
Resolution of Inflammation after Skeletal Muscle Ischemia–Reperfusion Injury: A Focus on the Lipid Mediators Lipoxins, Resolvins, Protectins and Maresins

 , , ,
, , ,
Abstract
:1. Introduction
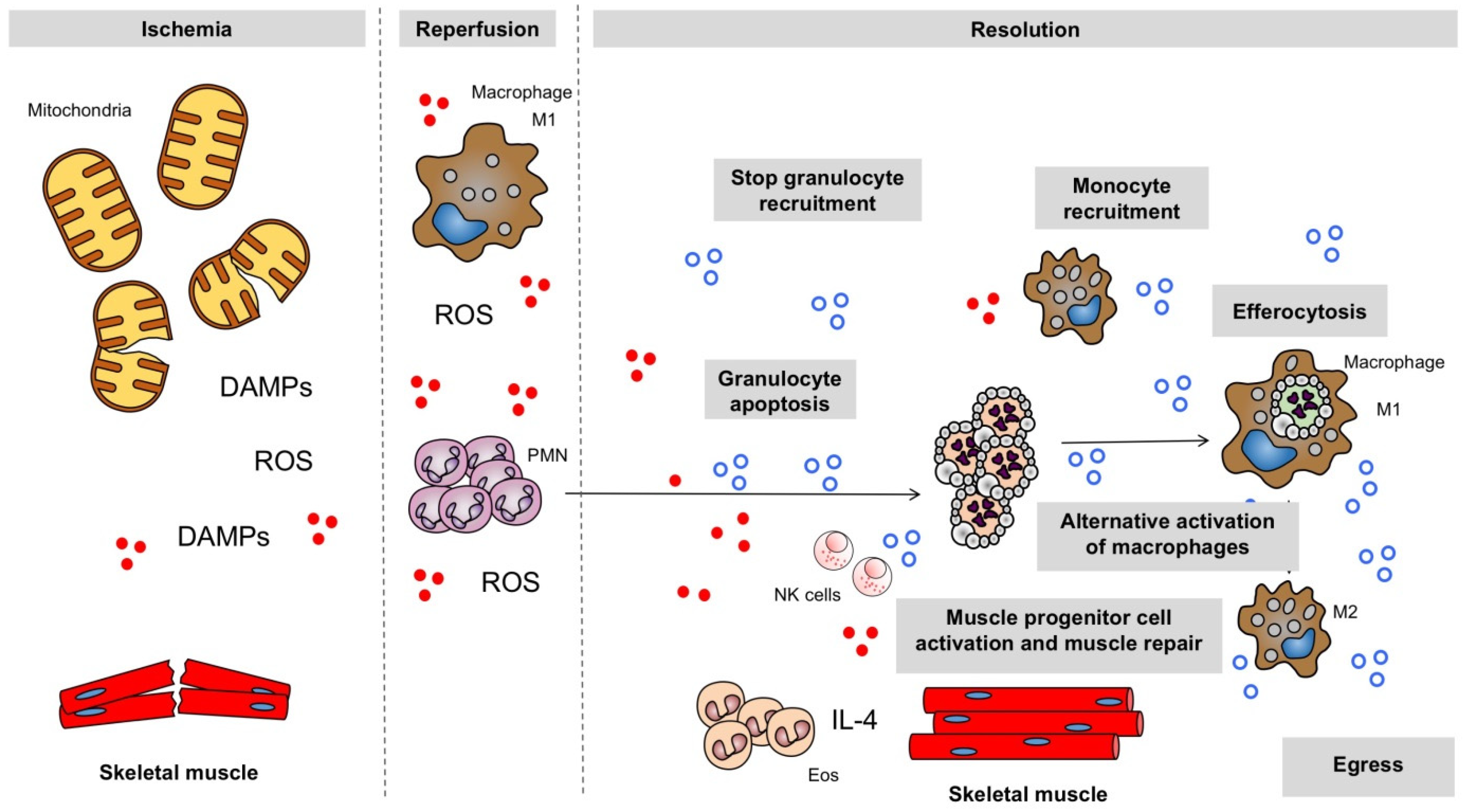
2. Inflammatory Response to Ischemia and Reperfusion in the Skeletal Muscle
2.1. Oxidative Stress
2.1.1. Deleterious Action of ROS
2.1.2. Sources of ROS
2.2. Mitochondrial Failure
2.3. Endogenous Danger Molecules
2.4. Chemokines and Cytokines
2.5. Neutrophils and Pro-Inflammatory Macrophages
2.6. Activation of Complement
3. Principles of Resolution of Inflammation
4. Specialized Pro-Resolving Lipid Mediators
5. Resolution Mechanisms in IRI: Involvement of Immune Cells and of Specialized Pro-Resolving Mediators
5.1. Macrophages
5.2. Eosinophils
5.3. Specialized Pro-Resolving Mediators (SPMs)
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Serhan, C.N. Treating Inflammation and Infection in the 21st Century: New Hints from Decoding Resolution Mediators and Mechanisms. FASEB J. 2017, 31, 1273–1288. [Google Scholar] [CrossRef] [Green Version]
- Pizzimenti, M.; Riou, M.; Charles, A.-L.; Talha, S.; Meyer, A.; Andres, E.; Chakfé, N.; Lejay, A.; Geny, B. The Rise of Mitochondria in Peripheral Arterial Disease Physiopathology: Experimental and Clinical Data. JCM 2019, 8, 2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merchant, S.H.; Gurule, D.M.; Larson, R.S. Amelioration of Ischemia-Reperfusion Injury with Cyclic Peptide Blockade of ICAM-1. Am. J. Physiol. Heart. Circ. Physiol. 2003, 284, H1260–H1268. [Google Scholar] [CrossRef] [PubMed]
- Defraigne, J.O.; Pincemail, J. Local and Systemic Consequences of Severe Ischemia and Reperfusion of the Skeletal Muscle. Physiopathology and Prevention. Acta Chir. Belg. 1998, 98, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Paradis, S.; Charles, A.-L.; Meyer, A.; Lejay, A.; Scholey, J.W.; Chakfé, N.; Zoll, J.; Geny, B. Chronology of Mitochondrial and Cellular Events during Skeletal Muscle Ischemia-Reperfusion. Am. J. Physiol. Cell Physiol. 2016, 310, C968–C982. [Google Scholar] [CrossRef] [Green Version]
- Larsen, G.L.; Henson, P.M. Mediators of Inflammation. Annu. Rev. Immunol. 1983, 1, 335–359. [Google Scholar] [CrossRef]
- Guillot, M.; Charles, A.-L.; Chamaraux-Tran, T.N.; Bouitbir, J.; Meyer, A.; Zoll, J.; Schneider, F.; Geny, B. Oxidative Stress Precedes Skeletal Muscle Mitochondrial Dysfunction during Experimental Aortic Cross-Clamping but Is Not Associated with Early Lung, Heart, Brain, Liver, or Kidney Mitochondrial Impairment. J. Vasc. Surg. 2014, 60, 1043–1051.e5. [Google Scholar] [CrossRef] [Green Version]
- Ghaly, A.; Marsh, D.R. Ischaemia-Reperfusion Modulates Inflammation and Fibrosis of Skeletal Muscle after Contusion Injury. Int. J. Exp. Pathol. 2010, 91, 244–255. [Google Scholar] [CrossRef]
- Yokoyama, H.; Tsujii, M.; Iino, T.; Nakamura, T.; Sudo, A. Inhibitory Effect of Edaravone on Systemic Inflammation and Local Damage in Skeletal Muscles Following Long-Term Ischemia to Murine Hind Limb. J. Orthop. Surg. 2019, 27, 2309499019874470. [Google Scholar] [CrossRef]
- Rodríguez-Lara, S.Q.; Trujillo-Rangel, W.A.; Castillo-Romero, A.; Totsuka-Sutto, S.E.; Garcia-Cobián, T.A.; Cardona-Muñoz, E.G.; Miranda-Díaz, A.G.; Ramírez-Lizardo, E.J.; García-Benavides, L. Effect of Telmisartan in the Oxidative Stress Components Induced by Ischemia Reperfusion in Rats. Oxid. Med. Cell. Longev. 2019, 2019, 1302985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lejay, A.; Paradis, S.; Lambert, A.; Charles, A.-L.; Talha, S.; Enache, I.; Thaveau, F.; Chakfe, N.; Geny, B. N-Acetyl Cysteine Restores Limb Function, Improves Mitochondrial Respiration, and Reduces Oxidative Stress in a Murine Model of Critical Limb Ischaemia. Eur. J. Vasc. Endovasc. Surg. 2018, 56, 730–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.K.; Grisham, M.B.; Granger, D.N.; Korthuis, R.J. Free Radical Defense Mechanisms and Neutrophil Infiltration in Postischemic Skeletal Muscle. Am. J. Physiol. 1989, 256, H789–H793. [Google Scholar] [CrossRef] [PubMed]
- Charles, A.-L.; Guilbert, A.-S.; Guillot, M.; Talha, S.; Lejay, A.; Meyer, A.; Kindo, M.; Wolff, V.; Bouitbir, J.; Zoll, J.; et al. Muscles Susceptibility to Ischemia-Reperfusion Injuries Depends on Fiber Type Specific Antioxidant Level. Front. Physiol. 2017, 8, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velayutham, M.; Hemann, C.; Zweier, J.L. Removal of H2O2 and Generation of Superoxide Radical: Role of Cytochrome c and NADH. Free Radic. Biol. Med. 2011, 51, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Tschopp, J.; Schroder, K. NLRP3 Inflammasome Activation: The Convergence of Multiple Signalling Pathways on ROS Production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D.; Goncalves, R.L.S.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Borch Jensen, M.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T.; et al. Suppressors of Superoxide-H2O2 Production at Site IQ of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell. Metab. 2016, 24, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, D.L.; Brookes, P.S. Oxygen Sensitivity of Mitochondrial Reactive Oxygen Species Generation Depends on Metabolic Conditions. J. Biol. Chem. 2009, 284, 16236–16245. [Google Scholar] [CrossRef] [Green Version]
- Charles, A.-L.; Guilbert, A.-S.; Bouitbir, J.; Goette-Di Marco, P.; Enache, I.; Zoll, J.; Piquard, F.; Geny, B. Effect of Postconditioning on Mitochondrial Dysfunction in Experimental Aortic Cross-Clamping. Br. J. Surg. 2011, 98, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Brandão, M.L.; Roselino, J.E.S.; Piccinato, C.E.; Cherri, J. Mitochondrial Alterations in Skeletal Muscle Submitted to Total Ischemia. J. Surg. Res. 2003, 110, 235–240. [Google Scholar] [CrossRef]
- Onukwufor, J.O.; Berry, B.J.; Wojtovich, A.P. Physiologic Implications of Reactive Oxygen Species Production by Mitochondrial Complex I Reverse Electron Transport. Antioxidants 2019, 8, 285. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, T.; Romaschin, A.; Walker, P.M. Free Radical Mediated Damage in Skeletal Muscle. Microcirc Endothel. Lymphat. 1989, 5, 157–170. [Google Scholar]
- Kvietys, P.R.; Granger, D.N. Role of Reactive Oxygen and Nitrogen Species in the Vascular Responses to Inflammation. Free Radic. Biol. Med. 2012, 52, 556–592. [Google Scholar] [CrossRef] [Green Version]
- Lejay, A.; Meyer, A.; Schlagowski, A.-I.; Charles, A.-L.; Singh, F.; Bouitbir, J.; Pottecher, J.; Chakfé, N.; Zoll, J.; Geny, B. Mitochondria: Mitochondrial Participation in Ischemia-Reperfusion Injury in Skeletal Muscle. Int. J. Biochem. Cell. Biol. 2014, 50, 101–105. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The Release of Damage-Associated Molecular Patterns and Its Physiological Relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Powers, K.A.; Szászi, K.; Khadaroo, R.G.; Tawadros, P.S.; Marshall, J.C.; Kapus, A.; Rotstein, O.D. Oxidative Stress Generated by Hemorrhagic Shock Recruits Toll-like Receptor 4 to the Plasma Membrane in Macrophages. J. Exp. Med. 2006, 203, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Laverny, G.; Bernardi, L.; Charles, A.L.; Alsaleh, G.; Pottecher, J.; Sibilia, J.; Geny, B. Mitochondria: An Organelle of Bacterial Origin Controlling Inflammation. Front. Immunol. 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Youker, K.A.; Rossen, R.D.; Gwechenberger, M.; Lindsey, M.H.; Mendoza, L.H.; Michael, L.H.; Ballantyne, C.M.; Smith, C.W.; Entman, M.L. Cytokines and the Microcirculation in Ischemia and Reperfusion. J. Mol. Cell. Cardiol. 1998, 30, 2567–2576. [Google Scholar] [CrossRef] [PubMed]
- Corrick, R.M.; Tu, H.; Zhang, D.; Barksdale, A.N.; Muelleman, R.L.; Wadman, M.C.; Li, Y.-L. Dexamethasone Protects Against Tourniquet-Induced Acute Ischemia-Reperfusion Injury in Mouse Hindlimb. Front. Physiol. 2018, 9, 244. [Google Scholar] [CrossRef] [Green Version]
- Ascer, E.; Mohan, C.; Gennaro, M.; Cupo, S. Interleukin-1 and Thromboxane Release after Skeletal Muscle Ischemia and Reperfusion. Ann. Vasc. Surg. 1992, 6, 69–73. [Google Scholar] [CrossRef]
- Seekamp, A.; Warren, J.S.; Remick, D.G.; Till, G.O.; Ward, P.A. Requirements for Tumor Necrosis Factor-Alpha and Interleukin-1 in Limb Ischemia/Reperfusion Injury and Associated Lung Injury. Am. J. Pathol. 1993, 143, 453–463. [Google Scholar]
- Zhang, F.; Hu, E.C.; Gerzenshtein, J.; Lei, M.-P.; Lineaweaver, W.C. The Expression of Proinflammatory Cytokines in the Rat Muscle Flap with Ischemia-Reperfusion Injury. Ann. Plast. Surg. 2005, 54, 313–317. [Google Scholar]
- Wang, J. Neutrophils in Tissue Injury and Repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Tidball, J.G. Inflammatory Cell Response to Acute Muscle Injury. Med. Sci. Sports Exerc. 1995, 27, 1022–1032. [Google Scholar] [CrossRef]
- Menger, M.D.; Pelikan, S.; Steiner, D.; Messmer, K. Microvascular Ischemia-Reperfusion Injury in Striated Muscle: Significance of “Reflow Paradox”. Am. J. Physiol. 1992, 263, H1901–H1906. [Google Scholar] [CrossRef]
- Zimmerman, B.J.; Guillory, D.J.; Grisham, M.B.; Gaginella, T.S.; Granger, D.N. Role of Leukotriene B4 in Granulocyte Infiltration into the Postischemic Feline Intestine. Gastroenterology 1990, 99, 1358–1363. [Google Scholar] [CrossRef]
- Smith, J.K.; Carden, D.L.; Korthuis, R.J. Activated Neutrophils Increase Microvascular Permeability in Skeletal Muscle: Role of Xanthine Oxidase. J. Appl. Physiol. 1991, 70, 2003–2009. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, M.; Anderson, G.; Vĕtvicka, V.; Justus, D.E.; Ross, G.D. Microvascular Effects of Complement Blockade with Soluble Recombinant CR1 on Ischemia/Reperfusion Injury of Skeletal Muscle. J. Immunol. 1993, 150, 5104–5113. [Google Scholar]
- Lehr, H.A.; Guhlmann, A.; Nolte, D.; Keppler, D.; Messmer, K. Preservation of Postischemic Capillary Perfusion by Selective Inhibition of Leukotriene Biosynthesis. Transpl. Proc. 1991, 23, 833–834. [Google Scholar]
- Carden, D.L.; Korthuis, R.J. Mechanisms of Postischemic Vascular Dysfunction in Skeletal Muscle: Implications for Therapeutic Intervention. Microcirc Endothel. Lymphat. 1989, 5, 277–298. [Google Scholar]
- Granger, D.N. Role of Xanthine Oxidase and Granulocytes in Ischemia-Reperfusion Injury. Am. J. Physiol. 1988, 255, H1269–H1275. [Google Scholar] [CrossRef]
- Korthuis, R.J.; Grisham, M.B.; Granger, D.N. Leukocyte Depletion Attenuates Vascular Injury in Postischemic Skeletal Muscle. Am. J. Physiol. 1988, 254, H823–H827. [Google Scholar] [CrossRef]
- Yokota, J.; Minei, J.P.; Fantini, G.A.; Shires, G.T. Role of Leukocytes in Reperfusion Injury of Skeletal Muscle after Partial Ischemia. Am. J. Physiol. 1989, 257, H1068–H1075. [Google Scholar] [CrossRef]
- Rubin, B.B.; Chang, G.; Liauw, S.; Young, A.; Romaschin, A.; Walker, P.M. Phospholipid Peroxidation Deacylation and Remodeling in Postischemic Skeletal Muscle. Am. J. Physiol. 1992, 263, H1695–H1702. [Google Scholar] [CrossRef]
- Walden, D.L.; McCutchan, H.J.; Enquist, E.G.; Schwappach, J.R.; Shanley, P.F.; Reiss, O.K.; Terada, L.S.; Leff, J.A.; Repine, J.E. Neutrophils Accumulate and Contribute to Skeletal Muscle Dysfunction after Ischemia-Reperfusion. Am. J. Physiol. 1990, 259, H1809–H1812. [Google Scholar] [CrossRef]
- Kyriakides, C.; Austen, W.; Wang, Y.; Favuzza, J.; Kobzik, L.; Moore, F.D.; Hechtman, H.B. Skeletal Muscle Reperfusion Injury Is Mediated by Neutrophils and the Complement Membrane Attack Complex. Am. J. Physiol. 1999, 277, C1263–C1268. [Google Scholar] [CrossRef] [PubMed]
- Formigli, L.; Lombardo, L.D.; Adembri, C.; Brunelleschi, S.; Ferrari, E.; Novelli, G.P. Neutrophils as Mediators of Human Skeletal Muscle Ischemia-Reperfusion Syndrome. Hum. Pathol. 1992, 23, 627–634. [Google Scholar] [CrossRef]
- Gute, D.C.; Ishida, T.; Yarimizu, K.; Korthuis, R.J. Inflammatory Responses to Ischemia and Reperfusion in Skeletal Muscle. Mol. Cell Biochem. 1998, 179, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.X.; Tidball, J.G. Interactions between Neutrophils and Macrophages Promote Macrophage Killing of Rat Muscle Cells In Vitro. J. Physiol. 2003, 547, 125–132. [Google Scholar] [CrossRef]
- Oklu, R.; Albadawi, H.; Jones, J.E.; Yoo, H.-J.; Watkins, M.T. Reduced Hind Limb Ischemia-Reperfusion Injury in Toll-like Receptor-4 Mutant Mice Is Associated with Decreased Neutrophil Extracellular Traps. J. Vasc. Surg. 2013, 58, 1627–1636. [Google Scholar] [CrossRef] [Green Version]
- Wright, D.G.; Gallin, J.I. A Functional Differentiation of Human Neutrophil Granules: Generation of C5a by a Specific (Secondary) Granule Product and Inactivation of C5a by Azurophil (Primary) Granule Products. J. Immunol. 1977, 119, 1068–1076. [Google Scholar]
- Tidball, J.G. Inflammatory Processes in Muscle Injury and Repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R345–R353. [Google Scholar] [CrossRef] [Green Version]
- Farrar, C.A.; Asgari, E.; Schwaeble, W.J.; Sacks, S.H. Which Pathways Trigger the Role of Complement in Ischaemia/Reperfusion Injury? Front. Immunol. 2012, 3, 341. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, E.W.; Mollnes, T.E.; Harlan, J.M.; Winn, R.K. C1-Inhibitor Reduces the Ischaemia-Reperfusion Injury of Skeletal Muscles in Mice after Aortic Cross-Clamping. Scand. J. Immunol. 2002, 56, 588–592. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Arumugam, T.V.; Shiels, I.A.; Reid, R.C.; Fairlie, D.P.; Taylor, S.M. Protective Effects of a Potent C5a Receptor Antagonist on Experimental Acute Limb Ischemia-Reperfusion in Rats. J. Surg. Res. 2004, 116, 81–90. [Google Scholar] [CrossRef]
- Groeneveld, A.B.; Raijmakers, P.G.; Rauwerda, J.A.; Hack, C.E. The Inflammatory Response to Vascular Surgery-Associated Ischaemia and Reperfusion in Man: Effect on Postoperative Pulmonary Function. Eur. J. Vasc. Endovasc. Surg. 1997, 14, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Heideman, M.; Norder-Hansson, B.; Bengtson, A.; Mollnes, T.E. Terminal Complement Complexes and Anaphylatoxins in Septic and Ischemic Patients. Arch. Surg. 1988, 123, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Farrar, C.A.; Abe, K.; Pratt, J.R.; Marsh, J.E.; Wang, Y.; Stahl, G.L.; Sacks, S.H. Predominant Role for C5b-9 in Renal Ischemia/Reperfusion Injury. J. Clin. Investig. 2000, 105, 1363–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Q.; Li, K.; Smyth, L.A.; Xing, G.; Wang, N.; Meader, L.; Lu, B.; Sacks, S.H.; Zhou, W. C3a and C5a Promote Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2012, 23, 1474–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Levy, B.D. Resolvins in Inflammation: Emergence of the pro-Resolving Superfamily of Mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Barnig, C.; Cernadas, M.; Dutile, S.; Liu, X.; Perrella, M.A.; Kazani, S.; Wechsler, M.E.; Israel, E.; Levy, B.D. Lipoxin A4 Regulates Natural Killer Cell and Type 2 Innate Lymphoid Cell Activation in Asthma. Sci. Transl. Med. 2013, 5, 174ra26. [Google Scholar] [CrossRef] [Green Version]
- Barnig, C.; Frossard, N.; Levy, B.D. Towards Targeting Resolution Pathways of Airway Inflammation in Asthma. Pharmacol. Ther. 2018, 186, 98–113. [Google Scholar] [CrossRef]
- Levy, B.D.; Serhan, C.N. Resolution of Acute Inflammation in the Lung. Annu. Rev. Physiol. 2014, 76, 467–492. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving Inflammation: Dual Anti-Inflammatory and pro-Resolution Lipid Mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Barnig, C.; Bezema, T.; Calder, P.C.; Charloux, A.; Frossard, N.; Garssen, J.; Haworth, O.; Dilevskaya, K.; Levi-Schaffer, F.; Lonsdorfer, E.; et al. Activation of Resolution Pathways to Prevent and Fight Chronic Inflammation: Lessons From Asthma and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 1699. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils Promote the Development of Reparative Macrophages Mediated by ROS to Orchestrate Liver Repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef] [Green Version]
- Sendama, W. The Effect of Ageing on the Resolution of Inflammation. Ageing Res. Rev. 2020, 57, 101000. [Google Scholar] [CrossRef] [PubMed]
- Fiore, S.; Serhan, C.N. Formation of Lipoxins and Leukotrienes during Receptor-Mediated Interactions of Human Platelets and Recombinant Human Granulocyte/Macrophage Colony-Stimulating Factor-Primed Neutrophils. J. Exp. Med. 1990, 172, 1451–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid Mediator Class Switching during Acute Inflammation: Signals in Resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Hamberg, M.; Samuelsson, B. Lipoxins: Novel Series of Biologically Active Compounds Formed from Arachidonic Acid in Human Leukocytes. Proc. Natl. Acad. Sci. USA 1984, 81, 5335–5339. [Google Scholar] [CrossRef] [Green Version]
- Chiang, N.; Serhan, C.N. Structural Elucidation and Physiologic Functions of Specialized Pro-Resolving Mediators and Their Receptors. Mol. Asp. Med. 2017, 58, 114–129. [Google Scholar] [CrossRef]
- Lee, S.; Huen, S.; Nishio, H.; Nishio, S.; Lee, H.K.; Choi, B.-S.; Ruhrberg, C.; Cantley, L.G. Distinct Macrophage Phenotypes Contribute to Kidney Injury and Repair. J. Am. Soc. Nephrol. 2011, 22, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zeng, Z.; Zhao, L.; Chen, P.; Xiao, W. Impaired Skeletal Muscle Regeneration Induced by Macrophage Depletion Could Be Partly Ameliorated by MGF Injection. Front. Physiol. 2019, 10, 601. [Google Scholar] [CrossRef] [Green Version]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory Monocytes Recruited after Skeletal Muscle Injury Switch into Antiinflammatory Macrophages to Support Myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Saclier, M.; Yacoub-Youssef, H.; Mackey, A.L.; Arnold, L.; Ardjoune, H.; Magnan, M.; Sailhan, F.; Chelly, J.; Pavlath, G.K.; Mounier, R.; et al. Differentially Activated Macrophages Orchestrate Myogenic Precursor Cell Fate during Human Skeletal Muscle Regeneration. Stem. Cells 2013, 31, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Honda, H.; Kimura, H.; Rostami, A. Demonstration and Phenotypic Characterization of Resident Macrophages in Rat Skeletal Muscle. Immunology 1990, 70, 272–277. [Google Scholar] [PubMed]
- Juban, G.; Chazaud, B. Efferocytosis during Skeletal Muscle Regeneration. Cells 2021, 10, 3267. [Google Scholar] [CrossRef]
- Grounds, M.D. Phagocytosis of Necrotic Muscle in Muscle Isografts Is Influenced by the Strain, Age, and Sex of Host Mice. J. Pathol. 1987, 153, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Hammers, D.W.; Rybalko, V.; Merscham-Banda, M.; Hsieh, P.-L.; Suggs, L.J.; Farrar, R.P. Anti-Inflammatory Macrophages Improve Skeletal Muscle Recovery from Ischemia-Reperfusion. J. Appl. Physiol. 2015, 118, 1067–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, P.-L.; Rybalko, V.; Baker, A.B.; Suggs, L.J.; Farrar, R.P. Recruitment and Therapeutic Application of Macrophages in Skeletal Muscles after Hind Limb Ischemia. J. Vasc. Surg. 2018, 67, 1908–1920.e1. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Villalta, S.A. Regulatory Interactions between Muscle and the Immune System during Muscle Regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1173–R1187. [Google Scholar] [CrossRef] [Green Version]
- Gondin, J.; Théret, M.; Duhamel, G.; Pegan, K.; Mathieu, J.R.R.; Peyssonnaux, C.; Cuvellier, S.; Latroche, C.; Chazaud, B.; Bendahan, D.; et al. Myeloid HIFs Are Dispensable for Resolution of Inflammation during Skeletal Muscle Regeneration. J. Immunol. 2015, 194, 3389–3399. [Google Scholar] [CrossRef] [Green Version]
- Heredia, J.E.; Mukundan, L.; Chen, F.M.; Mueller, A.A.; Deo, R.C.; Locksley, R.M.; Rando, T.A.; Chawla, A. Type 2 Innate Signals Stimulate Fibro/Adipogenic Progenitors to Facilitate Muscle Regeneration. Cell 2013, 153, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotfi, R.; Herzog, G.I.; DeMarco, R.A.; Beer-Stolz, D.; Lee, J.J.; Rubartelli, A.; Schrezenmeier, H.; Lotze, M.T. Eosinophils Oxidize Damage-Associated Molecular Pattern Molecules Derived from Stressed Cells. J. Immunol. 2009, 183, 5023–5031. [Google Scholar] [CrossRef]
- Puxeddu, I.; Berkman, N.; Ribatti, D.; Bader, R.; Haitchi, H.M.; Davies, D.E.; Howarth, P.H.; Levi-Schaffer, F. Osteopontin Is Expressed and Functional in Human Eosinophils. Allergy 2010, 65, 168–174. [Google Scholar] [CrossRef]
- Lee, J.J.; Jacobsen, E.A.; McGarry, M.P.; Schleimer, R.P.; Lee, N.A. Eosinophils in Health and Disease: The LIAR Hypothesis. Clin. Exp. Allergy 2010, 40, 563–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenceslau, C.F.; McCarthy, C.G.; Szasz, T.; Webb, R.C. Lipoxin A4 Mediates Aortic Contraction via RHOA/RHO Kinase, Endothelial Dysfunction and Reactive Oxygen Species. J. Vasc. Res. 2014, 51, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Zhai, H.; Wang, Y.; Li, L.; Wu, J.; Wang, F.; Sun, S.; Yao, S.; Shang, Y. Aspirin-Triggered Lipoxin A4 Attenuates Lipopolysaccharide-Induced Intracellular ROS in BV2 Microglia Cells by Inhibiting the Function of NADPH Oxidase. Neurochem. Res. 2012, 37, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- Börgeson, E.; Lönn, J.; Bergström, I.; Brodin, V.P.; Ramström, S.; Nayeri, F.; Särndahl, E.; Bengtsson, T. Lipoxin A4 Inhibits Porphyromonas Gingivalis-Induced Aggregation and Reactive Oxygen Species Production by Modulating Neutrophil-Platelet Interaction and CD11b Expression. Infect. Immun. 2011, 79, 1489–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, L.; Li, J.; Chen, X.; Chen, K.; Li, W.; Li, X.; Zhang, L.; Duan, W.; Lei, J.; Xu, Q.; et al. Lipoxin A4 Attenuates Cell Invasion by Inhibiting ROS/ERK/MMP Pathway in Pancreatic Cancer. Oxid. Med. Cell. Longev. 2016, 2016, 6815727. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Clish, C.B.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel Functional Sets of Lipid-Derived Mediators with Antiinflammatory Actions Generated from Omega-3 Fatty Acids via Cyclooxygenase 2-Nonsteroidal Antiinflammatory Drugs and Transcellular Processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef] [Green Version]
- Tjonahen, E.; Oh, S.F.; Siegelman, J.; Elangovan, S.; Percarpio, K.B.; Hong, S.; Arita, M.; Serhan, C.N. Resolvin E2: Identification and Anti-Inflammatory Actions: Pivotal Role of Human 5-Lipoxygenase in Resolvin E Series Biosynthesis. Chem. Biol. 2006, 13, 1193–1202. [Google Scholar] [CrossRef] [Green Version]
- Duffield, J.S.; Hong, S.; Vaidya, V.S.; Lu, Y.; Fredman, G.; Serhan, C.N.; Bonventre, J.V. Resolvin D Series and Protectin D1 Mitigate Acute Kidney Injury. J. Immunol. 2006, 177, 5902–5911. [Google Scholar] [CrossRef] [Green Version]
- Kasuga, K.; Yang, R.; Porter, T.F.; Agrawal, N.; Petasis, N.A.; Irimia, D.; Toner, M.; Serhan, C.N. Rapid Appearance of Resolvin Precursors in Inflammatory Exudates: Novel Mechanisms in Resolution. J. Immunol. 2008, 181, 8677–8687. [Google Scholar] [CrossRef] [Green Version]
- Chiang, N.; Dalli, J.; Colas, R.A.; Serhan, C.N. Identification of Resolvin D2 Receptor Mediating Resolution of Infections and Organ Protection. J. Exp. Med. 2015, 212, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Dufton, N.; Hannon, R.; Brancaleone, V.; Dalli, J.; Patel, H.B.; Gray, M.; D’Acquisto, F.; Buckingham, J.C.; Perretti, M.; Flower, R.J. Anti-Inflammatory Role of the Murine Formyl-Peptide Receptor 2: Ligand-Specific Effects on Leukocyte Responses and Experimental Inflammation. J. Immunol. 2010, 184, 2611–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, N.; Fredman, G.; Bäckhed, F.; Oh, S.F.; Vickery, T.; Schmidt, B.A.; Serhan, C.N. Infection Regulates Pro-Resolving Mediators That Lower Antibiotic Requirements. Nature 2012, 484, 524–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Xu, Z.; Niu, H.; Gao, N.; Guan, Y.; Li, C.; Dang, Y.; Cui, X.; Liu, X.L.; Duan, Y.; et al. An Injectable Oxygen Release System to Augment Cell Survival and Promote Cardiac Repair Following Myocardial Infarction. Sci. Rep. 2018, 8, 1371. [Google Scholar] [CrossRef] [PubMed]
- Borselli, C.; Storrie, H.; Benesch-Lee, F.; Shvartsman, D.; Cezar, C.; Lichtman, J.W.; Vandenburgh, H.H.; Mooney, D.J. Functional Muscle Regeneration with Combined Delivery of Angiogenesis and Myogenesis Factors. Proc. Natl. Acad. Sci. USA 2010, 107, 3287–3292. [Google Scholar] [CrossRef] [Green Version]
- Shvartsman, D.; Storrie-White, H.; Lee, K.; Kearney, C.; Brudno, Y.; Ho, N.; Cezar, C.; McCann, C.; Anderson, E.; Koullias, J.; et al. Sustained Delivery of VEGF Maintains Innervation and Promotes Reperfusion in Ischemic Skeletal Muscles via NGF/GDNF Signaling. Mol. Ther. 2014, 22, 1243–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Gao, N.; Niu, H.; Dang, Y.; Guan, J. Oxygen-Release Microspheres Capable of Releasing Oxygen in Response to Environmental Oxygen Level to Improve Stem Cell Survival and Tissue Regeneration in Ischemic Hindlimbs. J. Control. Release 2021, 331, 376–389. [Google Scholar] [CrossRef]
- Lance, K.D.; Chatterjee, A.; Wu, B.; Mottola, G.; Nuhn, H.; Lee, P.P.; Sansbury, B.E.; Spite, M.; Desai, T.A.; Conte, M.S. Unidirectional and Sustained Delivery of the Proresolving Lipid Mediator Resolvin D1 from a Biodegradable Thin Film Device. J. Biomed. Mater. Res. A 2017, 105, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wang, J.; Shao, J.; Kouwer, P.H.J.; Bronkhorst, E.M.; Jansen, J.A.; Walboomers, X.F.; Yang, F. A Tunable and Injectable Local Drug Delivery System for Personalized Periodontal Application. J. Control. Release 2020, 324, 134–145. [Google Scholar] [CrossRef]
- Sok, M.C.P.; Baker, N.; McClain, C.; Lim, H.S.; Turner, T.; Hymel, L.; Ogle, M.; Olingy, C.; Palacios, J.I.; Garcia, J.R.; et al. Dual Delivery of IL-10 and AT-RvD1 from PEG Hydrogels Polarize Immune Cells towards pro-Regenerative Phenotypes. Biomaterials 2021, 268, 120475. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, X.; Jiang, L.; Zhang, L.; Dong, Y.; Midgley, A.C.; Kong, D.; Wang, S. Regulation of the Inflammatory Response by Vascular Grafts Modified with Aspirin-Triggered Resolvin D1 Promotes Blood Vessel Regeneration. Acta Biomater. 2019, 97, 360–373. [Google Scholar] [CrossRef]
- Vasconcelos, D.P.; Costa, M.; Neves, N.; Teixeira, J.H.; Vasconcelos, D.M.; Santos, S.G.; Águas, A.P.; Barbosa, M.A.; Barbosa, J.N. Chitosan Porous 3D Scaffolds Embedded with Resolvin D1 to Improve in Vivo Bone Healing. J. Biomed. Mater. Res. A 2018, 106, 1626–1633. [Google Scholar] [CrossRef]
- Dalli, J.; Serhan, C.N. Specific Lipid Mediator Signatures of Human Phagocytes: Microparticles Stimulate Macrophage Efferocytosis and pro-Resolving Mediators. Blood 2012, 120, e60–e72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedirko, V.; McKeown-Eyssen, G.; Serhan, C.N.; Barry, E.L.; Sandler, R.S.; Figueiredo, J.C.; Ahnen, D.J.; Bresalier, R.S.; Robertson, D.J.; Anderson, C.W.; et al. Plasma Lipoxin A4 and Resolvin D1 Are Not Associated with Reduced Adenoma Risk in a Randomized Trial of Aspirin to Prevent Colon Adenomas. Mol. Carcinog. 2017, 56, 1977–1983. [Google Scholar] [CrossRef] [PubMed]
- Schebb, N.H.; Kühn, H.; Kahnt, A.S.; Rund, K.M.; O’Donnell, V.B.; Flamand, N.; Peters-Golden, M.; Jakobsson, P.-J.; Weylandt, K.H.; Rohwer, N.; et al. Formation, Signaling and Occurrence of Specialized Pro-Resolving Lipid Mediators-What Is the Evidence so Far? Front. Pharmacol. 2022, 13, 838782. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Yiang, G.-T.; Liao, W.-T.; Tsai, A.P.-Y.; Cheng, Y.-L.; Cheng, P.-W.; Li, C.-Y.; Li, C.-J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Dalli, J.; Colas, R.A.; Quintana, C.; Barragan-Bradford, D.; Hurwitz, S.; Levy, B.D.; Choi, A.M.; Serhan, C.N.; Baron, R.M. Human Sepsis Eicosanoid and Proresolving Lipid Mediator Temporal Profiles: Correlations With Survival and Clinical Outcomes. Crit. Care Med. 2017, 45, 58–68. [Google Scholar] [CrossRef]
- Uno, H.; Furukawa, K.; Suzuki, D.; Shimizu, H.; Ohtsuka, M.; Kato, A.; Yoshitomi, H.; Miyazaki, M. Immunonutrition Suppresses Acute Inflammatory Responses through Modulation of Resolvin E1 in Patients Undergoing Major Hepatobiliary Resection. Surgery 2016, 160, 228–236. [Google Scholar] [CrossRef]
- Dushianthan, A.; Cusack, R.; Burgess, V.A.; Grocott, M.P.; Calder, P.C. Immunonutrition for Acute Respiratory Distress Syndrome (ARDS) in Adults. Cochrane Database Syst. Rev. 2019, 1, CD012041. [Google Scholar] [CrossRef]
- Chiang, N.; Gronert, K.; Clish, C.B.; O’Brien, J.A.; Freeman, M.W.; Serhan, C.N. Leukotriene B4 Receptor Transgenic Mice Reveal Novel Protective Roles for Lipoxins and Aspirin-Triggered Lipoxins in Reperfusion. J. Clin. Investig. 1999, 104, 309–316. [Google Scholar] [CrossRef]
- Sansbury, B.E.; Li, X.; Wong, B.; Patsalos, A.; Giannakis, N.; Zhang, M.J.; Nagy, L.; Spite, M. Myeloid ALX/FPR2 Regulates Vascularization Following Tissue Injury. Proc. Natl. Acad. Sci. USA 2020, 117, 14354–14364. [Google Scholar] [CrossRef]
- Giannakis, N.; Sansbury, B.E.; Patsalos, A.; Hays, T.T.; Riley, C.O.; Han, X.; Spite, M.; Nagy, L. Dynamic Changes to Lipid Mediators Support Transitions among Macrophage Subtypes during Muscle Regeneration. Nat. Immunol. 2019, 20, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Scalia, R.; Gefen, J.; Petasis, N.A.; Serhan, C.N.; Lefer, A.M. Lipoxin A4 Stable Analogs Inhibit Leukocyte Rolling and Adherence in the Rat Mesenteric Microvasculature: Role of P-Selectin. Proc. Natl. Acad. Sci. USA 1997, 94, 9967–9972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Yao, W.; Liu, Z.; Li, H.; Zhang, Z.-J.; Hei, Z.; Xia, Z. Lipoxin A4 Preconditioning Attenuates Intestinal Ischemia Reperfusion Injury through Keap1/Nrf2 Pathway in a Lipoxin A4 Receptor Independent Manner. Oxid. Med. Cell. Longev. 2016, 2016, 9303606. [Google Scholar] [CrossRef] [Green Version]
- Keyes, K.T.; Ye, Y.; Lin, Y.; Zhang, C.; Perez-Polo, J.R.; Gjorstrup, P.; Birnbaum, Y. Resolvin E1 Protects the Rat Heart against Reperfusion Injury. Am. J. Physiol. Heart. Circ. Physiol. 2010, 299, H153–H164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kain, V.; Ingle, K.A.; Colas, R.A.; Dalli, J.; Prabhu, S.D.; Serhan, C.N.; Joshi, M.; Halade, G.V. Resolvin D1 Activates the Inflammation Resolving Response at Splenic and Ventricular Site Following Myocardial Infarction Leading to Improved Ventricular Function. J. Mol. Cell. Cardiol. 2015, 84, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Mediator | Disease Model | Action(s) | Ref |
|---|---|---|---|
| Lipoxin A2/ATL | Mouse/hind-limb IRI | Attenuate hind-limb IRI—induced lung injuryDetachment of adherent PMN in mesenteric IRI | [119] |
| Lipoxin A2 | Rat/hind-limb IRI | Decreases inflammatory response, oxidative stress and cell apoptosis. | [95] |
| Resolvin D1 | Mouse/ hind-limb IRI | Enhances perfusion recovery during ischemia, | [120] |
| Resolvin D1 | Mouse/hind-limb IRI | Reduces PMN recruitment into the lungs | [99] |
| Resolvin D2 | Mouse/hind-limb IRI | Reduces PMN recruitment into the lungs | [100] |
| Resolvin D2 | Mouse/CTX-induced muscle injury | Enhances macrophage M2 efferocytosis | [121] |
| Protectin D1 | Mouse/kidney IRI | Protects from ischemia–reperfusion-induced kidney damage and loss of function; Regulates macrophage M1 function | [98] |
| Mediator | Disease Model | Action(s) | Ref |
|---|---|---|---|
| Lipoxin A2 | Rat/superfusing of the mesentery | Inhibits PMN rolling and adherence in mesenteric circulation | [122] |
| Lipoxin A2 | Rat/Intestinal IRI | Attenuates intestinal ischemia–reperfusion injury | [123] |
| Resolvin E1 | Rat/Cardiac IRI | Cardioprotective; Limits infarct size | [124] |
| Resolvin D1 | Mouse/kidney IRI | Protects from ischemia–reperfusion-induced kidney damage and loss of function; Regulates macrophage M1 function | [98] |
| Resolvin D1 | Rat/Cardiac IRI | Reduces accumulation of PMN and fibrosis; Leads to improved cardiac function | [125] |
| Protectin D1 | Mouse/kidney IRI | Protects from ischemia–reperfusion-induced kidney damage and loss of function; Regulates macrophages | [98] |
| Resolvin D1 | Mouse/kidney IRI | Decreases PMN infiltration and tissue fibrosis; Organ protective | [98] |
| Protectin D1 | Mouse/kidney IRI | Protects from ischemia–reperfusion-induced kidney damage and loss of function; Regulates macrophage M1 function | [98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barnig, C.; Lutzweiler, G.; Giannini, M.; Lejay, A.; Charles, A.-L.; Meyer, A.; Geny, B. Resolution of Inflammation after Skeletal Muscle Ischemia–Reperfusion Injury: A Focus on the Lipid Mediators Lipoxins, Resolvins, Protectins and Maresins. Antioxidants 2022, 11, 1213. https://doi.org/10.3390/antiox11061213
Barnig C, Lutzweiler G, Giannini M, Lejay A, Charles A-L, Meyer A, Geny B. Resolution of Inflammation after Skeletal Muscle Ischemia–Reperfusion Injury: A Focus on the Lipid Mediators Lipoxins, Resolvins, Protectins and Maresins. Antioxidants. 2022; 11(6):1213. https://doi.org/10.3390/antiox11061213
Chicago/Turabian StyleBarnig, Cindy, Gaetan Lutzweiler, Margherita Giannini, Anne Lejay, Anne-Laure Charles, Alain Meyer, and Bernard Geny. 2022. "Resolution of Inflammation after Skeletal Muscle Ischemia–Reperfusion Injury: A Focus on the Lipid Mediators Lipoxins, Resolvins, Protectins and Maresins" Antioxidants 11, no. 6: 1213. https://doi.org/10.3390/antiox11061213
APA StyleBarnig, C., Lutzweiler, G., Giannini, M., Lejay, A., Charles, A.-L., Meyer, A., & Geny, B. (2022). Resolution of Inflammation after Skeletal Muscle Ischemia–Reperfusion Injury: A Focus on the Lipid Mediators Lipoxins, Resolvins, Protectins and Maresins. Antioxidants, 11(6), 1213. https://doi.org/10.3390/antiox11061213