD patients but not in the controls. Serum gamma-glutamyl transferase levels outweighed the aging effect on brain shrinkage in female patients | Prospective cohort study | [63] |
| Oxidative status in AD patients | To examine the relationship between AD and oxidative status | 47 | Serum MDA levels of AD patients were found to be significantly increased compared with the controls and decreased after abstinence; serum CAT did not return to normal status at week 2 after abstinence; the activity of CAT was significantly correlated with MDA levels | Prospective cohort study | [54] |
| Oxidoreductive homeostasis in AD male patients and the risk of alcohol drinking relapse in a 6 month follow up | To verify the hypothesis that oxidoreductive blood balance can also affect demand for energy substances, such as alcoholic beverages, in AD individuals as well as the severity of their AD and risk of drinking relapse | 77 | The risk of alcohol drinking relapse was lower in patients with an above-median initial blood concentration of nitric oxide metabolites and total antioxidant status; the oxidative stress parameters correlated with AD severity markers | Prospective cohort study | [57] |
| Alcohol-responsive genes in the frontal cortex and nucleus accumbens of human alcoholics | To compare the RNA expression profile of the nucleus accumbens and prefrontal cortex of the human brain from matched individual alcoholic and control cases | 14 | Downregulation of genes encoding essential proteins involved in vesicular transport and cellular architecture in nucleus accumbens of the alcoholic | Comparative postmortem study | [64] |
| MAO-A levels in brain regions in AD | To verify the hypothesis that the MAO-A level is elevated in the prefrontal cortex during AD as the cellular response to oxidative stress and mitochondrial toxicity | 32 | MAO-A was significantly greater in the prefrontal cortex and all brain regions analyzed in AD; an association between prefrontal and anterior cingulate cortex MAO-A and the severity of depressed mood was observed | Cohort study | [65] |
| Investigation of antioxidant activity of human serum | To conduct a comparative investigation of the total antioxidant activity of human serum | 30 | All applied methods revealed that the serum total antioxidant activity of the AD patients was lower than the total antioxidant activity of the control group | Cohort study | [52] |
| Concentrations of manganese SOD in the serum of AD patients | The quantitative determination of the plasma concentrations of oxidative stress-associated parameters (concentrations of lactoferrin, Cu, Zn-SOD, and Mn-SOD) in AD and controls | 35 | Increased oxidative stress was observed in AD patients | Cohort study | [53] |
| Aim of the Review | Significant Findings | Reference |
|---|---|---|
| To review principles in alcohol-induced neurodegeneration; the interplay between oxidative stress, neuroimmune response, and excitotoxicity | Alcohol-induced oxidative stress initiated the innate immune response; no direct link between an alcohol-induced hyperglutamatergic state and excitotoxicity in humans; neuroimmune response and excitotoxicity leading to neurodegeneration (i.e., apoptosis and necrosis); chronic alcohol intake has the potential for the development of neurodegenerative diseases; the interplay between oxidative stress, neuroimmune response, and excitotoxicity leading to neurodegeneration | [30] |
| To review ALDH2 polymorphism in disease, aging, alcohol addiction, and its potential as a therapeutic target | ALDH2 plays a vital role in the pathogenesis of human conditions such as AUD, cancer, cardiovascular diseases, diabetes mellitus, and neurodegenerative diseases; the clearance of endogenous aldehydes mediates its effect; animal model studies suggested an ALDH2 activator, Alda-1, may have a preventive effect against neurodegenerative diseases including AUD | [24] |
| To review the neurobiological basis for alcohol-induced aggression, impulsivity, and suicidal behavior | Oxidative stress plays a critical underlying role in alcohol toxicity and behavioral impairments; antioxidant therapy should be an integral part of acute alcohol intoxication and AUD treatment | [39] |
| To review connections between carnitine metabolism and the pathophysiology of the AUD | Alcohol use appears to impact carnitine metabolism, most clearly in the setting of alcoholic cirrhosis; an increase in plasma carnitine may be related to disordered fatty acid metabolism and oxidative stress in AUD; carnitine can be a supplementation in the treatment of AUD | [66] |
| To provide a rationale for using CBD to treat human subjects with AUD, based on the findings of experimental studies | CBD reduces alcohol-related steatosis and fibrosis in the liver by reducing lipid accumulation, stimulating autophagy, modulating inflammation, reducing oxidative stress, and inducing death of activated hepatic stellate cells; CBD reduces the level of alcohol drinking in animal models of AUD by reducing ethanol intake, motivation for ethanol, relapse, anxiety, and impulsivity; it reduces alcohol-related steatosis and fibrosis in the liver and reduces alcohol-related brain damage | [67] |
| To review the mechanisms of alcohol on the pathological relationships of neurodegeneration that cause permanent neuronal damage in AUD | Chronic alcohol abuse through oxidative reduction response and inflammatory activation leads to cytoskeletal destabilization of BBB integrity, which further activates astrocytes and, thus, finally causes BBB disruption and neuronal death | [68] |
| To review if anxiety disorders, depression, and AUD share oxidative stress in their etiologies | Animal and human studies confirm a link between oxidative stress and anxiety, depression, and AUD. Oxidative stress might also be involved in the etiology of neuropsychiatric diseases by causing accelerated telomere shortening, mitochondrial dysfunction, inflammation excitotoxicity, and influence neuronal signaling | [23] |
| To review how induction of neuroimmune genes by binge drinking increases neuronal excitability and oxidative stress, contributing to the neurobiology of AD | Ethanol-induced immune gene, NOX, catalyzes the formation of ROS and superoxide and thereby increases oxidative stress; oxidative stress, by inducing innate immune genes, significantly contributes to alcoholic brain damage and alcoholic neurodegeneration | [69] |
| To review the interrelationship between H2S signaling and cigarette smoking or alcohol drinking | The evidence from cellular and animal studies and also clinical observations identify H2S as a regulator of oxidative stress and inflammatory response in the pathogenesis of various diseases associated with cigarette smoking and alcohol drinking | [70] |
| To review neuroimmune factors, such as cytokines, Toll-like receptors (TLRs), and HMGB1, and the neuroimmune signaling influence of alcohol drinking habits | The findings support the hypothesis that adolescence is a period of risk for persistent and long-lasting increases in brain neuroimmune gene expression that promote persistent and long-term increases in alcohol consumption, neuroimmune gene induction, and neurodegeneration associated with AUD | [71] |
| To review the role of microglia in the regulation of ethanol neurotoxic action | Microglia, the immune cells of the central nervous system, play an essential role in modulating alcohol-induced neurotoxicity. Microglia are implicated in alcohol-induced neuroinflammation and alcohol-induced increases in oxidative stress | [72] |
| To investigate how alcohol abuse causes damage to and functional impairment of organs, such as the heart, stomach, liver, and nervous system, and its prenatal effects | The potential target of compounds that can be used to prevent therapies for alcohol abusers are listed (e.g., curcumin, resveratrol, piceatannol, S-adenosyl-l-methionine, and betaine) | [73] |
| To evaluate the role of glutathione and redox signaling in cocaine, methamphetamine, and alcohol addiction | Redox signaling through oxidation and reduction reactions plays an essential role in numerous cell-signaling cascades including those with opposing cellular consequences, proliferation, and apoptosis; oxidative damage in the early and late stages of AD is a contributing factor to brain damage induced by heavy alcohol consumption; chronic alcohol exposure in humans and rodents can decrease glutathione and glutathione reductase levels in the brain and alter GST in the brain, blood, and saliva; oxidative stress signaling induced by alcohol may not only contribute to cellular injury but also influences the motivational states that drive alcohol consumption | [50] |
| Genes and Genetic Variations Studied | Number of Subjects | Ethnicity | Genotyping Method | Outcomes | Reference |
|---|---|---|---|---|---|
| CAT rs1001179 | 201 patients and 97 controls | Slovenian | TaqMan | Association between rs1001179 and AD and AUDIT scores | [89] |
| CAT rs1001179 | 85 | African American | TaqMan | No association | [28] |
| GSTP1 rs1695 | 39 patients and 43 controls | Central Brazilians | Allele-specific PCR and sequencing | Association between rs1695 and AUD | [21] |
| GSTM1 (*1, *2), GSTT1 (*1, *2) | 200 patients and 400 controls | North Italian | PCR-RFLP | Association between the GSTM1 null genotype and ethanol intake | [90] |
| ADH1B (*1, *2), ALDH2 (*1, *2), GSTM1 (*1, *2), GSTT1 (*1, *2), | 121 patients and 145 controls | Central Indians | PCR-RFLP | Association between ALDH*2/*2 and the GSTT1 null genotype with alcohol consumption | [11] |
| ADH2 (*1, *2, *3), ADH3 (*1, *2), ALDH2 (*1, *2) | 80 AD patients and 144 controls | Han Chinese | PCR-RFLP | Protective role of ALDH*2/*2 against AD | [107] |
| ADH1B rs122994, ALDH2 rs671 | 34 AD patients and 121 controls | Japanese | PCR-RFLP | Associations between ADH1B rs122994, ALDH2 rs671, and AD | [58] |
| 9 polymorphisms in ALDH2 and 41 in ADH | 4597 | Northern European | Sequenom mass array | Association between ADH1B rs1042026 and alcohol intake; an association of ADH1C rs1693482 and ADH5 rs1230165 with alcohol consumption | [91] |
| ADH1B (*1, *2) | 152 | Jewish American | Enzymatic amplification followed by hybridization with allele-specific oligonucleotides | Association between ADH1B*2 and lower rates of alcohol consumption and more unpleasant reactions | [95] |
| ADH1A rs2276332, ADH1B rs1229984 and rs4147536, ADH1C rs11499823, rs4147541, and rs1693431, ALDH2 rs671, rs4646778, rs886205, rs4646775 | 5451 | Japanese | TaqMan SNP Genotyping Assays | Significant association between ADH1B rs1229984 and ALDH2 rs671 with drinking behavior | [93] |
| ADH2 (*1, *2, *3), ADH3 (*1, *2), ALDH2 (*1, *2) | 340 | Mission Indian | Allele-specific PCR | Association of ADH2*1 with AD and ADH2*3 with AD and a lower number of maximum drinks | [99] |
| ALDH1A1*2 and ALDH1A1*3 | 137 AD patients and 108 controls | Trinidadian and Toboggan (either Africans or Indians) | Sequencing | Association of ALDH1A1*2 with a lifetime diagnosis of AD and high levels of alcohol consumption | [104] |
| 175 SNPs in 17 alcohol-metabolizing genes | 1588 | European American | NA | Strong association between ADH1B rs1229984 and the DSM-IV symptom count and the maximum number of drinks | [94] |
| ADH2 (*1, *2), ADH3 (*1, *2), ALDH2 (*1, *2) | 34 AD patients and 92 controls | New Zealand (White, Asian, and Polynesian (New Zealand Maori and others)) | A newly developed DNA sequencing assay | Protective role of ADH2*2 allele against AUD | [100] |
| 110 SNPs in ADH7, ADH1C, ADH1B, ADH1A, ADH6, ADH4, ADH5 | NA | European American and African American | Sequenom mass array system | Significant associations between ADH4 rs1984362, rs4699718, rs3762894, rs4148886, rs4699714, rs7694646, rs1126672, DWSHpy188I, rs1042364, rs1042365, rs2602866, and rs2602846 and AUD | [101] |
| 103 SNPs in the ADH gene region | 812 | Northern and Southern European | Sequenom mass array system | Associations between ADH1B rs2018417, rs1229985, rs17033, and rs1789877; ADH1A rs931635, rs1229967, rs1618572, rs1230025, and rs2276332; rs3857224, and rs3762894 in the ADH6–ADH4 intergenic region and ADH4 rs1800759 with early and late stages of alcohol metabolism | [97] |
| ALDH2 (*1, *2) | 68 AUD patients and 232 controls | Korean | PCR-RFLP | Association between ALDH2*1 and AUD | [105] |
| ADH1B rs1159918, ADH1C rs1614972, ADH4 rs1042364, ALDH2 rs2238151 | 99 AUD patients and 100 controls | Central-West Brazilians | TaqMan assay | Statistically significant association between ADH4 rs1042364, ALDH2 rs2238151, and AUD | [102] |
| ADH2 (*1, *2), ALDH2 (*1, *2) | 153 AUD patients and 153 controls | Japanese | PCR-RFLP | Association between ALDH2*1/*1 and ADH2*1/*1 with AUD | [98] |
| ALDH1A1 rs348479, rs348472, rs610529, rs2288087, rs13959, rs348449, rs595958, rs918836 | 104 alcoholics and 201 controls | Finnish | TaqMan | Association between ALDH1A1 rs348479 and rs610529 with AD | [103] |
| ~750,000 genomic variants | 71 heavy drinkers and 126 controls | Taiwanese | High-density SNP arrays | Strong association between ALDH2 rs671 and drinking behavior | [106] |
| ADH1B rs1229984 | 5632 | European and African American | KASPar assays or Sequenom | Protective effect of ADH1B rs1229984 against AD | [92] |
| 43 SNPs | 808 AD patients and 1248 controls | European | Illumina 660 genome-wide SNP array, Illumina BeadXpress platform using a VeraCode SNP panel | Association between ADH1C rs1614972 and AD | [96] |
| Outcome | Number of Subjects | Ethnicity | Platform | References |
|---|---|---|---|---|
| Association between ADH1C rs1614972 and AD | 487 AD male patients and 1358 controls | Caucasian (German) | Human-Hap 550 BeadChip | [116] |
| Genome-wide significant association between alcohol intake and the number of drinks per week and ALDH2 rs671 in East Asians and ADH1B rs1229984 in non-Hispanic whites and Hispanic/Latino | 86,627 | Non-Hispanic Whites, Hispanic/Latinos, East Asians, and African Americans | Affymetrix Axiom arrays | [86] |
| Genome-wide significant associations between AD risk and ADH5, ADH4, ADH6, ADH1A, ADH1B, ADH7, and ALDH2 | 117 AD patients and 279 controls | Korean | Illumina Human660 W BeadChip | [123] |
| Genome-wide significant associations between ADH1B rs1229984 and AD and DSM-IV AD criterion count | 7418 (1121 families), 3175 (585 families) | European American and African American | Illumina Human1M array, Illumina Human OmniExpress 12V1 array, Illumina 2.5M array, Smokescreen genotyping array | [120] |
| Genome-wide significant associations with variants near ALDH2 | 1045 | Thai | Illumina Global Screening Array (GSA) and Illumina Multi-Ethnic Global Array (MEGA) | [122] |
| Identification of two significant loci that were associated with ADH1B rs2075633, ADH1B rs1229984, and ALDH2 rs671 | 533 males with AD and 2848 controls | Han Chinese | Illumina Global Screening Array-24 v1.0 BeadChip | [83] |
| Association between rs1229978 (near ADH1C) with AUDIT-C, and rs1154433 (near ADH1C) and ADH4 rs5860563 with AUD | 274,424 | European American, African American, Latino American, East, and South Asian American | Affymetrix Axiom Biobank Array | [9] |
| Association between ADH1C rs141973904 and AUD | 20,328 | European | Illumina HumanHap550+ BeadChip V1 V2, OmniExpress + BeadChip V3, Custom array V4 | [118] |
| Association between rs1789891 and AD | 1333 male patients with severe AD and 2168 controls | Caucasian (German) | Illumina Human610Quad or 660w Quad BeadChip (patients), Illumina HumanHap550 BeadChip (controls) | [3] |
| Association between rs34361428 and AD | 739 patients with ADS and 251 controls | English, Scottish, Welsh, or Irish | Illumina PsychArray | [119] |
| Association between AD and ADH1B rs1229984 in European Americans, and ADH1B rs2066702 and ADH1C rs1789882 in African Americans | 5697 | European American and African American | Illumina HumanOmni1-Quad v1.0 microarray | [85] |
| Association between ALDH2 rs671 and AD, flushing response, and maximum drinks in males | 313 | Han Chinese | Illumina Cyto12 array version 2-1 | [115] |
| Genome-wide significant associations between alcohol consumption and ADH5 rs29001570, ADH1C rs35081954, and rs145452708 located in the region between ADH1B and ADH1C, and rs193099203 located in an 4q23 intergenic region | 112,117 | White British | Affymetrix UK Biobank Axiom array, Affymetrix UK BiLEVE Axiom array | [117] |
| Association between alcohol consumption and ALDH2 rs671 and ADH1B rs1229984 | 733 cases and 729 controls | Japanese | Infinium HumanHap550 Bead Array (Illumina) | [124] |
| Strong association between ALDH2 rs671 and drinking behavior, evaluated using AUDIT | 71 heavy drinkers and 126 controls | Taiwanese | Affymetrix Axiom Genome-Wide TWB 2.0 array | [106] |
| Significant associations between the maximum number of drinks and ADH1B rs1229984 in European Americans and ADH1B rs2066702 in African Americans | 9500 | European American and African American | Illumina HumanOmnil-Quad v1.0 microarray | [121] |
| Topic of the Study | Aim of the Study | Number of Animals Included | Significant Findings, Safety, Disease Response, and Disease Control | Type of Study | Reference |
|---|---|---|---|---|---|
| Oxidative stress inhibition by resveratrol in AD mice | Administration of different dosages of resveratrol in alcoholic adult male mice and measuring oxygen radical levels and alteration of BDNF in the liver | 5 | Prolonged resveratrol consumption counteracts serum-free oxygen radical formation caused by chronic alcohol intake without influencing the natural free oxygen radical defense in a mouse model of alcohol addiction Resveratrol supplementation can counteract alcohol-induced BDNF elevation in the liver | Prospective animal study | [149] |
| Chronic voluntary alcohol drinking and anxiety-like behavior, thiamine deficiency, and brain damage in alcohol-preferring mice | To evaluate the effects of alcohol on neurobehavioral and neuropathological changes in a mouse model | 7 | Chronic voluntary drinking caused anxiety-like behaviors; alcohol increased the expression of neuroinflammation markers and caspase-3 and glial fibrillary acidic protein; alcohol inhibited the expression of thiamine transporters in the brain and reduced thiamine levels in the blood and caused oxidative stress and ER stress and stimulated neurogenesis | Prospective animal study | [48] |
| Lobeline as a potential treatment for drug abuse | To evaluate the possible anticonvulsant and neuroprotective activities of lobeline as a candidate in the treatment of alcohol addiction | 69 | Lobeline decreased CAT in the hippocampus; lobeline has anticonvulsant and neuroprotective actions that may be mediated by antioxidant-like mechanisms, indicating its potential as a candidate drug in alcoholism therapy | Prospective animal study | [153] |
| Effects of NAC on alcohol abstinence and alcohol-induced adverse effects in rats | To investigate the association of NAC intake, alcoholism, and alcohol abstinence on lipid profile, in vivo LDL oxidation, oxidative stress, and antioxidant status in the serum and liver of rats | 30 | Ethanol exposure enhanced serum in vivo oxidized-LDL as well as serum and hepatic oxidative stress | Prospective animal study | [142] |
| Potential role of clavulanic acid in ethanol withdrawal | To investigate the effect of clavulanic acid on the symptoms of ethanol withdrawal in rats | 126 | Clavulanic acid improved withdrawal-induced anxiety-like behavior and seizure vulnerability induced following ethanol withdrawal | Prospective animal study | [154] |
| Level of monoamine oxidase A activity and protein levels in rodent brain during acute withdrawal after chronic intermittent ethanol vapor exposure | To determine whether chronic alcohol vapor exposure causes upregulation of MAO-A activity or levels in the prefrontal and anterior cingulate cortex of rodents during acute withdrawal | 16 | Chronic ethanol vapor exposure significantly elevated MAO-A activity and protein levels in the prefrontal and anterior cingulate cortex at 24 h withdrawal | Prospective animal study | [131] |
| Olive polyphenol’s effects in a mouse model of chronic ethanol addiction | To determine whether polyphenols confer a protective potential against alcohol-induced oxidative stress | 40 | Alcoholic mice showed a worse oxidative status than nonalcoholic mice, but polyphenol supplementation partially counteracted the alcohol pro-oxidant effects | Prospective animal study | [152] |
| Ethanol-mediated upregulation of interferon-gamma, double-stranded RNA-activated protein kinase and p53 | To investigate the upregulation of the double-stranded RNA-activated protein kinase signaling pathway by ethanol | 54 | Chronic ethanol exposure activates the IFN-γ–PKR–p53 pathway in the frontal cortex of rodents. Double-stranded RNA-activated protein kinase expression was more significant in the brains of rodents exposed to ethanol at earlier ages compared to later in life, suggesting a mechanism by which young brains could be more susceptible to ethanol-related brain injury | Prospective animal study | [126] |
| Sensitivity of the prefrontal cortex and hippocampus to alcohol-induced toxicity | To gain a better understanding of the potential contribution of selective prefrontal cortex damage and one-carbon metabolism dysfunction to its alcohol-induced neurological impairments | 10 | The prefrontal cortex is more vulnerable to chronic alcohol-induced oxidative stress and neuronal cell death than the hippocampus | Prospective animal study | [135] |
| Genetic vulnerability to alcohol withdrawal and brain mitochondrial oxidative homeostasis | To elucidate the mechanisms involved in the actions of a QTL with a significant effect on genetic predisposition to alcohol withdrawal | 54 | Administration of NAC significantly reduces symptoms of alcohol withdrawal (i.e., convulsions) in mice | Prospective animal study | [143] |
| Examining the use of MAO inhibitors to prevent ethanol-induced brain injury | To investigate the ethanol-mediated KLF11-MAO cell death cascade in the frontal cortex of rats exposed to a modified binge ethanol model and control rats | 64 | The KLF11-MAO pathway is activated by binge ethanol exposure, and MAOIs are neuroprotective by preventing the binge ethanol-induced changes associated with this cell death cascade | Prospective animal study | [132] |
| Topic of the Study | Aim of the Study | Number of Patients Included | Significant Findings, Safety, Disease Response, and Disease Control | Type of Study | Reference |
|---|---|---|---|---|---|
| Baclofen in AD treatment | To examine brain metabolites following administration of baclofen or placebo in AD individuals | 31 | There were significant differences between baclofen and placebo on parietal concentrations of glutathione when controlling for recent drinking, with baclofen-treated participants demonstrating significantly higher levels of GSH/Cr ratios relative to placebo | Randomized placebo-controlled trial | [138] |
| Influence of neonatal vitamin K and vulnerability to AD | To test the hypothesis that vitamin K supplementation administered to newborns facilitates the synthesis of blood-clotting proteins that might reduce the development of AD later in life | 238 | Vitamin K treatment was associated with significantly lower rates of AD and fewer symptoms of problem drinking | Retrospective cohort study | [151] |
| Tetrahydrobiopterin and microvascular dysfunction in young adult binge drinkers | To examine microvascular dysfunction in an ex vivo experimental model (isolated arterioles) from young adults with a history of repeated binge drinking, moderate alcohol drinking, and alcohol abstention and the role of tetrahydrobiopterin | 36 | In young adult binge drinkers, microvascular dysfunction may be exacerbated with acute pathophysiological stimulus; these binge-induced dysfunctions may be reversed by tetrahydrobiopterin | Cohort study | [150] |
| Study Title | NCT Identifier | Number of Patients | Condition | Intervention | Endpoints | Study Status | Results |
|---|---|---|---|---|---|---|---|
| NAC for Adolescent AUD | NCT03707951 | 120 | AUD | NAC, placebo | Effects of NAC on a platform of weekly evidence-based brief alcohol intervention | Recruiting | NA |
| A Study of NAC for AUD | NCT04964843 | 50 | AUD | NAC, placebo | Assess the impact of NAC on AUD | Not yet recruiting | NA |
| Influence of NAC Maintenance on Alcohol Effects Completed | NCT03216954 | 14 | AUD | Alcohol, placebo, NAC | Evaluate the behavioral effects of alcohol during placebo and NAC maintenance | Complete | No statistically significant results |
| Topiramate Augmenting Strategies for the Treatment of AUD | NCT03120468 | 16 | Alcoholism | TPM and NAC, TPM and placebo | Evaluate the safety and tolerability of TPM + NAC versus TPM + placebo for AUD treatment | Active, not recruiting | NA |
| Clinical Trial for AUD and PTSD | NCT02966873 | 200 | Addiction, alcohol abuse | NAC, placebo, cognitive behavioral therapy | Evaluate the effects of NAC in reducing AUD severity and PTSD symptomatology | Active, not recruiting | NA |
| Melatonin Use for Sleep Problems in AD Patients | NCT03043443 | 60 | Alcohol-related disorders | Melatonin, placebo | Record sleeping problems | Completed | NA |
| NAC Treatment of AUD In Veterans With TBI | NCT02791945 | 30 | TBI, hazardous and harmful alcohol use | Medical management counseling, NAC, placebo | Assess the efficacy of NAC in reducing alcohol use and improving brain injury symptoms in veterans with mTBI who consume alcohol at hazardous or harmful levels | Completed | No statistically significant results |
| Imaging GABAergic/Glutamatergic Drugs in Bipolar Alcoholics | NCT03220776 | 81 | AUD, BD | NAC, gabapentin, placebo | Manipulate neurochemical Dysfunctions characteristic of individuals with co-occurring BD and AUD, using gabapentin and NAC, and evaluate medication-related changes in response inhibition and alcohol cue-reactivity fMRI tasks, as well as drinking and mood in individuals with AUD + BD | Recruiting | NA |
| NAC for Treating Comorbid PTSD and SUD | NCT02911285 | 90 | PTSD, AUD, SUD | NAC, placebo, cognitive behavioral therapy | Determine the benefits of NAC in treating AUD and comorbid PTSD | Completed | Changes in AUD and PTSD severity and alcohol craving |
| Herbal Supplements for Improvement of Liver Function in Participants with Alcoholic Liver Disease | NCT03503708 | 40 | Alcoholic liver disease | Livitol-70 | Study the efficacy of the herbal supplement to improve liver functioning of alcoholic liver disease patients | Not yet recruiting | NA |
| Effect of NAC on Alcohol and Cocaine Use Disorders: A Double-Blind Randomized Controlled Trial | NCT03018236 | 100 | Cocaine addiction, alcohol addiction | Alcohol NAC, alcohol placebo, cocaine NAC, cocaine placebo | Evaluate the use of NAC in the treatment of alcohol and cocaine use disorders | Unknown | NA |
| Antioxidant Replacement Therapy in Patients with Alcohol Abuse | NCT00936000 | 38 | Alcohol abuse | Protandim | Determine the safety and efficacy of in vivo antioxidant replacement therapy on alveolar–capillary barrier function in individuals with a history of chronic alcohol abuse | Completed | NA |
| A Study of Pleiotropic Pioglitazone Effects on the Alcoholic Lung (APPEAL Study) | NCT03060772 | 50 | Alcoholism | Pioglitazone | Measure the effect of pioglitazone | Terminated due to the fact of COVID-19 | NA |
| NAC in AD | NCT00568087 | 46 | Alcoholism | NAC, placebo | Find out if NAC reduces alcohol drinking and craving | Completed | Reduction in heavy drinking days for both groups |
| NAC plus Naltrexone for the Treatment of AD | NCT01214083 | 111 | Alcoholism | NAC plus high-dose naltrexone, high-dose naltrexone alone, low-dose naltrexone alone | Determine which of these combinations works better in reducing alcohol drinking | Completed | Reduction in heavy drinking days for all groups |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsermpini, E.E.; Plemenitaš Ilješ, A.; Dolžan, V. Alcohol-Induced Oxidative Stress and the Role of Antioxidants in Alcohol Use Disorder: A Systematic Review. Antioxidants 2022, 11, 1374. https://doi.org/10.3390/antiox11071374
Tsermpini EE, Plemenitaš Ilješ A, Dolžan V. Alcohol-Induced Oxidative Stress and the Role of Antioxidants in Alcohol Use Disorder: A Systematic Review. Antioxidants. 2022; 11(7):1374. https://doi.org/10.3390/antiox11071374
Chicago/Turabian StyleTsermpini, Evangelia Eirini, Anja Plemenitaš Ilješ, and Vita Dolžan. 2022. "Alcohol-Induced Oxidative Stress and the Role of Antioxidants in Alcohol Use Disorder: A Systematic Review" Antioxidants 11, no. 7: 1374. https://doi.org/10.3390/antiox11071374
APA StyleTsermpini, E. E., Plemenitaš Ilješ, A., & Dolžan, V. (2022). Alcohol-Induced Oxidative Stress and the Role of Antioxidants in Alcohol Use Disorder: A Systematic Review. Antioxidants, 11(7), 1374. https://doi.org/10.3390/antiox11071374