
The Flavonoid Hesperidin Methyl Chalcone Targets Cytokines and Oxidative Stress to Reduce Diclofenac-Induced Acute Renal Injury: Contribution of the Nrf2 Redox-Sensitive Pathway

 , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Compounds Used in the Study
2.3. Evaluation of Renal Function Markers
2.4. Ferric-Reducing Ability Potential (FRAP), 2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic Acid) (ABTS•+) Radical Cation, and Reduced Glutathione (GSH) Assays
2.5. Assessment of Thiobarbituric Acid-Reactive Substances (TBARS)
2.6. Evaluation of Cytokines and Neutrophil Gelatinase-Associated Lipocalin (NGAL) Production
2.7. Histopathological and Swelling Evaluations
2.8. Reverse Transcription and Quantitative Polymerase Chain Reaction (RT-qPCR) Assay
2.9. Immunofluorescence Assay in Confocal Microscopy
2.10. Statistical Methodology
3. Results
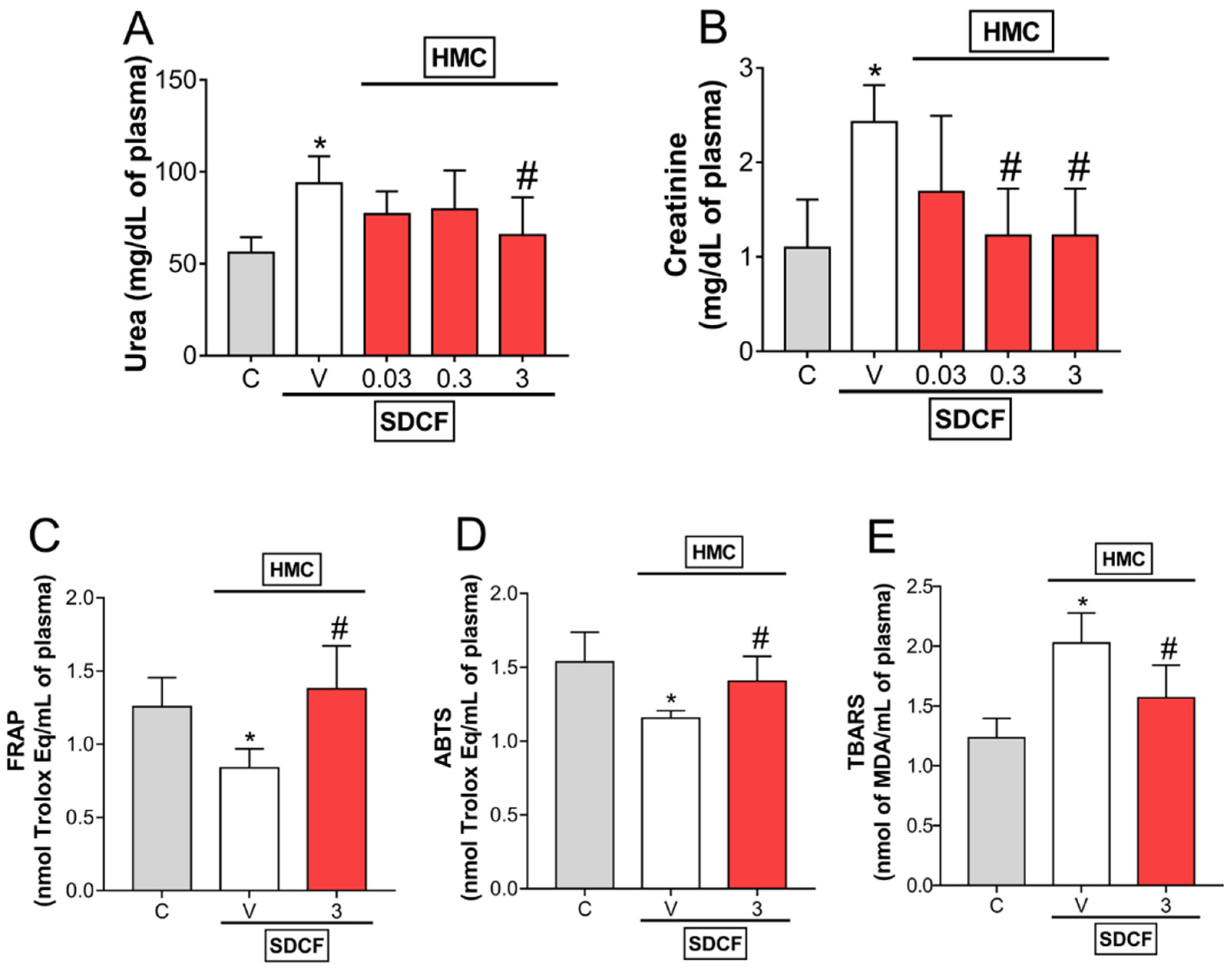
3.1. HMC Reduces SDCF-Triggered Renal Dysfunction: Urea and Creatinine Levels, and Oxidative Stress in Plasma
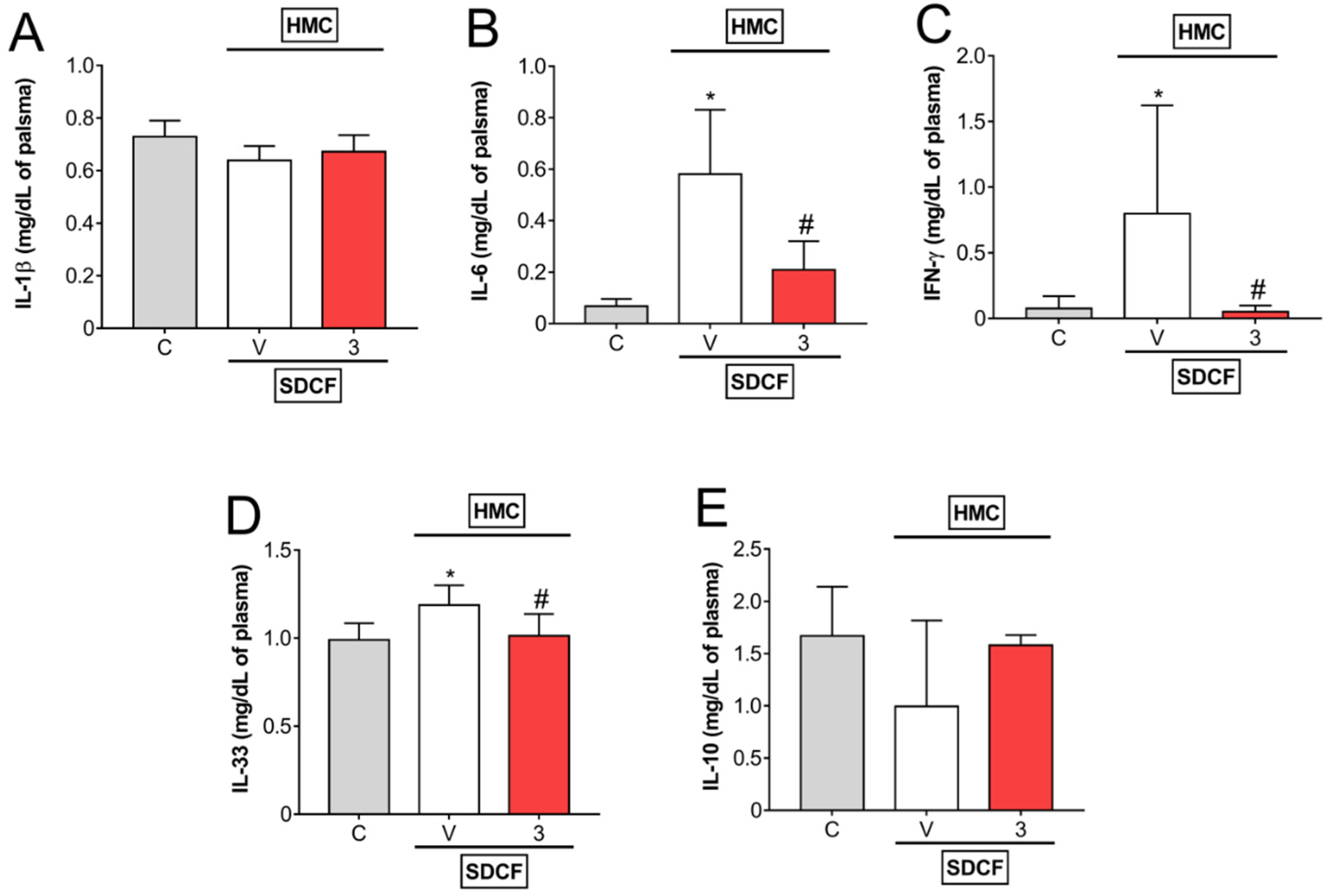
3.2. HMC Reduces IL-6, IFN-γ, and IL-33, but Does Not Modify IL-1β and IL-10 Levels in Plasma
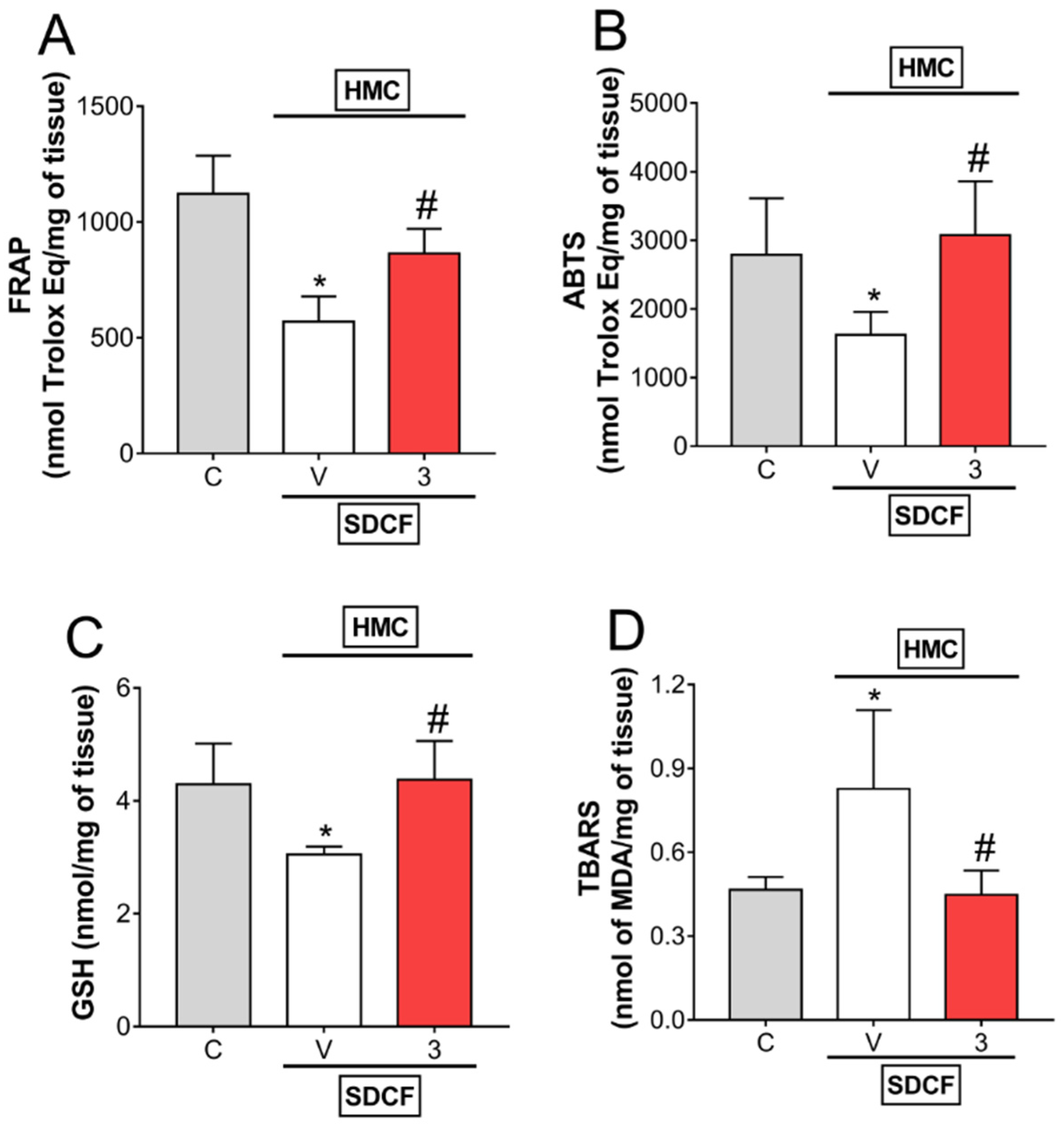
3.3. HMC Reduces Oxidative Stress in Renal Tissue
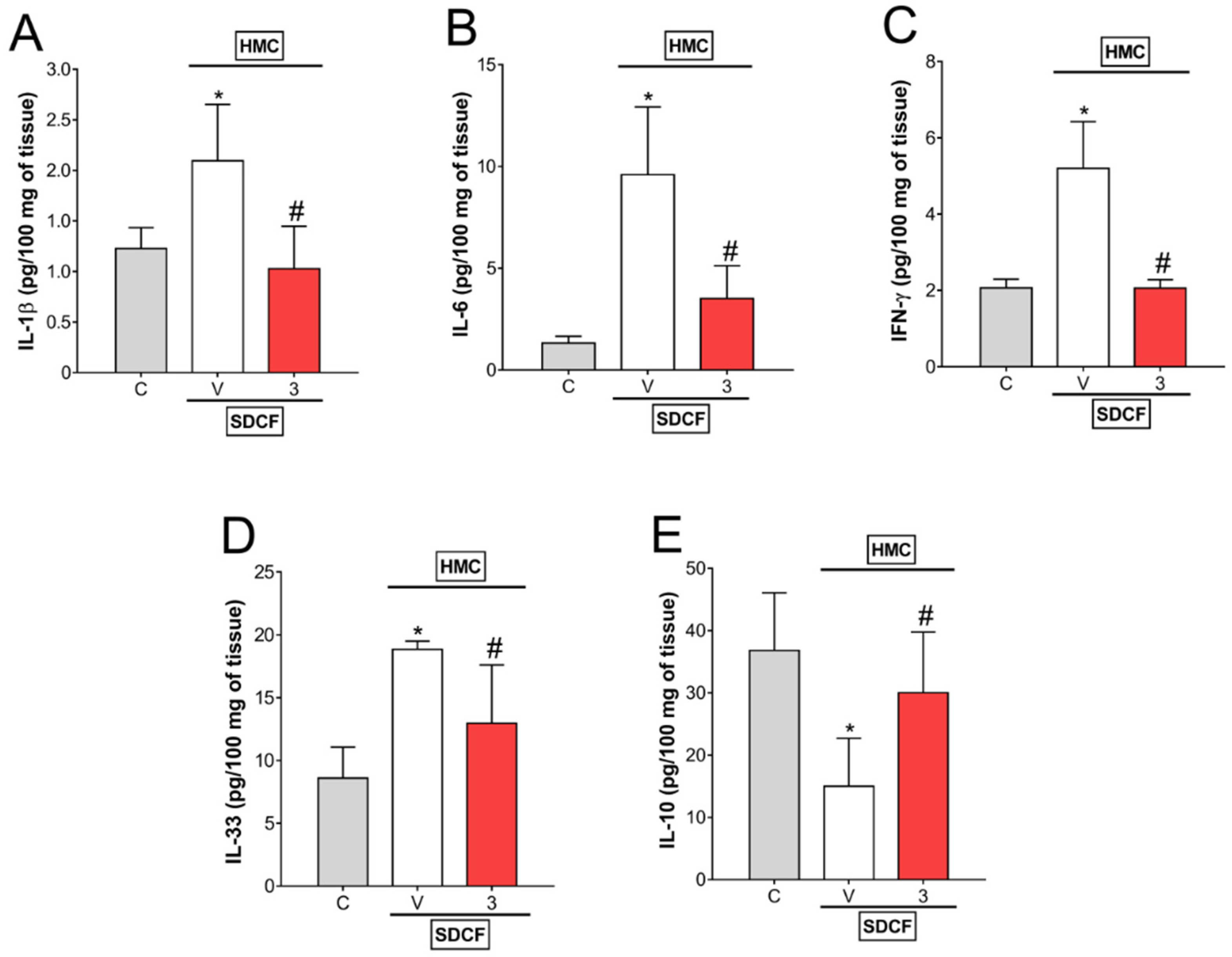
3.4. HMC Reduces IL-1β, IL-6, IFN-γ, and IL-33, as well as Increases IL-10 Levels in Renal Tissue
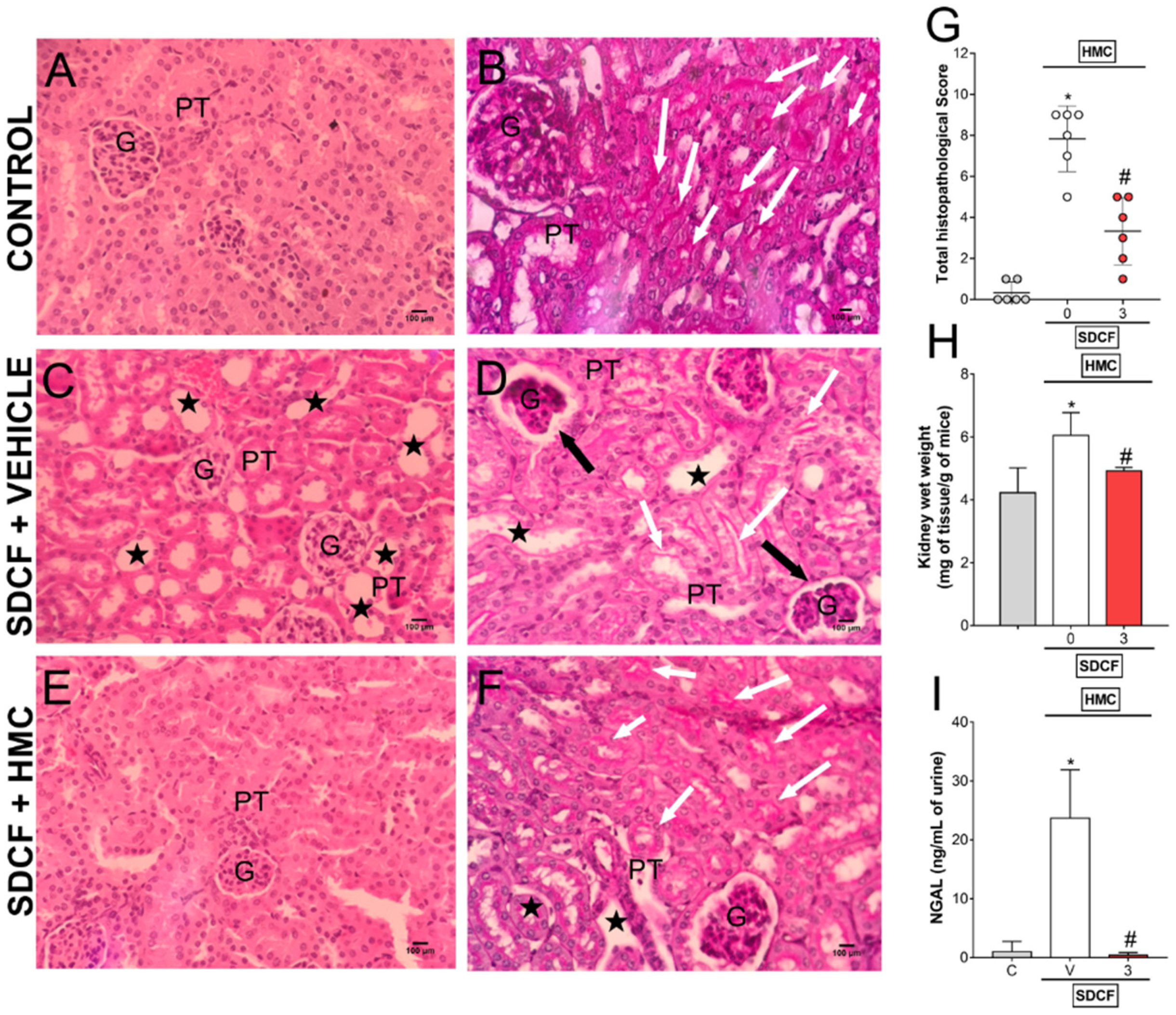
3.5. HMC Reduces SDCF-Induced Renal Histopathology, Swelling and Tubular Cells Cytotoxicity
3.6. HMC induces Nrf2 Signaling to Reduce SDCF-Induced AKI
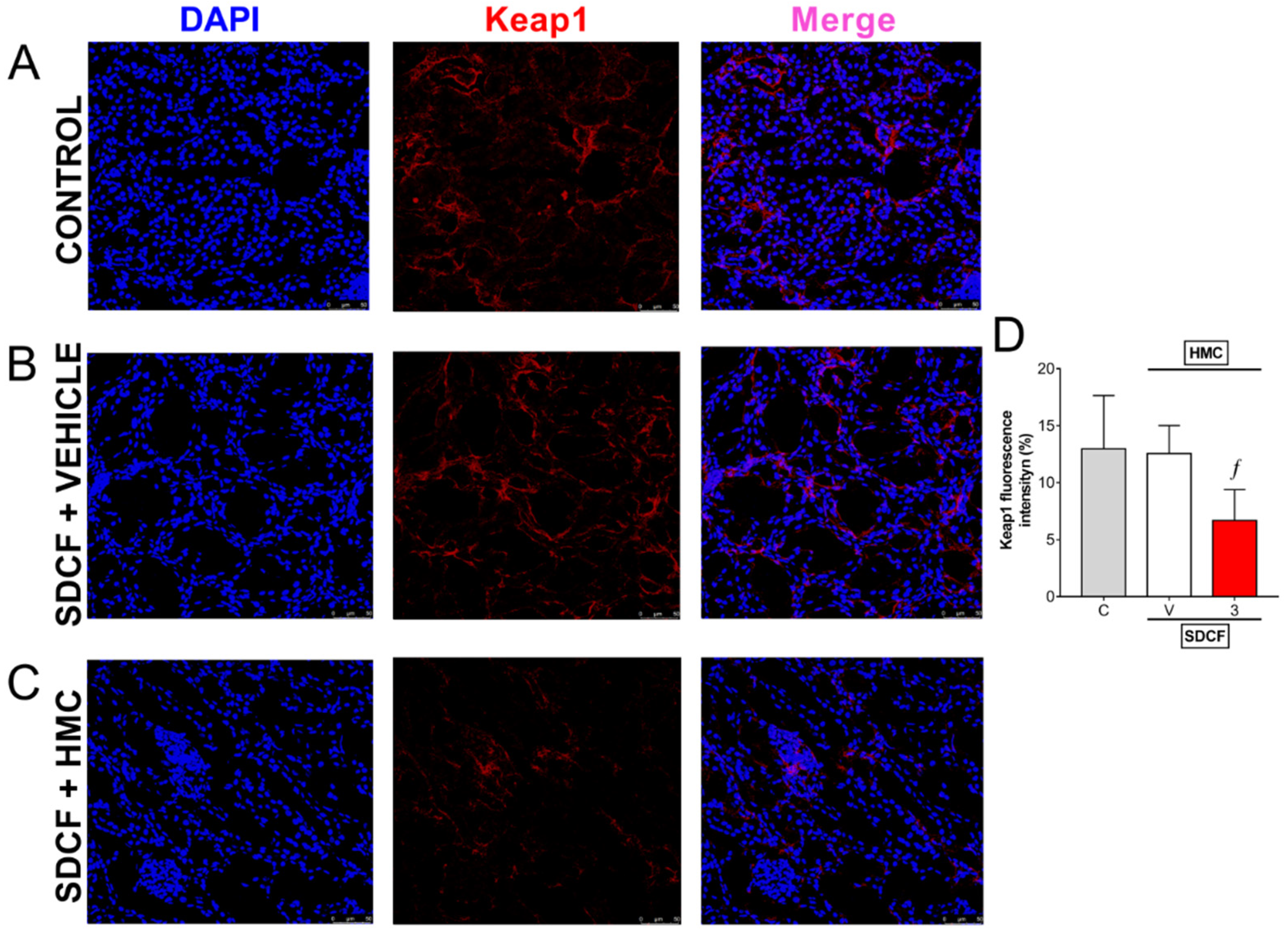
3.7. HMC Reduces Keap1 in the Kidney
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Borghi, S.M.; Fattori, V.; Ruiz-Miyazawa, K.W.; Bertozzi, M.M.; Lourenco-Gonzalez, Y.; Tatakihara, R.I.; Bussmann, A.J.C.; Mazzuco, T.L.; Casagrande, R.; Verri, W.A., Jr. Pyrrolidine dithiocarbamate inhibits mouse acute kidney injury induced by diclofenac by targeting oxidative damage, cytokines and NF-kappaB activity. Life Sci. 2018, 208, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Donnan, P.T.; Bell, S.; Guthrie, B. Non-steroidal anti-inflammatory drug induced acute kidney injury in the community dwelling general population and people with chronic kidney disease: Systematic review and meta-analysis. BMC Nephrol. 2017, 18, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungprasert, P.; Cheungpasitporn, W.; Crowson, C.S.; Matteson, E.L. Individual non-steroidal anti-inflammatory drugs and risk of acute kidney injury: A systematic review and meta-analysis of observational studies. Eur. J. Intern. Med. 2015, 26, 285–291. [Google Scholar] [CrossRef]
- Ejaz, P.; Bhojani, K.; Joshi, V.R. NSAIDs and kidney. J. Assoc. Physicians India 2004, 52, 632–640. [Google Scholar]
- Lucas, G.N.C.; Leitao, A.C.C.; Alencar, R.L.; Xavier, R.M.F.; Daher, E.F.; Silva Junior, G.B.D. Pathophysiological aspects of nephropathy caused by non-steroidal anti-inflammatory drugs. J. Bras. Nefrol. 2019, 41, 124–130. [Google Scholar] [CrossRef] [Green Version]
- McGettigan, P.; Henry, D. Use of non-steroidal anti-inflammatory drugs that elevate cardiovascular risk: An examination of sales and essential medicines lists in low-, middle-, and high-income countries. PLoS Med. 2013, 10, e1001388. [Google Scholar] [CrossRef]
- Altman, R.; Bosch, B.; Brune, K.; Patrignani, P.; Young, C. Advances in NSAID development: Evolution of diclofenac products using pharmaceutical technology. Drugs 2015, 75, 859–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, P.; Koncz, T.; Pan, S.; Lowry, S. Analgesic effectiveness of celecoxib and diclofenac in patients with osteoarthritis of the hip requiring joint replacement surgery: A 12-week, multicenter, randomized, double-blind, parallel-group, double-dummy, noninferiority study. Clin. Ther. 2008, 30, 70–83. [Google Scholar] [CrossRef]
- Fattori, V.; Borghi, S.M.; Guazelli, C.F.; Giroldo, A.C.; Crespigio, J.; Bussmann, A.J.; Coelho-Silva, L.; Ludwig, N.G.; Mazzuco, T.L.; Casagrande, R.; et al. Vinpocetine reduces diclofenac-induced acute kidney injury through inhibition of oxidative stress, apoptosis, cytokine production, and NF-kappaB activation in mice. Pharmacol. Res. 2017, 120, 10–22. [Google Scholar] [CrossRef]
- Dreiser, R.L.; Marty, M.; Ionescu, E.; Gold, M.; Liu, J.H. Relief of acute low back pain with diclofenac-K 12.5 mg tablets: A flexible dose, ibuprofen 200 mg and placebo-controlled clinical trial. Int. J. Clin. Pharmacol. Ther. 2003, 41, 375–385. [Google Scholar] [CrossRef]
- Schmidt, M.; Sorensen, H.T.; Pedersen, L. Diclofenac use and cardiovascular risks: Series of nationwide cohort studies. BMJ 2018, 362, k3426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.F.; Harris, R.C. Cyclooxygenases, the kidney, and hypertension. Hypertension 2004, 43, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Gan, T.J. Diclofenac: An update on its mechanism of action and safety profile. Curr. Med. Res. Opin. 2010, 26, 1715–1731. [Google Scholar] [CrossRef] [PubMed]
- Abiola, T.S.; Adebayo, O.C.; Babalola, O.O. Diclofenac-Induced Kidney Damage in Wistar Rats: Involvement of Antioxidant Mechanism. J. Biosci. Med. 2019, 7, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Alkuraishy, H.M.; Al-Gareeb, A.I.; Hussien, N.R. Diclofenac-induced acute kidney injury is linked with oxidative stress and pro-inflammatory changes in sprague-dawley rats. J. Contemp. Med. Sci. 2019, 5, 140–144. [Google Scholar] [CrossRef]
- Hickey, E.J.; Raje, R.R.; Reid, V.E.; Gross, S.M.; Ray, S.D. Diclofenac induced in vivo nephrotoxicity may involve oxidative stress-mediated massive genomic DNA fragmentation and apoptotic cell death. Free Radic. Biol. Med. 2001, 31, 139–152. [Google Scholar] [CrossRef]
- Nizamutdinova, I.T.; Jeong, J.J.; Xu, G.H.; Lee, S.H.; Kang, S.S.; Kim, Y.S.; Chang, K.C.; Kim, H.J. Hesperidin, hesperidin methyl chalone and phellopterin from Poncirus trifoliata (Rutaceae) differentially regulate the expression of adhesion molecules in tumor necrosis factor-alpha-stimulated human umbilical vein endothelial cells. Int. Immunopharmacol. 2008, 8, 670–678. [Google Scholar] [CrossRef]
- Ferraz, C.R.; Carvalho, T.T.; Manchope, M.F.; Artero, N.A.; Rasquel-Oliveira, F.S.; Fattori, V.; Casagrande, R.; Verri, W.A., Jr. Therapeutic Potential of Flavonoids in Pain and Inflammation: Mechanisms of Action, Pre-Clinical and Clinical Data, and Pharmaceutical Development. Molecules 2020, 25, 762. [Google Scholar] [CrossRef] [Green Version]
- Pinho-Ribeiro, F.A.; Hohmann, M.S.; Borghi, S.M.; Zarpelon, A.C.; Guazelli, C.F.; Manchope, M.F.; Casagrande, R.; Verri, W.A., Jr. Protective effects of the flavonoid hesperidin methyl chalcone in inflammation and pain in mice: Role of TRPV1, oxidative stress, cytokines and NF-kappaB. Chem. Biol. Interact. 2015, 228, 88–99. [Google Scholar] [CrossRef] [Green Version]
- Chanal, J.L.; Cousse, H.; Sicart, M.T.; Bonnaud, B.; Marignan, R. Absorption and elimination of (14C) hesperidin methylchalcone in the rat. Eur. J. Drug Metab. Pharmacokinet. 1981, 6, 171–177. [Google Scholar] [CrossRef]
- Gastillo, J.; Benavente, O.; Borrego, F. Analysis of commercial hesperidin methylchlcone by high performance liquid chromatography. J. Chromatogr. 1991, 555, 285–290. [Google Scholar] [CrossRef]
- Rasquel-Oliveira, F.S.; Manchope, M.F.; Staurengo-Ferrari, L.; Ferraz, C.R.; Saraiva-Santos, T.; Zaninelli, T.H.; Fattori, V.; Artero, N.A.; Badaro-Garcia, S.; de Freitas, A.; et al. Hesperidin methyl chalcone interacts with NFkappaB Ser276 and inhibits zymosan-induced joint pain and inflammation, and RAW 264.7 macrophage activation. Inflammopharmacology 2020, 28, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.M.; Pinho-Ribeiro, F.A.; Steffen, V.S.; Caviglione, C.V.; Pala, D.; Baracat, M.M.; Georgetti, S.R.; Verri, W.A.; Casagrande, R. Topical formulation containing hesperidin methyl chalcone inhibits skin oxidative stress and inflammation induced by ultraviolet B irradiation. Photochem. Photobiol. Sci. 2016, 15, 554–563. [Google Scholar] [CrossRef]
- Ruiz-Miyazawa, K.W.; Pinho-Ribeiro, F.A.; Borghi, S.M.; Staurengo-Ferrari, L.; Fattori, V.; Amaral, F.A.; Teixeira, M.M.; Alves-Filho, J.C.; Cunha, T.M.; Cunha, F.Q.; et al. Hesperidin Methylchalcone Suppresses Experimental Gout Arthritis in Mice by Inhibiting NF-kappaB Activation. J. Agric. Food Chem. 2018, 66, 6269–6280. [Google Scholar] [CrossRef]
- Jawien, A.; Bouskela, E.; Allaert, F.A.; Nicolaides, A.N. The place of Ruscus extract, hesperidin methyl chalcone, and vitamin C in the management of chronic venous disease. Int. Angiol. 2017, 36, 31–41. [Google Scholar] [CrossRef]
- Staurengo-Ferrari, L.; Badaro-Garcia, S.; Hohmann, M.S.N.; Manchope, M.F.; Zaninelli, T.H.; Casagrande, R.; Verri, W.A., Jr. Contribution of Nrf2 Modulation to the Mechanism of Action of Analgesic and Anti-inflammatory Drugs in Pre-clinical and Clinical Stages. Front. Pharmacol. 2018, 9, 1536. [Google Scholar] [CrossRef] [Green Version]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Kakkos, S.K.; Bouskela, E.; Jawien, A.; Nicolaides, A.N. New data on chronic venous disease: A new place for Cyclo 3® Fort. Int. Angiol. 2018, 37, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Allaert, F.A.; Hugue, C.; Cazaubon, M.; Renaudin, J.M.; Clavel, T.; Escourrou, P. Correlation between improvement in functional signs and plethysmographic parameters during venoactive treatment (Cyclo 3 Fort). Int. Angiol. 2011, 30, 272–277. [Google Scholar]
- Stoianova, V. Cyclo 3 fort—Alternative in chronic venous insufficiency. Akush. Ginekol. (Sofiia) 2006, 45 (Suppl. 3), 78–80. [Google Scholar]
- Beltramino, R.; Penenory, A.; Buceta, A.M. An open-label, randomized multicenter study comparing the efficacy and safety of Cyclo 3 Fort versus hydroxyethyl rutoside in chronic venous lymphatic insufficiency. Angiology 2000, 51, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Kirtley, W.R.; Peck, F.B. Administration of massive doses of vitamin P hesperidin methyl chalcone. Am. J. Med. Sci. 1948, 216, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Wood, R.C., 3rd; Wyatt, J.E.; Bullins, K.W.; Hanley, A.V.; Hanley, G.A.; Denham, J.W.; Panus, P.C.; Harirforoosh, S. Effects of rebamipide on nephrotoxicity associated with selected NSAIDs in rats. Eur. J. Pharmacol. 2013, 720, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Borghi, S.M.; Domiciano, T.P.; Rasquel-Oliveira, F.S.; Ferraz, C.R.; Bussmann, A.J.C.; Vignoli, J.A.; Camilios-Neto, D.; Ambrosio, S.R.; Arakawa, N.S.; Casagrande, R.; et al. Sphagneticola trilobata (L.) Pruski-derived kaurenoic acid prevents ovalbumin-induced asthma in mice: Effect on Th2 cytokines, STAT6/GATA-3 signaling, NFkappaB/Nrf2 redox sensitive pathways, and regulatory T cell phenotype markers. J. Ethnopharmacol. 2022, 283, 114708. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M.; Anderson, K.E. Clinical pharmacokinetics of diclofenac. Therapeutic insights and pitfalls. Clin. Pharmacokinet. 1997, 33, 184–213. [Google Scholar] [CrossRef]
- Huo, X.; Meng, Q.; Wang, C.; Wu, J.; Wang, C.; Zhu, Y.; Ma, X.; Sun, H.; Liu, K. Protective effect of cilastatin against diclofenac-induced nephrotoxicity through interaction with diclofenac acyl glucuronide via organic anion transporters. Br. J. Pharmacol. 2020, 177, 1933–1948. [Google Scholar] [CrossRef]
- Sivaraj, R.; Umarani, S. Diclofenac-induced biochemical changes in nephrotoxicity among male Albino rats. Int. J. Basic Clin. Pharmacol. 2018, 7, 640–643. [Google Scholar]
- Bickel, M.; Khaykin, P.; Stephan, C.; Schmidt, K.; Buettner, M.; Amann, K.; Lutz, T.; Gute, P.; Haberl, A.; Geiger, H.; et al. Acute kidney injury caused by tenofovir disoproxil fumarate and diclofenac co-administration. HIV Med. 2013, 14, 633–638. [Google Scholar] [CrossRef]
- Babladi, V.P.; Patil, N.; Manjunath, G.; Salimath, P.V.; Ninne, S.R.; Chary, K.M. A Case Report on Diclofenac Induced Chronic Kidney Disease. Indian J. Pharm. Pract. 2019, 12, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, R.S.; Lokhandwala, M.F.; Banday, A.A. Age-Related Mitochondrial Impairment and Renal Injury Is Ameliorated by Sulforaphane via Activation of Transcription Factor NRF2. Antioxidants 2022, 11, 156. [Google Scholar] [CrossRef]
- Van der Heijden, R.A.; Bijzet, J.; Meijers, W.C.; Yakala, G.K.; Kleemann, R.; Nguyen, T.Q.; de Boer, R.A.; Schalkwijk, C.G.; Hazenberg, B.P.; Tietge, U.J.; et al. Obesity-induced chronic inflammation in high fat diet challenged C57BL/6J mice is associated with acceleration of age-dependent renal amyloidosis. Sci. Rep. 2015, 5, 16474. [Google Scholar] [CrossRef]
- Guazelli, C.F.S.; Fattori, V.; Ferraz, C.R.; Borghi, S.M.; Casagrande, R.; Baracat, M.M.; Verri, W.A., Jr. Antioxidant and anti-inflammatory effects of hesperidin methyl chalcone in experimental ulcerative colitis. Chem. Biol. Interact. 2021, 333, 109315. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [PubMed] [Green Version]
- Edelstein, C.L. Biomarkers of acute kidney injury. Adv. Chronic Kidney Dis. 2008, 15, 222–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devarajan, P. Neutrophil gelatinase-associated lipocalin (NGAL): A new marker of kidney disease. Scand. J. Clin. Lab. Investig. Suppl. 2008, 241, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Yang, H.; Chen, H.; Zhang, M.; Ma, Q. High expression of neutrophil gelatinase-associated lipocalin (NGAL) in the kidney proximal tubules of diabetic rats. Adv. Med. Sci. 2015, 60, 133–138. [Google Scholar] [CrossRef]
- Kuwabara, T.; Mori, K.; Mukoyama, M.; Kasahara, M.; Yokoi, H.; Saito, Y.; Yoshioka, T.; Ogawa, Y.; Imamaki, H.; Kusakabe, T.; et al. Urinary neutrophil gelatinase-associated lipocalin levels reflect damage to glomeruli, proximal tubules, and distal nephrons. Kidney Int. 2009, 75, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Abdou, R.M.; El-Maadawy, W.H.; Hassan, M.; El-Dine, R.S.; Aboushousha, T.; El-Tanbouly, N.D.; El-Sayed, A.M. Nephroprotective activity of Aframomum melegueta seeds extract against diclofenac-induced acute kidney injury: A mechanistic study. J. Ethnopharmacol. 2021, 273, 113939. [Google Scholar] [CrossRef]
- Lee, D.W.; Faubel, S.; Edelstein, C.L. Cytokines in acute kidney injury (AKI). Clin. Nephrol. 2011, 76, 165–173. [Google Scholar] [CrossRef]
- Kinsey, G.R.; Okusa, M.D. Role of leukocytes in the pathogenesis of acute kidney injury. Crit. Care 2012, 16, 214. [Google Scholar] [CrossRef] [Green Version]
- Naito, Y.; Takagi, T.; Higashimura, Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch. Biochem. Biophys. 2014, 564, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sverrisson, K.; Axelsson, J.; Rippe, A.; Asgeirsson, D.; Rippe, B. Acute reactive oxygen species (ROS)-dependent effects of IL-1beta, TNF-alpha, and IL-6 on the glomerular filtration barrier (GFB) in vivo. Am. J. Physiol. Renal. Physiol. 2015, 309, F800–F806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, R.M.; Pinho-Ribeiro, F.A.; Steffen, V.S.; Caviglione, C.V.; Vignoli, J.A.; Baracat, M.M.; Georgetti, S.R.; Verri, W.A., Jr.; Casagrande, R. Hesperidin methyl chalcone inhibits oxidative stress and inflammation in a mouse model of ultraviolet B irradiation-induced skin damage. J. Photochem. Photobiol. B 2015, 148, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [Green Version]
- Hohmann, M.S.; Zaninelli, T.H.; Staurengo-Ferrari, L.; Manchope, M.F.; Badaro-Garcia, S.; de Freitas, A.; Casagrande, R.; Verri, W.A.J. Nrf2 in Immune Responses during Inflammation. In Nrf2 and Its Modulation in Inflammation; Deng, H., Ed.; Springer: Cham, Switzerland, 2020; pp. 23–50. [Google Scholar]
- Rockwell, C.E.; Zhang, M.; Fields, P.E.; Klaassen, C.D. Th2 skewing by activation of Nrf2 in CD4+ T cells. J. Immunol. 2012, 188, 1630–1637. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bussmann, A.J.C.; Zaninelli, T.H.; Saraiva-Santos, T.; Fattori, V.; Guazelli, C.F.S.; Bertozzi, M.M.; Andrade, K.C.; Ferraz, C.R.; Camilios-Neto, D.; Casella, A.M.B.; et al. The Flavonoid Hesperidin Methyl Chalcone Targets Cytokines and Oxidative Stress to Reduce Diclofenac-Induced Acute Renal Injury: Contribution of the Nrf2 Redox-Sensitive Pathway. Antioxidants 2022, 11, 1261. https://doi.org/10.3390/antiox11071261
Bussmann AJC, Zaninelli TH, Saraiva-Santos T, Fattori V, Guazelli CFS, Bertozzi MM, Andrade KC, Ferraz CR, Camilios-Neto D, Casella AMB, et al. The Flavonoid Hesperidin Methyl Chalcone Targets Cytokines and Oxidative Stress to Reduce Diclofenac-Induced Acute Renal Injury: Contribution of the Nrf2 Redox-Sensitive Pathway. Antioxidants. 2022; 11(7):1261. https://doi.org/10.3390/antiox11071261
Chicago/Turabian StyleBussmann, Allan J. C., Tiago H. Zaninelli, Telma Saraiva-Santos, Victor Fattori, Carla F. S. Guazelli, Mariana M. Bertozzi, Ketlem C. Andrade, Camila R. Ferraz, Doumit Camilios-Neto, Antônio M. B. Casella, and et al. 2022. "The Flavonoid Hesperidin Methyl Chalcone Targets Cytokines and Oxidative Stress to Reduce Diclofenac-Induced Acute Renal Injury: Contribution of the Nrf2 Redox-Sensitive Pathway" Antioxidants 11, no. 7: 1261. https://doi.org/10.3390/antiox11071261
APA StyleBussmann, A. J. C., Zaninelli, T. H., Saraiva-Santos, T., Fattori, V., Guazelli, C. F. S., Bertozzi, M. M., Andrade, K. C., Ferraz, C. R., Camilios-Neto, D., Casella, A. M. B., Casagrande, R., Borghi, S. M., & Verri, W. A., Jr. (2022). The Flavonoid Hesperidin Methyl Chalcone Targets Cytokines and Oxidative Stress to Reduce Diclofenac-Induced Acute Renal Injury: Contribution of the Nrf2 Redox-Sensitive Pathway. Antioxidants, 11(7), 1261. https://doi.org/10.3390/antiox11071261